
Abstract
Li–Fraumeni syndrome (LFS) is a hereditary tumor that exhibits autosomal dominant inheritance. LFS develops in individuals with a pathogenic germline variant of the cancer-suppressor gene, TP53 (individuals with TP53 pathogenic variant). The number of individuals with TP53 pathogenic variant among the general population is said to be 1 in 500 to 20,000. Meanwhile, it is found in 1.6% (median value, range of 0–6.7%) of patients with pediatric cancer and 0.2% of adult patients with cancer. LFS is diagnosed by the presence of germline TP53 pathogenic variants. However, patients can still be diagnosed with LFS even in the absence of a TP53 pathogenic variant if the familial history of cancers fit the classic LFS diagnostic criteria. It is recommended that TP53 genetic testing be promptly performed if LFS is suspected. Chompret criteria are widely used for the TP53 genetic test. However, as there are a certain number of cases of LFS that do not fit the criteria, if LFS is suspected, TP53 genetic testing should be performed regardless of the criteria. The probability of individuals with TP53 pathogenic variant developing cancer in their lifetime (penetrance) is 75% for men and almost 100% for women. The LFS core tumors (breast cancer, osteosarcoma, soft tissue sarcoma, brain tumor, and adrenocortical cancer) constitute the majority of cases; however, various types of cancers, such as hematological malignancy, epithelial cancer, and pediatric cancers, such as neuroblastoma, can also develop. Furthermore, approximately half of the cases develop simultaneous or metachronous multiple cancers. The types of TP53 pathogenic variants and factors that modify the functions of TP53 have an impact on the clinical presentation, although there are currently no definitive findings. There is currently no cancer preventive agent for individuals with TP53 pathogenic variant. Surgical treatments, such as risk-reducing bilateral mastectomy warrant further investigation. Theoretically, exposure to radiation could induce the onset of secondary cancer; therefore, imaging and treatments that use radiation should be avoided as much as possible. As a method to follow-up LFS, routine cancer surveillance comprising whole-body MRI scan, brain MRI scan, breast MRI scan, and abdominal ultrasonography (US) should be performed immediately after the diagnosis. However, the effectiveness of this surveillance is unknown, and there are problems, such as adverse events associated with a high rate of false positives, overdiagnosis, and sedation used during imaging as well as negative psychological impact. The detection rate of cancer through cancer surveillance is extremely high. Many cases are detected at an early stage, and treatments are low intensity; thus, cancer surveillance could contribute to an improvement in QOL, or at least, a reduction in complications associated with treatment. With the widespread use of genomic medicine, the diagnosis of LFS is unavoidable, and a comprehensive medical care system for LFS is necessary. Therefore, clinical trials that verify the feasibility and effectiveness of the program, comprising LFS registry, genetic counseling, and cancer surveillance, need to be prepared.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Li–Fraumeni syndrome (LFS) is a hereditary cancer predisposition syndrome, in which multiple cancerous tumors are likely to develop over a person’s lifetime. While concerns have long surrounded this disease, underlying LFS is often overlooked, as a diverse range of cancers develop across a wide range of age groups. In the past, LFS was suspected from medical and family histories, and was diagnosed with a TP53 genetic test. However, as cancer genomic medicine is becoming more widespread, an increasing number of cases are diagnosed as secondary findings of a cancer genetic panel test. However, no system has been established for following LFS in Japan, and there is no consistent approach in clinical settings.
Experts of hereditary tumors, including LFS, gathered in 2016 at the Childhood Cancer Predisposition Workshop, which was held as a subcommittee of American Association of Cancer Research (AACR). As a result, the optical surveillance and care standard for pediatric hereditary tumors was formulated based on precision genetics, and it was reported in 17 papers in the Clinical Cancer Research Journal in 2017 [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17]. A comprehensive medical care system for hereditary tumors with onset in childhood and adolescents and young adults (AYA) needs to be established in Japan. Thus, in 2017, the Health Science and Labor Research Grants “A study to implement cancer genomic medicine system for hereditary tumors onset in childhood” Group was formed, with the preparation of LFS medical guidelines as the research objective.
Basic items
What is LFS?
LFS is a hereditary tumor syndrome caused by a pathogenic variant of TP53, a tumor-suppressor gene, which leads to a high probability of cancer during a lifetime. LFS was first reported by Frederic Li and Joseph Fraumeni in 1969, when cancer with a unique spectrum was observed in four families with a proband with rhabdomyosarcoma [18]. In 1988, Li and Fraumeni defined the classic LFS diagnostic criteria based on the analysis of 24 families, in which individuals inherited early-onset cancers through autosomal dominant inheritance (Table 1) [19]. In addition, in 1990, Malkin et al. showed that the causal gene of LFS is the TP53 pathogenic variant [20].
Epidemiology
In the general public, the ratio of individuals with germline TP53 pathogenic variant (individuals with TP53 pathogenic variant) is said to be 1 in 5000 or 1 in 20,000 [21, 22]. Recently, about 64,000 people were randomly extracted from three databases of the US National Cancer Institute, and 131 were found to be individuals with TP53 pathogenic variant (0.2%, about 1 in 500) [23]. In Japan, 0.27% people in the database of Tohoku Medical Megabank Organization (2KJPN) were found to be individuals with TP53 pathogenic variant [24].
The incidence of cancer patients with the TP53 pathogenic variant has been reported by two large-scale studies on pediatric cancers [25, 26], and four clinical sequence studies on cancer [27,28,29,30]; both of which indicated that the incidence is about 2%. In adult patients with cancer, four of 1566 (0.2%) were found to be individuals with TP53 pathogenic variants in the clinical sequence performed by the US Memorial Sloan Kettering Cancer Center [31].
Diagnosis
Genetic diagnosis and classic LFS
The causal gene of LFS is TP53, and LFS is diagnosed by detecting TP53 pathogenic variants [20]. However, some people meet the classic LFS diagnostic criteria without the pathogenic TP53 variant, and such individuals are diagnosed with LFS [19].
Chompret criteria
The Chompret criteria are used to perform TP53 genetic testing for suspected LFS (Table 1). As TP53 was reported to be the causal gene of LFS, it was found that many patients with cancer had TP53 pathogenic variant despite not satisfying the classic LFS diagnostic criteria; thus, the Chompret criteria was proposed to avoid any cases of LFS. The Chompret criteria consist of items on family history, multiple cancers, rare cancers, and juvenile breast cancer based on the clinical data of LFS; since it was first proposed in 2001 [32], it has been revised in 2009 [33] and 2015 [34]. The Chompret criteria will be revised as needed as the clinical presentation of LFS is further elucidated.
Explanation
In terms of “rare cancer” as an item of the Chompret criteria, the ratio of individuals with TP53 pathogenic variant is high among patients with adrenocortical cancer and choroid plexus carcinoma, and its reproducibility in embryonal anaplastic rhabdomyosarcoma has not been reported. The ratio of individuals with TP53 pathogenic variant among patients with adrenocortical cancer is 50–100% for those aged < 18 years (median of 75%) and 4–33% for those aged ≥ 18 years (median of 13%) [21, 25, 26, 35,36,37,38,39,40,41]; thus, in adrenocortical cancer, LFS is suspected, regardless of the patient’s age. The ratio of individuals with TP53 pathogenic variant among patients with choroid plexus carcinoma was 25–100% (median of 45%), in which all patients aged < 18 years [21, 25, 42,43,44,45]. Furthermore, 11 of 15 embryonal anaplastic rhabdomyosarcoma cases (73%) were pathogenic TP53 variant carriers [46]; however, as there is only one report, further investigation is needed.
In terms of the Chompret criteria item “juvenile breast cancer,” the ratio of individuals with TP53 pathogenic variant in patients with normal breast cancer patients, breast cancer patients with premenopausal onset, and breast cancer patients with a family history of breast cancer was 0–1.0% [47,48,49,50,51,52], 0–3.8% [50, 53,54,55,56,57], and 1.0–2.9% [58, 59], respectively. In contrast, the ratios of individuals with TP53 pathogenic variant among patients with breast cancer aged < 31, 31–40, 41–50, and ≥ 51 years were 0–3.8%, 0–2.6%, 0–0.8%, and 0–0.2%, respectively. The ratio of individuals with TP53 pathogenic variant is higher among breast cancer patients with premenopausal onset or family history of breast cancer than in those with breast cancer; however, there is no clear basis for considering 31 years as a cut-off.
Secondary findings
As genomic medicine has become more widespread, TP53 pathogenic variants are occasionally detected as a secondary finding of genetic panel testing. When TP53 pathogenic variants are detected in an analysis of somatic line, many such pathogenic variants are acquired changes and do not exist in the germline, but some might; therefore, germline testing is necessary. Upon carefully determining the likelihood of finding the same pathogenic variants from somatic line analysis in the germline based on medical and family history, germline analysis should be performed as needed.
