
Abstract
Quinoa (Chenopodium quinoa Willd.) is a grain crop grown in the Andes renowned as a highly nutritious plant exhibiting tolerance to abiotic stress such as drought, cold and high salinity. Quinoa grows across a range of latitudes corresponding to differing day lengths, suggesting regional adaptations of flowering regulation. Improved understanding and subsequent modification of the flowering process, including flowering time, ensuring high yields, is one of the key factors behind expansion of cultivation zones and goals of the crop improvement programs worldwide. However, our understanding of the molecular basis of flower initiation and development in quinoa is limited. Here, we use a computational approach to perform genome-wide identification and analysis of 611 orthologues of the Arabidopsis thaliana flowering genes. Conservation of the genes belonging to the photoperiod, gibberellin and autonomous pathways was observed, while orthologues of the key genes found in the vernalisation pathway (FRI, FLC) were absent from the quinoa genome. Our analysis indicated that on average each Arabidopsis flowering gene has two orthologous copies in quinoa. Several genes including orthologues of MIF1, FT and TSF were identified as homologue-rich genes in quinoa. We also identified 459 quinoa-specific genes uniquely expressed in the flower and/or meristem, with no known orthologues in other species. The genes identified provide a resource and framework for further studies of flowering in quinoa and related species. It will serve as valuable resource for plant biologists, crop physiologists and breeders to facilitate further research and establishment of modern breeding programs for quinoa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Quinoa (Chenopodium quinoa Willd.), a grain crop grown in the Andes, is receiving worldwide attention as a highly nutritious plant with gluten free and low-glycaemic index seeds (Bazile et al. 2016b; Zurita-Silva et al. 2014). Its unique tolerance to abiotic stresses such as drought, severe cold and high salinity allows quinoa to be grown even in the most adverse conditions (Hariadi et al. 2011; Jacobsen et al. 2003). In 2017, the first chromosome-scale reference genome sequence of quinoa was published, and it has provided the much-needed resource for plant biologists to design molecular breeding and crop improvement programs for quinoa (Jarvis et al. 2017).
Crop improvement programs mainly aim to breed high-yielding varieties to ensure future food security (Massawe et al. 2016; Nie et al. 2016; Peng et al. 2015). The yield of a crop plant is majorly governed by flowering process as it is the first step towards seed formation. However, current knowledge of the molecular basis of quinoa flowering is limited and the details of flowering genes and associated pathways in quinoa remain elusive (Jarvis et al. 2017). It has been reported that quinoa cultivars grown in South America from Colombia to southern Chile exhibit photoperiodic adaptation to the latitude. For example, cultivars grown close to equator, mainly in Colombia, are more photoperiod-sensitive. However, the photoperiod sensitivity is not observed in the cultivars of highlands such as Peru, Bolivia and coastal parts of Chile (Curti et al. 2016). Whether quinoa is a short-day or day-neutral plant is still under debate (Curti et al. 2014; Tapia 1979). Although, the studies of quinoa floral morphology and the effects of heat and light on its flowering have been undertaken, the understanding of the underlying genetic framework is lacking (Bertero et al. 1996; Lesjak and Calderini 2017). The genome sequence of quinoa serves as an entry point for deciphering the complex molecular pathways that contribute to floral evocation. This knowledge in turn will aid development of breeding and crop improvement programs for expanding quinoa cultivation in other parts of the world. Its ability to grow and survive in harsh conditions makes quinoa an ideal food crop choice for ensuring future food security in changing climatic conditions.
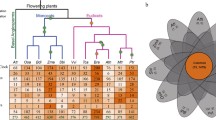
Flowering is governed by external and internal cues which are communicated to the plant through a complex network of flowering genes (Andrés and Coupland 2012). The flowering pathways of the model plant (Park et al. 1999) Arabidopsis thaliana and their corresponding flowering genes have been extensively characterised (The Arabidopsis Genome 2000). Several pathways control flowering process, and they form a complex network of genes regulating photoperiod, circadian clock function, vernalisation response, gibberellin signalling, autonomous signalling and plant ageing (reviewed in Mouradov et al. 2002b). These pathways receive signals from the environment, and fine-tune and co-ordinate them via flowering signal integrator genes (Moon et al. 2005). Finally, the output of this cross-talk leads to transition from vegetative phase to reproductive phase. However, Arabidopsis is a diploid species and quinoa an allotetraploid. The tetraploid quinoa genome was formed upon hybridisation of two diploid subgenomes; the A-genome from Chenopodium pallidicaule and the B-genome from Chenopodium suecicum (Brown et al. 2015; Štorchová et al. 2014; Walsh et al. 2015). The hybridisation event occurred an estimated 3.3–3.6 million years ago (Jarvis et al. 2017). Further, Arabidopsis is a long-day flowering plant (Park et al. 1999) and quinoa ecotypes show range of photoperiodic sensitivities (Curti et al. 2016). Chenopodium quinoa like Beta vulgaris, Chenopodium rubrum and Spinacia oleracea belongs to the Amaranthaceae family (Fig. 1a) that separated from A. thaliana ~ 140 million years ago, shortly after the monocot-eudicot split (Höft et al. 2017). Hence, the differences in the chromosomal organisation of Arabidopsis and quinoa as well as their unique developmental characteristics will likely be reflected in genic makeup of the flowering pathways. Presence of conserved and species-specific key flowering regulators has been reported for other crop plant species like the model legume soybean and sugar beet (Jung et al. 2012; Pin Pierre et al. 2012).

Distribution of the flowering genes identified. a Phylogenetic relationships between quinoa and related flowering plant species. Phylogenetic tree adapted from (Zou et al. 2017). b Overlap between genes identified using BLASTP only, Orthofinder and MCScanX (synteny). c Location of flowering genes along C. quinoa chromosomes (B – BLASTP, O – Orthofinder and S – synteny)
We have undertaken a genome-wide analysis to identify quinoa orthologues of the A. thaliana genes, particularly those involved in flowering. As a polyploid, quinoa contains duplicate copies of most genes and those could have undergone sub- or neo-functionalisation. We identified 611 orthologues of known genes involved in flowering and performed in silico gene expression analysis across six tissues. We also identified 459 ‘orphan genes’, which are unique to quinoa and highly expressed in shoot apical meristem and flower tissue, suggesting that they could constitute a pool of quinoa-specific flowering regulators. Our study provides an essential genomic resource for functional characterisation of the quinoa flowering pathways, opening new research avenues and facilitating future efforts into breeding robust, high-yielding crop types.
