
Abstract
Sustained angiogenesis stands as a hallmark of cancer. The intricate vascular tumor microenvironment fuels cancer progression and metastasis, fosters therapy resistance, and facilitates immune evasion. Therapeutic strategies targeting tumor vasculature have emerged as transformative for cancer treatment, encompassing anti-angiogenesis, vessel normalization, and endothelial reprogramming. Growing evidence suggests the dynamic regulation of tumor angiogenesis by infiltrating myeloid cells, such as macrophages, myeloid-derived suppressor cells (MDSCs), and neutrophils. Understanding these regulatory mechanisms is pivotal in paving the way for successful vasculature-targeted cancer treatments. Therapeutic interventions aimed to disrupt myeloid cell-mediated tumor angiogenesis may reshape tumor microenvironment and overcome tumor resistance to radio/chemotherapy and immunotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Angiogenesis, the formation of new blood vessels from pre-existing ones, is a hallmark of cancer. Tumor angiogenesis is a pivotal process that promotes cancer growth, progression, and metastasis and induces therapy resistance [1]. Over the past few decades, considerable efforts have been directed towards understanding the molecular and cellular mechanisms that underlie tumor angiogenesis. These lead to the development of promising anti-angiogenic therapeutic strategies that aim to inhibit overgrowth and sprouting of tumor endothelial cells (ECs). Beyond the traditional anti-angiogenic concept that focuses on vessel delivery function, recent advances have revealed that the interactions between the tumor vasculature and the immune system are critical for regulation of tumor vascularity and immunity [2, 3]. The tumor microenvironment (TME) is a complex milieu composed of various non-neoplastic cell types, including ECs, stromal cells, and a diverse array of immune cells. The dynamic interplay between these cellular components in the vascular TME has significant implications for tumor development, immune evasion and the efficacy of cancer therapies, particularly immunotherapies. Therefore, development of efficient therapeutic strategies that reprogram the vascular TME will offer exciting opportunities for cytotoxic radio/chemotherapy and T cell-based immunotherapy.
In this review, we discuss the emerging strategies for tumor vasculature-targeting therapy. We provide a comprehensive overview of the complex regulation of tumor angiogenesis by myeloid cells, including macrophages, myeloid-derived suppressor cells (MDSCs), and neutrophils within the TME. We discuss the impact of these immune cells on tumor angiogenesis. We highlight that myeloid cells interact with ECs to regulate tumor angiogenesis and create a specialized niche that induces immune evasion and promotes tumor growth, providing crucial targets for vasculature-targeting therapy. These approaches may have the potential to revolutionize cancer treatment, paving the way for more effective therapeutic strategies.
Tumor angiogenesis
Basic principle of aberrant tumor angiogenesis
Tumor angiogenesis is fundamental to cancer progression, metastasis, and therapy resistance. Tumor angiogenesis refers to the pathophysiological process where new blood vessels sprout from pre-existing ones to supply nutrients, oxygen, and cellular network for tumor growth [1, 4,5,6,7]. The intricate network of blood vessels also allows cancer cells to infiltrate the bloodstream and disseminate throughout the body, giving rise to metastasis. These collectively suggest anti-angiogenic therapy, a treatment that aims to inhibit EC overgrowth and sprouting, as a promising strategy for cancer treatment. Notably, the newly formed vessels are structurally and functionally abnormal—they are tortuous and leaky with a disorganized, haphazard architecture. This abnormal vasculature leads to a chaotic blood flow, which creates a heterogeneously hypoxic tumor microenvironment [8]. Such hostile conditions can foster cancer cells that are more aggressive and therapy resistant, further promoting tumor growth and metastasis. Furthermore, the abnormal vessels also form a barrier to the effective delivery of drugs to the tumor, thereby contributing to therapy resistance [9]. Vessel normalization has, therefore, joined anti-angiogenic treatment as promising strategies for solid tumor treatment.
Tumor angiogenesis is a complex process, subject to regulation by a balance between pro-angiogenic and anti-angiogenic factors within a solid tumor [10, 11]. When the equilibrium tilts toward pro-angiogenic factors, ECs are stimulated to proliferate and migrate towards the tumor, forming new blood vessels. The presence of excessive pro-angiogenic factors further stimulates vascular abnormalities [12, 13]. This imbalance drives both neovascularization and vascular aberrancy, serving as a critical therapeutic target for vessel normalization and cancer treatment. Finally, tumor ECs undergo genetic and metabolic alteration to acquire pro-tumor phenotypes including aberrant vessel structure and function, a rewired adhesome that reduces lymphocyte attachment, and local release of immunosuppressive molecules. Thus, anti-angiogenesis, vessel normalization, and endothelial programming serve as promising strategies for vasculature-targeting approaches for cancer treatment (Fig. 1).

Therapeutic strategies for vasculature-targeting anti-cancer treatment. Therapeutic strategies targeting tumor vasculature have emerged as transformative for cancer treatment, encompassing anti-angiogenesis, vessel normalization, and endothelial reprogramming
Anti-angiogenic therapy
Anti-VEGF/VEGFR
Anti-angiogenic therapy represents a promising strategy for cancer treatment by inhibiting the formation of new blood vessels that nourish tumors, thus depriving them of essential oxygen and nutrients for growth. Numerous anti-angiogenic agents targeting pro-angiogenic factors, such as vascular endothelial growth factor (VEGF, i.e., VEGF-A), fibroblast growth factors (FGF), and epidermal growth factor (EGF), have been extensively explored [14,15,16]. Among the most widely used anti-angiogenic agents are the monoclonal antibodies and tyrosine kinase inhibitors (TKIs) that target VEGF and the VEGF downstream kinases, respectively. VEGF plays a crucial role in both physiological and pathological angiogenesis [4]. In tumors, overexpression of VEGF, mainly driven by hypoxia-inducible factor (HIF)-1α, promotes abnormal blood vessel growth and acts as a vascular permeability factor [17]. VEGF usually binds to the tyrosine kinase receptor VEGFR2, in collaboration with neuropilin-1 and VEGFR3, and interacts with other modulating pathways such as Notch, angiopoietin/Tie2, and ephrin/Eph to facilitate vessel growth [18,19,20,21,22]. A number of anti-angiogenic agents have been approved by FDA for treating cancer, highlighting their role in current oncology therapeutics. For instance, bevacizumab (Avastin), a humanized monoclonal antibody that blocks VEGF, is a notable example of an anti-angiogenic agent demonstrating anti-tumor results in colon and kidney cancers [23]. Additionally, small-molecule pharmacological inhibitors of VEGF receptor tyrosine kinase, such as sunitinib and sorafenib, also offer a promising opportunity to cancer therapy [24, 25]. However, the overall efficacy of these anti-angiogenic therapies is often limited and does not produce long-term benefits in patients with most other cancer types, such as glioblastoma [26,27,28,29,30].Both intrinsic and acquired mechanisms contribute to tumor resistance to anti-angiogenic therapy, driven by the existence of redundant angiogenic pathways and the adaptive mechanisms that lead tumor cells to survive an avascular and hypoxic TME, respectively [6, 28].
Inhibition of vascular maturation
Inhibition of vascular maturation, a key aspect of functional vascularity, represents another therapeutic strategy for cancer. The EC growth factor signaling pathways composed of angiopoietin (Ang)-1/2 and their receptor, Tie2, play a critical role in this process. Ang-1, mainly secreted by pericytes and smooth muscle cells, promotes vascular remodeling and stabilization. Ang-1 overexpression is often observed in tumor vasculature, which enhances EC proliferation and pericyte-mediated vascular maturation, and increased vascular functions intensify the malignancy of various cancers [31]. Conversely, Ang-2 can induce angiogenesis and destabilize vasculature by binding to Tie2 and integrin receptors [32]. Given the role of angiopoietins in vascular biology, antibodies targeting these angiopoietins and dual inhibitors of Ang-2 and VEGF show promising results in various malignancies [20].
