
Abstract
This manuscript presents practical recommendations for managing acute attacks and implementing preventive immunotherapies for neuromyelitis optica spectrum disorders (NMOSD), a rare autoimmune disease that causes severe inflammation in the central nervous system (CNS), primarily affecting the optic nerves, spinal cord, and brainstem. The pillars of NMOSD therapy are attack treatment and attack prevention to minimize the accrual of neurological disability. Aquaporin-4 immunoglobulin G antibodies (AQP4-IgG) are a diagnostic marker of the disease and play a significant role in its pathogenicity. Recent advances in understanding NMOSD have led to the development of new therapies and the completion of randomized controlled trials. Four preventive immunotherapies have now been approved for AQP4-IgG-positive NMOSD in many regions of the world: eculizumab, ravulizumab - most recently-, inebilizumab, and satralizumab. These new drugs may potentially substitute rituximab and classical immunosuppressive therapies, which were as yet the mainstay of treatment for both, AQP4-IgG-positive and -negative NMOSD. Here, the Neuromyelitis Optica Study Group (NEMOS) provides an overview of the current state of knowledge on NMOSD treatments and offers statements and practical recommendations on the therapy management and use of all available immunotherapies for this disease. Unmet needs and AQP4-IgG-negative NMOSD are also discussed. The recommendations were developed using a Delphi-based consensus method among the core author group and at expert discussions at NEMOS meetings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a rare and severe inflammatory autoimmune disease of the central nervous system (CNS) that was identified as a distinct clinical entity with the discovery of aquaporin-4 immunoglobulin G antibodies (AQP4-IgG) [90, 124, 137]. The 2015 international consensus-based diagnostic criteria are currently used to diagnose NMOSD. Applying these criteria, AQP4-IgG-positive NMOSD appears relatively straightforward to diagnose, but identifying AQP4-IgG-negative patients with these criteria is also possible [245].
Without proper treatment, patients with NMOSD may develop significant disability over time due to the recurrence of attacks and insufficient recovery from severe disease attacks [92, 108, 111]. Currently, there is no known curative treatment for NMOSD; therefore, the main goals of therapy are to counteract acute attacks promptly and effectively and to prevent future attacks by initiating immunotherapy as soon as a definite diagnosis of NMOSD is established. Recently, several prospective randomized controlled trials (RCT) have led to FDA approval of the first three immunotherapies for patients with AQP4-IgG-positive NMOSD: eculizumab in June 2019, inebilizumab in June 2020, and satralizumab in August 2020 [51, 179, 223, 250]. In addition, rituximab was approved for NMOSD in Japan in June 2022 based on the results of an investigator-initiated phase II/III clinical study [216], and in May 2023, the EMA approved ravulizumab for the treatment of AQP4-IgG-positive NMOSD.
The Neuromyelitis Optica Study Group (NEMOS), founded in 2008 as a nationwide network of primary, secondary, and tertiary care centers in Germany, aims to improve the care of patients with NMOSD (www.nemos-net.de) [92, 225], and the group updated its recommendations on the diagnosis and differential diagnosis of NMOSD in 2023 [88]. Interestingly, an international panel of clinical experts has very recently published a Delphi consensus on the management of AQP4-IgG-positive NMOSD, which focuses on recommendations for eculizumab, inebilizumab, and satralizumab [176]. In this article, the NEMOS group now provides its updated practical recommendations for the treatment of NMOSD, which cover the management of acute attacks and preventing future attacks through immunotherapy, including approved and established off-label treatments. These recommendations are based on current literature and the expert opinions of clinical care providers specialized in NMOSD, who are all members of NEMOS. Myelin–oligodendrocyte–glycoprotein (MOG)-IgG-associated disease (MOGAD) is now considered a distinct disease entity; therefore, recommendations for therapy in patients with MOGAD will be addressed separately [20, 89]. Patients with NMOSD who are negative for both AQP4-IgG and MOG-IgG in serum are described as “double-negative” throughout the manuscript.
Methods
The initial version of this manuscript was prepared by TK, KG, and AB as part of a core working group of 24 German neurologists from 15 NEMOS centers, all specialized in the management of NMOSD, through discussions at NEMOS meetings and sessions. The manuscript was then reviewed and edited by representatives of the group, and specific recommendations were developed by the core group through the Delphi method [159]. The revised version of the manuscript, including the recommendations, was distributed to all members of NEMOS (see Appendix) for further feedback and revisions. Survey results are presented in the supplementary material.
AQP4-IgG-positive NMOSD: from pathophysiology to therapy
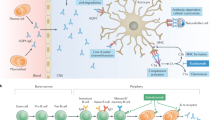
Autoantibodies against AQP4 are present in more than 80% of NMOSD patients and play a central role in disease pathogenesis [94]. An increased understanding of the pathophysiology of AQP4-IgG-positive NMOSD has led to the development of targeted therapeutic approaches. Lesions in AQP4-IgG-positive NMOSD patients are characterized by mixed immune cell infiltrates as well as IgG and complement (C9neo) deposition around blood vessels, associated with macrophage, neutrophil, and eosinophil infiltration [137, 138]. Complement-mediated enhancement of local inflammation and cytotoxicity are key elements in the disease process [125, 127, 128, 173]. These inflammatory reactions lead to loss of AQP4 expression on astrocytes, particularly at the blood–brain barrier, resulting in astrocytic loss and secondary damage to oligodendrocytes and neurons.
In addition to nonspecific immunosuppressants, such as oral glucocorticoids, azathioprine or mycophenolate mofetil, more tailored therapeutic approaches have been developed that specifically target the pathophysiology of the disease. Depletion of B cells through anti-CD20 antibodies such as rituximab has been successfully used as a treatment of NMOSD for more than 15 years [50], confirming the central role of B cells in the disease [48, 54, 244]. Recently, the monoclonal anti-CD19 antibody inebilizumab, which, in addition to B cells, also targets CD19-expressing antibody-producing plasmablasts, has been shown to be beneficial in AQP4-IgG-positive NMOSD [33, 209]. In addition, inhibiting the proinflammatory interleukin-6 (IL-6) pathway through drugs such as tocilizumab and satralizumab has also been effective in treating NMOSD by addressing blood–brain barrier dysfunction, CNS infiltration of immune cells, B cell differentiation into plasmablasts and plasma cells with increased AQP4-IgG secretion, and B cell survival [41, 70, 195, 219]. Moreover, inhibition of the terminal complement cascade by the C5 inhibitors eculizumab and ravulizumab has been effectively used for attack prevention, highlighting the central role of autoantibody-mediated complement activation in the final steps of NMOSD pathophysiology [14].
Treatment of NMOSD: general aspects and outcome measures
Outcome measures and attack definitions used in most studies of NMOSD have been developed for multiple sclerosis (MS) and have not been validated for NMOSD [240]. Since several features differentiate MS from NMOSD, such as the absence of progressive disease independent of acute attacks, more severe attacks, and mostly no informative changes on magnetic resonance imaging (MRI) over time, outcome measures used for MS are challenging to directly apply to NMOSD. As there are no evidence-based definitions of neuromyelitis optica attacks, elaborate definitions have been proposed in the context of placebo-controlled studies; however, they are yet to be standardized [49]. In addition, there is no validated method to evaluate attack severity. The expanded disability score (EDSS) and functional system scores (FSS) were adopted from MS to measure disability in NMOSD. Nevertheless, some dimensions, such as visual function, pain, fatigue, depression, cognition, and function of upper limbs are not adequately represented within the EDSS. Low contrast visual acuity as well as specific visual function questionnaires, such as the National Eye Institute-Visual Function Questionnaire, might be additional helpful tools to detect and monitor visual impairment [175]. Moreover, there is a lack of randomized controlled trials on the treatment of NMOSD attacks. While current definitions of NMOSD attacks may vary, they generally involve the emergence of new neurological symptoms and/or exacerbation of pre-existing ones that persist for more than 24 h, with or without new/enlarging or enhancing MRI lesions. Pseudo-attack, on the other hand, refers to a worsening of pre-existing neurological symptoms that can be attributed to other clinical factors, such as infections, fever, injury, comorbidities, adverse reactions to medications, change in mood, pain, or dysautonomia [100].
How to treat attacks?
Without treatment, the outcome of NMOSD attacks is often poor, with full recovery observed in only a minority of patients [92, 111]. However, recovery can be improved by prompt initiation of therapy in acute attacks and early escalation [5, 111]. Factors such as pre-existing disability, type and severity of an attack, age, and, most importantly, time interval to treatment initiation also contribute to recovery [19, 84, 110, 111, 131, 214].
The standard of care for acute attacks in both AQP4-IgG-positive and double-negative NMOSD are high-dose glucocorticoids and apheresis therapy. Methylprednisolone (MP) should be usually administered intravenously (i. v.) at a dose of 1000 mg per day for 3–5 days, followed by an oral MP taper (starting with either 1 mg/kg/day or 20–30 mg/day and then tapered to 10–15 mg/day within 2–3 weeks) in combination with proton pump inhibition and thrombosis prophylaxis. Low-dose oral glucocorticoids for up to 3–6 months are also considered beneficial for preventing subsequent early attacks, although there is a lack of prospective studies [232]. The duration of low-dose add-on glucocorticoid treatment depends on the AQP4-IgG serostatus, disease activity, mode of action, and the expected time to the attack-preventive effect of the subsequent immunotherapy.
If, within the first days, patients do not respond sufficiently to MP, rescue treatment with apheresis therapy, such as therapeutic plasma exchange (PE) or immunoadsorption (IA), should be administered early on [30, 111, 210, 241]. Most studies performed an average of 5 cycles daily or every other day, but up to 10 cycles may be applied [170]. IA has been used less frequently, and no clear difference has been established between PE and IA in terms of therapeutic outcomes, but data are limited, and more experience exists for PE [28, 110].