Results and discussion
Identification of quinoa orthologues of A. thaliana flowering genes
There are 355 protein coding genes which have been identified as involved in flowering in a model plant species A. thaliana (Lin et al. 2014; Nie et al. 2016). We used a combination of sequence similarity (BLASTP search of quinoa genes against A. thaliana with 50% sequence similarity and 50% query coverage by subject thresholds and a modified Orthofinder search, see “Materials and Methods”) and collinearity (MCScanX analysis)–based approaches to identify 611 putative orthologues of 278 A. thaliana genes in quinoa (Fig. 1c). In total, 689 orthologous gene pairs were identified using both sequence similarity and collinearity, 327 gene pairs were identified using sequence similarity only and 156 gene pairs were identified using collinearity only (Fig. 1b). One-third (38.6%) of the orthologous pairs identified based on sequence similarity were also collinear, which is consistent with previous estimates (Wang et al. 2012). While only 47.3% A. thaliana genes had orthologues in quinoa which met the similarity threshold (50% similarity, 50% query coverage by subject), 78.3% of A. thaliana genes had orthologues identified by either BLAST search with similarity thresholds, the Orthofinder search or collinearity. The Orthofinder and collinearity searches use less stringent similarity criteria (no sequence similarity or query coverage thresholds) and the results suggest that the majority of A. thaliana genes have candidate orthologues in quinoa, some of them displaying considerable sequence divergence (Additional File 1). However, since the probability of functional conservation increases with sequence conservation (Joshi and Xu 2007), the subsequent analysis focuses on the 533 genes identified using identity and coverage threshold and Orthofinder searches (Table 1).
We investigated the expression profiles of the 533 putative quinoa flowering genes across six tissues: apical meristem, lateral meristem, flower, whole seedling, leaf and stem. In total, 90.2% of the genes had evidence of expression (FPKM > 1.0) in at least one of the tissues (Additional File 1). The mean recorded expression was 33.5 FPKM (median 7.9 FPKM). While majority of the expressed genes (70.9%) showed evidence of expression in all tissues, the highest expression points were predominantly (75.7%) recorded in the flowering-related tissues (flower and meristem) (Fig. 2a, b).

Expression of the quinoa orthologues of A. thaliana flowering genes. a Tissue distribution of expressed putative quinoa flowering genes (FPKM > 1.0). b Expression levels recorded for the six tissues
Quinoa genome encodes genes belonging to the pathways controlling flowering in A. thaliana
In A. thaliana, the relationships between key flowering regulators are known. The plant senses input from multiple sources and integrates the signals received to initiate flowering. Several pathways are involved including photoperiod and circadian clock, vernalisation, gibberellin pathway and autonomous and age-related pathways, together with the floral integrators and meristem identity genes (Fig. 3). For all the pathways, orthologues of A. thaliana genes were identified in the quinoa genome.

Mapping of quinoa orthologues to A. thaliana flowering pathways. Red font – orthologues identified using BLASTP and/or Orthofinder. Blue font – orthologues identified using Orthofinder only
Photoperiod and circadian clock pathway
Light is one of the main environmental regulators of flowering in plants. Plant sense day length changes and uses the cues from the environment to control the timing of the onset of flowering. The photoperiod pathway is composed of three parts: light input, circadian clock and output (Shim et al. 2017). The light signal is perceived predominantly by the blue-light-perceiving cryptochromes (CRY) and the red-light-perceiving phytochromes (PHY), and the information is integrated into the innate photoperiodic timing governed by the circadian clock genes (Shim et al. 2017). Most of the clock genes are transcriptional regulators and act as repressors, which are expressed at precise time points during the day. CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) and LATE ELONGATED HYPOCOTYL (LHY) are expressed in the morning and repress evening-phased genes (Kamioka et al. 2016; Nagel et al. 2015). The PSEUDORESPONSE REGULATOR (PRR) genes are expressed during the day, repress transcription of CCA1 and LHY (Liu et al. 2016) and allow expression of evening-phased genes EARLY FLOWERING 3 (ELF3) and EARLY FLOWERING 4 (ELF4), which in turn repress PRR expression. Circadian clock is necessary for photoperiodic flowering because the key integrator gene CONSTANS (CO) is regulated by circadian clock and light signalling pathways (Shim et al. 2017). CO transcription is repressed by the family of proteins known as CYCLING DOF FACTOR (CDF) (Fornara et al. 2009), and CDF transcription is controlled by CCA1, LHY and PRRs. GIGANTEA (GI) and FLAVIN-BINDING KELCH REPEAT, F-BOX 1 form a complex which recognises CDFs as substrates for degradation. The orthologues of the key genes acting in the photoperiod pathway could be identified pointing to the pathway conservation. Orthologues of PHYA (AUR62017871, AUR62003557), PHYB (AUR62016209, AUR62039279), PHYC (AUR62015008, AUR62029114), CRY1 (AUR62027766, AUR62018922), CRY2 (AUR62002133, AUR62015577), LHY (AUR62022683, AUR62004570), ELF3 (AUR62040202, AUR62043053, AUR62009205), ELF4 (AUR62012247, AUR62022878, AUR62022877), GI (AUR62034894, AUR62026637), CDF2 (AUR62039802, AUR62034427), and CDF3 (AUR62021670, AUR62008425) were found. Most of the genes are present in two copies, which is consistent with the genome duplication history and the tetraploid nature of quinoa. The photoperiod and circadian clock genes were predominantly expressed in leaf and flower (Fig. 4).

Expression patterns of the genes found in different flowering pathways.
No orthologues of CO, but six orthologues of CONSTANS-like (COL), were identified. Similarly, COL genes have been identified in two related species: B. vulgaris and C. rubrum (Chia et al. 2008; Drabešová et al. 2014). In the current analysis, counterparts of the B. vulgaris and C. rubrum and orthologues of COL2 (BvCOL1, CrCOL1 and CrCOL2) and COL4 (BvCOL2), as well as COL5 and BvCOL3, were found. In C. rubrum, a short-day plant, CrCOL1 and CrCOL2 play a role in the regulation of the circadian clock and are downregulated during the dark–light transition together with FLOWERING LOCUS T (CrFT1) (Drabešová et al. 2014). The orthologues found in quinoa may play similar roles, but their expression patterns and roles need to be confirmed by further experiments.
A close relative of quinoa, sugar beet (B. vulgaris), can behave either as an annual or biennial crop, in which flowering is triggered by long days (annual) and long days and vernalisation (biennial). Biennial genotypes evolved from annual genotypes closer to the equator to adapt to northern latitudes (Hoft et al. 2017). In B. vulgaris, the bolting locus B (Abegg 1936) is a master switch distinguishing annuals from biennials. Bolting locus B encodes a pseudoresponse regulator gene BOLTING TIME CONTROL (BTC1) (Pin Pierre et al. 2012). BTC1 is a homologue of A. thaliana PSEUDORESPONSE REGULATOR 7 (PRR7) involved in the circadian clock pathway. However, unlike PRR7, BTC1 responds to photoperiod and vernalisation (Pin Pierre et al. 2012). Two potential orthologues of BTC1 (AUR62041615, AUR62036007) were identified in quinoa. Another PRR7 orthologue in sugar beet, BvPRR7, which responds to both vernalisation and circadian clock, has been identified (Omolade et al. 2016), but no orthologues in quinoa were found. As quinoa is reported not to require vernalisation, potential roles of BTC1 orthologues in quinoa require further investigation.