Targeting the development pathways of ECs
Tumor angiogenesis is tightly controlled by EC differentiation and growth. This process can be triggered by hypoxia [33], Notch [34], and Wnt signaling pathways [35]. Hypoxia, typically resulting from rapid tumor growth and disordered vasculature, initiates a survival response within tumors. To survive under these extreme conditions, tumors employ a host of mechanisms, primarily the activation of HIFs that induce transcription of hypoxia-adaptive metabolic enzyme and VEGF [36, 37]. Moreover, hypoxia can stimulate activation of mTOR, PI3K, and AKT through post-translational modifications of these proteins [38], which are central to EC metabolism, survival, and motility regulation in response to nutrient and oxygen depletion. Notch signaling emerges as a pivotal player in the orchestration of vessel sprouting, branching, and maturation. Aberrations in Notch signaling have been linked to tumor angiogenesis, positioning the Notch pathway as a potential target for anti-angiogenic cancer therapies [34]. A recent study on DII4-induced Notch signaling in EC growth and development shows that antibodies against Dll4 and VEGF had strikingly different effects on tumor blood vessels [39], suggesting differing mechanisms underlying Notch- and VEGF-mediated tumor angiogenesis. Dll4-driven Notch signaling appeared vital during active blood vessel formation, but less so for maintaining normal vessels [39]. Beside its established role in developmental angiogenesis and vascular differentiation, Wnt pathway has been implicated in tumor angiogenesis. The interaction between Wnt and Frizzled receptors activates varied signaling outcomes in both canonical and non-canonical pathways, contributing to regulation of EC functions. For example, canonical Wnt-frizzled signaling produces a β-catenin/Lef/TCF complex which triggers brain angiogenesis during development [40]. In the context of GBM, activation of Wnt/β-catenin signaling in ECs is associated with chemoresistance [41], highlighting a potential target in GBM treatment. On the other hand, in the non-canonical pathway, Ca2+/calmodulin-dependent protein kinase II (CAMKII) activation influences ventral cell fate [42], and other signaling cascades like JNK and Daam-1 drive EC proliferation and maintain cellular architecture [43,44,45].
Overall, these findings highlight multiple regulatory mechanisms, mediated through hypoxia, VEGF, Notch, and Wnt, for EC proliferation, migration, and differentiation during tumor angiogenesis. Understanding their dysregulation in cancer may help develop new targets for anti-angiogenic therapies.
Vessel normalization therapy
Anti-angiogenic therapy can adversely enhance tumor hypoxia and reduce drug delivery, resulting from destroyed tumor vasculature, leading to increased resistance of tumors to radio/chemotherapy and targeted molecular therapy. Considering structurally and functionally abnormal vascularity in cancer, i.e., tortuous, leaky vasculature due to dysfunctional EC sprouting and overgrowth, a different strategy, namely, vessel normalization, that aims to restore normal vessel function, has been exploited [11]. This could be achieved by re-balancing the pro- and anti-angiogenic factors presented in the TME, with reduced hypoxia, improved perfusion allowing for proper drug delivery, and enhanced immune cell infiltration [12, 13]. Previous preclinical studies show that vessel normalizing doses of anti-VEGF treatment improve T cell infiltration and enhance immunotherapy [46, 47], due to enhanced vessel delivery and reduced intratumoral hypoxia. Moreover, a recent clinical trial shows promising results for combining anti-VEGF bevacizumab with immune checkpoint blockade in liver cancer treatment [29].
Additional therapeutic strategies for vessel normalization include decreasing vascular leakiness, enhancing the structural integrity, increasing perfusion, and adding angiostatic factors, with multiple targets identified. For example, targeting regulator of G protein signaling 5 (Rgs5) protein leads to more typical vessel morphology and function in tumors, without reducing vessel density [48]. Inhibiting L1CAM, a neural adhesion protein in tumor ECs, results in pruning and fortification of vessels, thereby reducing tumor growth and metastases [49]. Inhibition of neuronal nitric oxide synthase (nNOS) in cancer cells restores proper NO gradients, leading to denser and more effective vessels for oxygen and drug delivery [50]. Restoring semaphorin-3A (SEMA3A) initially prunes immature vessel, and long-term application increases vessel maturation [51]. Activation of R-Ras or lysophosphatidic acid (LPA) in ECs promotes vascular normalization [52, 53]. Chloroquine, known for its antimalarial properties, also plays a role in vessel normalization through endosomal Notch1 trafficking and signaling in ECs [54]. Activation of transient receptor potential vanilloid-4 (TRPV4) in tumor ECs restores normal mechanosensitivity and increases drug delivery [55]. Further strategies include using thrombospondin-1 (TSP-1), an endogenous antiangiogenic factor, to normalize vessels, enhance drug delivery, and increase the effectiveness of treatments like cisplatin [56].
Another innovative approach in vessel normalization involves modulating various cells within the perivascular niche. For instance, eribulin, a chemotherapy agent, regulates endothelial-pericyte interactions to fortify vessels and improve treatment outcomes [57]. Desmoplasia, characterized by fibrotic tissue growth, impairs vascular function by compressing vessels [13]. Therefore, normalizing the extracellular matrix (ECM) is crucial, as it can improve both vascular function and treatment outcomes. Strategies targeting cancer-associated fibroblasts and the extracellular matrix, such as inhibiting TGF-β [58] or sonic hedgehog pathways [59], and using Nab-paclitaxel [60], show promise in reducing vessel compression. Additionally, altering metabolic pathways in pro-tumor macrophages leads to the formation of well-organized and fortified vessels, thereby enhancing oxygen delivery [61]. Antitumor CD4+ T cells also play a role in normalizing vessels by modulating angiogenic gene expression in tumors [62]. Inhibiting VEGF expression from these T cells further suggests their role in promoting abnormal tumor vessel phenotypes [63]. These findings highlight the complex interactions among ECs, other cells, and ECM in the TME, which may induce vessel abnormalities. Understanding and targeting these interactions can normalize tumor vasculature and improve cancer therapy outcomes.
Although these strategies hold promise, the benefits of vessel normalization monotherapy have often been small and transient. For example, administration of low-dose bevacizumab to control excessive EC growth has been a central method used in vessel normalization. However, vessel normalization anti-VEGF therapies often lead to a transient window that is potentially open for additional therapies, after which tumors become resistant [64, 65]. Furthermore, the timing and dosing of vessel normalization therapy needs to be further optimized when combined with immunotherapies and other conventional cytotoxic therapies, as tumor immunogenicity and vascularity change over tumor development and treatment exposure [6, 13].
Endothelial reprogramming therapy
EC plasticity has been well characterized during embryogenesis [66, 67]. In pathological settings including cardiac, renal, and liver fibrosis, ossifying myositis, pulmonary hypertension, and cerebral cavernous malformation, ECs can take endothelial mesenchymal transition (Endo-MT) de novo to generate fibroblasts and stem-like cells [68,69,70]. Notably, cell plasticity plays a central role in the EC transcriptomic alteration and aberrant vascular phenotypes in cancer [71, 72]. As an alternative process to angiogenesis and vascular abnormality driven by pro-angiogenic factor-induced vessel sprouting and outgrowth, ECs retain key endothelial functions but undergo cell plasticity-mediated genetic reprogramming to induce aberrant vascularity in the tumor microenvironment. For example, ECs acquire partial Endo-MT, also known as endothelial transformation, to promote their ability to proliferate, migrate and secrete [71,72,73]. These transformed ECs, unlike normal ECs, take transcriptomic alteration to drive distinct behaviors due to the influence of the TME, forming an abnormal architecture of tumor vasculature. This leads to poor perfusion and hypoxia within the TME, which fosters the selection of more aggressive, treatment-resistant cancer cells [74], and creates a physical barrier that shields tumor cells from immune cell attack and impedes the delivery of chemotherapeutic drugs, thereby inducing tumor resistance to chemo/radiotherapy and immunotherapy [7, 28]. The strategy for genetic reprogramming of tumor ECs, e.g., by targeting EC plasticity, aims to normalize these cells by reversing their abnormal traits of gene expression, making the vasculature resemble the normal one in structure and function, and, therefore, may eventually improve the efficacy of cytotoxic treatment and immunotherapy approaches [71]. In addition to transcriptomic alteration, tumor ECs also undergo metabolic changes in the TME [75]. Metabolic switches in tumor ECs are driven by genetic and epigenetic alteration of metabolism-associated genes in response to the cues in the TME, such as hypoxia. The adaptively rewired metabolism fosters EC survival and growth in the TME, contributing to aberrant tumor angiogenesis. Metabolic reprogramming of tumor ECs, therefore, serves as an additional strategy for vasculature-targeting cancer therapy [75].
