
Abstract
Chemical signaling strength during intercellular communication can be regulated by secretory cells through controlling the amount of signaling molecules that are released from a secretory vesicle during the exocytosis process. In addition, the chemical signal can also be influenced by the amount of neurotransmitters that is accumulated and stored inside the secretory vesicle compartment. Here, we present the development of analytical methodologies and cell model systems that have been applied in neuroscience research for gaining better insights into the biophysics and the molecular mechanisms, which are involved in the regulatory aspects of the exocytosis machinery affecting the output signal of chemical transmission at neuronal and neuroendocrine cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Secretory vesicles are central in neuroscience and endocrinology. These organelles accommodate high concentrations of various neurotransmitter molecules and hormones serving as units of chemical message. Fusion of neurotransmitter-filled vesicles with the cell plasma membrane through a Ca2+-dependent process called exocytosis result in a rapid release of signaling molecules into the extracellular space and is a key process in intercellular communication [46]. To promote vesicle fusion of secretory cells in our bodies relies on a highly conserved molecular machinery based on the soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNARE) protein. These SNARE proteins consist of v-SNAREs localized in the vesicle membrane that associates with target t-SNAREs at the inner leaflet of the cell plasma membrane to initiate vesicle docking, fusion, and vesicle content release. These secreted signaling molecules diffuse from the release site of the emitting cell and bind to specific receptors at the surface of target cells inducing specific physiological reactions. In synaptic transmission, depending on type of neurotransmitters released, binding to target receptors at neighboring neurons may for instance serve to either excite or inhibit signal transmission in neuronal pathways. Hence, the secretory vesicle is an essential organelle that directly is influencing our brain function such as cognition, emotions, learning, and memory. Therefore, many neurological drugs are aimed to affect the quantity of neurotransmitters released and residing in the extracellular space by influencing the potency of neurochemical signals. The signal strength in cellular communication can be tuned at target cells receiving the signal via the sensitivity and level of the receptors expression in the cell membrane. Often, the amount of neurotransmitters released from an exocytosis event do not succeed to fully saturate the post-synaptic receptors in a synapse [38, 57, 93, 96]. Consequently, it has been realized that modulation of chemical signaling strength, thus can be regulated by the secreting cell through varying the amount of neurotransmitters released owing to the different modes of exocytosis that can be triggered [10, 14, 71, 72, 82, 93].
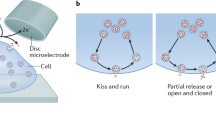
The maximal potency can be achieved from the classic view of exocytosis called “full exocytosis.” In this mode, the vesicle compartment upon fusion fully collapses into the plasma membrane releasing the entire vesicle content into the extracellular space [4, 11, 45, 66, 70, 77, 99]. In contrast to full vesicle content release, after vesicle fusion, the fusion pore can be controlled to stay open for a limited time and then close again. This allows a fraction of the vesicle neurotransmitter content to be released before the partly emptied vesicle is rapidly recycled while preserving its original shape [4, 78, 85]. The effectiveness of the chemical signal can thus be tuned by adjusting the pore size and the time it stays open [4, 14, 51, 60, 76]. The chemical signal can be minimized through the exocytosis mode “kiss and run,” where a transient 1–2 nm fusion pore that may also flicker, allows very small quantities of neurotransmitters to be secreted before the pore closes for vesicle reuse [3, 4, 12, 24, 34, 39, 66, 83, 91, 100]. A “moderate” signal can be delivered by means of a third mode of exocytosis where the transient fusion pore dilates into a larger pore extending the “kiss and run” in terms of fusion pore size and duration the pore is open, resulting in that more and yet not all of the vesicle content is expelled before the vesicle compartment is retrieved for recycling. This mode that more recently has been discovered and accepted has been termed many different names, e.g., “fuse-pinch-and-linger,” “extended kiss-and-run,” or “open and close exocytosis,” and seem to be the most prevalent mode of exocytosis used by for instance chromaffin cells [1, 11, 60, 61, 71, 78, 86, 89].
Depending on type of secretory cells and category of vesicles, quantification with carbon fiber microelectrode amperometry has estimated that about 30–60% of the vesicle content is released when this mode is triggered [51, 71, 72, 82]. Additionally, in chromaffin cells, fine-tuning of fusion pore size can serve as a molecular sieve, adding chemical selectivity of the compounds released by allowing molecules of a certain size to pass and others to be retained within the vesicle compartment [41, 69]. Hence, it is apparent that regulation of neurotransmitter release can directly be associated with mechanisms controlling the dynamics of the fusion pore affecting chemical signaling strength during intercellular communication and synaptic function. Therefore, to elucidate the regulatory mechanism and improve our understanding of what factors act to control fusion pore dynamics during exocytosis, it is important to identify the molecular drivers for vesicle-membrane interactions and the biophysics affecting that influences the exocytosis machinery. Many studies on secretion have, apart from neuronal cells also involved cell lines, simplified model systems and many major findings have been made through studies at chromaffin cells [15, 22, 53].
However, controversy still remains on the mechanisms for the regulatory aspect of exocytosis and the prevalence for different modes of exocytosis in secretory cells. This might relate to variance in the type of information provided by different analytical methods and limitations of instrumental techniques used in different experiments [69]. In addition, depending on the cell type and category of vesicles, the experimental conditions can vary widely. For instance, monitoring exocytosis release from small synaptic vesicles (40–60 nm in diameter) is a much greater challenge than from large dense-core vesicles (LDCVs) that vary in size from 80 to 300 nm in diameter [1, 82]. Therefore, in progress to clarify and verify results, development of new analytical tools improving sensitivity and spatiotemporal resolution for studying exocytosis is important. With individual methods varying in terms of different kinds of information achieved and technical limitations, an approach to improve the ability to overview the exocytosis process is to combine methodologies that provide complementary information. For instance, electrochemical methods offer quantitative and high temporally resolved detail information on fusion pore dynamics at the initial stages of exocytosis while reporting less on the later stages of exocytosis. In contrast, many imaging techniques have the capability to, with high spatial resolution, overview and track vesicle from docking, fusion, and vesicle content release and depending on the method used, also through vesicle recapture, but in comparison to electrochemical methods are limited in sensitivity and temporal resolution. Therefore, by merging electrochemical methods with imaging techniques offer the opportunity to simultaneously record the complementary information from both methods during secretion studies. For example, by combining high temporally resolved amperometry or electrophysiology, together with total internal reflection microscopy imaging, quantitative information of neurotransmitter release can be correlated with high spatial information on vesicle activity prior and post vesicle fusion, or by placing multiple lithographic electrodes at secreting cells electrochemical imaging capabilities of the exocytosis process can be created [42, 43, 49, 87, 92, 98, 101].