Several retrospective studies have shown that an early start of apheresis therapy is associated with better outcomes in NMOSD patients [29, 56, 110]. When apheresis therapy was started within 0–2 days of symptom onset without delay, up to 40% of patients showed complete remission; however, with a later treatment starting ≥ 7 days after symptom onset, only 3.7% of patients completely recovered [29, 56, 110]. Therefore, early adjunctive (add-on to glucocorticoids) apheresis therapy should be considered in patients with severe attacks. Using apheresis as the only first-line treatment without high-dose glucocorticoids was advantageous for patients with the presence of myelitis in one study [111]. A repeated treatment course has also been shown to improve outcomes and lower the number of non-responders [111]. Thus, patients may benefit from repeated therapy courses (apheresis and/or glucocorticoids) if they insufficiently responded or continued exhibiting functional deficits after the first or second treatment course for NMOSD attacks (Fig. 1). Other experimental therapy approaches, such as intravenous immunoglobulins (IVIG), early anti-CD20 therapy, and early anti-complement therapy, have been reported in single case series to possibly lead to favorable outcomes in acute attacks [65, 127, 132, 147]. Future therapies may also include antibodies against the neonatal Fc receptor, which are currently being studied in an open-label trial [236, ClinicalTrials.gov].

Attack therapy
How to prevent attacks?
The second main goal of NMOSD therapy is to prevent further attacks. Unlike MS, disability in NMOSD mainly results from poor and incomplete recovery from clinical attacks [56, 92, 111, 154, 247]. AQP4-IgG-positive NMOSD typically takes a relapsing course in almost all cases, with patients having a particularly high risk for attacks in the year following onset or any subsequent attack. However, some patients may experience several years and even decades without attacks [4, 242]. Risk factors for attacks and disability in AQP4-IgG-positive NMOSD include the age of onset, sex, ethnicity, onset attack phenotype, and treatment [171]. Studies have shown that the risk of attacks is similar between AQP4-IgG-positive and AQP4-IgG-negative patients, even when comparing strictly double-negative and AQP4-IgG-positive NMOSD [96, 206]. As every attack can leave a patient permanently disabled and worsen health-related quality of life, early initiation of immunotherapy is crucial to prevent further clinical episodes and avoid long-term neurological sequelae [25, 148].
Before 2019, recommendations on the long-term treatment of NMOSD, including ours, were mainly based on retrospective studies, case series, open-label trials, and expert opinions without any approved immunotherapies. The serostatus (AQP4-IgG-positive vs. double-negative) was not considered in treatment algorithms. Azathioprine, mycophenolate mofetil, or rituximab and in some countries also oral glucocorticoids were commonly used as first-line agents for both AQP4-IgG-positive and -negative patients, with the reduction of attack rates analyzed irrespective of the serostatus in retrospective studies. In multiple non-RCTs, rituximab was shown to be superior to mycophenolate mofetil and azathioprine, and it has become the preferred first-choice therapy in recent years [83, 149, 160]. The IL-6 receptor (IL-6-R) antibody tocilizumab has also been increasingly used as rescue therapy, showing benefits in several case series [12, 194, 195].
Four therapies, eculizumab, inebilizumab, and satralizumab and most recently ravulizumab have been approved for use in AQP4-IgG-positive NMOSD since 2019. Rituximab was also approved for NMOSD in Japan in 2022 following the positive results of the placebo-controlled RIN-1 trial [216]. Nevertheless, inclusion criteria and methods varied considerably between studies and indications for use do not fully reflect study populations. The order of preference for these therapies is yet unclear, and further comparative trials and real-world data are needed. In addition, outside of clinical studies, fatalities have been reported during treatment with these new therapies [193]. However, clinical data on these cases are not yet published, and causality and mortality risks are still unknown.
In the following section, we summarize and discuss specific therapies that have been widely used or recently approved and provide specific recommendations for choosing, initiating, and maintaining therapy regimes. For an overview, see also Table 1.
Specific therapies (see also Tables 1, 2, 3)
Classical immunosuppressants: azathioprine, mycophenolate mofetil, low-dose oral glucocorticoids
Oral glucocorticoid therapy broadly suppresses the immune system through both genomic and non-genomic mechanisms by binding to glucocorticoid receptors expressed on lymphocytes. On one hand, low-dose glucocorticoids are used long-term in AQP4-IgG-negative NMOSD patients (often in MOG-IgG-positive patients) as well as in patients with concomitant autoimmune disease, mostly as an adjuvant yet sometimes as monotherapy in countries, where monoclonal antibodies and/or other immunosuppressive therapies are not available. On the other hand, low-dose glucocorticoids are also used for a shorter period as a bridge after an attack until the full effect of subsequent immunotherapy is achieved. There are no controlled data on dosing or the optimal timing for tapering and stopping add-on glucocorticoid therapy. One study reported a safe and effective use of azathioprine combined with low-dose glucocorticoids in Chinese patients with AQP4-IgG-positive NMOSD [185]. Moreover, low-dose glucocorticoid monotherapy (5–15 mg) prevented attacks in some Asian patients with NMOSD in small retrospective studies, showing that attacks are more likely to occur with rapid tapering and doses below 10 mg/day [218, 237]. However, long-term use of glucocorticoids is associated with several side effects, including infections, diabetes mellitus, weight gain, Cushing’s syndrome, and osteoporosis [166] (Table 1). Therefore, a daily prednisolone dose of 7.5 mg or less should ideally be achieved if used as a long-term treatment.
Azathioprine and mycophenolate mofetil have been used as the standard of care in (adult and pediatric) NMOSD for more than 20 years, and they effectively reduce attack rates in both AQP4-IgG-positive and -negative patients, as shown in retrospective studies and non-RCTs [27, 213, 235]. They are widely available and affordable but not approved for NMOSD. Comparisons of effectiveness in the literature are inconsistent, with most studies suggesting that both compounds have comparable efficacy and are similarly inferior to rituximab [39, 74, 160, 183].
Azathioprine, a purine analogue, is used to inhibit the activation and differentiation of lymphocytes. Due to its presumably delayed onset of action, it is often combined with oral glucocorticoids for up to 6 months after initiation of therapy [47]. Thiopurine S-methyltransferase polymorphisms appear to modify the risk of side effects [73, 134]. Allopurinol and other xanthine oxidase inhibitors should be avoided as they may increase the plasma levels of active metabolites of azathioprine. Long-term use of azathioprine for neuroimmunological diseases, such as MS or NMOSD, has been studied for more than 20 years, and an increased risk of malignancies is known, such as skin cancer but also lymphoproliferative disorders [45, 122]. One case of progressive multifocal leukoencephalopathy (PML) in a patient with NMOSD under monotherapy with azathioprine has also been reported [67].
Mycophenolate mofetil is a prodrug of mycophenolic acid, which suppresses the proliferation of lymphocytes by inhibiting the inosine-5'-monophosphate dehydrogenase, thereby depleting guanosine nucleotides preferentially in T- and B-lymphocytes. It takes approximately 6–12 weeks for the full therapeutic effect to be seen. It is recommended that mycophenolate mofetil is combined with overlapping oral glucocorticoids for at least the first 3 months of therapy [156]. There is a possibly increased risk of malignancy associated with the use of mycophenolate mofetil, and it must be avoided during pregnancy.
B cell depletion
Rituximab
Rituximab is a chimeric monoclonal antibody that binds to and depletes CD20-positive B-cells. Its clinical effectiveness in NMOSD has been demonstrated in various retrospective and prospective studies, including AQP4-IgG-positive and -negative (adult and pediatric) patients, with a reduction of attack rates by over 80% [52, 53, 61, 74, 135, 253]. A recent randomized, double-blinded, placebo-controlled trial from Japan (RIN-1) confirmed the efficacy of rituximab in AQP4-IgG-positive NMOSD, although the sample size of the trial was small (Table 2) ([216]. Patients received 375 mg/m2 body surface of rituximab i. v. every week for 4 weeks, followed by 6-month interval dosing (1,000 mg every 2 weeks, at 24 weeks and 48 weeks). Based on the results of the trial, rituximab has been recently approved for the treatment of NMOSD in Japan but not in other regions of the world, including Europe and the United States, where it is widely used as an off-label therapy. Therefore, reimbursement by healthcare systems may be an issue in many countries.
The onset of action of B-cell depletion is expected within a few weeks; CD19/20-positive B-cells and CD27 memory cells may be used as surrogate markers for treatment monitoring and re-dosing [1, 22, 105, 217]. As efficient B-cell depletion is essential to avoid attacks and given that most patients exhibit depleted B-cells in the peripheral blood for at least 6 months after a rituximab infusion, re-dosing every 6 months is considered an adequate retreatment frequency. Although the optimal strategy for rituximab re-dosing in NMOSD has yet to be determined, a retreatment dosage of a single infusion at a dose of 500 mg or 1000 mg is often used in a real-world setting [103]. As there is evidence that attacks may occur within the initial 6 months after starting rituximab therapy [22, 37, 76, 207] and the time to full clinical effectiveness is presumed to be delayed, concomitant oral glucocorticoid tapering is recommended after therapy initiation. Anti-drug antibodies (ADA) may affect the effectiveness and should be considered in patients with failing B-cell depletion and ongoing disease activity during the disease course [165]. In addition, fragment c gamma receptor 3A (FCGR3A) polymorphisms (F/F variant) may also impact the response to rituximab [105].
While the use and experience of rituximab in NMOSD comprise more than 15 years, controlled safety data are limited. The safety profile of rituximab is generally acceptable but may depend on factors, such as age, gender, weight, disability, duration of therapy, and history of prior immunosuppressive therapies [22, 107, 228]. Most adverse events observed in the RIN-1 study and its open-label extension (OLE) RIN-2 study were mild and infusion-related (Table 1) [216, 217]. The risk of hypogammaglobulinemia and ensuing infections, which typically comprise infections of the upper respiratory and urinary tract, increases with treatment duration [15, 107, 141, 198, 228]. Serology testing for hepatitis B virus (HBV) is mandatory as there is a potentially life-threatening but avoidable risk of reactivation. Opportunistic infections are rare in rituximab monotherapy, and there are rare cases of PML reported in rituximab-treated cancer patients and patients with rheumatological diseases, such as systemic lupus erythematosus (SLE), as well as in patients receiving additional immunosuppressive therapies, but none in NMOSD [23, 155]. Rare cases of leukopenia and severe neutropenia, or a prolonged B-cell depletion lasting up to several years after rituximab use, have been described in NMOSD patients [182, 192, 205].