Please refer to “Recommendations for communication process in genomic medicine—part 1: cancer genetic panel test (2nd edition)” (Japan Agency for Medical Research and Development (AMED) Genomic Drug Promotion Research Project, research supervisor: Shinji Kosugi, https://www.amed.go.jp/content/000056785.pdf) for the interpretation of secondary findings from cancer genetic panel tests.
Clinical presentation
Cancer penetrance and LFS core tumors
Individuals with TP53 pathogenic variant have a high probability of developing cancer during their lifetime (penetrance) (75% for men and almost 100% for women) [34, 60]. Juvenile onset of cancer and multiple onsets and types of cancers in one patient are characteristics of LFS. Breast cancer, osteosarcoma, soft tissue sarcoma, brain tumor, and adrenocortical cancer are LFS core cancers, which have high incidence in patients with LFS. As mentioned earlier, breast cancer often has premenopausal onset, rhabdomyosarcoma is a common soft tissue sarcoma in children, and leiomyosarcoma is common in adults. Common brain tumors include choroid plexus carcinoma with early onset in infancy to childhood, medulloblastoma, and glioma. LFS is known to lead to the onset of various types of cancers including epithelial carcinoma, such as gastric cancer and colorectal cancer; hematological malignancies, such as leukemia and lymphoma; and pediatric cancers, such as neuroblastoma.
As the Chompret criteria state, patients with adrenocortical cancer, choroid plexus carcinoma, and anaplastic rhabdomyosarcoma have TP53 pathogenic variants at high probability. In recent years, it has been shown that sonic hedgehog (SHH) medulloblastoma and hypodiploid acute lymphocytic leukemia are most likely associated with LFS.
Explanations
Cancer penetrance: The cancer penetrance of individuals with TP53 pathogenic variant was 73.8% over an average follow-up period of 28 years [60]. Compared to the general population, SIR was 41.1 (95% CI: 29.9–55.0) [61] and RR was 4.0 (95% CI: 3.3–4.8) [62]; thus, carcinogenesis risk is significantly higher for individuals with TP53 pathogenic variant. Within the same family, the carcinogenesis risk of pathogenic TP53 variant carriers is higher; where RR was 43.8 (95% CI: 18.5–103.5) and 18.5 (95% CI: 8.3–41.3) [63] for women and men, respectively. Multivariate analysis indicated that the OR was 1,075 (95% CI: 358–3299) and 151 (95% CI: 60–380) [64], respectively.
Table 2 shows the comparison of LFS core cancer penetrance for individuals with TP53 pathogenic variant and the general population. When osteosarcomas are comprehensively analyzed, compared to general population, the OR was 1.69 (95% CI: 1.01–2.80) [65], while the HR was 15.7 (P < 0.0001) within the family [66]; thus, the penetrance of osteosarcoma is significantly higher for individuals with TP53 pathogenic variant. Furthermore, in southeastern Brazil, there is a high incidence of LFS with TP53 p.R337H, and the risk of adrenocortical cancer in this area is RR of 2047 (95% CI: 455–9212) compared to the general population [67]. The precision of adrenocortical cancer onset prediction is reported to have a sensitivity of 84.6% (95% CI: 54.6–98.1) and specificity of 99.7% (95% CI: 99.7–99.8%).
As for tumors other than LFS core cancers, individuals with TP53 pathogenic variant develop various types of cancers, such as gastrointestinal cancer, urogenital cancer, hematological malignancy, lung cancer, and malignant melanoma. But evidence suggesting that penetrance of these cancers is higher than those of the general public is limited [34, 60,61,62, 68].
Explanation
Age of cancer onset: An analysis of 415 individuals with TP53 pathogenic variant in 214 French families with LFS showed that the cancer penetrance for young people aged 0, 5, and 18 years were 4%, 22%, and 41%, respectively [34]. Furthermore, an analysis by the US National Cancer Institute of 286 pathogenic TP53 variants in 107 families with LFS showed that the 50% cumulative cancer onset age was 46 years for men and 31 years for women. An analysis of 145 individuals with TP53 pathogenic variant from 10 families by the University of Texas MD Anderson Cancer Center (MDACC) showed that when subjects were divided into young and old to maximize the relative risk for probands/parents/grandparents, the carcinogenesis risk of young individuals with TP53 pathogenic variant was 133- and 165-fold higher than that of the general public for men and women, respectively (older pathogenic TP53 variant carriers had 15- and 26-fold higher risks for men and women, respectively) [66].
Pathogenic TP53 variant genotype and phenotype and factors that influence clinical presentation
More than 250 types of TP53 pathogenic variants (pathogenic/likely pathogenic variant) have been reported; however, as many of TP53 variants are missense variants, the interpretation of pathological significance can be challenging and must be carefully interpreted by experts. In addition, to avoid different interpretations of pathological significance between health care providers and facilities, an inspection agency that performs interpretation of pathological significance based on the standardization, such as the US ACMG, or consultation by experts familiar with interpretation of pathogenic TP53 variants should be utilized.
About 70% of pathogenic TP53 variants are missense variants [35, 69], and abnormalities in splicing, intragenic deletions, frameshift variants, nonsense variants, in-frame insertions/deletions, and intron deletion have been reported [35]. There are six hotspots for pathogenic TP53 variants (p.R175H, p.G245S, p.R248Q, p.R248W, p.R273H, and p.R282W), and about 20% of LFS families have some hotspots for pathogenic germline variants [35]. The types of germline pathogenic variants are similar to pathogenic variants of somatic line for cancer that occurs in non-carriers of pathogenic TP53 variants. However, even for an abnormality in the same location, the resulting cancers are likely to vary widely [70]. Individuals with missense variants in the DNA binding site for TP53 tend to develop cancers with relatively high-grade malignancy, whereas carriers of missense variants in the non-DNA binding site tend to develop cancers with relatively low-grade malignancy [34, 71]. At present, the clinical presentation cannot be predicted from the genotype. Meanwhile, factors that modify functions of TP53 might influence the type of cancer and the age of onset.
Explanation
There are significant differences in the frequency at which individuals with missense variants in the DNA binding site of TP53 DNA develop rhabdomyosarcoma and osteosarcoma [65]. Moreover, the frequency of leiomyosarcoma is high when there are changes in tetramerization domains, such as TP53 codon 337 or 344 [65]; however, reproducibility has not been confirmed. In contrast, carriers of TP53 p.R337H, present at higher frequency in the southeastern part of Brazil have a high incidence of adrenocortical cancer. As for the age of cancer onset, individuals with missense variants, especially individuals with missense variants in the DNA binding site, tend to exhibit earlier onset of cancer; however, the reproducibility of such findings has not been demonstrated [34].
There are several reports on the involvement of a single-nucleotide polymorphism (SNP: rs2279744, SNP309) of MDM2, which regulates the degradation of p53, with juvenile onset of soft tissue sarcoma and breast cancer as well as several other cancers [72]. Indeed, in individuals with TP53 pathogenic variant, the age of cancer onset is significantly younger when the TP53 codon 72 has the Arg allele compared to individuals in which the same site exhibits a homozygous Pro allele. In cases exhibiting MDM2 SNP309 G allele, and TP53 codon 72 Arg allele in particular, the age of onset is clearly younger than when MDM2 SNP309 T and TP53 codon 72 Pro allele are homozygous [73]. In addition, it has been reported that polymorphism of TP53 intron 3 (PIN3) invokes juvenile onset of cancer [74, 75]. Patients with cancer have short telomere lengths, and the speed of telomere shortening is fast for individuals with TP53 pathogenic variant [76], and hypermethylation of miR-34A lowers the survival rate [77,78,79]. However, these studies have not been reproduced. Factors that modify functions of TP53 are related to the clinical presentation of LFS; therefore, basic medical research, such as multigenic analysis, is necessary.
Prevention and treatment
Clinical trials of chemoprophylaxis, such as metformin, are in progress as cancer preventive agents for pathogenic TP53 variant carriers, yet no drug has been confirmed to be effective. Therefore, in addition to usual cancer preventive measures, such as avoiding alcohol, smoking, and exposure to ultraviolet rays, radiation, and carcinogenic substances, the main measures are risk-reducing surgical treatment of LFS target organs, such as the breasts, and early detection and treatment through cancer surveillance. While clinical trials of molecular targeted drugs targeting the TP53 and TP53 pathway are in progress, their efficacy has not yet been proven. Although it has been reported that TP53 pathogenic variants are factors of poor prognosis for B-cell acute lymphoblastic leukemia [80], evidence of resistance of LFS-associated cancers to treatment are limited; thus, standard treatment of each cancer is prioritized. To avoid the onset of secondary cancer, it is recommended to avoid radiation exposure, irradiation, and alkylating agents as much as possible. However, such treatment methods will be used in cases where it is considered unavoidable, such as rhabdomyosarcoma [6]. One characteristic of LFS is multiple cancers, and surgical treatment during an early stage of the disease will avoid sequela of multiple treatments. From this perspective, early detection through surveillance is important.