Characterization of exocytosis with amperometry recording
Since the introduction of single-cell carbon fiber microelectrode recording of exocytosis activity at chromaffin cells by the Wightman lab [50, 90], amperometry has been a major analytical technique used in neurochemical research due to the simplicity of method implementation, the outstanding temporal resolution of sub-millisecond time scale and that this is the only existing method providing quantitative information of single-vesicle neurotransmitter release [63, 90]. In this electrochemical method, a carbon fiber microelectrode is placed in close contact to a secretory cell (as shown in Fig. 1a) and by applying an oxidation potential to the electrode, single vesicles release of signaling molecules that are electroactive, e.g., dopamine, adrenaline, noradrenaline, or serotonin, can be detected as individual current peaks at cells stimulated to exocytosis. By integrating the current versus time for each recorded amperometric peak, the total charge (Q) can be used to determine the number of neurotransmitter released from individual exocytosis events, by calculations using Faraday’s law expressed as Q = nNF, where n is the number of electrons transferred in the redox reaction (n = 2 for oxidation of catecholamines), N is the number of neurotransmitter molecules detected, and F is the Faraday constant (96,485 C mol−1). Typically, an amperometric current spike is marked by a rapid rise corresponding to a quick release of high-concentration neurotransmitter solution through the fusion pore as the pore dilates, followed by a slower decay representing the subsequent diffusion of molecules from the release site to the electrode surface [9, 63]. Analyzing the kinetic parameters from single current spikes present dynamic information on fusion pore formation, pore dilation and closing, which are important parameters for characterization of modes for exocytosis triggered at cells. Many amperometric peaks display a pre-spike feature called a “foot” signifying detection of neurotransmitter passing through an initial and stabilized narrow fusion pore, before the pore dilates releasing all or a fraction of the vesicle neurotransmitter content [5,6,7]. At partial vesicle release, a “post-spike foot” can sometimes be detected indicating that neurotransmitters are being released through a fusion pore that is closing for vesicle recapture and recycling [60]. As observed in both neuronal and chromaffin cells, a fusing vesicle can also be caught in a state of flickering that in amperometric recording can be displayed as a series of “stand-alone feet” signifying minuscule bursts of neurotransmitters release through a pore that by multiple rounds rapidly is opening and closing [63, 102]. The amplitude and lifetime of “foot features” portray information on fusion pore size, and kinetic information on fusion pore dynamics and stability. Hence, amperometry is a powerful analytical technique for quantitative single-cell exocytosis research and for characterization of mechanisms involved in regulation of neurochemical release.

Here illustrates a few examples how the exocytosis process can be studied using live cells and synthetic cell models. a A schematic of an experimental set-up for amperometric measurement of exocytosis at live single cell [37]. b A DIC image of a cell model from bleb plasma membrane of a chromaffin cell, scale bar equals 10 μm [60]. c A schematic of a protein-free cell model using DNA-zipper molecules mimicking the SNARE protein complex role in catalyzing vesicle fusion and neurotransmitter release [81]
Amperometry to characterize the biophysics of the exocytosis process
With the key features of amperometry, this technique has been applied in many studies regarding the biophysics and key molecules involved in vesicle fusion and controlling fusion pore dynamics. For instance, investigations on the role of components associated to the sophisticated fusion machinery composed by SNARE protein promoting vesicle membrane fusion and questions regarding the involvement of SNARE protein in modulating neurotransmitter release, quantitative and kinetic measurements using amperometry has been central, with much of the work performed at chromaffin cells [13, 20, 36, 44, 67]. It is still debated what is the energy needed in terms of the minimum number of SNAREs for merging of a vesicle with the plasma membranes to initiate fusion and what is the potential role for SNARE proteins in deforming membranes into a high-curvature narrow fusion pore, as well as the possibility by the SNARE proteins to influence subsequent fusion pore dynamics. By introducing nanolipoprotein discs, a system with reconstituted v-SNARE proteins designed for complex binding to t-SNAREs expressed at the plasma membrane of chromaffin cells, a model system to study the cooperative effect of SNARE proteins forming and controlling fusion pore dynamics has been developed. This system has shown that very few SNAREs are needed to induce fusion pore formation and by the presence of a larger number of SNAREs can upon complex formation trigger fusion pore dilation [17, 25, 94, 95].
To gain a better understanding on what prevents the vesicle from fully collapsing with the plasma membrane when partial exocytosis is triggered, studies on the biophysics of fusion pore dynamics have facilitated to overview parameters that affect size, stability, and life time of the fusion pore [3, 26, 28, 29, 40]. It has been debated whether the fusion pore is lipidic or dominated by protein, with recent studies indicating it might be a mixture of both [16, 80]. Consequently, incubating cells with phospholipids of various geometrical shapes, e.g., cylindrical shapes, cone, or inverted cone shape, which modifies the spontaneous curvature of the cell membrane and directly affects membrane energetics and membrane dynamics of the highly curved fusion pore. Depending on lipid shape, whether facilitating positive or negative membrane curvature, alterations in membrane composition has shown to affect fusion pore size and act to slow or speed up the rate of fusion pore dilation during exocytosis [8, 23, 60, 76, 88]. Modifying the biophysics in terms of membrane rigidity for instance by adjusting the membrane cholesterol alters fusion pore dynamics, with lower membrane cholesterol concentrations accelerating exocytotic release and higher concentrations stiffening the membrane, stabilizes the fusion pore structure by increasing pore life time [18, 27, 84, 97]. Membrane tension is another key biophysical force that in neuroendocrine cells have shown to be a key regulatory force affecting the mechanism for driving fusion pore dilation and when applied by ATP-driven actin assembly forces at the plasma membrane reduces the secretory vesicle size during the exocytosis release process [21, 73]. The influence of biophysics on the exocytosis process has also been studied in controlled systems by a simplified cell model system created from protein-free giant liposomes (as presented in Fig. 1c) or cell plasma membrane “blebs” of chromaffin cells, where membrane is kept at higher complexity and more similar to cell membranes in comparison to protein-free cell models (as shown in Fig. 1b) [22, 59, 65, 80]. Blebs are plasma membrane vesicles that can be achieved from subjecting cells in culture to different kinds of stress, for instance by chemical treatment using formaldehyde, which disrupt cytoskeletal attachment points to the plasma membrane and thereby induces formation of micron-sized cell plasma membrane protrusions at the cell that can serve as cell model membranes [79].