Inebilizumab
Inebilizumab is a humanized monoclonal antibody that binds to and depletes CD19-positive B-cells, including CD19-positive subpopulations of (auto)antibody-producing plasma cells. Data from a phase III randomized placebo-controlled study (N-Momentum) and its OLE [51, 142, 188] showed that inebilizumab significantly reduced the attack rate in patients with NMOSD compared to placebo, which led to a preponed end of the blinded phase of the trial [51] (Table 2). Moreover, the trial showed that the risk of an EDSS-based disability progression confirmed after 3 months was lower in patients who received inebilizumab [142]. During the OLE (median 4.5 years), the observed effects of inebilizumab were persistent and even enhanced, with most attacks occurring during the first year of treatment and without new safety concerns [188].
Inebilizumab was initially approved in 2020 in the United States for the treatment of adult patients with AQP4 IgG-positive NMOSD. Approvals have since been granted in many regions of the world, including Europe in 2022 (as monotherapy).The onset of action and clinical effectiveness is unclear, but 4 week post-induction B-cells and plasma cells are reduced to < 10% of baseline [24]. In addition, 6 months after treatment initiation, 70% of patients exhibit a depletion to ≤ 4 cells/µl [24]. Serum concentrations of glial fibrillary acidic protein (sGFAP) and of neurofilament light chain (sNfL) were significantly lower in patients treated with inebilizumab compared to placebo [6, 7]. ADAs were observed in a subset of participants but had no impact on the efficacy of inebilizumab [24]. The impact of the FCGR3A F/F variant in patients treated with inebilizumab and in the context of B-cell depletion needs to be explored in larger populations [24].
The side effects of inebilizumab treatment include infections and infusion-related reactions, which mainly occur with the first infusion and are mild to moderate in severity [48] (Table 1). No cases of opportunistic infections or reactivations of viral infections have been reported so far. Two patients died during the open-label period: one who had experienced severe pneumonia followed by an attack shortly before enrolment into the open-label period died from respiratory insufficiency shortly after the first dose of 300 mg inebilizumab, while the other developed new neurological symptoms and a large cerebral lesion after three doses of inebilizumab. PML was considered a differential diagnosis but was not confirmed. To date, no other cases of suspected PML or other opportunistic infections associated with inebilizumab therapy have been reported, although a PML warning is included in the product information.
Complement inhibition
Eculizumab
Eculizumab is a humanized monoclonal antibody that binds to the complement protein C5 and disrupts the terminal complement cascade. It was originally developed for rheumatological diseases and was initially approved in 2007 to treat paroxysmal nocturnal hemoglobinuria (PNH), followed by approval for other indications, including myasthenia gravis. A small pilot study (n = 14) [181] followed by the phase III randomized placebo-controlled study (PREVENT) [179, 246] evaluated the efficacy of eculizumab in NMOSD. These studies included only adult AQP4-IgG-positive patients with high disease activity (Table 2). The results of the PREVENT trial showed that eculizumab, as a monotherapy or add-on therapy, significantly reduced the risk of attacks and was effective across all subgroups compared to placebo [172, 180, 246]. Today, eculizumab is approved for the treatment of adult patients with AQP4-IgG-positive NMOSD in the United States (2019), Japan (2019), and other countries. In Europe, it was approved in 2019 for patients with a relapsing disease course (i.e., after a second attack).
Eculizumab has a rapid onset of action and provides continuous near-complete inhibition of C5 activity upon the first infusion. There have been occasional reports of the development of ADAs against eculizumab, but these do not affect the therapeutical efficacy of the drug [69]. An impact of genetic variants in the C5-encoding gene on the response to eculizumab in patients with PNH has been described [161], but their role in NMOSD remains unclear. Long-term effectiveness data for eculizumab in NMOSD are limited, and the OLE was terminated early in most countries after the drug approval [246]. A recent study of 55 patients treated with eculizumab in Germany and Austria reported that all patients were attack-free over a median observation time of 14.6 months (IQR 7.4–21.2) [193].
Long-term safety data for eculizumab use in NMOSD (i.e., treatment > 5 years) are primarily derived from other indications. Serious infections, including those with encapsulated bacteria, such as meningococcal and Neisseria gonorrhoea, have been reported—despite meningococcal vaccination [181, 246]. In the PREVENT trial, one patient receiving concomitant eculizumab and azathioprine treatment died from pleural empyema with positive streptococcal cultures. Infusion-related reactions and anaphylaxis are rare, and no NMOSD patient has discontinued therapy due to these reactions in the phase III trial [179].
Ravulizumab
Ravulizumab is a long-lasting monoclonal antibody that targets the complement factor C5. It has been engineered to exhibit altered intracellular antibody recycling and has a four times longer half-life than eculizumab. Therefore, it needs to be administered i. v. only once every 8 weeks. Today, it is approved for the treatment of PNH, aHUS and myasthenia gravis [132], and the EMA recently approved ravulizumab for the treatment of adult patients with AQP4-IgG. Due to its more straightforward application scheme, it may replace the current complement inhibitor eculizumab as in other indications.
The phase III trial investigating ravulizumab for AQP4-IgG-positive NMOSD has met its primary endpoint (CHAMPION–NMOSD) [178]; the OLE is still ongoing (Table 2). In this trial, ravulizumab was given open-label and compared to the placebo arm of the pivotal PREVENT as an external comparator, since a randomized placebo-controlled trial was deemed inappropriate from the perspective of research ethics. Alternatively, a non-inferiority trial comparing ravulizumab with eculizumab was not feasible: Due to the rarity of events (NMOSD attacks), the trial would have necessitated enrolling several thousands of patients.
After at least 50 weeks on drug, no attacks were observed with ravulizumab (compared to 20 in 47 PREVENT placebo patients in the same period of time). No differences in the treatment effect were observed between patients who had received rituximab before ravulizumab compared to those who did not. A sensitivity analysis using propensity score methods revealed that including the historic placebo arm as a comparator did not introduce significant bias [8]. In addition, analysis of ravulizumab’s pharmacodynamics showed a near identical inhibition of C5 as with eculizumab [168]. The onset of action is rapid, and near-complete complement inhibition is observed after the first infusion. The development of ADAs against ravulizumab was so far only observed in patients with myasthenia gravis and not in patients with NMOSD [229]. The side effects of ravulizumab are similar to those of eculizumab. Two meningococcal infections were reported during the CHAMPION trial (one patient was under monotherapy; the other was concomitantly treated with mycophenolate mofetil and prednisolone, had been exposed to rituximab 13 months before and still showed reduced CD19 B-cell counts); both resolved without sequelae. No deaths were reported [178].
Long-term data on ravulizumab are not available yet. Nevertheless, as the study design and efficacy data of both eculizumab and ravulizumab are based on the same comparator (the PREVENT placebo arm), both drugs may be considered closely related members of the same drug class, and recommendations on the use of ravulizumab can reasonably be adopted from the hitherto use of eculizumab.
In general, complement inhibition increases the risk of infections with encapsulated bacteria up to 2000-fold [144]. To prevent such infections, it is essential that patients complete meningococcal vaccination at least 2 weeks before starting on eculizumab or ravulizumab treatment. Alternatively, if therapy needs to be started immediately, such as in many cases of NMOSD, patients must receive antibiotic prophylaxis until at least 2 weeks after completed vaccination. There are no data on the vaccination response in NMOSD patients during or after immunotherapy (e.g., prior rituximab treatment) or shortly after attack treatment (e.g., after high-dose glucocorticoids). Recent reports have shown that NMOSD attacks can occur within 2 weeks of receiving meningococcal vaccination [193], and the European product information on eculizumab warns of disease activity in all eculizumab-treated indications following meningococcal vaccination. As a result, starting treatment with a complement inhibitor under antibiotic prophylaxis and postponing vaccination may be a preferred option. However, more studies are needed to determine the best vaccination timing with an adapted risk–benefit ratio in patients with active NMOSD. As vaccination does not reliably prevent meningococcal infection, patients must be comprehensively counselled about symptoms indicative of meningitis before treatment is started (Table 1).
Interleukin-6 receptor blockade
Tocilizumab
Retrospective and prospective case series since 2013 have reported that IL-6-R blockade with tocilizumab can effectively prevent attacks in NMOSD, mostly after standard immunotherapies, including rituximab [12, 16, 136, 194, 195]. The recent TANGO trial also showed that tocilizumab is more effective than azathioprine in preventing attacks in highly active NMOSD [255]. However, data on tocilizumab as a first-line therapy for NMOSD are scarce, and tocilizumab has not been granted regulatory approval for NMOSD. The onset of action can be expected after a few weeks. One study investigating tocilizumab treatment within 2 weeks after an NMOSD attack showed favorable effects on the disease course [63]. Nevertheless, data on the long-term use of tocilizumab in NMOSD remain scarce and mainly arise from other indications. Side effects are similar to satralizumab (see next paragraph and Table 1) but include the risk of diverticulitis and gastrointestinal perforations [97].
Satralizumab
Satralizumab, a humanized monoclonal IgG2 antibody, has been developed to have optimized antibody recycling and a longer half-life compared to tocilizumab. Two pivotal clinical trials, SAkuraStar and SAkuraSky, have been conducted to evaluate the efficacy of s. c. satralizumab in reducing the attack rate in patients with NMOSD, either as a monotherapy (SAkuraStar) or as an add-on treatment (SAkuraSky; Table 2). Satralizumab was effective in patients with AQP4-IgG-positive but not in patients with AQP4-IgG-negative NMOSD in these trials. However, they did not have sufficient power to draw definitive conclusions for AQP4-IgG-negative patients [223, 250].