Genetic reprogramming of ECs
The approach of genetic reprogramming of tumor ECs is initially termed as vascular de-transformation therapy, emphasizing its main target on EC plasticity [71]. Genetic reprogramming of tumor ECs would be expected to induce the formation of a stable, functionally normal, and structurally orderly vasculature, which reduces tumor hypoxia, improves drug delivery, and alleviates immunosuppression, thereby enhancing anti-tumor immune responses and the efficacy of other therapies [72]. Several strategies have been exploited for the genetic reprogramming of tumor ECs. HGF/c-Met is identified as a critical regulator of Endo-MT in cancer [73]. EC-specific c-Met knockout inhibits EC plasticity, reduces vascular aberrancy, and sensitizes tumor to chemotherapy [73]. Moreover, c-Met-mediated activation of Wnt signaling drives transformation of ECs into mesenchymal stem cell-like cells, leading to multidrug resistance in ECs and tumor chemoresistance [41]. Furthermore, platelet-derived growth factor (PDGF)-mediated EC plasticity controls VEGFR2 expression through Snail, which contributes to tumor resistance to anti-VEGF treatment [9]. Based on these results, combination of anti-PDGFR and anti-VEGFR therapy was explored in tumor, which shows promising synergistic anti-tumor effects [9]. A more recent whole kinome-wide screen identifies that p21-activating kinase 4 (PAK4) is a key driver of Endo-MT in cancer [76]. Inactivation of PAK4 reprograms transcriptome in ECs and normalizes tumor vasculature. Notably, genetic and pharmacological ablation of PAK4 in ECs reshapes the immune landscape within the TME, improving T-cell infiltration and sensitizing tumor to CAR-T cell therapy [76]. Furthermore, several additional targets have been identified for endothelial reprogramming, including ERG, Myct1, and Lrg1. Forced expression of ERG, a transcription factor essential for endothelial homeostasis, restores the angiogenic balance in tumor ECs, thereby inhibiting tumor growth and vascular abnormalities [77]. Interestingly, Myct1, a downstream protein of ETV2 and Myc, which is primarily expressed in ECs, plays a crucial role in mesenchymal-like transcriptional activation. Myct1 deficiency in mouse tumor models decreases angiogenesis and increases antitumor immunity, thereby limiting tumor growth [78]. Lrg1 is exclusively expressed on tumor ECs rather than normal ECs or pericytes. Deletion or antibody-based neutralization of Lrg1 results in vessel normalization and promotes the TME toward an anti-tumor, immune-active state, enhancing the efficacy of various cancer therapies [79].
In addition to structural abnormalities, tumor vasculature is also characterized by altered EC adhesiveness. Tumor ECs undergo genetic alteration, often with downregulated adhesion proteins, such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) that are necessary for immune cell attachment and extravasation [75, 80]. This leads to less T cell attachment to the endothelium, inhibiting T cell infiltration and contributing to tumor immune evasion. It is tempting to speculate that mesenchymal-like activation drives this dysfunctional adhesion in tumor ECs, induced by epithelial-mesenchymal transition (EMT)-associated transcriptional repressors including Snail, Slug, Twist-1/-2, and Zeb-1/-2. As such, inhibition of PAK4 reduces expression of Slug and Zeb-1, upregulating expression of VCAM-1 and Claudin-14 in tumor ECs, which eventually enhances T cell adhesion and improves CAR T cell immunotherapy [76]. Together, these findings underscore the potential of genetic reprogramming of tumor ECs as a promising approach for cancer treatment.
Epigenetic reprogramming of ECs
Epigenetic reprogramming in ECs represents another promising strategy for targeting tumor angiogenesis, considering tumor ECs undergo substantial epigenetic alterations to modulate their functionality in cancer. Acetylation of histone H3 has been well characterized in tumor ECs, which epigenetically regulates the expression of key genes essential for EC function and angiogenesis, including CLU, FBN1, TSPAN2, and ICAM1 [81]. The activity of histone deacetylases (HDACs), especially HDAC1, is central to this process: they regulate MMP14 and VCAM-1 expression, driving EC growth and the formation of vascular structures [82, 83]. Inhibitors targeting HDACs, such as trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA), hold promise in anti-angiogenesis therapy, as they modulate the transcription of several crucial pro-angiogenic signaling components, including receptors VEGFR1 and VEGFR2 [84], HIF-1α, and VEGF [85]. HDAC inhibitors not only exhibit anti-angiogenic properties across various cancer types but also enhance leukocyte adherence and movement within tumor vessels, primarily through the upregulation of ICAM-1 [86], underscoring their potential to boost the effectiveness of immunotherapy. Moreover, histone methylation is also critical for tumor angiogenesis. EZH2, a key histone methyltransferase, reduces trimethylation of histone H3 at lysine 27 (H3K27me3), a repressive epigenetic mark, during Endo-MT induced by IL-1β and TGF-β2 [87]. Conversely, JMJD2B, a histone demethylase,epigenetically modulates Endo-MT by promoting repressive H3K9me3 occurring at the promoters of mesenchymal and TGF-β signaling genes, such as calponin (CNN1), AKT serine/threonine kinase 3 (AKT3), and sulfatase 1 (SULF1) [88].
Beyond histone modifications, DNA methylation significantly influences the behavior of tumor ECs and, consequently, the immune profiles as well. For instance, deletion of DNA methyltransferase 1 (DNMT1) in ECs inhibits tumor growth and reshapes the immune environment, due to the increased expression of cytokines, chemokines, cell adhesion molecules in ECs, such as Cxcl9 and Cxcl10 that are crucial for infiltration of CD8+ T cells into the tumor [89]. DNMT1 silencing in ECs also enhances the expression of IL-33, Ccl21, and Ccl19 that are critical for neogenesis of high endothelial venule (HEV), a specialized postcapillary venule adapted for lymphocyte trafficking. Moreover, DNMT inhibitor treatment boosts leukocyte infiltration into tumors by upregulating ICAM1 expression in ECs [86]. Interestingly, proangiogenic factor FGF2 promotes ERK-mediated DNMT1 phosphorylation and nuclear translocation to repress Cxcl9 and Cxcl10 transcription [89], suggesting feedback loops that regulate angiogenic pathway activation and epigenetic regulation.
In summary, recent studies identifying an intricate network of epigenetic regulation in ECs during tumor angiogenesis provide profound insights into the mechanisms driving epigentic regulation of EC functions, and opens new avenues for developing therapeutic strategies targeting these epigenetic alterations to inhibit tumor growth and enhance immunotherapy outcomes.
Metabolic reprogramming of ECs
Given metabolic adaptation is required for cell proliferation and migration, such as EC outgrowth and sprouting, targeting endothelial metabolism has emerged as a promising strategy for modulating tumor angiogenesis [75, 90,91,92,93,94,95]. This strategy may not only rewire tumor vasculature by targeting EC sprouting, but also recondition the metabolic TME as it changes the EC-derived metabolites that are locally released. A key regulatory pathway of endothelial metabolism is glycolysis, a process critical for EC survival and proliferation in the hypoxic TME, as it generates necessary energy and metabolites anaerobically. For instance, disruption of glycolysis via PFKFB3 inhibition stabilizes the vascular barrier by improving pericyte adhesion, reduces metastasis, and enhances the efficacy of cancer chemotherapy [90, 95]. Furthermore, decreasing aerobic glycolysis in tumor ECs reduces vascular abnormalities, increases T cell infiltration, and overcomes tumor resistance to immunotherapy [96]. Notably, PHGDH, which diverts glycolysis into a specific serine biosynthetic pathway, promotes aberrant tumor angiogenesis, through its role in regulating nucleotide synthesis and maintaining the redox balance essential for endothelial proliferation [94]. Endothelial metabolism also contributes to the immunosuppressive TME by providing immunomodulatory metabolites produced by the vascular niche. As such, inhibition of serine metabolism in tumors ECs reduces their production of lactate and 2-hydroxyglutarate, two immunosuppressants in the TME, leading to activation of anti-tumor immunity [94]. Beyond glucose metabolism, other metabolic pathways in ECs are also being explored as therapeutic targets. Loss of endothelial Adrb2, which encodes the β2-adrenergic receptor, leads to angiogenesis inhibition through oxidative phosphorylation [97]. Similarly, disrupting fatty acid metabolism in ECs, as evidenced by that knockdown of fatty acid synthase (FASN) and the loss of CPT1A, a critical enzyme in fatty acid oxidation (FAO), limit vessel sprouting and proliferation through mTOR signaling and nucleotide synthesis, indicating the role of lipid metabolism in maintaining the physical structure of tumor vessels [91, 92]. Additionally, restricting glutamine metabolism through glutaminase 1 (GLS1) impairs vessel sprouting due to disrupted EC proliferation and migration [93], highlighting the importance of glutamine in sustaining macromolecule production necessary for angiogenesis. Collectively, the metabolic processes within ECs are fundamental not just for their energy and biosynthetic needs but also play a pivotal role in maintaining the structural and functional integrity of blood vessels in the TME. Understanding of these regulatory pathways offer key insights into how blood vessels adapt and grow in the TME, opening up new possibilities for targeted therapies aimed at modulating tumor angiogenesis.