In a protein-free cell model, systems showed that membrane rigidity highly influences the speed of exocytosis and that membrane dynamics alone can drive the later stages of exocytosis and influence the stability of the fusion pore before dilation [23, 65, 81]. In experiments using a bleb cell model demonstrated that a threshold in membrane tension can reversibly dictate if full or partial exocytosis was triggered [59], and by altering the membrane rigidity by changing the membrane cholesterol concentration, higher cholesterol levels significantly enhanced fusion pore stability and life time when partial exocytosis release was triggered [59, 65]. Although much of the work so far has been performed on gaining a better understanding of membrane biophysics of exocytosis, the biophysical properties of the secretory vesicle are still quite poor and therefore future studies with this focus might also serve to better explain many of the complex mechanisms involved in regulating neurotransmission.
Secretory vesicle quantal size
The limiting amount of neurochemicals that can be released at an exocytosis event is naturally also directly related to the maximum number of neurotransmitter molecules that can be stored in a single vesicle compartment in the first place. This is often referred to as the vesicle “quantal size” referring to the basic unit of neurotransmitters that can be released as a discrete uniform “quanta” per exocytotic event [33, 48]. The secretory vesicles in neuroendocrine and neuronal cells are classified into two main groups; one is small synaptic vesicles containing low-molecular weight neurotransmitters such as dopamine, serotonin, norepinephrine, acetylcholine, or glutamate and mediate fast synaptic transmission. The other group is large dense-core vesicles (LDCVs) that typically contain monoamines (dopamine, adrenaline, noradrenaline, histamine, serotonin) and/or neuropeptides [2, 61]. Vesicles very efficiently accumulate high concentration of neurotransmitters, driven by proton and/or electrical gradients across the vesicle membrane depending on the type of neurotransmitter that is loaded into the vesicle [2, 32, 68]. The concentration of neurotransmitters in vesicles of chromaffin cells from adrenal glands is estimated to about 0.5–1 M [2, 32, 47, 62]. How the vesicle can perform this act and maintain charge neutrality and osmotic balance is still debated [19, 31, 35, 56]. The quantal size can be tuned using pharmacology enhancing or blocking the passage of neurotransmitters by specific transporter protein into the vesicle compartment or by affecting the proton/electrical gradient across the vesicle membrane and consequently affect the amount of transmitters released into the synaptic cleft [30, 51, 58, 74]. For neuroscience and pharmacological research, to elucidate how quantal size is maintained under physiological conditions and monitoring the effect on vesicle neurotransmitter content in response to potential drugs, analytical tools that are able to quantify the absolute neurotransmitter content in vesicles are crucial. However, universal methods for quantitative analysis of secretory vesicle content remain a challenge due to the variation in content and size of secretory vesicles. Quantitative measurement from the smallest of these organelles, the synaptic vesicle thus requires more sensitive and faster analysis techniques than probing the quantal content of larger dense-core vesicles in for instance neuroendocrine cells. Although fluorescence imaging with fluorescent dyes reporting on alteration in pH and membrane potential can offer real-time measurement of relative changes in vesicle quantal size [75], the only quantitative methods that count the absolute number of neurotransmitters encapsulated inside single secretory vesicles compartments are amperometry-based techniques [32, 51, 72]. In comparison to imaging methods for studying vesicle neurotransmitter loading, amperometry methods are to date limited to analysis of vesicles filled with neurotransmitters that are electroactive and performing single point-in-time measurements in contrast to measurements of relative dynamic changes in vesicle neurotransmitter content over time [32, 51, 72, 75].