Satralizumab has been approved for the treatment of AQP4-IgG-positive NMOSD in adults and adolescents aged 12 and older in Canada (2020), the United States (2020), Japan (2020), South Korea (2021), and Europe (2021). However, there is limited experience using satralizumab as a first-line therapy for adolescents with NMOSD (n = 4 in SAkuraStar treated with satralizumab, no adolescents in SAkuraSky). The clinical onset of action for satralizumab is believed to be within 8–12 weeks, based on the results of the two clinical trials [223, 250]. Long-term effectiveness data are available from the OLE of the SAkuraSky and SAkuraStar trials, which included 111 AQP4-IgG-positive patients treated with satralizumab for a median of 4.4 years, and showed that > 70% of patients remained attack-free, 90% were free from severe attacks, and > 85% did not experience worsening of their EDSS score [113]. Long-term safety data from both pivotal trials, collected for up to 7 years with a median treatment exposure of 4 years, have shown no new safety concerns, and no opportunistic infections have been reported thus far [251].
ADAs have been observed in a significant number of patients in the pivotal trials of satralizumab, with rates of 41% and 71%. However, ADAs were found to have no effect on treatment efficacy. Another practical issue is the effect of satralizumab on several CYP450 enzymes, which may require dose adjustments for drugs with a narrow therapeutic index (e.g., warfarin and carbamazepine).
Other drugs and interventions
IVIG therapy (“high-dose”; up to 1 g per kg body weight every 4 weeks) may be beneficial in NMOSD and used particularly in children and patients with contraindications to other treatments, or as an add-on therapy. Individual case reports, and case series in AQP4-IgG-positive and -negative NMOSD have shown a reduction of attack frequencies [220, 227]. One study found that IVIG (0.4 g/kg/day every 1–3 months) effectively treated NMOSD as an add-on to azathioprine [130]. Yet, on the other hand, concomitant IVIG administration with monoclonal antibodies may lead to reduced antibody levels. For instance, when given concomitantly with eculizumab, IVIG increased the elimination of eculizumab due to neonatal fragment constant receptor (FcRn) saturation [243]. In addition, IVIG at lower doses (regimens vary from country to country) are substituted in patients who experience symptomatic hypogammaglobulinemia during anti-CD19/20 therapy.
Methotrexate may also be effective either as a monotherapy, a step-down, or an add-on approach [186]. It may be considered as a monotherapy when other drugs are unavailable or contraindicated or for patients with rheumatological comorbidities. In addition, in several retrospective studies, tacrolimus has shown beneficial effects, mainly in Asian NMOSD patients [38, 114].
Finally, intermittent PE or IA combined with or without concomitant immunosuppressive therapy has also been shown to prevent attacks in severe NMOSD in individual case reports [79, 116, 153].
Stem cell therapy
Autologous hematopoietic stem cell transplantation (HSCT) has been employed in cases of refractory NMOSD with varying treatment regimens and results, with some improvement in disease activity and disability reported [10, 34, 35, 75, 115]. The inclusion of rituximab in the conditioning regimen before HSCT may improve treatment outcomes. Specifically, administering rituximab twice, once before PE and HSCT and a second time 1 day afterwards, has been suggested to be beneficial [34]. A meta-analysis of 9 studies involving a total of 39 NMOSD patients who underwent autologous HSCT reported a progression-free survival rate of 69%. Another PRISMA-compliant meta-analysis reported a good safety profile with intermediate-intensity regimens [158, 256]. The onset of action of HSCT is prompt, but the procedure bears a risk of severe infections and prolonged immunosuppression, although, unlike allogeneic HSCT, it poses no risk of a graft-versus-host-reaction. Nevertheless, the data on the long-term effectiveness of HSCT in NMOSD remain very limited. Compared to other highly effective approved immunotherapies, HSCT is yet to be studied in randomized, actively controlled trials and—if at all—may be used for individual cases of refractory NMOSD. Other experimental approaches, such as allogeneic HSCT, autologous and allogeneic mesenchymal stem cell transplantation, and peptide-loaded tolerogenic dendritic cell transplantation, have been performed in selected NMOSD patients [115].
Ineffective drugs and drugs not recommended
Certain drugs that are approved for MS treatment, such as beta-interferons, glatirameroids, natalizumab, alemtuzumab, sphingosine 1-phosphate receptor modulators, or fumarates, should not be used in patients diagnosed with NMOSD, especially in AQP4-IgG-positive cases. Some agents, such as glatirameroids, are merely ineffective in preventing attacks [17, 174], while others, such as interferon beta, natalizumab, fingolimod, alemtuzumab and dimethyl fumarate, have also been reported to trigger severe attacks [18, 95, 112, 152]. Mitoxantrone may have some effects on NMOSD attack rate [106] but should no longer be used due to its unfavorable safety profile and the limited duration of treatment. Data on cyclophosphamide in NMOSD are conflicting, and its use is not recommended due to the limited total dose allowance and potentially severe side effects [26, 87, 233].
AQP4-IgG-positive NMOSD: which drug to choose and how to start?
The efficacy of therapeutic antibodies in treating AQP4-IgG-positive NMOSD is superior to classical immunosuppressants and makes them the drugs of choice. Eculizumab, ravulizumab, inebilizumab, rituximab, and satralizumab have all demonstrated efficacy in RCTs for AQP4-IgG-positive NMOSD, although data from eculizumab, ravulizumab, inebilizumab, and satralizumab trials for treatment initiation (“first-line” therapy) after diagnosis is more limited.
Unfortunately, no head-to-head studies between monoclonal antibodies, including rituximab, have been conducted to date. A network meta-analysis on the RCT data of eculizumab, inebilizumab and satralizumab, with time to a first attack as the efficacy outcome, suggests that complement inhibition with eculizumab may be more effective in preventing NMOSD attacks than treatment with inebilizumab or satralizumab [248]. However, these results are limited by several methodological differences across the trials, including study population, inclusion criteria and attack definitions.
Instead, the choice of first-line treatments for NMOSD has to rely on several factors, which include disease activity and severity, mode and onset of action, possibility to combine it with immunosuppressive drugs, effect on autoimmune and other comorbidities, gender (“family planning”), frequency and route of drug administration (intravenous vs. s. c.), side effects and safety profile, as well as drug availability and regulatory approval status (see also Table 3). Factors, such as costs and patient- and physician preferences, also influence treatment decision-making.
Age is another important factor to consider. Satralizumab is the only drug that is approved for adolescents (≥ 12 years). Rituximab, azathioprine, and mycophenolate mofetil have been used in NMOSD patients < 18 years; however, data on the treatment of pediatric NMOSD are scarce and beyond the scope of this review. The experience with immunotherapies in very late-onset NMOSD (≥ 70 years) is also limited, and only very few patients in this age group have been studied in the context of RCTs. When choosing immunotherapy in elderly NMOSD patients, immunosenescence and the higher risk of comorbidities and infections should be considered. Some decision-making can be made based on experiences with other diseases, such as myasthenia gravis or rheumatological diseases (Fig. 2).

Long-term therapy for AQP4-IgG-positive NMOSD
Does double-negative NMOSD exist, and how shall we treat it?
The existence of true double-negative NMOSD and whether it should be included in diagnostic criteria for NMOSD is currently a matter of debate. The pathology of double- negative NMOSD is less well-understood and many previous studies on AQP4-IgG-negative NMOSD did not test for anti-MOG-IgG as long as MOGAD was not considered a distinct disease and reliable testing was not available. Studies have shown that many patients previously considered seronegative actually harbor in up to 40% MOG-IgG, and the number of true double-negative patients is likely to be lower than previously assumed [46, 77, 91, 109, 187]. In these cases, potential differential diagnoses such as MS, other autoantibody-associated diseases (e.g., anti-GFAP-IgG, anti‐CV2/CRMP5 antibodies), or rare diseases (e.g., neurosarcoidosis) must be thoroughly excluded [66, 88, 93]. If no alternative diagnoses can be found, patients are considered to have double-negative NMOSD. At present, it is unclear if double-negative NMOSD represents a unique disease entity or pathogenetically heterogeneous disease conditions. Thus, previously reported experiences and data on antibody-negative NMOSD should be interpreted with caution.
Currently, there are no approved therapies for double-negative NMOSD. Eculizumab, ravulizumab, inebilizumab and satralizumab are approved only for use in AQP4-IgG-positive NMOSD. In the pivotal trials for satralizumab and inebilizumab, some AQP4-IgG-negative NMOSD patients were included (some of them tested positive for MOG-IgG), but the overall number of patients was too low to draw valid conclusions concerning efficacy. Therefore, treatment recommendations for these patients are still based on expert opinions and usually consist of classical immunosuppressive therapies and rituximab. In refractory cases, treatment with tocilizumab has been shown to reduce the risk of attacks in some patients [195, 252, 255]. The observation that attacks in double-negative NMOSD can be severe and result in incomplete remission [56], supports treatment initiation after a first attack. Yet, there are conflicting results regarding the risk of attacks and disability progression in double-negative NMOSD [146, 199, 213] (Fig. 3).

Long-term therapy for double-negative NMOSD
Therapy sequence and switching between immunotherapies
Immunotherapies may be switched in case of therapy failure (severe attack during therapy despite of sufficient dosing and sufficient time to expect full action) or due to serious treatment-related side effects or new relevant comorbidities. A clear definition what constitutes a “severe” attack is currently missing, yet, an attack is usually considered severe if a relevant functional deficit or impairment occurs. In addition, the impact of “mild” attacks without relevant functional deficits on the long-term disease course in NMOSD remains unclear and needs further investigation.
Studies on treatment sequences and switching between monoclonal antibodies for NMOSD are limited, with only a few RCTs reporting data on safety aspects. The PREVENT study and the CHAMPION–NMOSD trial both required a 3-month gap between the last administration of rituximab and the initiation of eculizumab/ravulizumab therapy [178, 179], and there is no controlled safety data on shorter intervals. However, a subgroup analysis in the PREVENT study of 26 patients with prior rituximab medication, out of a total of 96 receiving eculizumab, showed a similar safety profile, especially regarding infection rates [172, 246]. As a 3-month gap before starting eculizumab/ravulizumab in patients with therapy failure to one of the antibodies increases the risk of further attacks, prompt initiation of eculizumab/ravulizumab without a defined safety gap to previous therapies seems preferable. Approval studies for other indications have also required similar (atypical hemolytic uremic syndrome) [43] or even longer time intervals (myasthenia gravis) [82] before starting eculizumab. Theoretically, switching from eculizumab to another immunotherapy should not result in long-lasting overlapping effects due to the short half-life of eculizumab of about 12 days [243]. The mean terminal elimination half-life of ravulizumab is longer and around 50 days. The use of rituximab shortly after or concomitantly with eculizumab has only been described in single case reports for patients with other conditions; other data on therapies with monoclonal antibodies after eculizumab or ravulizumab are not available [157, 191]. Eculizumab inhibits the complement-dependent cytotoxic effects of rituximab and thus can reduce the expected pharmacological effects of rituximab [58]. The subsequent therapy's onset and mode of action should be considered to prevent disease activity from reoccurring after stopping eculizumab/ravulizumab, and a short time interval is preferred. Bridging with oral glucocorticoids may also be considered [181].