Cancer surveillance
1.6.1 Cancer surveillance and protocol
Cancer surveillance for individuals with TP53 pathogenic variant began with the “Toronto Protocol” implemented in Canada and the USA [81, 82]. As LFS-associated cancers are diverse and affect whole body, whole-body (WB) MRI, brain MRI, breast MRI (adult women only), ultrasonography (US), and endoscopy (adult only) are performed on a tight schedule. The results of the Toronto Protocol were reported in 2011 and 2016, and indicated that surveillance improves the survival rate of individuals with TP53 pathogenic variant. Subsequently, various countries initiated cancer surveillance programs, and at present, six countries are using 12 cancer surveillance programs [83]. The US National Comprehensive Cancer Network (NCCN) guideline also recommends similar cancer surveillance (NCCN Clinical Practice Guidelines in Oncology, Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Ver 1. 2020–December 4, 2019 can be downloaded from the NCCN website). As LFS has high cancer penetrance and often presents as multiple cancers, the detection rate of such surveillance is high, and increases in proportion to the follow-up period.
In 2016, a subcommittee of AACR, Childhood Cancer Predisposition Workshop, gathered experts of various fields from around the world, and formulated the standard for appropriate surveillance and care of hereditary tumors with childhood onset, including LFS. The results were published in 17 papers on Clinical Cancer Research in 2017 [6]. The recommended cancer surveillance method used the Toronto Protocol as the basic framework, and this protocol can be used a template for future surveillance protocols (Table 3).
Effectiveness and disadvantages of surveillance
The absolute indexes that show the efficacy of the surveillance are “decrease in the mortality rate of cancer” and “decrease in the incidence of advanced cancer,” while the relative index is shown as “sensitivity and specificity”. However, efficacy has not been verified, and the disadvantages include false positives, overdiagnosis, adverse events associated with the sedation used for the imaging, and psychological impact.
Explanation
In terms of efficacy of surveillance, the Toronto Protocol with the longest follow-up period divided individuals with TP53 pathogenic variant into the surveillance group (n = 40) and the non-surveillance group (n = 49) [82]. According to the report, transition from the non-surveillance group to the surveillance group was permitted, but according to the intention to treat analysis, the detection rate of cancer was notably lower in the surveillance group (17.5% vs 87.8%, relative risk of 0.30, 95% CI 0.17–0.56). Therefore, the relative risk of cancer-related death in the surveillance group was 0.11 (95% CI 0.03–0.45); thus, further follow-up survey might be necessary to prove the efficacy of the surveillance.
In the UK, hereditary breast cancer surveillance is conducted in high-risk subjects, such as individuals with BRCA1/2 or TP53 pathogenic variants [84]. This method assessed the sensitivity and specificity, as the alternative index of efficacy over a short period of time, and it was used as the national program. Moreover, it is becoming established as a surveillance method that can be used to suppress disadvantages as much as possible by limiting target organs, target ages, and sex (women only).
Explanation
The disadvantages of surveillance are shown below.
False positive: Calculation of accurate numbers was difficult, but as an alternative index, the positive rate was about 20–30% for WB MRI (Table 4) [82, 85,86,87,88,89]. In the meta-analysis of WB MRI discussed below, 78.7% of positive subjects had no abnormalities or had benign tumors [83]. Four studies that used MRI, US, and multiple other tests for surveillance were summarized [81, 82, 85,86,87], and when brain MRI, breast MRI, and abdominal ultrasound were added to 59.4% of WB MRIs overall, about 90% of cancers could be detected. However, it should be noted that the number of false positives also increases in proportion to the number of test items.
Overdiagnosis: Analysis of the above four studies revealed that 40.6% of the detected tumors were low-grade lesions, and a high percentage was noted in children. For pediatric cases, even if the tumor is low-grade, treatment may follow the protocol used for high-grade tumors. This cannot be simply referred to as overdiagnosis. Thyroid cancer and non-invasive breast cancer have been shown to be detected in adult cases. It is predicted that surveillance will increase the rate of overdiagnosis.
Adverse events associated with sedation used for imaging: Adverse respiratory and cardiovascular events are predicted, but no previous study has targeted individuals with TP53 pathogenic variant.
Psychological impact: described in “Psychological issues” section.
Precision of tests used for surveillance
LFS has high cancer penetrance and often leads to the development of multiple cancers. Therefore, the cancer detection rate is high with various tests, in particular, during the baseline screening. Though the detection rate decreases in the subsequent screenings, a certain level of cancer detection rate is likely maintained. Although the false positive rate is also high, it could be reduced by comparing findings to previous images, as the radiologists become more familiar with imaging and as the number of screenings increases.
Explanation
In a Dutch multi-facility study that conducted breast cancer surveillance in people with high risk of breast cancer, such as individuals with TP53 and BRCA1/2 pathogenic variant (MRISC, Dutch MRI Screening) [90], the breast cancer detection rate was high at 9.6% and 12.1% at the baseline and second MRI, respectively, and it remained high in the subsequent screenings: 6.7%, 3.4%, and 4.1%. In contrast, the positive rate (rate of subjects requiring further tests), the alternative index of false positive rate, was around 12% from the baseline to the third screenings, respectively, which decreased to 9.3% and 6.9% in the fourth and fifth screenings. Similar trends were found in cancer surveillance on LFS; thus, while maintaining a certain level of cancer detection, false positives, a disadvantage of surveillance, are expected to decrease as the number of screenings increases. We discussed the precision of each screening method below.
WB MRI: WB MRI is performed with shortened imaging time. Although the image resolution is low, the cancer detection rate is high compared to other testing methods. When limited to the first WB MRI, the cancer detection rate was 3.6–13.6% [82, 85,86,87,88,89]. In contrast, WB MRI detects many benign lesions. In a British SIGNFY study, healthy individuals were used as a control group to individuals with TP53 pathogenic variant, but in the baseline WB MRI, benign lesion was discovered in seven out of 44 healthy subjects (15.9%) [89]. According to the SIGNIFY study and US MDACC study [85], while it was difficult to determine the specificity of WB MRI, the sensitivity was 70%.
A meta-analysis reported the results of baseline WB MRI from 12 studies [83]. Among the 572 individuals with TP53 pathogenic variant, 173 were determined to require further tests (positive rate of 29.9%). A total of 35 localized new cancers were detected in 34 subjects (cancer detection rate of 5.9% and positive predictive value of 19.7%). All subjects were given curative treatments. The breakdown of false positive cases were as follows: 119 subjects with no abnormalities (68.8%), 19 with benign tumors (9.8%), and seven with recurrence or metastasis of existing cancer (4.0%). There are few records of false negative cases, and the sensitivity and specificity were not calculated. Although the positive rate was high, as discussed above, it might be decreased by comparing findings with previous images, as radiologists become more familiar with the technique, and as the duration of surveillance accumulates [90].
Brain MRI: Brain tumor detection cannot be substituted by WB MRI. Among three studies in which brain MRI was used [82, 85, 86], one was the Toronto Protocol, in which the cumulative cancer detection rate based on brain MRI was 13.6% (median follow-up period, 32 months; range, 12–87 months). If we limit to the baseline brain MRI, the positive rate was 4.3% (5/116) and 22.9% (10/35), and the cancer detection rate was 1.7% (2/116) and 8.6% (3/35). The report by MDACC calculated a sensitivity of 60% and specificity of 80%.
Breast MRI: There are three reports, and the Toronto Protocol did not detect breast cancer (median follow-up period, 32 months; range, 12–87 months) [82]. According to the report by the US National Cancer Institute, the breast cancer detection rate on baseline MRI was 9.1% (2/22) [86]. In UK, breast cancer surveillance in those with high risk of breast cancer was conducted (MARIBS study), wherein the breast cancer detection rate on baseline MRI was 5.6%, and the cumulative cancer detection rate was 11.1% (follow-up period, 52–120 months) [84]. Although the positive rate could not be identified, benign lesions were detected. No previous reports have detailed the sensitivity and specificity; therefore, further investigation is necessary.
Abdominal US: In southeastern Brazil where the number of individuals with TP53 pathogenic variant is high, TP53 genetic testing was performed in about 180,000 newborns and close relatives. Individuals with TP53 pathogenic variant were divided into the surveillance (n = 346) and non-surveillance groups (n = 391), wherein the former received routine abdominal US [67]. There were seven and eight subjects with adrenocortical cancer in the surveillance group and the non-surveillance group, respectively; the relative risk was 0.989 (95% CI 0.362–2.699), and showed no significant difference. As a result of the preemptive effect of the surveillance, all adrenocortical cancer cases detected in the surveillance group were stage I and could be treated with surgical resection only. In contrast, in the non-surveillance group, advanced cancer cases were common, which in addition to surgical treatment, required chemotherapy, and mortality was observed one. In the surveillance group, tumors, such as neuroblastoma, were detected; therefore, long-term follow-up may be able to reduce the incidence and mortality of advanced abdominal cancer.