In summary, genetic, epigenetic and metabolic reprogramming of tumor ECs represent promising advances in vasculature-targeting therapy, with the potential to improve the efficacy of conventional cytotoxic treatments and immunotherapies [75]. There are potential drugs that may be tested for endothelial reprogramming therapy (Table 1). In addition, a number of clinical trials are currently undergoing to evaluate the synergistic effects of combining conventional anti-angiogenic agents, such as Bevacizumab and axitinib, with immunotherapies, aiming to enhance treatment efficacy and patient outcomes (Table 2).
Regulation of tumor angiogenesis by myeloid cells
The vasculature is the avenue through which circulation-derived immune cells are recruited into the solid tumors. The infiltrating immune cells are exposed to the local vascular niche and interact with ECs mainly through paracrine mechanisms. The infiltrating immune cells locoregionally regulate vascularity, potentially modulating sprouting angiogenesis and vascular abnormalities. Here we discuss the regulatory mechanism underlying tumor angiogenesis by myeloid cells, which may serve as key therapeutic targets for vasculature-based cancer treatment.
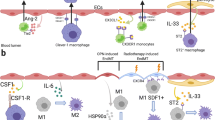
Tumor-infiltrating myeloid cells, mainly including macrophages, MDSCs and neutrophils, regulate tumor angiogenesis by secretion of a variety of pro-angiogenic factors. For instance, TAMs and MDSCs are known to secrete pro-angiogenic factors that stimulate EC proliferation and sprouting, leading to tumor angiogenesis and progression [98,99,100].Neutrophils serve as an additional source of released pro-angiogenic factors that regulate tumor growth and metastasis [101,102,103]. Myeloid cells can also indirectly enhance tumor angiogenesis by expressing matrix proteases and mesenchymal-associated factors that facilitate EC migration and vascular remodeling and maturation. In addition, myeloid cells, particularly perivascular macrophages, also contribute to dynamic vascular permeability in tumor [104]. Therefore, myeloid cells can regulate tumor angiogenesis through both direct secretion of pro-angiogenic factors and indirect modulation of the TME with multiple mechanisms potentially involved (Fig. 2).

Regulation of tumor angiogenesis by myeloid cells. Tumor-infiltrating myeloid cells, including macrophages, MDSCs, and neutrophils, interact with ECs and modulate tumor angiogenesis through secreted factors
Macrophages
Macrophage-produced pro-angiogenic factors
Macrophages are a major cellular component of solid tumors [105]. TAMs promote tumor angiogenesis by secreting a plethora of pro-angiogenic growth factors, cytokines, and chemokines that induce EC proliferation and migration, including EGF[106], FGF-2/bFGF [107], platelet-activating factor (PAF) [108], PDGF [109], VEGF [110,111,112], TNF-α [113], IL-1 [114, 115], IL-8/CXCL8 [116, 117], and CCL18 [118]. TAMs undergo alternative polarization in the TME to stimulate tumor angiogenesis [107, 119], which is characterized by elevated expression of these pro-angiogenic factors [120]. Macrophages are a major source of pro-angiogenic factors, particularly VEGF, that are present in tumors. Macrophages employ diverse mechanisms to express VEGF, mainly induced by hypoxia through HIFs-mediated transcriptional activation and further stimulated by multiple cytokines like IL-1β [121,122,123] and CCL18 [118]. Moreover, TAMs significantly contribute to production of proteases, particularly matrix metalloproteinase (MMP)-9, presented in the TME [124], which directly facilitates EC overgrowth and sprouting by remolding ECM and indirectly activates ECs by providing the active form of VEGF as a result of cleaving VEGF and releasing it from the binding to ECM [125,126,127,128,129]. Bone marrow-derived, MMP-9-expressing macrophages also participate in tumor neovascularization together with vascular endothelial progenitor cells [130], providing an additional mechanism for tumor angiogenesis. TAMs also release various additional factors that have pro-angiogenic activity, such as adrenomedullin (ADM), PGE2, Sema-4D, thymidine phosphorylase (TP), urokinase-type plasminogen activator (uPA), and YKL-40. For instance, ADM induces EC proliferation and tumor angiogenesis and growth [131], PGE2 enhances EC motility and survival, contributing to tumor angiogenesis [132], Sema-4D binds to its receptor Plexin-B1 on ECs to induce tumor angiogenesis [133], TP stimulates EC migration [134], uPA promotes ECM degradation and vascular invasion [135], and YKL-40 activates MAPK signaling in ECs, leading to increased expression of VEGFR-2 that facilitates vessel sprouting [136]. Targeting tumor macrophage-released pro-angiogenic factors represent a promising strategy for therapeutic modulation of tumor angiogenesis.
Perivascular macrophages
Macrophages expressing Tie2 receptor (also known as Tek) often reside near vasculature and exhibit high angiogenic potential, playing a significant role in physiological and pathological angiogenesis [137]. A subpopulation of Tie2+ macrophages show a pro-angiogenic activity during embryogenesis [138]. These Tie2+ tissue macrophages release VEGF-C and soluble VEGFR1 to bridge between EC tip cells and modulate vessel branching in development [139, 140]. Similar to these findings, bone marrow-derived TAMs cluster around tumor vasculature and co-express Tie2 and CD206, showing more robust pro-angiogenic activity than Tie2– monocytes or macrophages [141, 142]. Tie2+ monocytes express a number of tumor-promoting genes including Mmp9, Vegfa, Cxcl12, Tlr4, Nrp1, and Pdgfb at a high level [143], and their pro-angiogenic potential could be further stimulated by EC-derived factors in the perivascular niche [144]. The presence of Tie2+ macrophages in tumor regions has been linked to increased tumor microvascular density, enhanced tumor grade and distant metastasis, and reduced survival rates in human patients [145,146,147]. Perivascular macrophages accumulate in the tumor microenvironment following chemotherapy, radiotherapy, and anti-angiogenic therapy, contributing to vascular reconstruction, and potentially leading to tumor relapse [112, 148,149,150,151]. These macrophages originate from the precursors of a subset of Tie2+ circulating monocytes and are attracted to tumors by chemotaxis, induced by EC-derived angiopoietin-2 (ANG-2), a ligand of Tie2 [141, 144, 152]. Interaction with ECs stimulates Tie2 expression in TAMs and enhances the production of pro-angiogenic factors by these macrophages [144]. Ang-2 also drives Tie2+ macrophages to express IL-10 and CCL17, which inhibit T-cell proliferation and disrupt vascular homeostasis [153].
Recent studies have shed light on the importance of perivascular macrophages in the TME. In addition to promoting angiogenesis, these macrophages, residing near blood vessels, promote the formation of the vascular niche that contributes to tumor progression. Activation of these macrophages by extracellular matrix proteins, such as TNC (tenascin-C), through toll-like receptor 4 (TLR4) signaling, leads to the secretion of nitric oxide (NO) and TNF-α [154]. These factors, in turn, induce the expression of niche components in ECs, facilitating the establishment of a supportive TME for tumor growth and metastasis. These macrophages usually acquire anti-inflammatory phenotypes, contributing to spatially restrict immunosuppression in the vascular niche. As such, tumor EC-derived IL-6 induces alternative polarization and immunosuppressive phenotypes in perivascular macrophages [155]. In addition, Lyve-1+ macrophages have a critical role in creating a pro-angiogenic TME through maintaining and expanding a perivascular mesenchymal cell population, ultimately establishing a specialized niche that supports tumor progression [156]. Macrophage-derived TNF-α and endothelial TNF receptor are identified as crucial components of this regulatory mechanism. Perivascular macrophages, activated via TNC and TLR4 to induce the formation of pro-tumor vascular niche that drives tumor metastasis [154]. The spatial interaction between macrophages and ECs provides strong evidence for the intricate crosstalk that stimulates angiogenesis and tumor progression, metastasis, and therapy resistance [109].