Regarding inebilizumab, the initial phase I study in MS required a 12-month gap from any previous monoclonal antibody medication [3]. The N-MOmentum trial required a 6-month gap to rituximab and a 3-month gap to eculizumab or tocilizumab, but this study included only a small number of patients who had undergone these previous therapies [51]. The trial included 17 patients with a history of rituximab therapy, and among them, 13 were randomized to inebilizumab. Seven of them had a previous history of attacks during rituximab therapy, and none of them had an attack during inebilizumab treatment [68], but it remains unclear whether switching from anti-CD20 therapy to anti-CD19 therapy in case of disease activity is rational. Overall, 16 participants (94%) with prior use of rituximab experienced infections, compared to 70% without prior use of rituximab, and a greater proportion of patients with prior rituximab had IgG levels of < 500 mg/dL. There is no data on switching from inebilizumab to another monoclonal antibody. The half-life of inebilizumab is around 18 days, and B-cell depletion below the lower limit of normal is maintained in 94% of patients for at least 6 months following treatment. The exact time for B-cell repletion is not known. Extrapolation from the rituximab data should be handled with care as inebilizumab depletes a broader range of cells.
Trials with IL6-R blockers have similar time gaps to prior antibody medication. The TANGO trial showed a good safety profile for tocilizumab use more than 6 months after rituximab therapy [255]. A case series including three patients receiving rituximab up until < 6 months before and one patient right until the initiation of tocilizumab reported no severe infections [194]. The SAkura trials required a gap of 6 months from prior rituximab or eculizumab use [223, 250]. The ongoing SAkuraBONSAI trial is investigating the efficacy and safety of satralizumab in patients treated unsuccessfully with rituximab within the last 6 months [222]. From other indications, there are no reported infectious complications after the immediate switch from rituximab to tocilizumab, but some patients may develop neutropenia grade 3 [123, 195]. Data on patients with rheumatoid arthritis (n = 27) with tocilizumab to rituximab sequential therapy without a gap revealed no new safety signals after switching to rituximab [62]. The half-life of tocilizumab is 8–18 days depending on the dosage and route of administration. Due to the prolonged terminal half-life of satralizumab (approximately 30 days), the effect of satralizumab may persist for several weeks after stopping treatment. So far, no published data exist on switching from satralizumab to eculizumab, inebilizumab or rituximab.
In general, it is essential to be aware of the potential risk factors for severe infections with prior immunosuppressive treatments, pre-existing low IgG levels, older age, and concomitant glucocorticoid therapy [215]. The ongoing risks while continuing therapy must be weighed against the risk of re-occurring disease activity in case of cessation. In individual cases, long-lasting therapies (B-cell-directed therapies) may be switched to short-lasting therapy. Whether patients should switch between monoclonal antibody therapies due to potential side effects and whether a “prophylactic” switch may prevent severe side effects remains unanswered. For example, it remains unclear whether patients should switch from B-cell therapies to another monoclonal antibody or reduce dosage in case of increasing and severe hypogammaglobulinemia before experiencing relevant infections. Currently, there is a lack of data to support either approach.
Combination therapies
Except for inebilizumab, other monoclonal antibody therapies (eculizumab, ravulizumab, satralizumab, rituximab) for NMOSD have been evaluated in combination with immunosuppressive therapies, including azathioprine, mycophenolate mofetil and oral glucocorticoids. Thus far, studies have not shown a clear benefit in using combination therapies (i.e., antibody therapy combined with immunosuppressive therapies) over monotherapy with monoclonal antibodies for NMOSD. For patients suffering exclusively from NMOSD, monotherapy with monoclonal antibodies may be favored as the first-line treatment, as there is a risk of additional adverse events with combination therapy. However, immunosuppressive therapy may be offered as add-on therapy to patients refractory after treatment with > 2 monoclonal antibodies in succession—or in countries where alternative monoclonal antibodies are not available. Immunosuppressive drugs may also be added if necessary to treat comorbidities, such as SLE and Sjögren’s syndrome. In countries where monoclonal antibodies are not available at all, a combination of azathioprine/mycophenolate mofetil and glucocorticoids may be used. Inebilizumab has not been tested in combination with other immunosuppressants and is approved by the European Medicines Agency only as a monotherapy. If it is combined with another immunosuppressive therapy, the potential for increased immunosuppressive effects and side effects should be considered.
On the other hand, in patients with breakthrough disease during a course of classical immunosuppressive drugs, it is recommended to switch to antibody therapy immediately and continue immunosuppressive treatment at least temporarily or to bridge with oral glucocorticoids, to minimize the risk of a (severe) attack during the transition. Nevertheless, due to a lack of data, it remains a matter of debate when and how to stop background immunosuppressive therapies in patients who stabilize after starting antibody therapy. Data from the OLE of eculizumab showed that 37% of patients were able to stop or decrease their immunosuppressive therapies and remain stable [246], while data from the OLE of the SAkuraSky trial evaluating satralizumab in combination with immunosuppressive therapies showed that 16 out of 36 patients were able to taper their glucocorticoid dose, 3 stopped it entirely and remained attack-free, while 2 experienced three attacks during tapering [249]. Interactions between the various treatment modalities need to be considered. As mentioned above, for example co-treatment with IVIG can diminish the efficacy of eculizumab.
Treatment monitoring in NMOSD
All medications, including off-label and approved therapies, carry potential risks, and long-term post-marketing experience for registered drugs is limited. Therefore, thorough monitoring and patient education are necessary to minimize risks. Infectious risks may be increased during immunosuppression, and the risk may accumulate with the duration of treatment. Older patients likely are at higher risk due to immunosenescence and the occurrence of comorbidities [78, 151, 204]. Different therapies alter the patient’s immune system and immune cells to a variable extent, hence specific monitoring is required. Potential therapy-related complications, including gastrointestinal side effects, liver toxicity, as well as cardiovascular and cancer risks, should be considered (see Table 3). Practical monitoring recommendations also include performing brain and spinal cord MRI for safety surveillance and to reassure the diagnosis during the initial phase of the disease, at the time of acute attacks, at the therapy switch, and in the case of a suspected CNS infection (e.g., PML).
It is essential to continuously monitor the disease activity to detect treatment failure through therapy management and regular follow-up visits. Treatment failure is mainly defined as the occurrence of a new attack (see above severe vs. mild attack). The NEDA concept of “No Evidence of Disease Activity” applied in patients with MS is not established in NMOSD yet, and the significance of asymptomatic brain and spinal cord lesions regarding long-term disability remains unclear. In addition, the relevance of neurodegeneration (brain and spinal cord atrophy as well as retinal fiber loss) in the context of immunotherapies needs to be further investigated. Structured neurological examinations (including EDSS and FSS since generally accepted NMOSD-specific scales do not exist thus far) should be performed at the beginning and switch of therapy, in case new symptoms occur and, in the absence of clinically apparent attacks, every 6–12 months. Additional tests and scores, such as opticospinal impairment score, Hauser Ambulation Index, 9-Hole-Peg-Test (9HPT), Timed-25-Foot-Walk (T25FW) as well as quality of life measures may be used. More recently, a strong association between EDSS and quality of life dimensions measured by EuroQol 5 has been demonstrated [126]. Evoked potentials, MRI, and optical coherence tomography (OCT) can also be used to help monitor the long-term evolution of atrophy [36, 40, 117, 162,163,164, 169] and distinguish attacks from pseudo-attacks [100, 197].
There is no clear correlation between AQP4-IgG titers and disease activity, and monitoring of antibody levels for attack prediction is, therefore, not recommended in general [87, 133, 203]. However, some patients may become at least temporarily antibody-negative with B-cell-directed therapies or anti-IL-6-R therapy over time. Recently one large study (933 initially AQP4-gG positive patients) demonstrated that seroreversion is rare (11%), occurs predominantly in younger patients (age < 20 years) and in patients with initially low AQP4-IgG titers and is often transient [139]. Patients who serorevert are still considered AQP4-IgG-positive NMOSD cases, as in the RIN-1 trial. Overall the significance of transient as well as sustained seroreversion is currently unknown but testing antibody levels at least every 1–2 years could help increase knowledge on this topic and aid in future therapy guidance.
Duration of therapy
Long-term immunotherapy for NMOSD is associated with a potentially increased risk for side effects, especially for adolescents and young adults who may be on therapy for decades. There is limited data on the cessation or “de-escalation” of immunosuppressive therapies in NMOSD. A recent French study showed that rituximab de-escalation (including increased infusion intervals or switching to oral therapies) and discontinuation in AQP4-IgG-positive and double-negative NMOSD patients is associated with an increased risk for attacks in the following 12 months [55]. One retrospective study from Korea found that 14 out of 17 AQP4-IgG-positive NMOSD patients (82%) suffered an attack at a median interval of 6 months after discontinuation of immunosuppressive therapies (including azathioprine, mycophenolate mofetil, rituximab), despite being stable beforehand for a median of 5 years [104]. In that study, 5 (29%) were AQP4-IgG-negative at the time of discontinuation, but 4 (80%) of them reverted from AQP4-IgG-negative to AQP4-IgG-positive after treatment cessation. Another study reported a high attack ratio (77.5%; n = 100) after discontinuing immunosuppressive drugs (azathioprine > mycophenolate mofetil > rituximab), with a higher rate in patients with a previous history of longitudinally extending transverse myelitis (LETM) or shorter disease duration. Reasons for discontinuation included the patient’s decision, side effects, and desire for pregnancy [129]. In another small case series (n = 4; 3 adolescents), two patients remained stable after ceasing rituximab treatment, one turned AQP4-IgG-negative and showed no further attack for up to 10.5 years, and two experienced attacks after 3 and 5 years, respectively [239]. As a single attack can result in significant disability, stopping immunotherapy is currently not recommended in AQP4-IgG-positive patients. Data on treatment discontinuation in double-negative NMOSD is scarce, making it even more challenging to provide recommendations on treatment duration. However, accounting for the more heterogeneous aetiology and mixed prognosis of this group of patients, stopping therapy may be an option in those who were stable for at least 5 years and have had only few and mild attacks.