BvBBX19 a B-box zinc finger transcription factor is another bolting time regulator, belonging to the CO-like gene family (Dally et al. 2014). BvBBX19 acts epistatically with BvBTC1 (Dally et al. 2014) and has two orthologues in quinoa (AUR62016828, AUR62010390). Both BvBTC1 and BvBBX19 act upstream of sugar beet FLOWERING LOCUS T (FT) orthologues BvFT1 and BvFT2 and are suggested to play CO function (Dally et al. 2014). Presence of both BvBBX19 and BvBTC1 orthologues in quinoa opens new avenues for the functional characterisation of these genes and detailed investigation of their roles in flowering induction in quinoa.
Vernalisation pathway
Many plants growing in temperate climates require vernalisation (prolonged exposure to cool temperatures) to flower (Kim et al. 2009). In contrast to cold acclimation, vernalisation requires a long-term exposure to cold (i.e. during winter) to establish flowering competency the following spring. In A. thaliana, the vernalisation requirement is mainly due to two dominant genes: FRIGIDA (FRI) and FLOWERING LOCUS C (FLC) (Lee et al. 1994; Michaels and Amasino 1999; Michaels and Amasino 2001). In fact, naturally occurring mutations in FRI are responsible for flowering without vernalisation in some Arabidopsis accessions (Johanson et al. 2000; Strange et al. 2011). The main function of FRI is upregulation of FLC transcription, which results in the vernalisation requirement (Michaels and Amasino 2001). Additional genes including FRIGIDA-ESSENTIAL 1 (FES1) and SUPPRESSOR OF FRIGIDA 4 (SUF4), both containing zinc finger domains and putative transcription factor activity, are required for FRI functionality (Kim and Sung 2014). FLC is a negative regulator of flowering and its repression is key for flowering induction. Several genes including VERNALIZATION 2 (VRN2), CURLY LEAF (CLF), VERNALIZATION 1 (VRN1) and VERNALIZATION INSENSITIVE 3 (VIN3) are involved in vernalisation mediated repression of FLC. Finally, several of the genes controlling FLC expression including the transcription factor ENHANCER OF AG-4 2 (HUA2) (Doyle et al. 2004) and histone demethylase REF6 are known to act in both vernalisation and autonomous pathways.
Some of the genes involved in the vernalisation pathway of A. thaliana were identified in quinoa including FES1 (AUR62017047, AUR62014852), SUF4 (AUR62017849, AUR62003535), HUA2 (AUR62017698, AUR62015236), REF6 (AUR62001265, AUR62025745) and VRN2 (AUR62000472, AUR62006814). The vernalisation pathway genes were expressed predominantly in meristems (Fig. 4). A potential FLC orthologue (AUR62005643) similar to the B. vulgaris BvFL1 gene has been identified (Reeves et al. 2007). However, BLAST sequence similarity results suggest that it could be an orthologue of several FLC-related MADS box genes and not a true FLC orthologue. Additionally, BvFL1 which is the closest orthologue of FLC in sugar beet was shown not to be a major regulator of vernalisation response in biennial beets (Vogt et al. 2014), suggesting that AUR62005643 may play other roles. Although quinoa is reported not to require vernalisation to flower (Jarvis et al. 2017), its genome encodes several vernalisation-related genes. Interestingly, a similar observation was made in soybean, a short-day, vernalisation non-requiring crop plant (Jung et al. 2012). Quinoa is a frost-resistant crop grown in the Andean Altiplano (Jacobsen et al. 2007) where average temperatures during night times are at around 0 °C and frosts are common. Although many plant species in the temperate climate require vernalisation (Kim and Sung 2014), it should be noted that the two key genes (FLC, FRI) responsible for vernalisation response in A. thaliana are absent in quinoa. Additionally, the orthologues of genes belonging to the vernalisation pathway found in quinoa are mostly chromatin-modifying enzymes which could have acquired additional functions during evolution. The presence of several orthologues of vernalisation genes, including BTC1 and BvFL1 found in B. vulgaris, suggests that potential role of vernalisation should not be entirely discounted, especially in the genotypes adapted to cold climate and winter conditions. Results of the analysis performed in this study suggest that, if there are vernalisation-requiring quinoa varieties, the molecular mechanism is more likely to resemble that of sugar beet rather than the one of A. thaliana (Fig. 5).

Proposed model of quinoa flowering induction as compared to A. thaliana. The model is based on presence and absence of A. thaliana orthologues in quinoa. a Flowering induction in A. thaliana. b Model for flowering induction in C. quinoa
Gibberellin pathway
Gibberellins (GAs) are plant hormones which regulate various developmental processes, including dormancy, germination, stem elongation, flowering and flower development. They function not only to stimulate growth of plant organs but also to induce developmental phase transitions (Mutasa-Göttgens and Hedden 2009). Interestingly, GA is not a universal flowering stimulus and while in some species like Arabidopsis GAs promote flowering, in others, for instance pea (Pisum sativum), increasing GA content inhibits flowering (Plackett and Wilson 2017; Reinecke et al. 2013). In A. thaliana, GAs have a relatively minor influence on flowering time in long-day (LD) conditions, while in short days (SD), GA pathway becomes essential (Mutasa-Göttgens and Hedden 2009). GAs activate expression of key floral integrators (SOC1, LFY and FT) (Mutasa-Göttgens and Hedden 2009). In sugar beet, the role of GAs in bolting has been reported but their involvement in floral transition remains unconfirmed (Mutasa-Göttgens et al. 2010). The genes involved in the GA pathway encode both proteins involved in GA metabolism and signal transduction factors (Mouradov et al. 2002a). GAs promote flowering by initiation of degradation of DELLA proteins including REPRESSOR OF GA (RGA), GIBBERELLIC ACID INSENSITIVE (GAI) and RGA-LIKE (RGL). GAs are perceived by soluble nuclear-localised receptor GA INSENSITIVE DWARF (GID1), which undergo conformational change allowing them to interact with the DELLA proteins. DELLA proteins in turn activate GAMYB33 expression which promotes expression of floral integrator LFY (Conti 2017; Mutasa-Göttgens and Hedden 2009). Orthologues of the key genes involved in the gibberellin pathway including GID1B (AUR62044726, AUR62036678, AUR62034591), GID1C (AUR62000900), RGA1 (AUR62014191, AUR62039523) and MYB33 (AUR62000244, AUR62006590) are present in quinoa. The results point to conservation of the gibberellin pathway in quinoa. The genes identified in this analysis can be a starting point for further molecular analysis and elucidation of the role of the gibberellin pathway in quinoa flowering as well as other biological processes.