PET/CT Radiation on individuals with TP53 pathogenic variant is not recommended because it could cause secondary cancer; however, there are two case reports [91, 92], in which, the positive rates were 20.0% (6/30) and 33.3% (5/15), and the cancer detection rates were 10% (3/30) and 20% (3/15).
Psychological issues
Patients and their family members experience various emotions and thoughts as a result of discussions regarding suspected LFS, TP53 genetic testing, and treatment and surveillance for prevention after being diagnosed with LFS. Some might hope for a negative genetic test result, or some might wish for no negative findings in surveillance. Patients may be anxious about bad results, although some may have positive attitudes. Moreover, patients may want to know the situation, whether good or bad, to work on prevention, or want to be useful for their family members. Despair and fear, the psychological burden of talking to their family, and many other emotions are often described. Such psychological situations vary depending on whether the person has cancer or not, type of cancer they have or family has, the treatment conditions, the duration since diagnosis, age, sex, personality, or if the person being tested is themselves or their child. Furthermore, factors that modify such thoughts and emotions include medical conditions, cost, concern of the impact on life insurance, financial situation, school and work, marriage and child-rearing, and living with family.
It is difficult for others to understand the complex thoughts and emotions of each person, but it is significant in terms of psychological support for health care providers to offer opportunities to listen and show empathy and understanding. It is important to understand that it is natural for people facing serious health situations to feel anxiety, concerns, grief, anger, despair, and anguish, and to confirm these emotions without denying them. While most people are able to face the facts and deal with their emotions over time, it takes time to psychologically adapt to situations. Thus, rather than trying to quickly ease or resolve anxieties, health care providers should try to understand the patient’s psychological adaptation, listen to them without denying their feelings, and calmly be there for them. In addition, healthcare providers should accurately explain the latest information about LFS to subjects and family as many times as it takes, and carefully answer patients’ questions, as this will be a major support, and help them to calmly face the situation.
LFS treatment guidelines
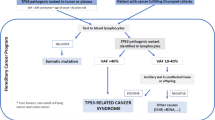
(CQ1) Are the Chompret criteria useful as criteria to perform TP53 genetic testing for suspected LFS?
Recommendations
The Chompret criteria allow the detection of individuals with TP53 pathogenic variant; however, as there are some patients with LFS who do not meet this criteria, it is not necessary to strictly adhere to this criteria when LFS is suspected (consensus rate* of 96%, 22/23).
* Consensus rates were determined by the votes of the 23 members of the study group, as described in the “Recommendation” paragraph of “Preparation method of the present treatment guidelines”.
Explanation
There is one report on the latest Chompret criteria (2015 version) [93], where the sensitivity was 75% (probability of not missing individuals with TP53 pathogenic variant), and the specificity was not very high at 64.5% (probability to accurately diagnose subjects who are not carriers of TP53 pathogenic variant). However, as it is important not to miss any individual with TP53 pathogenic variant when diagnosing LFS, the Chompret criteria are useful for identifying individuals with TP53 pathogenic variant. However, there is a certain number of individuals with TP53 pathogenic variant who do not meet the Chompret criteria (false negative rate of 25%). For pediatric patients with cancer, family members are also often young and therefore might not still have developed cancer, which makes it difficult to obtain an accurate family history. It has been reported that 25% of cases are de novo LFS; therefore, when LFS is suspected from a history of juvenile onset of cancer, family history, or multiple cancer, TP53 genetic testing is performed. While the classic LFS has high specificity (91.0–98.1%), its sensitivity is low (about 25.0–40.0%) [21, 62, 93].
Although TP53 genetic testing is not covered by health insurance in Japan, it can be performed by laboratories at the patient’s own expense.
(CQ2) Timing for implementing TP53 genetic testing
Recommendations
TP53 genetic testing is performed immediately when LFS is suspected. However, there are various ethical, legal, and social issues (ELSI) surrounding patients with LFS, and the test should only be performed after the subject and family have carefully considered the situation (consensus rate of 96%, 22/23).
Explanation
AACR recommends that TP53 genetic testing should be performed promptly when LFS is suspected [6], but when performing the test, it must be explained to subjects during genetic counseling that a diagnosis of LFS will have an effect on families and relatives, genetic testing and subsequent cancer surveillance are not covered by health insurance in Japan, and there are various ELSI (Table 5). In addition, it should be explained that missense variants are common in TP53 germline variants, which could make the interpretation of pathological significance difficult.
It has been indicated that treating LFS with radiation therapy and alkylating agents can damage DNA and increase the risk of secondary cancer onset. Therefore, when LFS is suspected in patients with cancer, it is highly significant to determine whether the subject has the germline TP53 pathogenic variant [6].
LFS has a peak of onset for adrenocortical cancer and choroid plexus carcinoma in infancy [35, 94]. Specifically, the latter, which is a brain tumor, can cause severe sequalae if diagnosis is delayed. Therefore, when individuals with TP53 pathogenic variant or their spouse is delivering a baby, performing the genetic test immediately after the delivery and knowing whether the child has LFS or not will lead to countermeasures for cancers that develop during infancy.
(CQ3) Should radiation exposure and irradiation of individuals with TP53 pathogenic variant be avoided?
Recommendations
Epidemiological reports on increased incidence of secondary cancers in individuals with TP53 pathogenic variant due to radiation exposure and irradiation are limited. However, theoretically, it is possible that cancer can develop; and thus, radiation should be avoided if there are other options (consensus rate of 91%, 21/23).
Explanation
TP53 is induced in response to cellular stress that causes damage to DNA. TP53 is called “the guardian of the genome,” and is at the center of the pathway that guides DNA repair, growth arrest, senescence, and apoptosis [95]. Therefore, theoretically, cells with weakened or disappeared TP53 function have aberrant pathways, which may induce carcinogenesis. While reports describing secondary cancer being induced by radiation and irradiation in pathogenic TP53 variant carriers are limited, it is recommended that radiation exposure from imaging tests, such as CT and PET-CT, and for treatment should be avoided as much as possible [6]. However, as there may be no other option for routine treatment, irradiation is allowed if the risk benefit balance indicates its utility.
Among individuals with TP53 pathogenic variant who developed choroid plexus carcinoma, three out of 11 who received radiation therapy developed a different cancer, while only one out of 17 who did not receive radiation therapy developed cancer [96]. Cancers that developed following radiation therapy presented as hematological malignancies, and solid tumors in locations that were not irradiated. At the US St. Jude Children’s Hospital, 3006 patients who underwent whole-genome sequencing 5 years after the primary cancer were analyzed [97]. Patients with hereditary tumor with germline pathogenic variants of at least one of 60 cancer susceptibility genes, including TP53, had a significantly higher risk of sarcoma onset due to radiation therapy than non-carriers (breast cancer RR 13.9; 95% CI 6.0–32.2, sarcoma RR 10.6; 95% CI 4.3–26.3). However, in this report, 175 patients were diagnosed with hereditary tumors but only 10 were individuals with TP53 pathogenic variant. The impact of radiation therapy on individuals with TP53 pathogenic variant was not analyzed by category.
(CQ4) Should cancer surveillance be conducted on individuals with TP53 pathogenic variant?
Recommendations
Cancer surveillance is recommended. However, the efficacy of cancer surveillance is still being studied, and it should be presented to subjects as one of the options (consensus rate of 96%, 22/23).
Explanation
Most cancer surveillances in progress overseas have short follow-up periods, and LFS is a rare disease with small number of subjects. Thus, the efficacy of cancer surveillance (reduced mortality and incidence of advanced cancer) has not yet been verified [67, 82, 83, 85,86,87,88,89]. However, most cancers detected by cancer surveillance are localized, and curative therapy is performed [83]. With a reduction in the therapeutic intensity, complications associated with therapy will be reduced and can lead to improved OQL. WB MRI used for cancer surveillance can only be performed a limited number of facilities in Japan, the false positive rate is high, and overdiagnosis is common. Moreover, in infancy, there are many disadvantages such as adverse events [98,99,100,101,102,103,104,105,106,107,108,109,110,111,112] associated with sedation (see “Effectiveness and disadvantage of surveillance” and “Precision of tests used for surveillance” sections). Not all psychological impacts of cancer surveillance on subjects are positive (see “Psychological issues”) [82, 88, 113,114,115,116]. Thus, it is recommended cancer surveillance should be suggested as an option and implemented in accordance with the wishes of the patient and their family. At this time, surveillance should be implemented on a clinical trial basis as much as possible, and nationwide data should be collected to promote the establishment of a standard surveillance program.
(CQ5) Who are subjects of cancer surveillance?
Recommendations
Individuals with germline TP53 pathogenic variant and those who meet the classic LFS diagnostic criteria (consensus rate of 100%, 23/23).