Serum GFAP levels have been found to be indicative of disease activity in AQP4-IgG-positive NMOSD and are positively correlated with the EDSS [7, 101]. However, further research is needed to determine the utility of serum GFAP levels as a biomarker for assessing attack risk and monitoring in AQP4-IgG-positive or seroconverted patients who discontinue therapy [202].
Pregnancy and NMOSD
NMOSD primarily affects women, with a male-to-female ratio among AQP4-IgG-positive patients of approximately 1:10 [31, 92]. AQP4 is highly expressed in the human placenta, and women with active disease have an increased risk for spontaneous abortions and eclampsia [189, 190, 201, 208]. Although AQP4-IgG can pass the placenta, it has not been shown to have a pathogenic effect on the newborn in the few cases reported thus far [196].
In 2020, therapeutic considerations on pregnancy-related treatment issues in NMOSD were published [140], and more recently, a detailed review and consensus-based recommendations on pregnancy and NMOSD have been proposed by the French MS society [230]. For detailed information on drugs used for the treatment of NMOSD and related risks, we refer to those reviews. Pregnancy is associated with an increased risk of attacks postpartum, potentially also during the third trimester of pregnancy [140]. Factors that increase the risk of disease activity during pregnancy and/or postpartum include a positive AQP4-IgG serostatus, young age, disease activity before pregnancy, and the withdrawal of immunotherapy. There have been numerous reports of attacks occurring in patients who stopped immunotherapy in preparation for pregnancy [44, 57, 102, 120, 234]. Therefore, it is important for women with NMOSD to plan their pregnancy and be advised ahead of time carefully.
Medications, such as mycophenolate mofetil and methotrexate, which are teratogenic, must be discontinued before pregnancy (Table 3). By contrast, in one study (N = 81), treatment with rituximab shortly prior to pregnancy has been found to reduce the attack risk without causing severe side effects in newborns [120]. Therapy with rituximab may offer the advantage of a continued long-lasting effect during pregnancy. In general, IgG1 monoclonal antibodies do not cross the foetoplacental barrier in the first trimester but increasingly transfer in the second and third trimesters [72]. In case rituximab is administered during pregnancy due to disease activity, patients must be counseled and advised that data on its safety and efficacy in pregnant women with NMOSD are limited. Importantly, in babies of mothers treated with B-cell therapies, live or attenuated live vaccines (but not non-live vaccines) should be given only after repletion of B cells. If infusions are paused during pregnancy, they should be restarted soon after delivery.
Another treatment option considered relatively safe for pregnant women with NMOSD is azathioprine, although recent meta-analyses and a retrospective cohort study on women with inflammatory bowel disease reported an increased risk of preterm birth and a twofold increased rate for stillbirth (1% vs. 0.5% in untreated) [150, 221, 257]. Female patients with a stable disease during azathioprine therapy who become pregnant should continue treatment but must be thoroughly counseled on potential risks.
Eculizumab exposure during pregnancy was found to be safe in a small retrospective registry study (n = 75) of women with atypical hemolytic uremic syndrome. Another observational study of 24 pregnancies in women with PNH showed that eculizumab exposure during pregnancy resulted in 85% live births, with no observed malformations [99, 200]. Female patients who are pregnant or planning to become pregnant while receiving eculizumab or ravulizumab may continue therapy after carefully weighing risks and benefits, although data on its safety and efficacy in pregnant women with NMOSD are limited. Tocilizumab has been used during pregnancy in women with rheumatoid arthritis, and no increased risk of malformations has been reported thus far, although the risk for spontaneous abortion and preterm birth may be slightly elevated [80, 238]. Data on pregnancy outcomes under satralizumab and inebilizumab are very limited and have not yet been published. Female patients who are pregnant or are planning to become pregnant while receiving immunotherapy with IL-6-R blockade may continue the therapy if the assumed benefits clearly outweigh the risks, but the potential risk of spontaneous abortion or preterm birth and the paucity of data must be discussed with the patient.
Oral glucocorticoids may increase the risk of cleft palate formation if administered during the first trimester and have also been associated with growth retardation and premature birth. Therefore, they should only be used after a careful risk–benefit assessment [140]. Fluorinated glucocorticoids (e.g., dexamethasone) should be avoided during the entire pregnancy, since they clearly increase the risk of congenital anomalies, intrauterine growth retardation and behavioural disorders in the offspring.
Treatment options for attacks during pregnancy include high-dose glucocorticoids and apheresis therapy (preferably with IA). The choice of treatment will depend on the severity of the attack and the stage of gestation [118]. Although glucocorticoids can transfer to breastmilk, they can be administered during breastfeeding as the infant’s exposure to the drug is minimal and even lower if breastfeeding is delayed for 2–4 h after glucocorticoid infusion [32].
The transfer of drugs into breast milk is influenced by various factors, including the molecule size and lipophilicity, as well as the stage of the breast milk [231]. For instance, the amount of drug present in the colostrum is generally higher than in mature milk. Methotrexate and mycophenolate mofetil should not be given during breastfeeding. Monoclonal antibodies are a viable option during breastfeeding as the amount that transfers into the mature breastmilk is minimal, and when ingested orally by the newborn, the pharmacologically effective amount of antibody in the baby’s serum is further minimized [42, 119, 121]. Although data are limited, there is some experience with rituximab and eculizumab during breastfeeding, suggesting that treatment with rituximab and eculizumab during lactation might be safe [42, 99]. In general, an interdisciplinary approach involving neonatologists and gynaecologists is important when considering therapies during pregnancy and postpartum.
Limited data are available on the effects of NMOSD on fertility. AQP4-IgG may potentially affect fertility as AQP4 is expressed in the paraventricular hypothalamus [9, 226]. Azathioprine and monoclonal antibodies appear to have only minimal effects on fertility. However, drugs such as methotrexate and mitoxantrone have been shown to decrease fertility in women and affect ovarian reserve and sperm count [140].
What role do vaccinations play?
There have been several case reports of first occurrences and flare-ups of NMOSD in temporal association with vaccinations, including COVID-19 vaccines [60, 85, 86, 145, 211]. However, no causal link could be established so far. Moreover, infections (including COVID-19) may contribute to the development of NMOSD and often precede NMOSD attacks, rendering vaccinations potentially important in NMOSD [258]. Furthermore, vaccinations are an essential element in increasing the safety of immunotherapeutic treatments for NMOSD. For example, meningococcal vaccination and/or antibiotic prophylaxis are required before starting eculizumab therapy and should be repeated at least every 2–3 years [143]. It is also important to check vaccination status before starting B-cell-directed therapies, as these treatments may reduce the humoral immune response [11, 167]. Due to the risk of hepatitis B reactivation and severe hepatitis B infection during B-cell-directed therapies, screening for anti-HBV titers should be performed, and vaccinations should be updated accordingly [13, 177]. At the same time, postponing immunotherapy due to an incomplete vaccination status bears the risk of attacks during the vaccination period [193]. Moreover, the impact of prior or ongoing immunotherapies on vaccination responses varies. In general, it is important to update all vaccinations according to national guidelines in a standard and timely manner, considering individual timing, disease activity, the benefits of vaccinations and the fact that (attenuated) live vaccines must not be used in immunosuppressed individuals.
Future therapies
Chimeric antigen receptor (CAR) T-cell therapy targeting B-cell antigens, such as CD19, is being investigated for autoimmune diseases, including NMOSD [2]. A phase I trial of CAR T-cell therapy targeting B-cell maturation antigen (BCMA) in 12 AQP4-IgG-positive NMOSD patients showed no attacks in 11 patients during a follow-up of 5.5 months (median), with hematologic toxic side effects and cytokine release syndrome being reported as potential side effects [184].
Other new drugs and new B-cell- or plasma-cell-directed therapies, such as ublituximab, T4406F telitacicept (B-lymphocyte stimulator blocker and proliferation-inducing ligand), bortezomib (proteasome inhibitor) and Bruton’s tyrosine kinase inhibitor SHR1459 [59, 147, 254] (clinicaltrials.gov), are being actively investigated for the prevention of attacks in AQP4-IgG-seropositive NMOSD. FcRn-inhibiting agents, which have shown efficacy in treating antibody-mediated autoimmune diseases, are also being investigated as potential treatments for NMOSD [71, 81]. The antihistamine drug cetirizine, which blocks eosinophils, has shown promising results as an add-on to other immunotherapies in a pilot trial [98].
Approaches to restoring immune tolerance and potentially inducing permanent remission in NMOSD include DNA or T-cell vaccination and the administration of AQP4 peptide-loaded autologous tolerogenic dendritic cells [21, 212, 259]. Aquaporumab, an antibody against AQP4, has been shown to have beneficial effects in a NMOSD mouse model but has not yet been tested in human trials [64, 224]. Such treatments might turn out to be beneficial for AQP4-IgG-positive patients by stopping disease activity and promoting tissue repair, potentially helping patients to avoid chronic long-term immunosuppressive therapies. However, reliable evidence is missing thus far.
Conclusions
Recent insights into the pathogenesis of NMOSD have led to the development of novel targeted and highly effective therapies for patients with AQP4-IgG-positive NMOSD. These therapies provide a more personalized approach to treatment, considering factors, such as disease activity, age, comorbidities, family planning, side effects, route of administration, patient choice, availability, and costs. They also allow for switching between therapies in case of side effects or insufficient treatment response. However, long-term experience and therapy sequences, as well as general risk management for potential lifelong therapies, are still being developed. To gather these “real-world” data, registries and platform trials should be established, and a standardized approach to data collection should be adopted.