Autonomous and age-related pathway
In A. thaliana, the autonomous pathway accelerates flowering independently of photoperiod and temperature by repression of the flowering regulator FLC (Cheng et al. 2017). Autonomous pathway genes operate mainly via chromatin epigenetic modification and post-transcriptional regulation (Cheng et al. 2017). For example FLOWERING CONTROL LOCUS A (FCA) and FPA are RNA-recognition proteins which regulate 3′-end formation, whereas FLOWERING LOCUS D (FLD) and FVE are involved in chromatin modification (Cheng et al. 2017). The key genes involved in the autonomous and age-related pathway of A. thaliana were identified in quinoa pointing to overall pathway conservation. The genes identified in quinoa include the following: FCA (AUR62039258, AUR62016232), FPA (AUR62023840, AUR62002349), FY (AUR62037193, AUR62026878, AUR62037187, AUR62037192), FLK (AUR62001059, AUR62004893), PCFS4 (AUR62021990, AUR62012664), FVE (AUR62038133, AUR62029071, AUR62009821, AUR62033965), FLD (AUR62020695, AUR62028030), LD (AUR62033197, AUR62043270) and SPL9 (AUR62024322, AUR62012061). The autonomous pathway genes were expressed predominantly in apical meristem (Fig. 4). Interestingly, although the key components of the autonomous pathway are conserved, their target FLC is missing from the quinoa genome. The age pathway is controlled by microRNA156 (miR156), which targets SQUAMOSA PROMOTER BINDING-LIKE (SPL) transcription factors (Wang 2014; Xing et al. 2010). In A. thaliana, the SPL genes can be divided into two major groups represented by SPL3 and SPL9. The SPL proteins promote flowering through activation of LEAFY (LFY), FRUITFULL (FUL) and SUPPRESSION OF CONSTANS OVEREXPRESSION 1 (SOC1) (Wang 2014). Orthologues of SPL9 but not SPL3 were identified in quinoa. The results are consistent with previous findings where SPL9 but not SPL3 orthologues were identified in monocots, suggesting that SPL3 might be evolutionarily younger (Wang and Wang 2015). Although to our knowledge no miRNA studies have been performed in quinoa, the presence of SPL9, a known miR156 target, and the evolutionary conservation of miR156 across monocots and dicots (Wang 2014) suggest possible roles of miRNAs in the control of flowering in quinoa.
Flowering pathway integrators
Flowering pathway integrators constitute central hubs, which integrate signals from several pathways and coordinate the precise timing of flowering. In A. thaliana, FLOWERING LOCUS C (FLC), SUPPRESSION OF CONSTANS OVEREXPRESSION 1 (SOC1), LEAFY (LFY) and FLOWERING LOCUS T (FT) are the main genes integrating the flowering pathways (Andrés and Coupland 2012). Additionally, TSF (TWIN SISTER OF FT) acts redundantly with FT. In the current study, six (AUR62000271, AUR62006619, AUR62033889, AUR62010060, AUR62013052, AUR62026435) and five (AUR62026237, AUR62026245, AUR62026433, AUR62026436, AUR62026437) potential orthologues of FT and TSF were identified, together with one potential orthologue of FD (AUR62018275), three of LFY (AUR62043310, AUR62044212, AUR62032216) and two of SOC1 (AUR62004274, AUR62033383). The high number of FT orthologues is consistent with previous analyses (Jarvis et al. 2017).
The putative orthologues of FT identified show divergent expression patterns (Additional File 2), suggesting possible divergence of function, specialisation or neo-functionalisation. Two of the FT homologues (AUR62006619, AUR62000271) had peak expression in leaf and two (AUR62010060, AUR62013052) had peak expression in flower. The remaining two homologues (AUR62026435, AUR62033889) did not reach the expression threshold (> 1 FPKM) in the tissues recorded. Five of the homologues are similar to the B. vulgaris BvFT1 and BvFT2 and the corresponding C. rubrum CrFTL2 and CrFTL1 genes. BvFT1 (CrFTL2) and BvFT2 (CrFTL1) have antagonistic functions. BvFT1 is a flowering repressor expressed in plants not competent to flower (Pin et al. 2010), while BvFT2 induces flowering and is upregulated by BTC1 (Pin Pierre et al. 2012). In C. rubrum, the CrFTL1 gene was highly upregulated after a 12-h period of darkness, resulting in flowering induction, but the function of the CrFTL2 gene was not identified (Cháb et al. 2008). The functions of the orthologues of FT genes found in quinoa require further investigation but the sequence similarity analysis suggests that they could have the antagonistic functions described for the FT genes identified in B. vulgaris.
In the genus Chenopodium, natural variation of the FT gene was identified owing to presence of indels in the third intron, depending on the genotype’s origin (Storchova et al. 2015). It is known that the natural variation of FT homologues facilitated regional adaptation of crop species (Wickland and Hanzawa 2015) and has been implicated in domestication of crops including rice, sunflower and soybean (Blackman et al. 2010; Ogiso-Tanaka et al. 2013; Wu et al. 2017). Further studies of FT locus diversity could help understand adaptation of quinoa to growth across a range of latitudes.
Master regulators of floral initiation
Meristem identity genes are activated by the upstream pathways and initiate floral development by triggering vegetative to reproductive phase transition in shoot apical meristem. The A. thaliana genes involved in this role include APETALA1 (AP1), CAULIFLOWER (CAL), FUL and UFO, which promote flower meristem identity and TFL, which represses floral identity (Pidkowich et al. 1999). In quinoa, orthologues of AP1 (AUR62007887, AUR62003073, AUR62023162), FUL (AUR62039674, AUR62039675, AUR62032129), UFO (AUR62019478, AUR62036157) and TFL (AUR62038028, AUR62014507) were identified, but the analysis could not detect orthologues of CAL in the quinoa genome (Fig. 3).
Several orthologues of A. thaliana flowering regulators are homologue-rich in quinoa
Comparison of the number of the gene copies found in the genome relative to what is expected based on the genome duplication history can help identify genes present in the higher/lower copy number. Gene copy number expansion followed by sequence and functional diversification is considered an important mechanism of adaptation (Lespinet et al. 2002). Quinoa is a tetraploid formed upon hybridisation of two ancestral genomes, the A-genome C. pallidicaule and the B-genome diploid C. suecicum. We hypothesised that certain gene families which might have undergone expansion could contribute to the evolution and adaptation of quinoa. The median number of quinoa orthologues found for every A. thaliana gene is two. We searched for the genes which depart from that ratio. The maximum copy number observed was 24. The copy number counts had the following distribution: 24 copies – one gene, seven copies – two genes, six copies – two genes, five copies – 12 genes, four copies – 12 genes, three copies – 19 genes, two copies – 144 genes and one copy – 30 genes. We focused on the genes with copy number greater or equal to five (Table 2). The genes with increased copy number were involved in photoperiod pathway, flower development, gibberellin pathway, vernalisation pathway, signal integration and other functions (Table 2). The photoperiod genes include ALTERED TRYPTOPHAN REGULATION 4 (ATR4), CONSTITUTIVE PHOTOMORPHOGENIC 1 (COP1), TWIN SISTER OF FT (TSF), PHYTOCHROME A SIGNAL TRANSDUCTION 1 (PAT1) and LIGHT INSENSITIVE PHOTOPERIOD 1 (LIP1). The flower development genes include MINI ZINC FINGER (MIF1) (Fig. 6a), TRANSLATIONALLY CONTROLLED TUMOUR PROTEIN (TCTP) and CENTRORADIALIS (ATC). The two genes classified as other are AGAMOUS-LIKE 16 (AGL16) (Fig. 6b) and HEXOKINASE (HXK1). The highest number of genes with expended copy number belonged to the photoperiod pathway, suggesting that addition of new homologues to the gene repertoire of quinoa could have contributed to the adaptation of quinoa to growth across a range of latitudes (Risi and Galwey 1989). In soybean, two patterns of expression following gene duplication could be observed. Duplicated and ancestral genes either retain similar expression patterns and the expression patterns mirror phylogenetic relationships which implies conservation of function or the expression patterns diverge, which points to potential neofunctionalisation of duplicated genes (Jung et al. 2012). We investigated sequence-based phylogenetic relationships and the corresponding expression profiles of duplicated genes in quinoa and both patterns were observed. For example, duplicated homologues of MINI ZINC FINGER (MIF1) (Fig. 6a) retained similar expression patterns, whereas duplicated homologues of AGL16 (Fig. 6b) show more divergent expression patterns, which do not follow inferred sequence similarity-based phylogenetic relationships. MIF1 is involved in integration of different hormonal signals, and its constitutive overexpression results in serious developmental defects including dwarfing, reduced apical dominance and altered flower morphology (Hu and Ma 2006). Duplication of the MIF1 genes could contribute to the morphological diversity of quinoa (Risi and Galwey 1989). In A. thaliana, AGL16 controls flowering and its effect is dependent on the presence of FLC and FT (Hu et al. 2014). The divergent expression patterns of AGL16, as well as lack of strong putative FLC homologue in quinoa, suggests that AGL16 homologues might have acquired new functions.