Macrophage-mediated vascular maturation
Macrophages regulate vascular maturation under physiological and pathological conditions. As a resident macrophage population in brain, microglia maintain the integrity of blood–brain barrier that mainly consist of tightly associated ECs [157, 158]. Loss of NG2 proteoglycan in myeloid-specific and pericyte-specific cells leads to significant reductions in early-stage intracranial tumor growth [159, 160]. Myeloid-specific NG2 loss-induced vascular deficits, characterized by poor pericyte coverage on ECs and immature vessel, result in smaller vessel diameter, lower patency, increased leakiness, inefficient blood flow in tumor vasculature, and elevated intratumoral hypoxia [159]. TAMs promote pericyte coverage and stabilize tumor vasculature through the secretion of PDGF-B, contributing to vascular maturation [161]. Adenosine deaminase 2 (CECR1) is highly expressed by TAMs, contributing to tumor angiogenesis [161]. Increased CECR1 expression correlates with higher microvascular density in GBM tissues. Inhibition of CECR1 reduces new vessel formation, while CECR1 stimulation promotes vascular maturation through paracrine activation of pericytes via PDGFB-PDGFRβ signaling [161].
Macrophage-mediated vascular permeability
VEGF was originally identified as vascular permeability factor (VPF) as a result of its potent ability to enhance vessel permeability, resulting in vascular leakage [162]. TAM-derived VEGF-A may, therefore, induce local vascular permeability in tumors. Consistent with this hypothesis, real-time intravital imaging reveals that dynamic vascular permeability occurs concurrently with cancer cell invasion and Tie2+ macrophage infiltration in the perivascular niche [163]. Genetic deletion of VEGF in TAMs reverses vascular permeability and cancer cell intravasation [163], suggesting a role of TAMs for regulation of vascular permeability. TAMs regulate vascular permeability through VEGF-induced downregulation of vascular junction proteins ZO-1 and VE-cadherin and through VLA4-mediated disruption of vascular adhesion proteins VCAM1 in ECs [163, 164]. In addition, M2-like polarized macrophage-derived exosomes containing miR-23a, miR-155 and miR-221 induces angiogenesis and vessel leakiness [165, 166], serving as an alternative mechanism for regulating tumor vascular permeability.
MDSCs
MDSCs are pathologically activated granulocytes (granulocytic or polymorphonuclear MDSCs, PMN-MDSCs) and monocytes (monocytic MDSCs, M-MDSCs) with potent immunosuppressive activity [167, 168]. MDSCs regulate immune responses in physiological and pathological conditions, including pregnancy, cancer, chronic infection, sepsis and autoimmunity [169]. In addition to their well-established role for direct suppression of lymphocyte activity, MDSCs secrete various pro-angiogenic molecules to induce tumor angiogenesis [100, 170, 171]. Tumor-associated Gr1+CD11b+ mouse MDSCs produce MMP-9 and release VEGF-A to promote angiogenesis [172]. Consistent with these findings, tumor-infiltrating MDSCs express MMP-2,-13,-14 at a high level [173], and overexpression of MMP inhibitor TIMP-2 reduces MDSC infiltration and vascular density in tumor [174], suggesting a critical role of protease for MDSC-mediated tumor angiogenesis. Moreover, G-CSF stimulates Stat3-dependent MDSC expression of Bv8 [175], a potent driver of myeloid cell-dependent tumor angiogenesis [176]. MDSCs also express FGF-2 [171], PDGF [177], IL-1β [178], IL-28/IFN-λ [179, 180], TGFβ, EGF, and HGF [181] that can stimulate EC proliferation and migration, contributing to tumor angiogenesis [182]. In addition, MDSCs could directly differentiate into ECs [172] and induce tumor cell formation of vascular mimicry (VM) [183], serving as alternative processes to sprouting angiogenesis.
Neutrophils
Neutrophils are the most abundant innate immune cells in bone marrow and peripheral blood [184]. Neutrophils have emerged as an important component of the TME, but their functional role in cancer is still controversial [185]. In accordance with their critical functions in developmental angiogenesis [186, 187], neutrophils modulate tumor angiogenesis by providing pro-angiogenic factors in a time- and tumor context-dependent manner, contributing to tumor growth and metastasis [101, 102]. Tumor-associated neutrophils secrete a plethora of pro-angiogenic molecules including VEGF [188, 189], FGF-2 [190], Bv8 [191, 192], IL-17 [193], and MMP-9 [188, 194]. Neutrophil-derived oncostatin M also up-regulates the secretion of VEGF [195], and reprograms and degrades the ECM which then primes the environment for angiogenesis [195]. MMP-9 released by neutrophils promotes the activation of VEGF and subsequent angiogenesis and tumor progression [129, 194, 196]. Neutrophils also carry an intracellular pool of VEGF and mediate its rapid secretion [197]. Interestingly, IFN-β inhibits the infiltration of proangiogenic neutrophils that express VEGF, MMP-9, and CXCR4 and reduces tumor growth, suggesting a potential therapeutic approach for targeting neutrophil-mediated tumor angiogenesis [188]. These findings together suggest that neutrophils support the pro-angiogenic switch during cancer development [194]. As such, neutrophils display different states based on the expression of markers such as HIF-1α, arginase 1, and MMP-9, in which HIF1α+ neutrophils significantly correlate with greater angiogenesis and worse overall survival [198]. In addition to their role in directly driving pro-angiogenic functions, neutrophils can also indirectly promote angiogenesis by activating pro-angiogenic functions of other immune cells [103]. For example, neutrophils reprogram T cells to acquire regulatory-like phenotypes and support their expression of IL-10, IL-17, and VEGF to promote angiogenesis [199].
Neutrophils also contribute to tumor vascularization through several non-angiogenic mechanisms, such as neutrophil extracellular trap (NET) formation [174, 175], vessel co-option, and VM mechanisms [200, 201]. NETs, the release of web-like DNA structures, constitute an important mechanism by which neutrophils prevent pathogen dissemination or deal with microorganisms of larger size [202]. Cancer cells can induce NET formation by neutrophils, leading to tumor angiogenesis [203, 204]. NET-associated myeloperoxidase produces H2O2 released to ECM and activates NF-κB-mediated signaling in ECs, resulting in enhanced EC proliferation and migration [205]. Angiopoietins (ANG-1/-2) also induce NETs formation and promote neutrophil adhesion to endothelium and stimulated EC proliferation [206]. Finally, VM structures provide vascular channels for neutrophil infiltration and activation, leading to their expression of arginase, CCL2, CXCR4, and MMP-9 to promote angiogenesis and evade anti-angiogenic therapy [201], collectively suggest a critical role of neutrophils for tumor angiogenesis.
Conclusion remarks and future perspectives
Anti-angiogenesis, vessel normalization, and endothelial reprogramming stand out as promising strategies for targeting the vasculature in cancer treatment. They hold significant potential when combined with various anti-cancer approaches including, but not limited to, radiotherapy, chemotherapy, molecular targeted therapy, and immunotherapy. The application of these strategies in clinical settings might require optimization based on factors like tumor type, size, stage, location, and pathology to achieve the maximal efficacy in combination therapy. Particularly, genetic, epigenetic and metabolic reprogramming of tumor ECs may offer unique opportunities to empower T cell-based immunotherapy, considering that endothelial reprogramming could (1) inhibit excessive angiogenesis and suppress vascular aberrancy, leading to increase vessel delivery function to improve lymphocyte infiltration as well as to relieve intratumoral hypoxia and to activate these lymphocytes, (2) regulate adhesion molecule expression on ECs to promote lymphocyte attachment to endothelium and their recruitment to the tumors, and (3) reverse pro-tumor immunity that is induced by locally EC-derived immunosuppressive molecules, facilitating lymphocyte activation.