Unmet needs in NMOSD therapy include understanding the long-term disease course, determining optimal immunotherapy durations, developing strategies for treatment cessation and de-escalation, and searching for biomarkers indicating attack risk. Double-negative NMOSD, which affects a minority of patients, should also be addressed in international and collaborative research. Finally, NMOSD is a global disease, and patients and caregivers worldwide should have equal and affordable access to available therapies and therapeutic knowledge.
Change history
13 October 2023
The section “Inebilizumab” has been updated within Box 3 instead of processing it separately in the XML and it has been updated
05 April 2024
A Correction to this paper has been published: https://doi.org/10.1007/s00415-024-12288-2
Abbreviations
- ADA:
-
Anti-drug antibodies
- AQP4:
-
Aquaporin-4
- AQP4-IgG:
-
Aquaporin-4 immunoglobulin G
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- EDSS:
-
Expanded Disability Status Scale
- FCGR3A:
-
Fragment c gamma receptor 3A
- FSS:
-
Functional system scores
- GFAP:
-
Glial fibrillary acidic protein
- HSCT:
-
Hematopoietic stem cell transplantation
- HBV:
-
Hepatitis B virus
- IA:
-
Immunoadsorption
- IL-6:
-
Interleukin-6
- IL-6-R:
-
Interleukin-6 receptor
- i. v.:
-
Intravenously
- IVIG:
-
Intravenous immunoglobulins
- MRI:
-
Magnetic resonance imaging
- MP:
-
Methylprednisolone
- MOG:
-
Myelin–oligodendrocyte–glycoprotein
- MOGAD:
-
MOG-IgG-associated disease
- MS:
-
Multiple sclerosis
- FcRn:
-
Neonatal fragment constant receptor
- NfL:
-
Neurofilament light chain
- NMOSD:
-
Neuromyelitis optica spectrum disorder
- NEMOS:
-
Neuromyelitis Optica Study Group
- OLE:
-
Open-label extension
- PNH:
-
Paroxysmal nocturnal hemoglobinuria
- PE:
-
Plasma exchange
- PML:
-
Progressive multifocal leukenzephalopathy
- RCT:
-
Randomized controlled trial
- SLE:
-
Systemic lupus erythematosus
- s. c.:
-
Subcutaneous
- T25FW:
-
Timed-25-Foot-Walk
- 9HPT:
-
9-Hole-Peg-Test
References
Abbadessa G, Miele G, Maida E, Minervini G, Lavorgna L, Bonavita S (2022) Optimal retreatment schedule of rituximab for neuromyelitis optica spectrum disorder: a systematic review. Mult Scler Relat Disord 63:103926
Aghajanian H, Rurik JG, Epstein JA (2022) CAR-based therapies: opportunities for immuno-medicine beyond cancer. Nat Metab 4:163–169
Agius MA, Klodowska-Duda G, Maciejowski M, Potemkowski A, Li J, Patra K, Wesley J, Madani S, Barron G, Katz E, Flor A (2019) Safety and tolerability of inebilizumab (MEDI- 551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult Scler J 25:235–245
Akaishi T, Nakashima I, Takahashi T, Abe M, Ishii T, Aoki M (2020) Neuromyelitis optica spectrum disorders with unevenly clustered attack occurrence. Neurol Neuroimmunol Neuroinflamm 7:e640
Akaishi T, Takeshita T, Himori N, Takahashi T, Misu T, Ogawa R, Kaneko K, Fujimori J, Abe M, Ishii T, Fujihara K, Aoki M, Nakazawa T, Nakashima I (2020) Rapid administration of high-dose intravenous methylprednisolone improves visual outcomes after optic neuritis in patients with AQP4-IgG-positive NMOSD. Front Neurol 11:932
Aktas O, Hartung HP, Smith MA, Rees WA, Fujihara K, Paul F, Marignier R, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, Cutter G, She D, Gunsior M, Cimbora D, Katz E, Cree BA, Investigators NMs (2023) Serum neurofilament light chain levels at attack predict post-attack disability worsening and are mitigated by inebilizumab: analysis of four potential biomarkers in neuromyelitis optica spectrum disorder. J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2022-330412
Aktas O, Smith MA, Rees WA, Bennett JL, She D, Katz E, Cree BAC, Group NMs, the NMsi (2021) Serum glial fibrillary acidic protein: a neuromyelitis optica spectrum disorder biomarker. Ann Neurol 89:895–910
Allen K, Pittock S, Levy M, Palace J, Oreja-Guevara C, Nakashima I, Berthele A, Bennett J, de Seze J, Barnett M, Paul F, Pozzilli C, Mashhoon Y, Yountz M (2022) Kim H (2022) Sensitivity analysis using propensity score methods for primary efficacy outcome in the CHAMPION-NMOSD trial. ECTRIMS 28:138–140
Altintas A, Dargvainiene J, Schneider-Gold C, Asgari N, Ayzenberg I, Ciplea AI, Junker R, Leypoldt F, Wandinger KP, Hellwig K (2020) Gender issues of antibody-mediated diseases in neurology: (NMOSD/autoimmune encephalitis/MG). Ther Adv Neurol Disord 13:1756286420949808
Aouad P, Li J, Arthur C, Burt R, Fernando S, Parratt J (2015) Resolution of aquaporin-4 antibodies in a woman with neuromyelitis optica treated with human autologous stem cell transplant. J Clin Neurosci 22:1215–1217
Apostolidis SA, Kakara M, Painter MM, Goel RR, Mathew D, Lenzi K, Rezk A, Patterson KR, Espinoza DA, Kadri JC, Markowitz DM, Mexhitaj I, Jacobs D, Babb A, Betts MR, Prak ETL, Weiskopf D, Grifoni A, Lundgreen KA, Gouma S, Sette A, Bates P, Hensley SE, Greenplate AR, Wherry EJ, Li R, Bar-Or A (2021) Cellular and humoral immune responses following SARS-CoV-2 mRNA vaccination in patients with multiple sclerosis on anti-CD20 therapy. Nat Med 27:1990–2001
Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M, Miyake S, Aranami T, Yamamura T (2014) Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica. Neurology 82:1302–1306
Araujo-Neto JM, Guimarães GS, Fernandes FF, Soares MA (2022) Hepatitis B surface antibody (Anti-HBs) kinetics during rituximab chemotherapy and performance of hepatitis B vaccine before immunosuppression: two prospective studies. Viruses 14:1780
Asavapanumas N, Tradtrantip L, Verkman AS (2021) Targeting the complement system in neuromyelitis optica spectrum disorder. Expert Opin Biol Ther 21:1073–1086
Avouac A, Maarouf A, Stellmann JP, Rico A, Boutiere C, Demortiere S, Marignier R, Pelletier J, Audoin B (2021) Rituximab-induced hypogammaglobulinemia and infections in AQP4 and MOG antibody-associated diseases. Neurol Neuroimmunol Neuroinflamm 8:e977
Ayzenberg I, Kleiter I, Schroder A, Hellwig K, Chan A, Yamamura T, Gold R (2013) Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol 70:394–397
Ayzenberg I, Schollhammer J, Hoepner R, Hellwig K, Ringelstein M, Aktas O, Kumpfel T, Krumbholz M, Trebst C, Paul F, Pache F, Obermann M, Zeltner L, Schwab M, Berthele A, Jarius S, Kleiter I, Neuromyelitis Optica Study G (2016) Efficacy of glatiramer acetate in neuromyelitis optica spectrum disorder: a multicenter retrospective study. J Neurol 263:575–582
Azzopardi L, Cox AL, McCarthy CL, Jones JL, Coles AJ (2016) Alemtuzumab use in neuromyelitis optica spectrum disorders: a brief case series. J Neurol 263:25–29
Banerjee A, Ng J, Coleman J, Ospina JP, Mealy M, Levy M (2019) Outcomes from acute attacks of neuromyelitis optica spectrum disorder correlate with severity of attack, age and delay to treatment. Mult Scler Relat Disord 28:60–63
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, Ramanathan S, Waters P, Tenembaum S, Graves JS, Chitnis T, Brandt AU, Hemingway C, Neuteboom R, Pandit L, Reindl M, Saiz A, Sato DK, Rostasy K, Paul F, Pittock SJ, Fujihara K, Palace J (2023) Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD panel proposed criteria. Lancet Neurol 22:268–282
Bar-Or A, Steinman L, Behne JM, Benitez-Ribas D, Chin PS, Clare-Salzler M, Healey D, Kim JI, Kranz DM, Lutterotti A, Martin R, Schippling S, Villoslada P, Wei CH, Weiner HL, Zamvil SS, Smith TJ, Yeaman MR (2016) Restoring immune tolerance in neuromyelitis optica: part II. Neurol Neuroimmunol Neuroinflamm 3:e277
Barreras P, Vasileiou ES, Filippatou AG, Fitzgerald KC, Levy M, Pardo CA, Newsome SD, Mowry EM, Calabresi PA, Sotirchos ES (2022) Long-term effectiveness and safety of rituximab in neuromyelitis optica spectrum disorder and MOG antibody disease. Neurology 99:e2504–e2516
Bennett CL, Focosi D, Socal MP, Bian JC, Nabhan C, Hrushesky WJ, Bennett AC, Schoen MW, Berger JR, Armitage JO (2021) Progressive multifocal leukoencephalopathy in patients treated with rituximab: a 20-year review from the Southern Network on Adverse Reactions. Lancet Haematol 8:e593–e604
Bennett JL, Aktas O, Rees WA, Smith MA, Gunsior M, Yan L, She D, Cimbora D, Pittock SJ, Weinshenker BG, Paul F, Marignier R, Wingerchuk D, Cutter G, Green A, Hartung H-P, Kim HJ, Fujihara K, Levy M, Katz E, Cree BAC (2022) Association between B-cell depletion and attack risk in neuromyelitis optica spectrum disorder: An exploratory analysis from N-MOmentum, a double-blind, randomised, placebo-controlled, multicentre phase 2/3 trial. EBioMedicine 86:104321