Relationships between genes present in higher copy number and their homologues in other species. a MIF1 gene. b AGL16 gene. The evolutionary history was inferred by using the maximum likelihood method based on the JTT matrix-based model. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Bootstrapping was performed with 100 replicates. The tree was rooted using A. thaliana gene branch
Quinoa genome encodes several hundred orphan genes potentially involved in flowering
Orphan genes are defined as genes with protein sequences unique to a given species. Although the function of a vast majority of the newly evolved orphan genes is unknown, some were shown to be essential (Arendsee et al. 2014) and a high proportion of orphan genes in A. thaliana has been linked to abiotic stress tolerance (Luhua et al. 2013). Orphan genes have also been shown to be recruited to the lineage-specific structures like nodules in legumes and storage vacuoles in grasses (Arendsee et al. 2014). Quinoa is grain crop with a unique floral structure. The inflorescence is a profusely branched panicle and the flowers lack petals (Bertero et al. 1996; Bhargava et al. 2007). The genotypes display considerable variation with regard to floral morphology and duration of developmental phases leading to inflorescence formation and anthesis (Risi and Galwey 1989). Flowers of quinoa can be unisexual or hermaphrodite and vary in size and arrangement (Bhargava et al. 2007). Genes which are found uniquely in quinoa and are expressed in flowering-related tissues and structures could be contributing to the quinoa-specific floral morphology and physiology. We focused on the quinoa genes which are expressed specifically in flower and/or meristems (genes expressed in lateral meristem only were not included)—3369 genes in total. We then identified genes which are potentially Chenopodium-specific, i.e. genes found in Chenopodium species (C. quinoa, C. pallidicaule and C. suecicum) but had no orthologues in other species (A. thaliana, B. vulgaris, Spinacia oleracea and Glycine max, as identified by Orthofinder). Out of 3369 genes, 459 were Chenopodium-specific (Additional File3). The highest number of putative Chenopodium-specific flowering genes show apical meristem specific expression (269) genes (Fig. 7a). Twenty-seven of those genes were putative transcription factors, belonging to several families including NAC, B3 and MYB (Fig. 7b). The genes and especially transcription factors identified could contribute to Chenopodium-specific flowering characteristics.

Quinoa-specific flowering regulators. a Tissue distribution of quinoa-specific genes expressed in flower and meristem only (FPKM>1.0). b Gene families represented by quinoa-specific transcription factors expressed in flower and meristem only
Conclusions
To date, the potential of quinoa as a crop plant is still untapped, and its properties remain underutilised (Bazile et al. 2016a). Our study took advantage of the recently available quinoa reference genome and identified several hundred orthologues of known flowering regulators. The results point to the overall conservation of the flowering pathways with vernalisation pathway being the most affected by the absence of orthologues of key genes. The results also point to an increased copy number of genes encoding FT and TSF genes which could contribute to the wide range of photoperiodic responses and local adaptation. The results provide a framework for the future studies of molecular mechanism of flowering in quinoa. Our identification and characterisation of flowering genes in quinoa will aid in designing studies that aim to study adaptation of quinoa to a range of latitudinal variations.
Materials and methods
Datasets used
C. quinoa (Cq_PI614886_gene_V1), C. pallidicaule (C_pallidicaule_gene_V1), C. suecicum (C_suecicum_gene_V1): http://www.cbrc.kaust.edu.sa/chenopodiumdb/
G. max: Gmax_275_Wm82.a2.v1, https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Gmax
B. vulgaris: BeetSet-2, http://bvseq.molgen.mpg.de/
S. oleracea: spinach_gene_v1, http://www.spinachbase.org/
A. thaliana: Athaliana_167_TAIR10, https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Athaliana
Orthologous genes identification
BLASTP
BLASTP v2.6.0 (blastp -evalue 1e-5 -outfmt ‘6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore qcovs qlen slen’) was used to perform comparison of the proteomes of C. quinoa, G. max, B. vulgaris and S. oleracea against the proteome of A. thaliana. For each query, one best matching subject (lowest e-value) was retained. Then, the results were filtered to retain high scoring pairs (HSPs) with sequence similarity and query coverage by subject exceeding 50%.
Orthofinder
Orthofinder v2.1.2 with default parameters was used to assign the genes from all the species (C. quinoa, C. pallidicaule, C. suecicum, G. max, B. vulgaris, S. oleracea, A. thaliana) to orthologous gene clusters. The results were parsed to identify all the species represented by each cluster. In order to keep the most confident assignments only, a previously developed procedure was applied (Golicz et al. 2016). For the gene to be identified as orthologue, the C. quinoa and A. thaliana genes had to be found within the same cluster and constitute best matches in all vs all comparison of C. quinoa genes against A. thaliana genes.
Collinearity
Collinearity between the genomes of species for which pseudomolecules were available (C. quinoa, G. max, B. vulgaris, S. oleracea, A. thaliana) was established using MCScanX with default parameters. All vs all comparisons of proteomes were performed using DIAMOND v0.8.25 (-evalue 1e-5 -max-target-seqs 20).
Expression analysis
The RNASeq datasets for six tissues were downloaded from NCBI Sequence Read Archive (SRA, Accessions: https://www.ncbi.nlm.nih.gov/sra/?term=SRR3938279, https://www.ncbi.nlm.nih.gov/sra/?term=SRR3938280, https://www.ncbi.nlm.nih.gov/sra/?term=SRR3938281, https://www.ncbi.nlm.nih.gov/sra/?term=SRR3938282, https://www.ncbi.nlm.nih.gov/sra/?term=SRR3938283, https://www.ncbi.nlm.nih.gov/sra/?term=SRR3938284). The reads were trimmed to remove low-quality sequence and adapter contamination using Trimmomatic v0.38 (ILLUMINACLIP:all.adapters.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:80). The reads were mapped to the C. quinoa genome assembly using Hisat2 v2.1.0 (--min-intronlen 20 --max-intronlen 1000). The reads mapping to the annotated genes were quantified using featureCounts v1.6.2 (-p -B -P -d 1 -D 1000 -t exon -g gene_id).