Among these innovative strategies, the induction of HEV neogenesis has emerged as a promising strategy to augment anti-tumor immunity and vessel functionality. HEVs play a vital role in lymphocyte trafficking and activation, serving as a critical target for therapeutic modulation of immunocyte infiltration [207]. Recent single-cell RNA-seq analyses suggest their significant involvement in upregulated expression of EC activation markers and co-stimulatory molecules that regulate dendritic cell function and T cell activation [208]. Activation of lymphotoxin β receptor (LTβR) signaling induces the formation of HEVs and T cell activation, and thereby sensitizes tumors to anti-angiogenic and anti-PD-L1 therapy [209], collectively suggesting that better understanding the immunostimulatory functions of HEVs may open new avenues for immunotherapeutic interventions in the future.
Despite these advancements, there are still several major limitations and challenges that restrict vascular-targeting therapy in cancer, due to treatment toxicity, vascular heterogeneity in cancer, and the lack of reliable biomarkers hinder the effectiveness and applicability of current treatments. Notably, tumors can develop resistance to traditional anti-angiogenic treatments by compensatory activation of additional pro-angiogenic pathways to sustain tumor vascularization and by activating HIF-1α to support tumor growth and progression in low-oxygen conditions. One particular challenge is to develop a vascular-targeting strategy to selectively promote the infiltration of cytotoxic T cells and/or NK cells, but not immunosuppressive myeloid cells. Future spatiotemporal analysis of tumor specimens at single-cell transcriptome, epigenome, and metabolome levels will help address these challenges, as they need a deep understanding of the complex interplay between tumor vasculature and immune system in the tumor microenvironment.
Further research into the interaction of infiltrating myeloid cells with ECs regulates tumor angiogenesis, providing insights into the resolution of vascular formation, maturation, and aberrancy in cancer. Understanding of the underlying regulatory mechanism for tumor angiogenesis may lead to identification of new therapeutic targets for anti-vascular therapy, contributing to development of more efficient approaches for anti-angiogenesis, vessel normalization, and endothelial reprogramming therapy. It also remains largely unclear how ECs spatiotemporally regulate the immunity of these macrophages, MDSCs, and neutrophils in the vascular niche. New knowledge filling this gap may help understand tumor immunosuppression and lead to development of new myeloid cell-based immunotherapy for cancer treatment.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed for this review article.
References
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473(7347):298–307
Fridman WH et al (2012) The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12(4):298–306
Tian L et al (2017) Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 544(7649):250–254
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285(21):1182–1186
Weis SM, Cheresh DA (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 17(11):1359–1370
Cao Y, Langer R, Ferrara N (2023) Targeting angiogenesis in oncology, ophthalmology and beyond. Nat Rev Drug Discov 22(6):476–495
Ebos JM, Kerbel RS (2011) Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 8(4):210–221
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307(5706):58–62
Liu T et al (2018) PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat Commun 9(1):3439
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3(6):401–410
Jain RK (2001) Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med 7(9):987–989
Carmeliet P, Jain RK (2011) Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 10(6):417–427
Martin JD, Seano G, Jain RK (2019) Normalizing function of tumor vessels: progress, opportunities, and challenges. Annu Rev Physiol 81:505–534
Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 8(8):579–591
Turner N, Grose R (2010) Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 10(2):116–129
Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358(11):1160–1174
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3(10):721–732
Lee P et al (2002) Neuropilin-1 is required for vascular development and is a mediator of VEGF-dependent angiogenesis in zebrafish. Proc Natl Acad Sci USA 99(16):10470–10475
Benedito R et al (2012) Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 484(7392):110–114
Daly C et al (2013) Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res 73(1):108–118
Lobov IB, Brooks PC, Lang RA (2002) Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci USA 99(17):11205–11210
Wang Y et al (2010) Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465(7297):483–486
Ferrara N et al (2004) Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 3(5):391–400
Faivre S et al (2006) Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol 24(1):25–35
Keating GM (2017) Sorafenib: a review in hepatocellular carcinoma. Target Oncol 12(2):243–253
Wick W et al (2017) Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med 377(20):1954–1963
Gilbert MR et al (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370(8):699–708
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8(8):592–603
Finn RS et al (2020) Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 382(20):1894–1905
Batchelor TT et al (2013) Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol 31(26):3212–3218
Thomas M, Augustin HG (2009) The role of the Angiopoietins in vascular morphogenesis. Angiogenesis 12(2):125–137
Maisonpierre PC et al (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277(5322):55–60
Harris AL (2002) Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer 2(1):38–47
Li JL, Harris AL (2005) Notch signaling from tumor cells: a new mechanism of angiogenesis. Cancer Cell 8(1):1–3
Zerlin M, Julius MA, Kitajewski J (2008) Wnt/Frizzled signaling in angiogenesis. Angiogenesis 11(1):63–69
Keith B, Johnson RS, Simon MC (2011) HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12(1):9–22
Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9(6):669–676
Wouters BG, Koritzinsky M (2008) Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8(11):851–864
Ridgway J et al (2006) Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444(7122):1083–1087
Stenman JM et al (2008) Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322(5905):1247–1250
Huang M et al (2020) Wnt-mediated endothelial transformation into mesenchymal stem cell-like cells induces chemoresistance in glioblastoma. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aay7522
Kühl M et al (2000) Ca(2+)/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzled homologs and promotes ventral cell fates in Xenopus. J Biol Chem 275(17):12701–12711
Habas R, Kato Y, He X (2001) Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell 107(7):843–854
Yamanaka H et al (2002) JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep 3(1):69–75
Cirone P et al (2008) A role for planar cell polarity signaling in angiogenesis. Angiogenesis 11(4):347–360
Huang Y et al (2012) Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA 109(43):17561–17566
Dong X et al (2023) Anti-VEGF therapy improves EGFR-vIII-CAR-T cell delivery and efficacy in syngeneic glioblastoma models in mice. J Immunother Cancer 11(3):e005583
Hamzah J et al (2008) Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 453(7193):410–414
Magrini E et al (2014) Endothelial deficiency of L1 reduces tumor angiogenesis and promotes vessel normalization. J Clin Invest 124(10):4335–4350
Goel S et al (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91(3):1071–1121
Maione F et al (2009) Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J Clin Invest 119(11):3356–3372
Sawada J et al (2012) Small GTPase R-Ras regulates integrity and functionality of tumor blood vessels. Cancer Cell 22(2):235–249
Takara K et al (2017) Lysophosphatidic acid receptor 4 activation augments drug delivery in tumors by tightening endothelial cell-cell contact. Cell Rep 20(9):2072–2086
Maes H et al (2014) Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26(2):190–206
Adapala RK et al (2016) Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene 35(3):314–322
Campbell NE et al (2010) The thrombospondin-1 mimetic ABT-510 increases the uptake and effectiveness of cisplatin and paclitaxel in a mouse model of epithelial ovarian cancer. Neoplasia 12(3):275–283
Funahashi Y et al (2014) Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci 105(10):1334–1342
Chauhan VP et al (2013) Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun 4:2516
Stylianopoulos T et al (2012) Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci USA 109(38):15101–15108
Alvarez R et al (2013) Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer 109(4):926–933
Wenes M et al (2016) Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab 24(5):701–715
De Palma M, Jain RK (2017) CD4(+) T cell activation and vascular normalization: two sides of the same coin? Immunity 46(5):773–775
Voron T et al (2015) VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 212(2):139–148
Willett CG et al (2004) Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 10(2):145–147