Explanation
In the Toronto Protocol, regardless of the onset, all individuals with TP53 pathogenic variant are considered subjects [81, 82]; however, according to the recommended protocol of AACR (the AACR protocol), subjects are individuals with TP53 pathogenic variant without cancer onset, and those who meet the classic LFS diagnostic criteria even if they do not have TP53 pathogenic varients [6]. Families of the latter group also become subjects after taking the TP53 genetic test. Many protocols target those who are in remission for a certain period of time following a cancer treatment, in addition to those without cancer onset [85,86,87,88,89]. This is because detection of recurrence and metastasis from existing onset has an impact on the determination of the cancer surveillance effect. However, since it has been indicated that about half of patients with LFS develop cancer within 5 years [34], it is valid to conduct surveillance even during cancer treatment or immediately after such treatment. In clinical trials, these cases need to be categories for analysis.
(CQ6) When should the cancer surveillance of individuals with TP53 pathogenic variant be started?
Recommendations
Cancer surveillance is started promptly after the diagnosis of LFS. However, when performing examination of children, which requires sedation, sufficient considerations need to be given to adverse events such as respiratory and circulatory suppression. The surveillance of adrenocortical cancer begins in childhood, and the surveillance of breast cancer and gastrointestinal cancer begins in adulthood (consensus rate of 100%, 23/23).
Explanation
The AACR protocol recommends that cancer surveillance begins immediately after the diagnosis [6]. Some overseas surveillance programs begin once sedation is no longer necessary. When sedation is difficult, examinations and tests can be used in place of surveillance until sedation is no longer necessary. Adrenocortical cancer, brain tumor, and soft tissue sarcoma should be especially observed as they often have childhood onset, but it needs to be known that a surveillance based on examinations and blood has limitations. In terms of sedation, sufficient discussion should be held with the family regarding the possibility of adverse events, such as respiratory and circulatory suppression, and surveillance should be implemented while carefully managing the patient.
In the AACR protocol, since adrenocortical cancer has high penetrance in childhood, US is performed frequently in children aged < 18 years, and surveillance on breast cancer and gastrointestinal cancer begins at 18 (for women) and 25 years, respectively [6]. However, since susceptible age and frequent locations for other LFS core tumors, osteosarcoma, soft tissue sarcoma, and brain tumor (especially glioma), have not been identified, surveillance should begin promptly following the diagnosis of LFS, and should continue over the lifetime of the patient.
Discussion
Among pediatric patients with cancer, 1–2% are estimated to be individuals with TP53 pathogenic variant [25,26,27,28,29,30]. In Japan, about 2,000 children develop cancer every year, 20–40 of them are assumed to have cancer onset due to LFS. According to the present questionnaire survey, only 36 children with LFS were receiving medical care at pediatric cancer facilities in 2017 [117]. Thus, a significant number of patients with LFS might be being overlooked. Alternatively, actual the rate of cancer onset during their lifetime may be lower than assumed, although it is assumed that almost all individuals with TP53 pathogenic variant are going to develop cancer at least once in their lifetime; thus, the clinical presentation of LFS is still not elucidated. Indeed, the clinical presentation of LFS (types of cancers patients with LFS develop) is affected by molecular biology, such as genotype, but it has been suggested that race also plays a role in these differences. Clarifying the clinical presentation of LFS in Japan will lead to simplification and individualization of health management and cancer surveillance for patients with LFS. With presumption of creating a registration and follow-up system such as registry for individuals with TP53 pathogenic variant, and providing appropriate follow-up and care, information should be gathered and updated, shared with registrants, and then shared globally.
Patients with LFS are exposed to various environmental factors such as ELSI (Table 5) [118], with some people arguing that a diagnosis of LFS should be proactively avoided. However, since we are entering the era of genomic medicine, the utility of genomic medicine is clear, and diagnosis of LFS is not considered to be a disadvantage. Also, not all of the psychological impacts of LFS are negative [82, 88, 113,114,115,116]. Indeed, learning about their genetic background, might make the patient feel motivated to protect their health as well as that of their children and family. We must have a “receptacle” ready to receive these patients with LFS. In other countries, once accurate genetic testing is secured, comprehensive medical management of LFS is steadily being formulated and established. This includes the avoidance of radiation and alkylating agents, cancer prevention and treatment, such as cancer surveillance, measures that lead to early detection of cancer, information update through genetic counseling, and psychological care [6]. In light of such international trends, the option of not diagnosing LFS or not informing the patients of an LFS diagnosis is ethically problematic. Thus, a medical care system needs to be prepared promptly to handle such issues.
The systematic review implemented by our study group did not find any evidence that cancer surveillance would improve the prognosis of individuals with TP53 pathogenic variant, but the cancer detection rate of cancer surveillance is high, and detected cancers often did not have metastasis. Detecting cancers in the early stage does not equal elimination of cancer death, but detecting the cancer before it invades into surrounding tissues or metastasizes would at least allow for less treatments such, as avoiding radiation therapy, which in turn reduces complications associated with treatments, improves QOL, and leads to prevention of secondary cancer. Prognosis for LFS core sarcomas, such as rhabdomyosarcoma and osteosarcoma are clearly influenced by stage of the cancer at the time of onset, and may be improved as a result of cancer surveillance. Cancer surveillance has various disadvantages, such as false positive, overdiagnosis, and adverse events associated with sedation. However, as the performance of diagnostic imaging machines is rapidly improving with the introduction of AI technology, more accurate and swifter diagnosis will become possible. Furthermore, recent developments in next-generation sequencing technology have improved the precision of molecular biology tests, such as liquid biopsy, which leads to simpler, more accurate, and earlier detection of cancer as an auxiliary test to imaging, or as an alternative test. Given these developments, we can now work to develop a medical care system that will enable cancer surveillance as a management option for patients with LFS. However, the financial burden of genetic tests and surveillance on subjects is enormous. Clinical trials should be publicly subsidized and the results used as the foundation to create a comprehensive treatment system for patients with LFS and hereditary tumor.
Policy recommendations
In the age of genomic medicine, diagnosis of hereditary tumors such as LFS cannot be avoided. Hereditary tumor diagnosis affects not only the proband but also their close relatives. Clinical trial should be prepared so that those diagnosed with hereditary tumors can participate whenever they need, regardless of the onset of cancer, and based on the result, comprehensive medical care system should be developed. We present following policy recommendations to enable clinical trial implementation:
-
(1)
Development of a hereditary tumor registry
-
(2)
Development of genetic counselors specialized in hereditary tumors
-
(3)
Public subsidies for the cost of genetic tests and cancer surveillance for cancer susceptibility genes such as TP53
Preparation method of the present treatment guidelines
Preparation of clinical questions (CQs)
We formed a “guideline preparation committee” within our study group, consisting of physicians, genetic counselors, and ethics researchers with expertise in hereditary tumors, and created an analytical framework consisting of medical practices, such as diagnosis of LFS, prevention and treatment, and follow-up, and CQs. For the clinical presentation of LFS, we created CQs outside of the analytical framework. The draft plan for the CQs was prepared based on PICO (P: patients, problem, population, I: interventions, C: comparisons, controls, comparators, O: outcomes), and after thorough discussion involving the whole committee, the guideline preparation committee determined the CQs.
Systematic review and preparation of the evidence report
We formed a “literature review committee” consisting of physicians who were not experts on hereditary tumors, and were independent of the guideline preparation committee in our study group. These physicians examined literature using PubMed, Embase, and Ichushi-Web, and classified approximately 10,000 papers based on the CQs, conducted abstract review, and full paper review, and determined papers to be evaluated. Furthermore, following data extraction, the evidences were and summarized, and an evidence report was published.
Recommendations
At the beginning of the study, the aim of the guideline preparation committee was to evaluate advantages and disadvantages based on the evidence report and prepare recommendations for the CQs. However, during the preparation of the evidence report, it became clear that since LFS is a rare disease, there were not many reports that targeted multiple cases, and not many reports that confirmed the standardization and reproducibility of study methods. Furthermore, for many CQs, it was difficult to determine advantages and disadvantages along with the summarization of evidence. Thus, in the present treatment guidelines, we employed expert opinions as needed, while also using the evidence report as much as possible. Whether the recommendation should be consented to or not was determined by votes of the 26 members of the study group consisting of multiple occupations, such as expert physicians of hereditary tumors and genetic counselors, non-expert physicians (pediatricians, pediatric surgeons, radiologists, orthopedic surgeons), ethics researchers, and patient group representative; 23 votes were affirmative. Members of the guideline preparation committee and the literature review committee were excluded from voting. The level of recommendation was not identified since the expert opinions were adopted throughout the guideline; therefore, the consensus rate was calculated from votes and noted.
External evaluations
The present guidelines were evaluated by the following:
Public comment from The Japanese Society of Pediatric Hematology/Oncology (JSPHO).
Public comment from the Japanese Society for Hereditary Tumors.
As the present guideline adopted expert opinions throughout, it was determined that evaluation by AGREE II would be difficult.
Revisions
The guideline is to be revised every 3 years (the next revision is scheduled for 2022).