Transcription factor identification
Transcription factors were identified by PlantTFDB ‘Transcription Factor Prediction’ tool (http://planttfdb.cbi.pku.edu.cn/prediction.php, accessed on 25.05.2018).
Change history
18 November 2019
The above article was published online with incorrect Fig. 5 legend. The legend for Fig. 4 was repeated in Fig. 5.
References
Abegg FA (1936) A genetic factor for the annual habit in beets and linkage relationship. J Agric Res 53:493–511
Andrés F, Coupland G (2012) The genetic basis of flowering responses to seasonal cues. Nat Rev Genet 13:627–639. https://doi.org/10.1038/nrg3291
Arendsee ZW, Li L, Wurtele ES (2014) Coming of age: orphan genes in plants. Trends Plant Sci 19:698–708. https://doi.org/10.1016/j.tplants.2014.07.003
Bazile D, Jacobsen S-E, Verniau A (2016a) The global expansion of quinoa: trends and limits. Front Plant Sci 7:622. https://doi.org/10.3389/fpls.2016.00622
Bazile D, Pulvento C, Verniau A, al-Nusairi MS, Ba D, Breidy J, Hassan L, Mohammed MI, Mambetov O, Otambekova M, Sepahvand NA, Shams A, Souici D, Miri K, Padulosi S (2016b) Worldwide evaluations of quinoa: preliminary results from post international year of quinoa FAO projects in nine countries. Front Plant Sci 7. https://doi.org/10.3389/fpls.2016.00850
Bertero D, Medan D, Hall AJ (1996) Changes in apical morphology during floral initiation and reproductive development in quinoa (Chenopodium quinoa Willd.). Ann Bot 78:317–324. https://doi.org/10.1006/anbo.1996.0126
Bhargava A, Shukla S, Ohri D (2007) Gynomonoecy in Chenopodium quinoa (Chenopodiaceae): variation in inflorescence and floral types in some accessions. Biologia 62:19–23. https://doi.org/10.2478/s11756-007-0001-z
Blackman BK, Strasburg JL, Raduski AR, Michaels SD, Rieseberg LH (2010) The role of recently derived FT paralogs in sunflower domestication. Curr Biol : CB 20:629–635. https://doi.org/10.1016/j.cub.2010.01.059
Brown DC, Cepeda-Cornejo V, Maughan PJ, Jellen EN (2015) Characterization of the granule-bound starch synthase I gene in Chenopodium. Plant Genome 8
Cháb D, Kolář J, Olson MS, Štorchová H (2008) Two FLOWERING LOCUS T (FT) homologs in Chenopodium rubrum differ in expression patterns. Planta 228:929–940. https://doi.org/10.1007/s00425-008-0792-3
Cheng J-Z, Zhou Y-P, Lv T-X, Xie C-P, Tian C-E (2017) Research progress on the autonomous flowering time pathway in Arabidopsis. Physiol Mol Biol Plants 23:477–485. https://doi.org/10.1007/s12298-017-0458-3
Chia TYP, Müller A, Jung C, Mutasa-Göttgens ES (2008) Sugar beet contains a large CONSTANS-LIKE gene family including a CO homologue that is independent of the early-bolting (B) gene locus. J Exp Bot 59:2735–2748. https://doi.org/10.1093/jxb/ern129
Conti L (2017) Hormonal control of the floral transition: can one catch them all? Dev Biol 430:288–301. https://doi.org/10.1016/j.ydbio.2017.03.024
Curti RN, de la Vega AJ, Andrade AJ, Bramardi SJ, Bertero HD (2014) Multi-environmental evaluation for grain yield and its physiological determinants of quinoa genotypes across Northwest Argentina. Field Crop Res 166:46–57. https://doi.org/10.1016/j.fcr.2014.06.011
Curti R, de la Vega A, Andrade A, Bramardi SJ, Bertero D (2016) Adaptive responses of quinoa to diverse agro-ecological environments along an altitudinal gradient in North West Argentina. Field Crops Res 189:10–18
Dally N, Xiao K, Holtgräwe D, Jung C (2014) The B2 flowering time locus of beet encodes a zinc finger transcription factor. Proc Natl Acad Sci 111:10365–10370
Doyle MR, Bizzell CM, Keller MR, Michaels SD, Song J, Noh Y-S, Amasino RM (2004) HUA2 is required for the expression of floral repressors in Arabidopsis thaliana. Plant J 41:376–385. https://doi.org/10.1111/j.1365-313X.2004.02300.x
Drabešová J, Cháb D, Kolář J, Haškovcová K, Štorchová H (2014) A dark–light transition triggers expression of the floral promoter CrFTL1 and downregulates CONSTANS-like genes in a short-day plant Chenopodium rubrum. J Exp Bot 65:2137–2146. https://doi.org/10.1093/jxb/eru073
Fornara F, Panigrahi KCS, Gissot L, Sauerbrunn N, Rühl M, Jarillo JA, Coupland G (2009) Arabidopsis DOF transcription factors act redundantly to reduce CONSTANS expression and are essential for a photoperiodic flowering response. Dev Cell 17:75–86. https://doi.org/10.1016/j.devcel.2009.06.015
Golicz AA, Bayer PE, Barker GC, Edger PP, Kim HR, Martinez PA, Chan CKK, Severn-Ellis A, McCombie WR, Parkin IAP, Paterson AH, Pires JC, Sharpe AG, Tang H, Teakle GR, Town CD, Batley J, Edwards D (2016) The pangenome of an agronomically important crop plant Brassica oleracea. Nat Commun 7:13390. https://doi.org/10.1038/ncomms13390
Hariadi Y, Marandon K, Tian Y, Jacobsen SE, Shabala S (2011) Ionic and osmotic relations in quinoa (Chenopodium quinoa Willd.) plants grown at various salinity levels. J Exp Bot 62:185–193. https://doi.org/10.1093/jxb/erq257
Höft N, Dally N, Hasler M, Jung C (2017) Haplotype variation of flowering time genes of sugar beet and its wild relatives and the impact on life cycle regimes frontiers in plant. Science 8:2211. https://doi.org/10.3389/fpls.2017.02211
Hu W, Ma H (2006) Characterization of a novel putative zinc finger gene MIF1: involvement in multiple hormonal regulation of Arabidopsis development. Plant J 45:399–422. https://doi.org/10.1111/j.1365-313X.2005.02626.x
Hu J-Y, Zhou Y, He F, Dong X, Liu LY, Coupland G, Turck F, de Meaux J (2014) miR824-regulated AGAMOUS-LIKE16 contributes to flowering time repression in Arabidopsis. Plant Cell 26:2024–2037. https://doi.org/10.1105/tpc.114.124685
Jacobsen SE, Mujica A, Jensen CR (2003) The resistance of quinoa (Chenopodium quinoa Willd.) to adverse abiotic factors. Food Rev Int 19:99–109. https://doi.org/10.1081/FRI-120018872
Jacobsen SE, Monteros C, Corcuera LJ, Bravo LA, Christiansen JL, Mujica A (2007) Frost resistance mechanisms in quinoa (Chenopodium quinoa Willd.). Eur J Agron 26:471–475. https://doi.org/10.1016/j.eja.2007.01.006