Kerbel RS (2008) Tumor angiogenesis. N Engl J Med 358(19):2039–2049
Bertrand JY et al (2010) Haematopoietic stem cells derive directly from aortic endothelium during development. Nature 464(7285):108–111
Kissa K, Herbomel P (2010) Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 464(7285):112–115
Zeisberg EM et al (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13(8):952–961
Maddaluno L et al (2013) EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498(7455):492–496
Zeisberg EM et al (2007) Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 67(21):10123–10128
Fan Y (2019) Vascular detransformation for cancer therapy. Trends Cancer 5(8):460–463
Lamplugh Z, Fan Y (2021) Vascular microenvironment, tumor immunity and immunotherapy. Front Immunol 12:811485
Huang M et al (2016) c-Met-mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J Clin Invest 126(5):1801–1814
Amersfoort J, Eelen G, Carmeliet P (2022) Immunomodulation by endothelial cells—partnering up with the immune system? Nat Rev Immunol 22(9):576–588
Cleveland AH, Fan Y (2023) Reprogramming endothelial cells to empower cancer immunotherapy. Trends Mol Med. https://doi.org/10.1016/j.molmed.2023.11.002
Ma W et al (2021) Targeting PAK4 to reprogram the vascular microenvironment and improve CAR-T immunotherapy for glioblastoma. Nat Cancer 2(1):83–97
Birdsey GM et al (2015) The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/β-catenin signaling. Dev Cell 32(1):82–96
Kabir AU et al (2021) Dual role of endothelial Myct1 in tumor angiogenesis and tumor immunity. Sci Transl Med. https://doi.org/10.1126/scitranslmed.abb6731
O’Connor MN et al (2021) LRG1 destabilizes tumor vessels and restricts immunotherapeutic potency. Med 2(11):1231-1252.e10
Huijbers EJM et al (2022) Tumors resurrect an embryonic vascular program to escape immunity. Sci Immunol 7(67):eabm6388
Hellebrekers DM et al (2007) Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res 67(9):4138–4148
Bazou D et al (2016) Flow-induced HDAC1 phosphorylation and nuclear export in angiogenic sprouting. Sci Rep 6:34046
Hu C et al (2021) HDAC1 and 2 regulate endothelial VCAM-1 expression and atherogenesis by suppressing methylation of the GATA6 promoter. Theranostics 11(11):5605–5619
Deroanne CF et al (2002) Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 21(3):427–436
Kim MS et al (2001) Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med 7(4):437–443
Hellebrekers DM et al (2006) Epigenetic regulation of tumor endothelial cell anergy: silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res 66(22):10770–10777
Maleszewska M et al (2015) Enhancer of zeste homolog-2 (EZH2) methyltransferase regulates transgelin/smooth muscle-22α expression in endothelial cells in response to interleukin-1β and transforming growth factor-β2. Cell Signal 27(8):1589–1596
Glaser SF et al (2020) The histone demethylase JMJD2B regulates endothelial-to-mesenchymal transition. Proc Natl Acad Sci USA 117(8):4180–4187
Kim DJ et al (2023) Priming a vascular-selective cytokine response permits CD8(+) T-cell entry into tumors. Nat Commun 14(1):2122
De Bock K et al (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154(3):651–663
Bruning U et al (2018) Impairment of angiogenesis by fatty acid synthase inhibition involves mTOR malonylation. Cell Metab 28(6):866-880.e15
Schoors S et al (2015) Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520(7546):192–197
Huang H et al (2017) Role of glutamine and interlinked asparagine metabolism in vessel formation. Embo J 36(16):2334–2352
Zhang D et al (2023) PHGDH-mediated endothelial metabolism drives glioblastoma resistance to chimeric antigen receptor T cell immunotherapy. Cell Metab 35(3):517-534.e8
Cantelmo AR et al (2016) Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 30(6):968–985
Shan Y et al (2022) Targeting tumor endothelial hyperglycolysis enhances immunotherapy through remodeling tumor microenvironment. Acta Pharm Sin B 12(4):1825–1839
Zahalka AH et al (2017) Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 358(6361):321–326
Riabov V et al (2014) Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol 5:75
Ribatti D et al (2007) Macrophages and tumor angiogenesis. Leukemia 21(10):2085–2089
Vetsika EK, Koukos A, Kotsakis A (2019) Myeloid-derived suppressor cells: major figures that shape the immunosuppressive and angiogenic network in cancer. Cells 8(12):1647
Tazzyman S, Lewis CE, Murdoch C (2009) Neutrophils: key mediators of tumour angiogenesis. Int J Exp Pathol 90(3):222–231
Liang W, Ferrara N (2016) The complex role of neutrophils in tumor angiogenesis and metastasis. Cancer Immunol Res 4(2):83–91
Ozel I et al (2022) The good, the bad, and the ugly: neutrophils, angiogenesis, and cancer. Cancers (Basel) 14(3):536
Lewis CE, Harney AS, Pollard JW (2016) The multifaceted role of perivascular macrophages in tumors. Cancer Cell 30(1):18–25
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4(1):71–78
Yin M et al (2016) Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest 126(11):4157–4173
Jetten N et al (2014) Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 17(1):109–118
Seo KH et al (2004) Essential role for platelet-activating factor-induced NF-kappaB activation in macrophage-derived angiogenesis. Eur J Immunol 34(8):2129–2137
Opzoomer JW et al (2021) Macrophages orchestrate the expansion of a proangiogenic perivascular niche during cancer progression. Sci Adv 7(45):eabg9518
Stockmann C et al (2008) Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 456(7223):814–818
Barbera-Guillem E et al (2002) Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res 62(23):7042–7049
Hughes R et al (2015) Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res 75(17):3479–3491
Leibovich SJ et al (1987) Macrophage-induced angiogenesis is mediated by tumour necrosis factor-alpha. Nature 329(6140):630–632
Voronov E, Carmi Y, Apte RN (2014) The role IL-1 in tumor-mediated angiogenesis. Front Physiol 5:114
Carmi Y et al (2009) The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J Immunol 183(7):4705–4714
Koch AE et al (1992) Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 258(5089):1798–1801
Kimura YN et al (2007) Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci 98(12):2009–2018
Lin L et al (2015) CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget 6(33):34758–34773
Lin EY et al (2006) Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 66(23):11238–11246
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122(3):787–795
Mantsounga CS et al (2022) Macrophage IL-1β promotes arteriogenesis by autocrine STAT3- and NF-κB-mediated transcription of pro-angiogenic VEGF-A. Cell Rep 38(5):110309
Nam EH, Park SR, Kim PH (2010) TGF-beta1 induces mouse dendritic cells to express VEGF and its receptor (Flt-1) under hypoxic conditions. Exp Mol Med 42(9):606–613
Fang HY et al (2009) Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 114(4):844–859
Afik R et al (2016) Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med 213(11):2315–2331
Egeblad M, Werb Z (2002) New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2(3):161–174
Arroyo AG, Iruela-Arispe ML (2010) Extracellular matrix, inflammation, and the angiogenic response. Cardiovasc Res 86(2):226–235
Vu TH et al (1998) MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93(3):411–422
Bergers G et al (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2(10):737–744
Coussens LM et al (2000) MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103(3):481–490
Du R et al (2008) HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13(3):206–220
Zhao Y et al (1998) PCR display identifies tamoxifen induction of the novel angiogenic factor adrenomedullin by a non estrogenic mechanism in the human endometrium. Oncogene 16(3):409–415
Kamiyama M et al (2006) EP2, a receptor for PGE2, regulates tumor angiogenesis through direct effects on endothelial cell motility and survival. Oncogene 25(53):7019–7028
Basile JR et al (2007) MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J Biol Chem 282(9):6899–6905
Akiyama S et al (2004) The role of thymidine phosphorylase, an angiogenic enzyme, in tumor progression. Cancer Sci 95(11):851–857
Stepanova V et al (2016) Urokinase-type plasminogen activator (uPA) promotes angiogenesis by attenuating proline-rich homeodomain protein (PRH) transcription factor activity and de-repressing vascular endothelial growth factor (VEGF) receptor expression. J Biol Chem 291(29):15029–15045
Shao R (2013) YKL-40 acts as an angiogenic factor to promote tumor angiogenesis. Front Physiol 4:122
Baer C et al (2013) Reciprocal interactions between endothelial cells and macrophages in angiogenic vascular niches. Exp Cell Res 319(11):1626–1634
Fantin A et al (2010) Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 116(5):829–840
Tammela T et al (2011) VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol 13(10):1202–1213
Stefater JA 3rd et al (2011) Regulation of angiogenesis by a non-canonical Wnt-Flt1 pathway in myeloid cells. Nature 474(7352):511–515
De Palma M et al (2005) Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8(3):211–226
De Palma M et al (2003) Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med 9(6):789–795
Pucci F et al (2009) A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 114(4):901–914