Amperometry techniques for quantification of vesicle quantal size
To present answers to the central question on what regulates secretory vesicle quantal size and for gaining a better understanding on how cells may tune their signals through triggering different modes of exocytosis during neurotransmission, methods that can provide both quantitative information on the absolute neurotransmitter content of vesicles and to be complemented with methods monitoring the amount neurotransmitters released through secretion are needed. Therefore, to complement amperometry measurements of exocytosis that count the number of neurotransmitters released during an exocytosis event, development of methods for monitoring the total vesicle neurotransmitter content has been key, and was first employed by combining cell-attached patch clamp capacitance measurement together with amperometry recording [4]. Here, quantification was achieved by correlating the amperometric current transients from neurotransmitter release with increases in capacitance steps as a result of single vesicle fusion with the cell plasma membrane. The quantitative analysis from these experiments revealed partial release as an alternative mode of exocytosis. Subsequently, this method was further developed by placing an amperometric carbon fiber microelectrode inside a patch pipette to provide dual detection of changes in membrane capacitance from single-vesicle exocytotic fusion events together with amperometric quantitative information on the number of neurotransmitters molecules secreted from each single vesicle into the patch pipette [2]. By comparing the vesicle membrane surface area from the patch clamp recording and correlating to the simultaneous amperometric recording of these measurements, the neurotransmitter concentration of single vesicles was calculated. The patch amperometry technique was further used to determine quantal release from different modes of exocytosis [3]. However, for quantal size analysis, it requires that the full vesicle content be secreted during exocytosis and this technique has a low throughput in the number of vesicle that can be analyzed per measurement. Thereafter, for vesicle content analysis, an amperometric method based on separation of isolated vesicles using capillary electrophoresis with amperometric detection [70,71,72]. This technique referred to as “electrochemical cytometry” might suggest by the name that this method involves chemical analysis of single cells, but is rather a method for analysis of subcellular structures and here aimed for neurotransmitter content analysis of secretory vesicles. In this method, a sample of isolated vesicles is injected into a quartz capillary and by applying an electric field, the vesicles migrate through the capillary and elute one-by-one onto a microfluidic platform. Here, individual vesicles subsequently are subjected to a lysis buffer, which induce vesicles to rupture, resulting in a current spike for each vesicle lysed that discharge the vesicle content of neurotransmitters at the surface of an amperometric carbon fiber electrode. From Faraday’s law, the total charge detected for each current spike is used to calculate the quantal size of each single vesicle. Using this method, one single sample of isolated vesicles injected onto the capillary provides quantitative measurement for a very large number of vesicles and therefore provide both very good throughput and statistical data for each measurement. Experiments using this technique, revealed that, by comparing the average quantal size of synaptic dopamine vesicles from striatum or from LDCVs in PC12 cells, and comparing to the amount neurotransmitters that is secreted at exocytosis, indicated that only a part of the vesicle content was released at these cells where previously full exocytosis was assumed to be the most prevalent mode of exocytosis [70, 71]. Hence, the electrochemical cytometry method in combination with amperometry recording of exocytosis at single cells offers a means for studying regulatory mechanisms by secretory cells and to give potential answers to the debate on the prevalence and what triggers the different modes of exocytosis at various cell types. The electrochemical cytometry method was recently further simplified by the discovery that placing an amperometric carbon fiber microelectrode in the solution of isolated large dense-core vesicles from chromaffin cells (shown in Fig. 2), vesicles collide, adsorb, and subsequently stochastically rupture and thereby releasing the vesicle content at the electrode surface for amperometric detection and quantification [32]. The frequency of vesicle rupture using this method is dependent on the applied potential at the carbon fiber electrode and is enhanced when the applied potential is increased from + 0.7 to + 0.9 V at the electrode surface [55]. Moreover, rupturability can also be amplified by excitation of augmented fluorophore-labeled lipids in the vesicle membrane or by modification of membrane permeability by incubation of vesicles with dimethyl sulfoxide [64]. Although by catalyzing the efficiency in vesicle rupture during measurements can alter current spike kinetics, by influencing the speed neurotransmitters are release from a vesicle, the total charge detected per vesicle is not altered and hence does not affect vesicle quantal size analysis. A mechanism for this methodology has been suggested that after vesicle adsorption to the electrode surface, the applied redox potential at the electrode surface induces electroporation of the vesicle membrane if the vesicle is in close enough to the electrode surface and that causes an irreversible pore dilation leading to vesicle rupture. Although the detection efficiency of this method has not yet experimentally been verified, the consistency in vesicle quantal size at various experimental conditions tested, e.g., applying various amperometric potentials or excitation of fluorophores in the vesicle membrane, in conjunction with the fact that a larger number of neurotransmitters are detected to be accumulated in the vesicle compartment compared to the amount neurotransmitters that is expelled at exocytosis, indicates that if not all, a major part of the vesicle neurotransmitter content is detected by this method [52, 55]. These new electrochemical cytometry tools provide the ability to quantify changes in quantal size of isolated vesicles at controlled experimental conditions and to study how vesicle responds to changes in chemical and physical environments as well as in response to pharmacological agents.