Research organizations
Guideline preparation committee
- Tadashi Kumamoto:
-
Department of Pediatric Oncology, National Cancer Center Hospital, LFS Expert Group, The Japanese Society for Hereditary Tumors
- Yukiko Tsunematsu:
-
Department of Pediatrics, Juntendo University School of Medicine, LFS Expert Group, The Japanese Society for Hereditary Tumors
- Yoshiko Nakano:
-
Division of Brain Tumor Translational Research, National Cancer Center Research Institute, Department of Pediatrics, The University of Tokyo Hospital
- Hiroyoshi Hattori:
-
Department of Clinical Genetics, National Hospital Organization Nagoya Medical Center
- Chieko Tamura:
-
Medical Information and Genetic Counseling Division, FMC Tokyo Clinic, LFS Expert Group, The Japanese Society for Hereditary Tumors
- Shimon Tashiro:
-
Department of Sociology, Graduate School of Arts and Letters, Tohoku University
Literature review committee
- Chisato Hamashima:
-
Department of Nursing, Faculty of Medical Technology, Teikyo University
- Teruhiko Terasawa:
-
Department of Emergency and General Internal Medicine, Fujita Health University School of Medicine
- Fumito Yamazaki:
-
Department of Pediatrics, Keio University School of Medicine
- Takafumi Katayama:
-
Department of Statistics and Information, College of Nursing Art and Science, University of Hyogo
Guideline preparation collaborators and consensus voters (not comprehensive)
- Akira Nakagawara:
-
Saga International Heavy Ion Cancer Radiation Therapy Center
- Yasuhiko Kaneko:
-
Research Institute for Clinical Oncology, Saitama Cancer Center
- Shigenobu Suzuki:
-
Department of Ophthalmic Oncology, National Cancer Center Hospital
- Akira Kawai:
-
Department of Musculoskeletal Oncology and Rehabilitation Medicine, National Cancer Center Hospital
- Tatsuro Tajiri:
-
Department of Pediatric Surgery, Kyoto Prefectural University of Medicine
- Atsushi Manabe:
-
Department of Pediatrics, Hokkaido University Graduate School of Medicine
- Masatoshi Takagi:
-
Department of Pediatrics and Developmental Biology, Tokyo Medical and Dental University Graduate school of Medical and Dental Science
- Mikiko Miyasaka:
-
Department of Radiology, National Center for Child Health and Development
- Taiki Nozaki:
-
Department of Radiology, St.Luke's International Hospital
- Junko Takita:
-
Department of Pediatrics, Graduate School of Medicine, Kyoto University
- Michinori Funato:
-
Department of Pediatrics, National Hospital Organization, Nagara Medical Center
- Michiya Ito:
-
Division of Health Administration and Policy, Tohoku Medical and Pharmaceutical University
- Naoko Kakee:
-
Division of Bioethics/Division of Information for Specific Pediatric Chronic Diseases, National Center for Child Health and Development
- Tomoro Hishiki:
-
Department of Pediatric Surgery, Chiba University Graduate School of Medicine
- Shinsuke Hirabayashi:
-
Department of Pediatrics, Hokkaido University Graduate School of Medicine
- Tomoaki Mori:
-
Department of Musculoskeletal Oncology and Rehabilitation, National Cancer Center Hospital
- Akihiro Sakurai:
-
Department of Medical Genetics and Genomics, Sapporo Medical University School of Medicine
- Katsunori Fujii:
-
Department of Pediatrics, Chiba University Graduate School of Medicine
- Hiroshi Yagata (deceased):
-
Department of Breast Care, Saitama Medical Center
- Akira Shimada:
-
Department of Pediatric Hematology/Oncology, Okayama University Hospital
- Eizou Hiyama:
-
Department of Pediatric Surgery, Hiroshima University Hospital
- Hideki Muramatsu:
-
Department of Pediatrics, Nagoya University Graduate School of Medicine
- Masahiro Yao:
-
Department of Urology, Yokohama City University Graduate School of Medicine
- Masayoshi Tsutsumi:
-
Japan Registered Clinical Laboratories Association
- Naonori Kawakubo:
-
Department of pediatric surgery, faculty of medical sciences, Kyushu University
- Teruhiko Yoshida:
-
Department of Genetic Medicine and Service, National Cancer Center Hospital
- Takeshi Nakajima:
-
Department of Clinical Genetics, Cancer Institute Hospital
- Yoshiko Nakayama:
-
Department of Pediatrics, Shinshu University School of Medicine
- Akiko Higuchi:
-
Children’s Cancer Association of Japan
- Hiroko Kondo:
-
Children’s Cancer Association of Japan
- Asako Katayama:
-
Children’s Cancer Association of Japan
Change history
03 December 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10147-021-02086-5
Abbreviations
- AACR:
-
American Association of Cancer Research
- AYA:
-
Adolescent and young adult
- ELSI:
-
Ethical, legal, and social issues
- GI:
-
Gastro-intestinal
- HR:
-
Hezard ratio
- LFS:
-
Li–Fraumeni syndrome
- NCCN:
-
National Comprehensive Cancer Network
- NCI:
-
National Cancer Institute
- NGS:
-
Next-generation sequencing
- OR:
-
Odds ratio
- QOL:
-
Quality of life
- RR:
-
Relative risk
- SHH:
-
Sonic hedge hog
- SIR:
-
Standardized incidence ratio
- US:
-
Ultrasonography
- WB MRI:
-
Whole-body MRI
References
Brodeur GM, Nichols KE, Plon SE et al (2017) Pediatric cancer predisposition and surveillance: an overview, and a tribute to Alfred G. Knudson Jr. Clin Cancer Res 23:e1–e5
Greer M-LC, Voss SD, States LJ (2017) Pediatric cancer predisposition imaging: focus on whole-body MRI. Clin Cancer Res 23:e6–e13
Porter CC, Druley TE, Erez A et al (2017) Recommendations for surveillance for children with leukemia-predisposing conditions. Clin Cancer Res 23:e14–e22
Walsh MF, Chang VY, Kohlmann WK et al (2017) Recommendations for childhood cancer screening and surveillance in DNA repair disorders. Clin Cancer Res 23:e23–e31
Tabori U, Hansford JR, Achatz MI et al (2017) Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin Cancer Res 23:e32–e37
Kratz CP, Achatz MI, Brugières L et al (2017) Cancer screening recommendations for individuals with Li–Fraumeni syndrome. Clin Cancer Res 23:e38–e45
Evans DGR, Salvador H, Chang VY et al (2017) Cancer and central nervous system tumor surveillance in pediatric neurofibromatosis 1. Clin Cancer Res 23:e46–e53
Evans DGR, Salvador H, Chang VY et al (2017) Cancer and central nervous system tumor surveillance in pediatric neurofibromatosis 2 and related disorders. Clin Cancer Res 23:e54–e61
Foulkes WD, Kamihara J, Evans DGR et al (2017) Cancer surveillance in Gorlin syndrome and rhabdoid tumor predisposition syndrome. Clin Cancer Res 23:e62–e67
Rednam SP, Erez A, Druker H et al (2017) Von Hippel-Lindau and hereditary pheochromocytoma/paraganglioma syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res 23:e68–e75
Schultz KAP, Rednam SP, Kamihara J et al (2017) PTEN, DICER1, FH, and their associated tumor susceptibility syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res 23:e76–e82
Villani A, Greer MC, Kalish JM et al (2017) Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clin Cancer Res 23:e83–e90
Druker H, Zelley K, McGee RB et al (2017) Genetic counselor recommendations for cancer predisposition evaluation and surveillance in the pediatric oncology patient. Clin Cancer Res 23:e91–e97
Kamihara J, Bourdeaut F, Foulkes WD et al (2017) Retinoblastoma and neuroblastoma predisposition and surveillance. Clin Cancer Res 23:e98–e106
Achatz MI, Porter CC, Brugieres L et al (2017) Cancer screening recommendations and clinical management of inherited gastrointestinal cancer syndromes in childhood. Clin Cancer Res 23:e107–e114
Kalish JM, Doros L, Helman LJ et al (2017) Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin Cancer Res 23:e115–e122
Wasserman JD, Tomlinson GE, Druker H et al (2017) Multiple endocrine neoplasia and hyperparathyroid-jaw tumor syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res 23:e123–e132
Li FP, Fraumeni JF Jr (1969) Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst 43:1365–1373
Li FP, Fraumeni JF Jr, Mulvihill JJ et al (1988) A cancer family syndrome in twenty-four kindreds. Cancer Res 48:5358–5362
Malkin D, Li FP, Strong LC et al (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233–1238
Gonzalez KD, Noltner KA, Buzin CH et al (2009) Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 27:1250–1256
Lalloo F, Varley J, Ellis D et al (2003) Prediction of pathogenic mutations in patients with early-onset breast cancer by family history. Lancet 361:1101–1102
de Andrade KC, Mirabello L, Stewart DR et al (2017) Higher-than-expected population prevalence of potentially pathogenic germline TP53 variants in individuals unselected for cancer history. Hum Mutat 38:1723–1730