Coffelt SB et al (2010) Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res 70(13):5270–5280
Ji J et al (2013) The frequency of tumor-infiltrating Tie-2-expressing monocytes in renal cell carcinoma: its relationship to angiogenesis and progression. Urology 82(4):974.e9–13
Du S et al (2022) Tumor cell-derived exosomes deliver TIE2 protein to macrophages to promote angiogenesis in cervical cancer. Cancer Lett 529:168–179
Matsubara T et al (2013) TIE2-expressing monocytes as a diagnostic marker for hepatocellular carcinoma correlates with angiogenesis. Hepatology 57(4):1416–1425
Chen L et al (2016) Tie2 expression on macrophages is required for blood vessel reconstruction and tumor relapse after chemotherapy. Cancer Res 76(23):6828–6838
Kioi M et al (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 120(3):694–705
Kozin SV et al (2010) Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res 70(14):5679–5685
Welford AF et al (2011) TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest 121(5):1969–1973
Venneri MA et al (2007) Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood 109(12):5276–5285
Coffelt SB et al (2011) Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J Immunol 186(7):4183–4190
Hongu T et al (2022) Perivascular tenascin C triggers sequential activation of macrophages and endothelial cells to generate a pro-metastatic vascular niche in the lungs. Nat Cancer 3(4):486–504
Yang F et al (2021) Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat Commun 12(1):3424
Ren X et al (2023) Macrophage-endothelial cell crosstalk orchestrates neutrophil recruitment in inflamed mucosa. J Clin Invest. https://doi.org/10.1172/JCI170733
Li Q, Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18(4):225–242
Kisler K, Nikolakopoulou AM, Zlokovic BV (2021) Microglia have a grip on brain microvasculature. Nat Commun 12(1):5290
Yotsumoto F et al (2015) NG2 proteoglycan-dependent recruitment of tumor macrophages promotes pericyte-endothelial cell interactions required for brain tumor vascularization. Oncoimmunology 4(4):e1001204
Thurgur H, Pinteaux E (2019) Microglia in the neurovascular unit: blood-brain barrier-microglia interactions after central nervous system disorders. Neuroscience 405:55–67
Zhu C et al (2017) CECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma. Oncogene 36(38):5356–5368
Senger DR et al (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219(4587):983–985
Harney AS et al (2015) Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov 5(9):932–943
Zhang S et al (2021) Macrophage-mediated vascular permeability via VLA4/VCAM1 pathway dictates ascites development in ovarian cancer. J Clin Invest. https://doi.org/10.1172/JCI140315
Yang Y et al (2021) M2 macrophage-derived exosomes promote angiogenesis and growth of pancreatic ductal adenocarcinoma by targeting E2F2. Mol Ther 29(3):1226–1238
Lu Y et al (2023) M2 macrophage-secreted exosomes promote metastasis and increase vascular permeability in hepatocellular carcinoma. Cell Commun Signal 21(1):299
Veglia F, Sanseviero E, Gabrilovich DI (2021) Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 21(8):485–498
Hegde S, Leader AM, Merad M (2021) MDSC: markers, development, states, and unaddressed complexity. Immunity 54(5):875–884
Veglia F, Perego M, Gabrilovich D (2018) Myeloid-derived suppressor cells coming of age. Nat Immunol 19(2):108–119
Binsfeld M et al (2016) Granulocytic myeloid-derived suppressor cells promote angiogenesis in the context of multiple myeloma. Oncotarget 7(25):37931–37943
Kujawski M et al (2008) Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest 118(10):3367–3377
Yang L et al (2004) Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 6(4):409–421
Yang L et al (2008) Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 13(1):23–35
Guedez L et al (2012) TIMP-2 targets tumor-associated myeloid suppressor cells with effects in cancer immune dysfunction and angiogenesis. J Immunother 35(6):502–512
Qu X et al (2012) Induction of Bv8 expression by granulocyte colony-stimulating factor in CD11b+Gr1+ cells: key role of Stat3 signaling. J Biol Chem 287(23):19574–19584
Shojaei F et al (2007) Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450(7171):825–831
Hsu YL et al (2019) CXCL17-derived CD11b(+)Gr-1(+) myeloid-derived suppressor cells contribute to lung metastasis of breast cancer through platelet-derived growth factor-BB. Breast Cancer Res 21(1):23
Shi H et al (2017) Recruited monocytic myeloid-derived suppressor cells promote the arrest of tumor cells in the premetastatic niche through an IL-1β-mediated increase in E-selectin expression. Int J Cancer 140(6):1370–1383
Mucha J et al (2014) MDSCs mediate angiogenesis and predispose canine mammary tumor cells for metastasis via IL-28/IL-28RA (IFN-λ) signaling. PLoS ONE 9(7):e103249
Pingwara R et al (2017) Interferon lambda 2 promotes mammary tumor metastasis via angiogenesis extension and stimulation of cancer cell migration. J Physiol Pharmacol 68(4):573–583
Toh B et al (2011) Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol 9(9):e1001162
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12(4):253–268
Li Y et al (2021) Targeting myeloid-derived suppressor cells to attenuate vasculogenic mimicry and synergistically enhance the anti-tumor effect of PD-1 inhibitor. iScience 24(12):103392
Amulic B et al (2012) Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489
Jaillon S et al (2020) Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer 20(9):485–503
Mueller MD et al (2000) Neutrophils infiltrating the endometrium express vascular endothelial growth factor: potential role in endometrial angiogenesis. Fertil Steril 74(1):107–112
Heryanto B, Girling JE, Rogers PA (2004) Intravascular neutrophils partially mediate the endometrial endothelial cell proliferative response to oestrogen in ovariectomised mice. Reproduction 127(5):613–620
Jablonska J et al (2010) Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest 120(4):1151–1164
Scapini P et al (2004) CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J Immunol 172(8):5034–5040
Gordon-Weeks AN et al (2017) Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2-dependent angiogenesis in mice. Hepatology 65(6):1920–1935
Shojaei F et al (2008) Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc Natl Acad Sci USA 105(7):2640–2645
Bordbari S et al (2022) SIRT1-mediated deacetylation of FOXO3a transcription factor supports pro-angiogenic activity of interferon-deficient tumor-associated neutrophils. Int J Cancer 150(7):1198–1211
Numasaki M et al (2005) IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol 175(9):6177–6189
Nozawa H, Chiu C, Hanahan D (2006) Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA 103(33):12493–12498
Queen MM et al (2005) Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res 65(19):8896–8904
Ardi VC et al (2007) Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci USA 104(51):20262–20267
Gaudry M et al (1997) Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood 90(10):4153–4161
Sorin M et al (2023) Single-cell spatial landscapes of the lung tumour immune microenvironment. Nature 614(7948):548–554
Nadkarni S et al (2016) Neutrophils induce proangiogenic T cells with a regulatory phenotype in pregnancy. Proc Natl Acad Sci USA 113(52):E8415-e8424
Teuwen LA et al (2021) Tumor vessel co-option probed by single-cell analysis. Cell Rep 35(11):109253
Tsai YM et al (2021) Cooperation between cancer and fibroblasts in vascular mimicry and N2-type neutrophil recruitment via Notch2-Jagged1 interaction in lung cancer. Front Oncol 11:696931
Papayannopoulos V (2018) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18(2):134–147
Jung HS et al (2019) Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS ONE 14(4):e0216055
Yang S et al (2023) Neutrophil extracellular traps promote angiogenesis in gastric cancer. Cell Commun Signal 21(1):176
Aldabbous L et al (2016) Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol 36(10):2078–2087
Lavoie SS et al (2018) Synthesis of human neutrophil extracellular traps contributes to angiopoietin-mediated in vitro proinflammatory and proangiogenic activities. J Immunol 200(11):3801–3813
Browning JL et al (2005) Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity 23(5):539–550
Veerman K et al (2019) Single-cell analysis reveals heterogeneity of high endothelial venules and different regulation of genes controlling lymphocyte entry to lymph nodes. Cell Rep 26(11):3116-3131.e5
Allen E et al (2017) Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aak9679
Funding
This work was partly financially supported by American Association for Cancer Research Judah Folkman Award (to Y.F.) and American Heart Association Innovation Project Award (to Y.F.).
Author information
Authors and Affiliations
Contributions
FY and YF wrote the manuscript and produced the figures. FY performed literature research. GL contributed to the concepts and produced the tables. YF conceived the concepts described in this review. All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, F., Lee, G. & Fan, Y. Navigating tumor angiogenesis: therapeutic perspectives and myeloid cell regulation mechanism. Angiogenesis (2024). https://doi.org/10.1007/s10456-024-09913-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10456-024-09913-z