Jarvis DE, Ho YS, Lightfoot DJ, Schmöckel SM, Li B, Borm TJA, Ohyanagi H, Mineta K, Michell CT, Saber N, Kharbatia NM, Rupper RR, Sharp AR, Dally N, Boughton BA, Woo YH, Gao G, Schijlen EGWM, Guo X, Momin AA, Negrão S, al-Babili S, Gehring C, Roessner U, Jung C, Murphy K, Arold ST, Gojobori T, Linden CG, van Loo EN, Jellen EN, Maughan PJ, Tester M (2017) The genome of Chenopodium quinoa. Nature 542:307–312. https://doi.org/10.1038/nature21370
Johanson U, West J, Lister C, Michaels S, Amasino R, Dean C (2000) Molecular analysis of FRIGIDA a major determinant of natural variation in Arabidopsis flowering time. Science 290:344–347
Joshi T, Xu D (2007) Quantitative assessment of relationship between sequence similarity and function similarity. BMC Genomics 8:222–222. https://doi.org/10.1186/1471-2164-8-222
Jung C-H, Wong CE, Singh MB, Bhalla PL (2012) Comparative genomic analysis of soybean flowering genes. PLoS One 7:e38250. https://doi.org/10.1371/journal.pone.0038250
Kamioka M, Takao S, Suzuki T, Taki K, Higashiyama T, Kinoshita T, Nakamichi N (2016) Direct repression of evening genes by CIRCADIAN CLOCK-ASSOCIATED1 in the Arabidopsis circadian clock. Plant Cell 28:696–711
Kim D-H, Sung S (2014) Genetic and epigenetic mechanisms underlying vernalization. The arabidopsis book 12:e0171–e0171. https://doi.org/10.1199/tab.0171
Kim D-H, Doyle MR, Sung S, Amasino RM (2009) Vernalization: winter and the timing of flowering in plants. Annu Rev Cell Dev Biol 25:277–299. https://doi.org/10.1146/annurev.cellbio.042308.113411
Lee I, Michaels SD, Masshardt AS, Amasino RM (1994) The late-flowering phenotype of FRIGIDA and mutations in LUMINIDEPENDENS is suppressed in the Landsberg erecta strain of Arabidopsis. Plant J 6:903–909. https://doi.org/10.1046/j.1365-313X.1994.6060903.x
Lesjak J, Calderini DF (2017) Increased night temperature negatively affects grain yield, biomass and grain number in Chilean quinoa. Front Plant Sci 8:352. https://doi.org/10.3389/fpls.2017.00352
Lespinet O, Wolf YI, Koonin EV, Aravind L (2002) The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res 12:1048–1059
Lin K, Zhang N, Severing EI, Nijveen H, Cheng F, Visser RGF, Wang X, de Ridder D, Bonnema G (2014) Beyond genomic variation - comparison and functional annotation of three Brassica rapagenomes: a turnip, a rapid cycling and a Chinese cabbage. BMC Genomics 15:250. https://doi.org/10.1186/1471-2164-15-250
Liu TL, Newton L, Liu M-J, Shiu S-H, Farré EM (2016) A G-box-like motif is necessary for transcriptional regulation by circadian pseudo-response regulators in Arabidopsis. Plant Physiol 170:528–539
Luhua S, Hegie A, Suzuki N, Shulaev E, Luo X, Cenariu D, Ma V, Kao S, Lim J, Gunay MB, Oosumi T, Lee SC, Harper J, Cushman J, Gollery M, Girke T, Bailey-Serres J, Stevenson RA, Zhu JK, Mittler R (2013) Linking genes of unknown function with abiotic stress responses by high-throughput phenotype screening. Physiol Plant 148:322–333. https://doi.org/10.1111/ppl.12013
Massawe F, Mayes S, Cheng A (2016) Crop diversity: an unexploited treasure trove for food security. Trends Plant Sci 21:365–368. https://doi.org/10.1016/j.tplants.2016.02.006
Michaels SD, Amasino RM (1999) FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11:949–956
Michaels SD, Amasino RM (2001) Loss of FLOWERING LOCUS C activity eliminates the late-flowering phenotype of FRIGIDA and autonomous pathway mutations but not responsiveness to vernalization. Plant Cell 13:935–941
Moon J, Lee H, Kim M, Lee I (2005) Analysis of flowering pathway integrators in Arabidopsis plant and cell physiology. 46:292–299. https://doi.org/10.1093/pcp/pci024
Mouradov A, Cremer F, Coupland G (2002a) Control of flowering time: interacting pathways as a basis for diversity. Plant Cell 14:s111–s130. https://doi.org/10.1105/tpc.001362
Mouradov A, Cremer F, Coupland G (2002b) Control of flowering time: interacting pathways as a basis for diversity. Plant Cell 14:S111–S130
Mutasa-Göttgens E, Hedden P (2009) Gibberellin as a factor in floral regulatory networks. J Exp Bot 60:1979–1989. https://doi.org/10.1093/jxb/erp040
Mutasa-Göttgens ES, Qi A, Zhang W, Schulze-Buxloh G, Jennings A, Hohmann U, Müller AE, Hedden P (2010) Bolting and flowering control in sugar beet: relationships and effects of gibberellin, the bolting gene B and vernalization. AoB PLANTS 2010:plq012-plq012. https://doi.org/10.1093/aobpla/plq012
Nagel DH, Doherty CJ, Pruneda-Paz JL, Schmitz RJ, Ecker JR, Kay SA (2015) Genome-wide identification of CCA1 targets uncovers an expanded clock network in <em>Arabidopsis</em>. Proc Natl Acad Sci 112:E4802
Nie S, Li C, Xu L, Wang Y, Huang D, Muleke EM, Sun X, Xie Y, Liu L (2016) De novo transcriptome analysis in radish (Raphanus sativus L.) and identification of critical genes involved in bolting and flowering. BMC Genomics 17:389. https://doi.org/10.1186/s12864-016-2633-2
Ogiso-Tanaka E, Matsubara K, Yamamoto SI, Nonoue Y, Wu J, Fujisawa H, Ishikubo H, Tanaka T, Ando T, Matsumoto T, Yano M (2013) Natural variation of the RICE FLOWERING LOCUS T 1 contributes to flowering time divergence in rice. PLoS One 8:e75959-e75959. https://doi.org/10.1371/journal.pone.0075959
Omolade O, Müller AE, Jung C, Melzer S (2016) BvPRR7 is a cold responsive gene with a clock function in beet. Biol Plant 60:95–104. https://doi.org/10.1007/s10535-015-0568-0
Park DH, Somers DE, Kim YS, Choy YH, Lim HK, Soh MS, Kim HJ, Kay SA, Nam HG (1999) Control of circadian rhythms and photoperiodic flowering by the Arabidopsis GIGANTEA. Gene Sci 285:1579–1582. https://doi.org/10.1126/science.285.5433.1579
Peng FY, Hu Z, Yang R-C (2015) Genome-wide comparative analysis of flowering-related genes in Arabidopsis, wheat, and barley. Int J Plant Genomics 2015:874361–874361. https://doi.org/10.1155/2015/874361