a A schematic of experimental set-up for electrochemical cytometry of isolated secretory vesicles. b Scanning electron microscopy (SEM) image of a 33-micrometer carbon fiber disc electrode after exposing to LDCVs sample from PC12 cells. c Magnified SEM image illustrating fixed isolated vesicles adsorbed to the surface of a carbon disc electrode
Intracellular electrochemical cytometry
The secretory vesicle responds to transport and changes in gradients of analytes across the vesicle membrane, where components from the cytosol and the vesicle are shuttled by specific membrane transport proteins that are controlled and driven by proton or potential gradients across the vesicle membrane. Hence, the quantal size of an isolated vesicle might be affected when isolated into a defined isosmotic sample solution in comparison to when situated in the native environment of the cell cytoplasm, where the vesicles quickly can respond to a myriad of biochemical reactions in the surroundings. For instance, in the cytoplasm, a constant access of ATP feed the ATPase proton pump keeping a proton gradient across the vesicle membrane. From a constant synthesis, neurotransmitters are also actively pumped from the cytosol and into the vesicle compartment. To be able to study quantal size of vesicles in their native environment, the electrochemical cytometry method was further developed for in situ analysis of live single cells [51]. By flame etching a carbon fiber microelectrode into a tens of micrometers long sharp tip, with the tip size about 100 nm, this spear-shaped electrode was placed into the cytoplasm of live cells through applying a gentle mechanical force (as shown in Fig. 3). Similar to the electrochemical cytometry method, by applying an oxidation potential to the electrode surface, vesicles in the cytoplasm collide, adsorb, rupture, and release the vesicle content at the surface of the carbon electrode allowing quantification of vesicle neurotransmitter content in situ of live cells. In comparison to electrochemical cytometry of isolated vesicles, here, the sample matrix is very different to bulk conditions, as the electrode is placed in a highly crowded environment and the etched electrode provides a surface area with high curvature shape as opposed to using a disc microelectrode. Hence, it is important to keep in mind that electrode sensitivity differs between these two methods and therefore comparison with absolute measures is preferentially performed using the same type of electrode. For instance, an amperometric flame-etched carbon fiber nanotip electrode was placed at the cell surface of a secreting PC12 cells treated with L-3,4-dihydroxyphenylalanine (L-DOPA) compared to vesicle quantal size using the same electrode for intracellular electrochemical recording [51]. As L-DOPA is a precursor for dopamine production in cells, which consequently upon transformation into dopamine is selectively and rapidly transported by the vesicular monoamine transporter protein (VMAT) into the vesicle compartments for storage, treatment of cells with this drug increased vesicle quantal size and showed in these experiments that also an increased amount of neurotransmitters released was observed. These measurements also confirmed that only part of the vesicle content is released from PC12 cells at exocytosis and that the ratio of neurotransmitter released by L-DOPA-treated cells was similar to untreated cells. Therefore, this suggests that the exocytosis mode used by these cells is preserve during drug treatment [51]. Intracellular cytometry was also used to shed some light on the affect of osmotic pressure on secretory vesicles. Previously, it was observed that secretory cells respond to hypertonic stress by reducing exocytosis activity and the amount neurotransmitters released at exocytosis. Assumptions were made that alteration in membrane biophysical properties and cytosolic molecular crowding caused the reduction in the amount neurotransmitters released and in exocytosis activity, while assuming that secretory vesicles in the cytoplasm of cells do not sense or respond to extracellular osmotic stress. To provide better understanding regarding the effect of osmotic stress on vesicle quantal size at vesicles in their native environment, the total content of neurotransmitter in vesicles was probed directly in the cytoplasm of live cells before and after exposure to osmotic stress [37]. These quantitative measures of alteration in quantal size were compared to the amount neurotransmitters released during exocytosis at chromaffin cells and vesicle size measurements using transmission electron microscopy (TEM) imaging were all performed under the same different experimental conditions. As the result, the ability to compare relative changes in quantal size, both before and after exocytosis at different osmotic conditions, showed that vesicles respond to osmotic stress by shrinking and reducing the quantal size with the aim to maintain a constant vesicle neurotransmitter concentration and therefore less neurotransmitters are released by cells during osmotic stress [37]. Intracellular electrochemical cytometry was also used to complement nano scale secondary ion mass spectroscopy (NanoSIMS) that together with TEM imaging analysis was used to correlate chemical imaging of single vesicle granules and studying kinetics and mechanism for catecholamine loading into dense-core vesicles from chromaffin cells, where pharmacological treatment with isotope-labeled L-DOPA at shorter and longer times and together with the VMAT blocker reserpine was applied. Combining all three methods, an overview picture was created how catecholamine quickly loads into the halo solution of these vesicles from where a certain permeability for catecholamine exists over the vesicle membrane and that catecholamines with slower kinetics bind to the dense core protein that serve as a matrix to accumulate a neurotransmitters at high concentration in these vesicles while reducing the osmotic pressure inside the vesicle compartment [54].