Yamaguchi-Kabata Y, Yasuda J, Tanabe O et al (2018) Evaluation of reported pathogenic variants and their frequencies in a Japanese population based on a whole-genome reference panel of 2049 individuals. J Hum Genet 63:213–230
Zhang J, Walsh MF, Wu G et al (2015) Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 373:2336–2346
Grobner SN, Worst BC, Weischenfeldt J et al (2018) The landscape of genomic alterations across childhood cancers. Nature 555:321–327
Parsons DW, Roy A, Yang Y et al (2016) Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol 2:616–624
Harris MH, DuBois SG, Glade Bender JL et al (2016) Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the individualized cancer therapy (iCat) study. JAMA Oncol 2:608–615
Worst BC, van Tilburg CM, Balasubramanian GP et al (2016) Next-generation personalised medicine for high-risk paediatric cancer patients—the INFORM pilot study. Eur J Cancer 65:91–101
Mody RJ, Wu YM, Lonigro RJ et al (2015) Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA 314:913–925
Schrader KA, Cheng DT, Joseph V et al (2016) Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol 2:104–111
Chompret A, Abel A, Stoppa-Lyonnet D et al (2001) Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet 38:43–47
Tinat J, Bougeard G, Baert-Desurmont S et al (2009) version of the Chompret criteria for Li–Fraumeni syndrome. J Clin Oncol 27:e108–109; author reply e10
Bougeard G, Renaux-Petel M, Flaman JM et al (2015) Revisiting Li–Fraumeni Syndrome from TP53 mutation carriers. J Clin Oncol 33:2345–2352
Wasserman JD, Novokmet A, Eichler-Jonsson C et al (2015) Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: a children’s oncology group study. J Clin Oncol 33:602–609
Raymond VM, Else T, Everett JN et al (2013) Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J Clin Endocrinol Metab 98:E119–E125
Sandrini F, Villani DP, Tucci S, Moreira AC, de Castro M, Elias LL (2005) Inheritance of R337H p53 gene mutation in children with sporadic adrenocortical tumor. Horm Metab Res 37:231–235
Pinto EM, Chen X, Easton J et al (2015) Genomic landscape of paediatric adrenocortical tumours. Nat Commun 6:6302
Herrmann LJ, Heinze B, Fassnacht M et al (2012) TP53 germline mutations in adult patients with adrenocortical carcinoma. J Clin Endocrinol Metab 97:E476–E485
Varley JM, McGown G, Thorncroft M et al (1999) Are there low-penetrance TP53 alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet 65:995–1006
Mastellaro MJ, Seidinger AL, Kang G et al (2017) Contribution of the TP53 R337H mutation to the cancer burden in southern Brazil: insights from the study of 55 families of children with adrenocortical tumors. Cancer 123:3150–3158
Seidinger AL, Mastellaro MJ, Paschoal Fortes F et al (2011) Association of the highly prevalent TP53 R337H mutation with pediatric choroid plexus carcinoma and osteosarcoma in southeast Brazil. Cancer 117:2228–2235
Custodio G, Taques GR, Figueiredo BC et al (2011) Increased incidence of choroid plexus carcinoma due to the germline TP53 R337H mutation in southern Brazil. PLoS One 6:e18015
Gozali AE, Britt B, Shane L et al (2012) Choroid plexus tumors; management, outcome, and association with the Li–Fraumeni syndrome: the Children’s Hospital Los Angeles (CHLA) experience, 1991–2010. Pediatr Blood Cancer 58:905–909
Tabori U, Shlien A, Baskin B et al (2010) TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol 28:1995–2001
Hettmer S, Archer NM, Somers GR et al (2014) Anaplastic rhabdomyosarcoma in TP53 germline mutation carriers. Cancer 120:1068–1075
Momozawa Y, Iwasaki Y, Parsons MT et al (2018) Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat Commun 9:4083
Yang XR, Devi BCR, Sung H et al (2017) Prevalence and spectrum of germline rare variants in BRCA1/2 and PALB2 among breast cancer cases in Sarawak, Malaysia. Breast Cancer Res Treat 165:687–697
Lolas Hamameh S, Renbaum P, Kamal L et al (2017) Genomic analysis of inherited breast cancer among Palestinian women: genetic heterogeneity and a founder mutation in TP53. Int J Cancer 141:750–756
Tung N, Lin NU, Kidd J et al (2016) Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol 34:1460–1468
Gomes MC, Kotsopoulos J, de Almeida GL et al (2012) The R337H mutation in TP53 and breast cancer in Brazil. Hered Cancer Clin Pract 10:3
Sidransky D, Tokino T, Helzlsouer K et al (1992) Inherited p53 gene mutations in breast cancer. Cancer Res 52:2984–2986
Rummel SK, Lovejoy L, Shriver CD, Ellsworth RE (2017) Contribution of germline mutations in cancer predisposition genes to tumor etiology in young women diagnosed with invasive breast cancer. Breast Cancer Res Treat 164:593–601
Giacomazzi J, Correia RL, Palmero EI et al (2014) The Brazilian founder mutation TP53 p.R337H is uncommon in Portuguese women diagnosed with breast cancer. Breast J 20:534–546
Carraro DM, Koike Folgueira MA, Garcia Lisboa BC et al (2013) Comprehensive analysis of BRCA1, BRCA2 and TP53 germline mutation and tumor characterization: a portrait of early-onset breast cancer in Brazil. PLoS One 8:e57581
Mouchawar J, Korch C, Byers T et al (2010) Population-based estimate of the contribution of TP53 mutations to subgroups of early-onset breast cancer: Australian Breast Cancer Family Study. Cancer Res 70:4795–4800
Ginsburg OM, Akbari MR, Aziz Z et al (2009) The prevalence of germ-line TP53 mutations in women diagnosed with breast cancer before age 30. Fam Cancer 8:563–567
Yang X, Wu J, Lu J et al (2015) Identification of a comprehensive spectrum of genetic factors for hereditary breast cancer in a Chinese population by next-generation sequencing. PLoS One 10:e0125571
Walsh T, Casadei S, Coats KH et al (2006) Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 295:1379–1388
Mai PL, Best AF, Peters JA et al (2016) Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 122:3673–3681
Hwang SJ, Lozano G, Amos CI, Strong LC (2003) Germline p53 mutations in a cohort with childhood sarcoma: sex differences in cancer risk. Am J Hum Genet 72:975–983
Ruijs MW, Verhoef S, Rookus MA et al (2010) TP53 germline mutation testing in 180 families suspected of Li–Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet 47:421–428
Wu CC, Krahe R, Lozano G et al (2011) Joint effects of germ-line TP53 mutation, MDM2 SNP309, and gender on cancer risk in family studies of Li–Fraumeni syndrome. Hum Genet 129:663–673
Wu CC, Shete S, Amos CI, Strong LC (2006) Joint effects of germ-line p53 mutation and sex on cancer risk in Li–Fraumeni syndrome. Cancer Res 66:8287–8292
Ognjanovic S, Olivier M, Bergemann TL, Hainaut P (2012) Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer 118:1387–1396
Brown BW, Costello TJ, Hwang SJ, Strong LC (2005) Generation or birth cohort effect on cancer risk in Li–Fraumeni syndrome. Hum Genet 118:489–498
Custodio G, Parise GA, Kiesel Filho N et al (2013) Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J Clin Oncol 31:2619–2626
Renaux-Petel M, Sesboue R, Baert-Desurmont S et al (2014) The MDM2 285G–309G haplotype is associated with an earlier age of tumour onset in patients with Li–Fraumeni syndrome. Fam Cancer 13:127–130
Olivier M, Goldgar DE, Sodha N et al (2003) Li–Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 63:6643–6650
Zhou R, Xu A, Gingold J, Strong LC, Zhao R, Lee DF (2017) Li–Fraumeni syndrome disease model: a platform to develop precision cancer therapy targeting oncogenic p53. Trends Pharmacol Sci 38:908–927
Nichols KE, Malkin D (2015) Genotype versus phenotype: the Yin and Yang of germline TP53 mutations in Li–Fraumeni Syndrome. J Clin Oncol 33:2331–2333
Bond GL, Hu W, Bond EE et al (2004) A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 119:591–602
Bougeard G, Baert-Desurmont S, Tournier I et al (2006) Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li–Fraumeni syndrome. J Med Genet 43:531–533
Sagne C, Marcel V, Bota M et al (2014) Age at cancer onset in germline TP53 mutation carriers: association with polymorphisms in predicted G-quadruplex structures. Carcinogenesis 35:807–815
Gemignani F, Moreno V, Landi S et al (2004) A TP53 polymorphism is associated with increased risk of colorectal cancer and with reduced levels of TP53 mRNA. Oncogene 23:1954–1956