Pidkowich MS, Klenz JE, Haughn GW (1999) The making of a flower: control of floral meristem identity in Arabidopsis. Trends Plant Sci 4:64–70. https://doi.org/10.1016/S1360-1385(98)01369-7
Pin Pierre A et al (2012) The role of a pseudo-response regulator gene in life cycle adaptation and domestication of beet. Curr Biol 22:1095–1101. https://doi.org/10.1016/j.cub.2012.04.007
Pin PA, Benlloch R, Bonnet D, Wremerth-Weich E, Kraft T, Gielen JJL, Nilsson O (2010) An antagonistic pair of FT homologs mediates the control of flowering time in sugar beet. Science 330:1397–1400
Plackett ARG, Wilson ZA (2017) Gibberellins and plant reproduction. In: Annual plant reviews online. https://doi.org/10.1002/9781119312994.apr0540
Reeves PA, He Y, Schmitz RJ, Amasino RM, Panella LW, Richards CM (2007) Evolutionary conservation of the FLOWERING LOCUS C-mediated vernalization response: evidence from the sugar beet (Beta vulgaris). Genetics 176:295–307. https://doi.org/10.1534/genetics.106.069336
Reinecke DM, Wickramarathna AD, Ozga JA, Kurepin LV, Jin AL, Good AG, Pharis RP (2013) Gibberellin 3-oxidase gene expression patterns influence gibberellin biosynthesis, growth, and development in pea. Plant Physiol 163:929–945
Risi JC, Galwey NW (1989) The pattern of genetic diversity in the Andean grain crop quinoa (Chenopodium quinoa Willd). I. Associations between characteristics. Euphytica 41:147–162. https://doi.org/10.1007/BF00022424
Shim JS, Kubota A, Imaizumi T (2017) Circadian clock and photoperiodic flowering in Arabidopsis: CONSTANS is a hub for signal integration. Plant Physiol 173:5–15
Štorchová H, Drabešová J, Cháb D, Kolář J, Jellen EN (2014) The introns in FLOWERING LOCUS T-LIKE (FTL) genes are useful markers for tracking paternity in tetraploid Chenopodium quinoa Willd. Genet Resour Crop Evol 62:913–925. https://doi.org/10.1007/s10722-014-0200-8
Strange A, Li P, Lister C, Anderson J, Warthmann N, Shindo C, Irwin J, Nordborg M, Dean C (2011) Major-effect alleles at relatively few loci underlie distinct vernalization and flowering variation in Arabidopsis accessions. PLoS One 6:e19949. https://doi.org/10.1371/journal.pone.0019949
Tapia M (1979) Quinua y Kaniwa. Cultivos Andinos Centro Internacional de Investigación para el Desarrollo (CIID) Instituto Interamericano de Ciencias Agrícolas (IICA)
The Arabidopsis Genome I (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815. https://doi.org/10.1038/35048692
Vogt SH, Weyens G, Lefèbvre M, Bork B, Schechert A, Müller AE (2014) The FLC-like gene BvFL1 is not a major regulator of vernalization response in biennial beets. Front Plant Sci 5:146. https://doi.org/10.3389/fpls.2014.00146
Walsh BM, Adhikary D, Maughan PJ, Emshwiller E, Jellen EN (2015) Chenopodium polyploidy inferences from salt overly sensitive 1 (SOS1) data. Am J Bot 102:533–543. https://doi.org/10.3732/ajb.1400344
Wang J-W (2014) Regulation of flowering time by the miR156-mediated age pathway. J Exp Bot 65:4723–4730. https://doi.org/10.1093/jxb/eru246
Wang H, Wang H (2015) The miR156/SPL module, a regulatory hub and versatile toolbox, gears up crops for enhanced agronomic traits. Mol Plant 8:677–688. https://doi.org/10.1016/j.molp.2015.01.008
Wang Y, Tang H, DeBarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, Kissinger JC, Paterson AH (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40:e49–e49. https://doi.org/10.1093/nar/gkr1293
Wickland DP, Hanzawa Y (2015) The FLOWERING LOCUS T/TERMINAL FLOWER 1 gene family: functional evolution and molecular mechanisms. Mol Plant 8:983–997. https://doi.org/10.1016/j.molp.2015.01.007
Wu F, Sedivy EJ, Price WB, Haider W, Hanzawa Y (2017) Evolutionary trajectories of duplicated FT homologues and their roles in soybean domestication. Plant J 90:941–953. https://doi.org/10.1111/tpj.13521
Xing S, Salinas M, Höhmann S, Berndtgen R, Huijser P (2010) miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 22:3935–3950. https://doi.org/10.1105/tpc.110.079343
Zou C, Chen A, Xiao L, Muller HM, Ache P, Haberer G, Zhang M, Jia W, Deng P, Huang R, Lang D, Li F, Zhan D, Wu X, Zhang H, Bohm J, Liu R, Shabala S, Hedrich R, Zhu JK, Zhang H (2017) A high-quality genome assembly of quinoa provides insights into the molecular basis of salt bladder-based salinity tolerance and the exceptional nutritional value. Cell Res 27:1327–1340. https://doi.org/10.1038/cr.2017.124
Zurita-Silva A, Fuentes F, Zamora P, Jacobsen S-E, Schwember AR (2014) Breeding quinoa (Chenopodium quinoa Willd.): potential and perspectives. Mol Breed 34:13–30. https://doi.org/10.1007/s11032-014-0023-5
Availability of data and materials
All the necessary data and materials can be found within the manuscript and the Additional Files.
Funding
This work was supported by the Concurso Nacional de Proyectos Fondecyt (ID: 11170226) and the University of Melbourne McKenzie Fellowship.
Author information
Authors and Affiliations
Contributions
AAG conceived the study, performed the analysis and wrote the manuscript; US conceived the study, helped analyse the data and wrote the manuscript; HA wrote parts of the manuscript; PLB assisted with editing of the manuscript; MBS assisted with editing of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: Owing to an error in the production process the above article was published online with incorrect legends for Figures 5 to 7. The legend for Fig. 4 was repeated in Fig. 5, causing all the subsequent figure legends to be incorrect.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Golicz, A.A., Steinfort, U., Arya, H. et al. Analysis of the quinoa genome reveals conservation and divergence of the flowering pathways. Funct Integr Genomics 20, 245–258 (2020). https://doi.org/10.1007/s10142-019-00711-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-019-00711-1