a A schematic of an intracellular electrochemical cytometry experiment showing placement of a carbon fiber electrode in the cytoplasm of a cell. b A SEM image of a nanotip conical carbon fiber microelectrode with a tip size of electrode about 100 nm and a tip length of 30–100 μm. c A representative amperometric trace of intracellular cytometry recording from a chromaffin cell [37]
Conclusions
It has been realized that emitting cells have the ability to take part in regulation of signaling strength during intercellular communication both through regulation of content release by the mode of exocytosis triggered and by the amount of signaling molecule that is stored in secretory vesicles. In gaining a better overview in how this regulation is accomplished, analytical tools that can provide methods to study molecular mechanisms controlling fusion pore dynamics and factors influence loading and storage of neurotransmitter in secretory vesicle compartments are crucial. This is also important for better understanding of how drug treatment and disease might act and affect the regulatory aspects of neurochemical release, as well as characterizing the physiological effects in response to regulatory neurotransmission. In this quest, to study these dynamic processes in cells and secretory vesicles, cell model systems and analytical techniques that can provide sufficient sensitivity and temporal resolution to probe these ultra-small systems at physiological relevant conditions will be of great importance to clarify the essential regulatory aspects of exocytosis.
References
Alabi AA, Tsien RW (2013) Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu Rev Physiol 75:393–422
Albillos A, Dernick G, Horstmann H, Almers W, Alvarez de Toledo G, Lindau M (1997) The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 389:509
Alés E, Tabares L, Poyato JM, Valero V, Lindau M, Alvarez de Toledo G (1999) High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat Cell Biol 1:40–44
Alvarez DT, Fernandez-Chacon R, Fernandez J (1993) Release of secretory products during transient vesicle fusion. Nature 363:554–558
Amatore C, Arbault S, Bonifas I, Bouret Y, Erard M, Ewing AG, Sombers LA (2005) Correlation between vesicle quantal size and fusion pore release in chromaffin cell exocytosis. Biophys J 88:4411–4420
Amatore C, Arbault S, Bonifas I, Guille M (2009) Quantitative investigations of amperometric spike feet suggest different controlling factors of the fusion pore in exocytosis at chromaffin cells. Biophys Chem 143:124–131
Amatore C, Arbault S, Bonifas I, Guille M, Lemaître F, Verchier Y (2007) Relationship between amperometric pre-spike feet and secretion granule composition in chromaffin cells: an overview. Biophys Chem 129:181–189
Amatore C, Arbault S, Bouret Y, Guille M, Lemaître F, Verchier Y (2006) Regulation of exocytosis in chromaffin cells by trans-insertion of lysophosphatidylcholine and arachidonic acid into the outer leaflet of the cell membrane. Chembiochem 7:1998–2003
Amatore C, Arbault S, Lemaître F, Verchier Y (2007) Comparison of apex and bottom secretion efficiency at chromaffin cells as measured by amperometry. Biophys Chem 127:165–171
Amatore C, Bouret Y, Travis ER, Wightman RM (2000) Interplay between membrane dynamics, diffusion and swelling pressure governs individual vesicular exocytotic events during release of adrenaline by chromaffin cells. Biochimie 82:481–496
Amatore C, Oleinick AI, Svir I (2010) Reconstruction of aperture functions during full fusion in vesicular exocytosis of neurotransmitters. ChemPhysChem 11:159–174
Aravanis A, Pyle J, Tsien R (2003) Single synaptic vesicles fusing transiently and successively without loss of identity. Nature 423:643–647
Archer DA, Graham ME, Burgoyne RD (2002) Complexin regulates the closure of the fusion pore during regulated vesicle exocytosis. J Biol Chem 277:18249–18252
Artalejo CR, Elhamdani A, Palfrey HC (1998) Secretion: dense-core vesicles can kiss-and-run too. Curr Biol 8:R62–R65
Bader M, Holz RW, Kumakura K, Vitale N (2002) Exocytosis: the chromaffin cell as a model system. Ann N Y Acad Sci 971:178–183
Bao H, Goldschen-Ohm M, Jeggle P, Chanda B, Edwardson JM, Chapman ER (2016) Exocytotic fusion pores are composed of both lipids and proteins. Nat Struct Mol Biol 23:67–73
Bello OD, Auclair SM, Rothman JE, Krishnakumar SS (2016) Using ApoE nanolipoprotein particles to analyze SNARE-induced fusion pores. Langmuir 32:3015–3023
Biswas S, Yin S-R, Blank PS, Zimmerberg J (2008) Cholesterol promotes hemifusion and pore widening in membrane fusion induced by influenza hemagglutinin. J Gen Physiol 131:503–513
Borges R, Pereda D, Beltrán B, Prunell M, Rodríguez M, Machado JD (2010) Intravesicular factors controlling exocytosis in chromaffin cells. Cell Mol Neurobiol 30:1359–1364
Borisovska M, Zhao Y, Tsytsyura Y, Glyvuk N, Takamori S, Matti U, Rettig J, Südhof T, Bruns D (2005) v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J 24:2114–2126
Bretou M, Jouannot O, Fanget I, Pierobon P, Larochette N, Gestraud P, Guillon M, Emiliani V, Gasman S, Desnos C, Lennon-Duménil AM, Darchen F (2014) Cdc42 controls the dilation of the exocytotic fusion pore by regulating membrane tension. Mol Biol Cell 25:3195–3209
Burgoyne RD, Morgan A (1998) Analysis of regulated exocytosis in adrenal chromaffin cells: insights into NSF/SNAP/SNARE function. BioEssays 20:328–335
Cans A-S, Wittenberg N, Karlsson R, Sombers L, Karlsson M, Orwar O, Ewing A (2003) Artificial cells: unique insights into exocytosis using liposomes and lipid nanotubes. Proc Natl Acad Sci 100:400–404
Chen X-K, Xiong Y-F, Zhou Z (2006) “Kiss-and-run” exocytosis in astrocytes. Neuroscientist 12:375–378
Chen YA, Scales SJ, Patel SM, Doung Y-C, Scheller RH (1999) SNARE complex formation is triggered by Ca 2+ and drives membrane fusion. Cell 97:165–174
Chernomordik LV, Kozlov MM (2003) Protein-lipid interplay in fusion and fission of biological membranes. Annu Rev Biochem 72:175–207
Chintagari NR, Jin N, Wang P, Narasaraju TA, Chen J, Liu L (2006) Effect of cholesterol depletion on exocytosis of alveolar type II cells. Am J Respir Cell Mol Biol 34:677–687
Chizmadzhev YA, Kuzmin PI, Kumenko DA, Zimmerberg J, Cohen FS (2000) Dynamics of fusion pores connecting membranes of different tensions. Biophys J 78:2241–2256
Cohen F, Melikyan G (2004) The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J Membr Biol 199:1–14
Colliver T, Pyott S, Achalabun M, Ewing AG (2000) VMAT-mediated changes in quantal size and vesicular volume. J Neurosci 20:5276–5282