Tabori U, Nanda S, Druker H, Lees J, Malkin D (2007) Younger age of cancer initiation is associated with shorter telomere length in Li–Fraumeni syndrome. Cancer Res 67:1415–1418
Id Said B, Kim H, Tran J, Novokmet A, Malkin D (2016) Super-transactivation TP53 variant in the germline of a family with Li–Fraumeni syndrome. Hum Mutat 37:889–892
Samuel N, Wilson G, Lemire M et al (2016) Genome-wide DNA methylation analysis reveals epigenetic dysregulation of microRNA-34A in TP53-associated cancer susceptibility. J Clin Oncol 34(30):3697–3704
Samuel N, Wilson G, Id Said B et al (2016) Transcriptome-wide characterization of the endogenous miR-34A-p53 tumor suppressor network. Oncotarget 7:49611–49622
Qian M, Cao X, Devidas M et al (2018) TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J Clin Oncol 36:591–599
Villani A, Tabori U, Schiffman J et al (2011) Biochemical and imaging surveillance in germline TP53 mutation carriers with Li–Fraumeni syndrome: a prospective observational study. Lancet Oncol 12:559–567
Villani A, Shore A, Wasserman JD et al (2016) Biochemical and imaging surveillance in germline TP53 mutation carriers with Li–Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 17:1295–1305
Ballinger ML, Best A, Mai PL et al (2017) Baseline surveillance in Li–Fraumeni syndrome using whole-body magnetic resonance imaging: a meta-analysis. JAMA Oncol 3:1634–1639
Leach MO, Boggis CR, Dixon AK et al (2005) Screening with magnetic resonance imaging and mammography of a UK population at high familial risk of breast cancer: a prospective multicentre cohort study (MARIBS). Lancet 365:1769–1778
Bojadzieva J, Amini B, Day SF et al (2018) Whole body magnetic resonance imaging (WB-MRI) and brain MRI baseline surveillance in TP53 germline mutation carriers: experience from the Li–Fraumeni Syndrome Education and Early Detection (LEAD) clinic. Fam Cancer 17:287–294
Mai PL, Khincha PP, Loud JT et al (2017) Prevalence of cancer at baseline screening in the National Cancer Institute Li–Fraumeni Syndrome Cohort. JAMA Oncol 3(12):1640–1645
O'Neill AF, Voss SD, Jagannathan JP et al (2018) Screening with whole-body magnetic resonance imaging in pediatric subjects with Li–Fraumeni syndrome: a single institution pilot study. Pediatr Blood Cancer 65
Paixao D, Guimaraes MD, de Andrade KC, Nobrega AF, Chojniak R, Achatz MI (2018) Whole-body magnetic resonance imaging of Li–Fraumeni syndrome patients: observations from a two rounds screening of Brazilian patients. Cancer Imaging 18:27
Saya S, Killick E, Thomas S et al (2017) Baseline results from the UK SIGNIFY study: a whole-body MRI screening study in TP53 mutation carriers and matched controls. Fam Cancer 16:433–440
Kriege M, Brekelmans CT, Boetes C et al (2006) Differences between first and subsequent rounds of the MRISC breast cancer screening program for women with a familial or genetic predisposition. Cancer 106:2318–2326
Nogueira ST, Lima EN, Nobrega AF et al (2015) (18)F-FDG PET-CT for surveillance of Brazilian patients with Li–Fraumeni syndrome. Front Oncol 5:38
Masciari S, Van den Abbeele AD, Diller LR et al (2008) F18-fluorodeoxyglucose-positron emission tomography/computed tomography screening in Li–Fraumeni syndrome. JAMA 299:1315–1319
Andrade RC, Dos Santos AC, de Aguirre Neto JC, Nevado J, Lapunzina P, Vargas FR (2017) TP53 and CDKN1A mutation analysis in families with Li–Fraumeni and Li–Fraumeni like syndromes. Fam Cancer 16:243–248
Orr BA, Clay MR, Pinto EM, Kesserwan C (2019) An update on the central nervous system manifestations of Li–Fraumeni syndrome. Acta Neuropathol 139(4):669–687
Aubrey BJ, Strasser A, Kelly GL (2016) Tumor-suppressor functions of the TP53 pathway. Cold Spring Harb Perspect Med 6:a026062
Bahar M, Kordes U, Tekautz T, Wolff J (2015) Radiation therapy for choroid plexus carcinoma patients with Li–Fraumeni syndrome: advantageous or detrimental? Anticancer Res 35:3013–3017
Wang Z, Wilson CL, Easton J et al (2018) Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol 36:2078–2087
Bluemke DA, Breiter SN (2000) Sedation procedures in MR imaging: safety, effectiveness, and nursing effect on examinations. Radiology 216:645–652
Bracken J, Heaslip I, Ryan S (2012) Chloral hydrate sedation in radiology: retrospective audit of reduced dose. Pediatr Radiol 42:349–354
Delgado J, Toro R, Rascovsky S et al (2015) Chloral hydrate in pediatric magnetic resonance imaging: evaluation of a 10-year sedation experience administered by radiologists. Pediatr Radiol 45:108–114
Glasier CM, Stark JE, Brown R et al (1995) Rectal thiopental sodium for sedation of pediatric patients undergoing MR and other imaging studies. AJNR Am J Neuroradiol 16:111–114
Greenberg SB, Faerber EN, Aspinall CL et al (1993) High-dose chloral hydrate sedation for children undergoing MR imaging: safety and efficacy in relation to age. AJR Am J Roentgenol 161:639–641
Hubbard AM, Markowitz RI, Kimmel B et al (1992) Sedation for pediatric patients undergoing CT and MRI. J Comput Assist Tomogr 16:3–6
Kang R, Shin YH, Gil NS et al (2017) A comparison of the use of propofol alone and propofol with midazolam for pediatric magnetic resonance imaging sedation - a retrospective cohort study. BMC Anesthesiol 17:138
Lee YJ, Kim DK, Kwak YH et al (2012) Analysis of the appropriate age and weight for pediatric patient sedation for magnetic resonance imaging. Am J Emerg Med 30:1189–1195
Malviya S, Voepel-Lewis T, Eldevik OP et al (2000) Sedation and general anaesthesia in children undergoing MRI and CT: adverse events and outcomes. Br J Anaesth 84:743–748
Mason KP, Turner DP, Houle TT et al (2014) Hemodynamic response to fluid management in children undergoing dexmedetomidine sedation for MRI. AJR Am J Roentgenol 202:W574–W579
Mason KP, Zurakowski D, Zgleszewski S et al (2010) Incidence and predictors of hypertension during high-dose dexmedetomidine sedation for pediatric MRI. Paediatr Anaesth 20:516–523
Merola C, Albarracin C, Lebowitz P et al (1995) An audit of adverse events in children sedated with chloral hydrate or propofol during imaging studies. Paediatr Anaesth 5:375–378
Nguyen MT, Greenberg SB, Fitzhugh KR et al (2001) Pediatric imaging: sedation with an injection formulation modified for rectal administration. Radiology 221:760–762
Ronchera-Oms CL, Casillas C, Marti-Bonmati L et al (1994) Oral chloral hydrate provides effective and safe sedation in paediatric magnetic resonance imaging. J Clin Pharm Ther 19:239–243
Sanborn PA, Michna E, Zurakowski D et al (2005) Adverse cardiovascular and respiratory events during sedation of pediatric patients for imaging examinations. Radiology 237:288–294
Jhaveri AP, Bale A, Lovick N et al (2015) The benefit and burden of cancer screening in Li–Fraumeni syndrome: a case report. Yale J Biol Med 88:181–185
Lammens CR, Bleiker EM, Aaronson NK et al (2010) Regular surveillance for Li–Fraumeni syndrome: advice, adherence and perceived benefits. Fam Cancer 9:647–654
McBride KA, Ballinger ML, Schlub TE et al (2017) Psychosocial morbidity in TP53 mutation carriers: is whole-body cancer screening beneficial? Fam Cancer 16:423–432
Ross J, Bojadzieva J, Peterson S et al (2017) The psychosocial effects of the Li–Fraumeni Education and Early Detection (LEAD) program on individuals with Li–Fraumeni syndrome. Genet Med 19:1064–1070
Nakano Y, Tsunematsu Y, Yamazaki F et al (2019) Pediatric patients with cancer predisposition in Japan: results of a questionnaire survey. Pediatr Blood Cancer 66(10):e27937
Tsunematsu Y (2015) Ethical legal and social implication of familial tumor. Nippon Rinsho 73:594–600
Funding
Funding for this guideline was provided by the Health and Labor Science Research Grant Program (Research Project to Promote Cancer Countermeasures) “A study to implement cancer genomic medicine system for hereditary tumors onset in childhood” (research supervisor, Tadashi Kumamoto).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The members of the research group confirmed that they have no conflicts of interest relating to this guideline.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to a retrospective Open Access order.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kumamoto, T., Yamazaki, F., Nakano, Y. et al. Medical guidelines for Li–Fraumeni syndrome 2019, version 1.1. Int J Clin Oncol 26, 2161–2178 (2021). https://doi.org/10.1007/s10147-021-02011-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10147-021-02011-w