Daniels A, Williams R, Wright P (1978) The character of the stored molecules in chromaffin granules of the adrenal medulla: a nuclear magnetic resonance study. Neuroscience 3:573–585
Dunevall J, Fathali H, Najafinobar N, Lovric J, Wigström J, Cans A-S, Ewing AG (2015) Characterizing the catecholamine content of single mammalian vesicles by collision–adsorption events at an electrode. J Am Chem Soc 137:4344–4346
Edwards RH (2007) The neurotransmitter cycle and quantal size. Neuron 55:835–858
Elhamdani A, Azizi F, Artalejo CR (2006) Double patch clamp reveals that transient fusion (kiss-and-run) is a major mechanism of secretion in calf adrenal chromaffin cells: high calcium shifts the mechanism from kiss-and-run to complete fusion. J Neurosci 26:3030–3036
Estévez-Herrera J, Domínguez N, Pardo MR, González-Santana A, Westhead EW, Borges R, Machado JD (2016) ATP: the crucial component of secretory vesicles. Proc Natl Acad Sci U S A 113:E4098–E4106
Fang Q, Zhao Y, Lindau M (2013) Juxtamembrane tryptophans of synaptobrevin 2 control the process of membrane fusion. FEBS Lett 587:67–72
Fathali H, Dunevall J, Majdi S, Cans A-S (2017) Extracellular osmotic stress reduces the vesicle size while keeping a constant neurotransmitter concentration. ACS Chem Neurosci 8:368–375
Fatt P, Katz B (1952) Spontaneous subthreshold activity at motor nerve endings. J Physiol 117:109–128
Fesce R, Grohovaz F, Valtorta F, Meldolesi J (1994) Neurotransmitter release: fusion or ‘kiss-and-run’? Trends Cell Biol 4:1–4
Frolov VA, Lizunov VA, Dunina-Barkovskaya AY, Samsonov AV, Zimmerberg J (2003) Shape bistability of a membrane neck: a toggle switch to control vesicle content release. Proc Natl Acad Sci U S A 100:8698–8703
Fulop T, Radabaugh S, Smith C (2005) Activity-dependent differential transmitter release in mouse adrenal chromaffin cells. J Neurosci 25:7324–7332
Gosso S, Turturici M, Franchino C, Colombo E, Pasquarelli A, Carbone E, Carabelli V (2014) Heterogeneous distribution of exocytotic microdomains in adrenal chromaffin cells resolved by high-density diamond ultra-microelectrode arrays. J Physiol 592:3215–3230
Hafez I, Kisler K, Berberian K, Dernick G, Valero V, Yong MG, Craighead HG, Lindau M (2005) Electrochemical imaging of fusion pore openings by electrochemical detector arrays. Proc Natl Acad Sci U S A 102:13879–13884
Han X, Wang C-T, Bai J, Chapman ER, Jackson MB (2004) Transmembrane segments of syntaxin line the fusion pore of Ca2+−triggered exocytosis. Science 304:289–292
Heuser J, Reese T (1981) Structural changes after transmitter release at the frog neuromuscular junction. J Cell Biol 88:564–580
Jahn R, Sudhof TC (1994) Synaptic vesicles and exocytosis. Annu Rev Neurosci 17:219–246
Johnson R (1988) Accumulation of biological amines into chromaffin granules: a model for hormone and neurotransmitter transport. Physiol Rev 68:232–307
Katz B (1969) The release of neural transmitter substances. Thomas, Springfield
Kisler E (2009) Electrochemical detection and Total internal reflection fluorescence microscopy: illuminating the exocytotic mechanism, PhD Thesis. Cornell University, NY
Leszczyszyn DJ, Jankowski JA, Viveros OH, Diliberto EJ, Near JA, Wightman RM (1990) Nicotinic receptor-mediated catecholamine secretion from individual chromaffin cells. Chemical evidence for exocytosis. J Biol Chem 265:14736–14737
Li X, Majdi S, Dunevall J, Fathali H, Ewing AG (2015) Quantitative measurement of transmitters in individual vesicles in the cytoplasm of single cells with nanotip electrodes. Angew Chem Int Ed 54:11978–11982
Li X, Mohammadi AS, Ewing AG (2016) Single cell amperometry reveals curcuminoids modulate the release of neurotransmitters during exocytosis from PC12 cells. J Electroanal Chem 781:30–35
Lledo P-M (1997) Exocytosis in excitable cells: a conserved molecular machinery from yeast to neuron. Eur J Endocrinol 137:1–9
Lovrić J, Dunevall J, Larsson A, Ren L, Andersson S, Meibom A, Malmberg P, Kurczy ME, Ewing AG (2016) Nano secondary ion mass spectrometry imaging of dopamine distribution across nanometer vesicles. ACS Nano 11:3446–3455
Lovrić J, Najafinobar N, Dunevall J, Majdi S, Svir I, Oleinick A, Amatore C, Ewing AG (2016) On the mechanism of electrochemical vesicle cytometry: chromaffin cell vesicles and liposomes. Faraday Discuss 193:65–79
Machado JD, Camacho M, Alvarez J, Borges R (2009) On the role of intravesicular calcium in the motion and exocytosis of secretory organelles. Commun Integr Biol 2:71–73
Mainen ZF, Malinow R, Svoboda K (1999) Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature 399:151–155
Markov D, Mosharov EV, Setlik W, Gershon MD, Sulzer D (2008) Secretory vesicle rebound hyperacidification and increased quantal size resulting from prolonged methamphetamine exposure. J Neurochem 107:1709–1721
Mellander LJ, Kurczy ME, Najafinobar N, Dunevall J, Ewing AG, Cans A-S (2014) Two modes of exocytosis in an artificial cell. Sci Rep 4:3847
Mellander LJ, Trouillon R, Svensson MI, Ewing AG (2012) Amperometric post spike feet reveal most exocytosis is via extended kiss-and-run fusion. Sci Rep 2:907
Montesinos MS, Machado JD, Camacho M, Diaz J, Morales YG, de la Rosa DA, Carmona E, Castaneyra A, Viveros OH, O’Connor DT (2008) The crucial role of chromogranins in storage and exocytosis revealed using chromaffin cells from chromogranin a null mouse. J Neurosci 28:3350–3358
Morris S, Schultens H, Schober R (1977) An osmometer model for changes in the buoyant density of chromaffin granules. Biophys J 20:33–48
Mosharov EV, Sulzer D (2005) Analysis of exocytotic events recorded by amperometry. Nat Methods 2:651–658
Najafinobar N, Lovrić J, Majdi S, Dunevall J, Cans AS, Ewing A (2016) Excited fluorophores enhance the opening of vesicles at electrode surfaces in vesicle electrochemical cytometry. Angew Chem 128:15305–15309
Najafinobar N, Mellander LJ, Kurczy ME, Dunevall J, Angerer TB, Fletcher JS, Cans A-S (2016) Cholesterol alters the dynamics of release in protein independent cell models for exocytosis. Sci Rep 6:33702
Neher E, Marty A (1982) Discrete changes of cell membrane capacitance observed under conditions of enhanced secretion in bovine adrenal chromaffin cells. Proc Natl Acad Sci U S A 79:6712–6716
Ngatchou AN, Kisler K, Fang Q, Walter AM, Zhao Y, Bruns D, Sørensen JB, Lindau M (2010) Role of the synaptobrevin C terminus in fusion pore formation. Proc Natl Acad Sci U S A 107:18463–18468
Njus D, Kelley PM, Harnadek GJ (1986) Bioenergetics of secretory vesicles. Biochim Biophys Acta Rev Bioenerg 853:237–265
Oleinick A, Hu R, Ren B, Tian Z-Q, Svir I, Amatore C (2016) Theoretical model of neurotransmitter release during in vivo vesicular exocytosis based on a grainy biphasic nano-structuration of chromogranins within dense core matrixes. J Electrochem Soc 163:H3014–H3024
Omiatek DM, Bressler AJ, Cans A-S, Andrews AM, Heien ML, Ewing AG (2013) The real catecholamine content of secretory vesicles in the CNS revealed by electrochemical cytometry. Sci Rep 3:1447
Omiatek DM, Dong Y, Heien ML, Ewing AG (2010) Only a fraction of quantal content is released during exocytosis as revealed by electrochemical cytometry of secretory vesicles. ACS Chem Neurosci 1:234–245
Omiatek DM, Santillo MF, Heien ML, Ewing AG (2009) Hybrid capillary-microfluidic device for the separation, lysis, and electrochemical detection of vesicles. Anal Chem 81:2294–2302
Papadopulos A (2017) Membrane shaping by actin and myosin during regulated exocytosis. Mol Cell Neurosci S1044–7431
Pothos EN, Larsen KE, Krantz DE, Liu Y-J, Haycock JW, Setlik W, Gershon MD, Edwards RH, Sulzer D (2000) Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J Neurosci 20:7297–7306
Preobraschenski J, Zander J-F, Suzuki T, Ahnert-Hilger G, Jahn R (2014) Vesicular glutamate transporters use flexible anion and cation binding sites for efficient accumulation of neurotransmitter. Neuron 84:1287–1301
Ren L, Mellander LJ, Keighron J, Cans A-S, Kurczy ME, Svir I, Oleinick A, Amatore C, Ewing AG (2016) The evidence for open and closed exocytosis as the primary release mechanism. Q Rev Biophys 49:e12
Robitaille R, Tremblay J (1987) Non-uniform release at the frog neuromuscular junction: evidence of morphological and physiological plasticity. Brain Res Rev 12:95–116
Ryan TA (2003) Kiss-and-run, fuse-pinch-and-linger, fuse-and-collapse: the life and times of a neurosecretory granule. Proc Natl Acad Sci U S A 100:2171–2173
Scott RE (1976) Plasma membrane vesiculation: a new technique for isolation of plasma membranes. Science 194:743–745
Sharma S, Lindau M (2016) The mystery of the fusion pore. Nat Struct Mol Biol 23:5
Simonsson L, Kurczy ME, Trouillon R, Hook F, Cans A-S (2012) A functioning artificial secretory cell. Sci Rep 2:824
Staal RG, Mosharov EV, Sulzer D (2004) Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci 7:341–346
Stevens CF, Williams JH (2000) “Kiss and run” exocytosis at hippocampal synapses. Proc Natl Acad Sci U S A 97:12828–12833
Stratton BS, Warner JM, Wu Z, Nikolaus J, Wei G, Wagnon E, Baddeley D, Karatekin E, O’Shaughnessy B (2016) Cholesterol increases the openness of SNARE-mediated flickering fusion pores. Biophys J 110:1538–1550
Taraska JW, Perrais D, Ohara-Imaizumi M, Nagamatsu S, Almers W (2003) Secretory granules are recaptured largely intact after stimulated exocytosis in cultured endocrine cells. Proc Natl Acad Sci U S A 100:2070–2075
Trouillon R, Ewing AG (2014) Actin controls the vesicular fraction of dopamine released during extended kiss and run exocytosis. ACS Chem Biol 9:812–820
Trouillon R, Passarelli MK, Wang J, Kurczy ME, Ewing AG (2012) Chemical analysis of single cells. Anal Chem 85:522–542
Uchiyama Y, Maxson MM, Sawada T, Nakano A, Ewing AG (2007) Phospholipid mediated plasticity in exocytosis observed in PC12 cells. Brain Res 1151:46–54
van Kempen GTH, van den Berg R, Eilers P, Westerink R (2011) Three distinct modes of exocytosis revealed by amperometry in neuroendocrine cells. Biophys J 100:968–977
Wightman R, Jankowski J, Kennedy R, Kawagoe K, Schroeder T, Leszczyszyn D, Near J, Diliberto E, Viveros O (1991) Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc Natl Acad Sci U S A 88:10754–10758
Wightman RM, Haynes CL (2004) Synaptic vesicles really do kiss and run. Nat Neurosci 7:321–322
Wigström J, Dunevall J, Najafinobar N, Lovrić J, Wang J, Ewing AG, Cans A-S (2016) Lithographic microfabrication of a 16-electrode array on a probe tip for high spatial resolution electrochemical localization of exocytosis. Anal Chem 88:2080–2087
Wu X-S, Wu L-G (2014) The yin and yang of calcium effects on synaptic vesicle endocytosis. J Neurosci 34:2652–2659
Wu Z, Auclair SM, Bello O, Vennekate W, Dudzinski NR, Krishnakumar SS, Karatekin E (2016) Nanodisc-cell fusion: control of fusion pore nucleation and lifetimes by SNARE protein transmembrane domains. Sci Rep 6:27287
Wu Z, Bello OD, Thiyagarajan S, Auclair SM, Vennekate W, Krishnakumar SS, O’Shaughnessy B, Karatekin E (2017) Dilation of fusion pores by crowding of SNARE proteins. elife 6:e22964
Yamashita T, Tohyama M (2003) The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci 6:461–467
Yang S-T, Kreutzberger AJ, Lee J, Kiessling V, Tamm LK (2016) The role of cholesterol in membrane fusion. Chem Phys Lipids 199:136–143
Zhang B, Adams KL, Luber SJ, Eves DJ, Heien ML, Ewing AG (2008) Spatially and temporally resolved single-cell exocytosis utilizing individually addressable carbon microelectrode arrays. Anal Chem 80:1394–1400
Zhang Q, Cao Y-Q, Tsien RW (2007) Quantum dots provide an optical signal specific to full collapse fusion of synaptic vesicles. Proc Natl Acad Sci U S A 104:17843–17848
Zhang Q, Li Y, Tsien RW (2009) The dynamic control of kiss-and-run and vesicular reuse probed with single nanoparticles. Science 323:1448–1453
Zhao Y, Fang Q, Herbst AD, Berberian KN, Almers W, Lindau M (2013) Rapid structural change in synaptosomal-associated protein 25 (SNAP25) precedes the fusion of single vesicles with the plasma membrane in live chromaffin cells. Proc Natl Acad Sci U S A 110:14249–14254
Zhou Z, Misler S, Chow RH (1996) Rapid fluctuations in transmitter release from single vesicles in bovine adrenal chromaffin cells. Biophys J 70:1543–1552
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the special issue on the function of chromaffin cells in honor of Professor Antonio Garcia
This article is part of the special issue on Chromaffin Cells in Pflügers Archiv—European Journal of Physiology
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fathali, H., Cans, AS. Amperometry methods for monitoring vesicular quantal size and regulation of exocytosis release. Pflugers Arch - Eur J Physiol 470, 125–134 (2018). https://doi.org/10.1007/s00424-017-2069-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-017-2069-9