Berthele A, Levy M, Wingerchuk DM, Pittock SJ, Shang S, Kielhorn A, Royston M, Sabatella G, Palace J (2023) A single relapse induces worsening of disability and health-related quality of life in patients with neuromyelitis optica spectrum disorder. Front Neurol 14:1099376
Bichuetti DB, Oliveira EML, Boulos FdC, Gabbai AA (2012) Lack of response to pulse cyclophosphamide in neuromyelitis optica: evaluation of 7 patients. Arch Neurol 69:938–939
Bichuetti DB, Perin MMM, Souza NA, Oliveira EML (2019) Treating neuromyelitis optica with azathioprine: 20-year clinical practice. Mult Scler (Houndmills, Basingstoke, Engl) 25:1150–1161
Boedecker SC, Luessi F, Engel S, Kraus D, Klimpke P, Holtz S, Meinek M, Marczynski P, Weinmann A, Weinmann-Menke J (2022) Immunoadsorption and plasma exchange-efficient treatment options for neurological autoimmune diseases. J Clin Apher 37:70–81
Bonnan M, Valentino R, Debeugny S, Merle H, Ferge JL, Mehdaoui H, Cabre P (2018) Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry 89:346–351
Bonnan M, Valentino R, Olindo S, Mehdaoui H, Smadja D, Cabre P (2009) Plasma exchange in severe spinal attacks associated with neuromyelitis optica spectrum disorder. Mult Scler (Houndmills, Basingstoke, Engl) 15:487–492
Borisow N, Hellwig K, Paul F (2018) Neuromyelitis optica spectrum disorders and pregnancy: relapse-preventive measures and personalized treatment strategies. EPMA J 9:249–256
Boz C, Terzi M, Karahan SZ, Sen S, Sarac Y, Mavis ME (2018) Safety of IV pulse methylprednisolone therapy during breastfeeding in patients with multiple sclerosis. Mult Scler J 24:1205–1211
Brod SA (2020) Review of approved NMO therapies based on mechanism of action, efficacy and long-term effects. Mult Scler Relat Disord 46:102538
Burt RK, Balabanov R, Han X, Burns C, Gastala J, Jovanovic B, Helenowski I, Jitprapaikulsan J, Fryer JP, Pittock SJ (2019) Autologous nonmyeloablative hematopoietic stem cell transplantation for neuromyelitis optica. Neurology 93:e1732–e1741
Burton JM, Duggan P, Costello F, Metz L, Storek J (2021) A pilot trial of autologous hematopoietic stem cell transplant in neuromyelitis optic spectrum disorder. Mult Scler Relat Disord 53:102990
Cacciaguerra L, Valsasina P, Mesaros S, Martinelli V, Drulovic J, Filippi M, Rocca MA (2020) Spinal cord atrophy in neuromyelitis optica spectrum disorders is spatially related to cord lesions and disability. Radiology 297:154–163
Capobianco M, Malucchi S, di Sapio A, Gilli F, Sala A, Bottero R, Marnetto F, Doriguzzi Bozzo C, Bertolotto A (2007) Variable responses to rituximab treatment in neuromyelitis optica (Devic’s disease). Neurol Sci 28:209–211
Chen B, Wu Q, Ke G, Bu B (2017) Efficacy and safety of tacrolimus treatment for neuromyelitis optica spectrum disorder. Sci Rep 7:831
Chen H, Qiu W, Zhang Q, Wang J, Shi Z, Liu J, Lian Z, Feng H, Miao X, Zhou H (2017) Comparisons of the efficacy and tolerability of mycophenolate mofetil and azathioprine as treatments for neuromyelitis optica and neuromyelitis optica spectrum disorder. Eur J Neurol 24:219–226
Chien C, Brandt AU, Schmidt F, Bellmann-Strobl J, Ruprecht K, Paul F, Scheel M (2018) MRI-based methods for spinal cord atrophy evaluation: a comparison of cervical cord cross-sectional area, cervical cord volume, and full spinal cord volume in patients with aquaporin-4 antibody seropositive neuromyelitis optica spectrum disorders. AJNR Am J Neuroradiol 39:1362–1368
Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, Ogawa M, Toda T, Yamamura T (2011) Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A 108:3701–3706
Ciplea AI, Langer-Gould A, de Vries A, Schaap T, Thiel S, Ringelstein M, Gold R, Hellwig K (2020) Monoclonal antibody treatment during pregnancy and/or lactation in women with MS or neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 7:e723
Cofiell R, Kukreja A, Bedard K, Yan Y, Mickle AP, Ogawa M, Bedrosian CL, Faas SJ (2015) Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood 125:3253–3262
Collongues N, Alves Do Rego C, Bourre B, Biotti D, Marignier R, da Silva AM, Santos E, Maillart E, Papeix C, Palace J, Leite MIS, De Seze J (2021) Pregnancy in patients With AQP4-Ab, MOG-Ab, or double-negative neuromyelitis optica disorder. Neurology 96:e2006–e2015
Confavreux C, Saddier P, Grimaud J, Moreau T, Adeleine P, Aimard G (1996) Risk of cancer from azathioprine therapy in multiple sclerosis: a case-control study. Neurology 46:1607–1612
Contentti EC, Lopez PA, Pettinicchi JP, Criniti J, Pappolla A, Miguez J, Patrucco L, Carnero Contentti E, Liwacki S, Tkachuk V, Balbuena ME, Vrech C, Deri N, Correale J, Marrodan M, Ysrraelit MC, Leguizamon F, Luetic G, Menichini ML, Tavolini D, Mainella C, Zanga G, Burgos M, Hryb J, Barboza A, Lazaro L, Alonso R, Liguori NF, Nadur D, Chercoff A, Alonso Serena M, Caride A, Paul F, Rojas JI (2021) Assessing attacks and treatment response rates among adult patients with NMOSD and MOGAD: Data from a nationwide registry in Argentina. Mult Scler J Exp Transl Clin 7:20552173211032336
Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J, Thapa P, McKeon A (2011) Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology 77:659–666
Cotzomi E, Stathopoulos P, Lee CS, Ritchie AM, Soltys JN, Delmotte FR, Oe T, Sng J, Jiang R, Ma AK, Vander Heiden JA, Kleinstein SH, Levy M, Bennett JL, Meffre E, O’Connor KC (2019) Early B cell tolerance defects in neuromyelitis optica favour anti-AQP4 autoantibody production. Brain 142:1598–1615
Cree BA, Bennett JL, Sheehan M, Cohen J, Hartung HP, Aktas O, Kim HJ, Paul F, Pittock S, Weinshenker B, Wingerchuk D, Fujihara K, Cutter G, Patra K, Flor A, Barron G, Madani S, Ratchford JN, Katz E (2016) Placebo-controlled study in neuromyelitis optica-ethical and design considerations. Mult Scler (Houndmills, Basingstoke, Engl) 22:862–872
Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C (2005) An open label study of the effects of rituximab in neuromyelitis optica. Neurology 64:1270–1272
Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, Fujihara K, Paul F, Cutter GR, Marignier R, Green AJ, Aktas O, Hartung H-P, Lublin FD, Drappa J, Barron G, Madani S, Ratchford JN, She D, Cimbora D, Katz E (2019) Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. The Lancet 394:1352–1363
Dale RC, Brilot F, Duffy LV, Twilt M, Waldman AT, Narula S, Muscal E, Deiva K, Andersen E, Eyre MR, Eleftheriou D, Brogan PA, Kneen R, Alper G, Anlar B, Wassmer E, Heineman K, Hemingway C, Riney CJ, Kornberg A, Tardieu M, Stocco A, Banwell B, Gorman MP, Benseler SM, Lim M (2014) Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology 83:142–150
Damato V, Evoli A, Iorio R (2016) Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: a systematic review and meta-analysis. JAMA Neurol 73:1342–1348
Damato V, Theorell J, Al-Diwani A, Kienzler AK, Makuch M, Sun B, Handel A, Akdeniz D, Berretta A, Ramanathan S, Fower A, Whittam D, Gibbons E, McGlashan N, Green E, Huda S, Woodhall M, Palace J, Sheerin F, Waters P, Leite MI, Jacob A, Irani SR (2022) Rituximab abrogates aquaporin-4-specific germinal center activity in patients with neuromyelitis optica spectrum disorders. Proc Natl Acad Sci U S A 119:e2121804119
Demuth S, Collongues N, Audoin B, Ayrignac X, Bourre B, Ciron J, Cohen M, Deschamps R, Durand-Dubief F, Maillart E, Papeix C, Ruet A, Zephir H, Marignier R, De Seze J (2023) Rituximab de-escalation in patients with neuromyelitis optica spectrum disorder. Neurology. https://doi.org/10.1212/WNL.0000000000207443
Demuth S, Guillaume M, Bourre B, Ciron J, Zephir H, Sirejacob Y, Kerbrat A, Lebrun-Frenay C, Papeix C, Michel L, Laplaud D, Vukusic S, Maillart E, Cohen M, Audoin B, Marignier R, Collongues N, Group NS (2022) Treatment regimens for neuromyelitis optica spectrum disorder attacks: a retrospective cohort study. J Neuroinflamm 19:62
Deng S, Lei Q, Lu W (2022) Pregnancy-related attack in neuromyelitis optica spectrum disorder with AQP4-IgG: a single-center study and meta-analysis. Front Immunol 12:800666
Di Gaetano N, Cittera E, Nota R, Vecchi A, Grieco V, Scanziani E, Botto M, Introna M, Golay J (2003) Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol 171:1581–1587
Ding J, Jiang X, Cai Y, Pan S, Deng Y, Gao M, Lin Y, Zhao N, Wang Z, Yu H, Qiu H, Jin Y, Xue J, Guo Q, Ni L, Zhang Y, Hao Y, Guan Y (2022) Telitacicept following plasma exchange in the treatment of subjects with recurrent neuromyelitis optica spectrum disorders: a single-center, single-arm, open-label study. CNS Neurosci Ther 28:1613–1623
Dinoto A, Sechi E, Ferrari S, Gajofatto A, Orlandi R, Solla P, Maccabeo A, Maniscalco GT, Andreone V, Sartori A, Manganotti P, Rasia S, Capra R, Mancinelli CR, Mariotto S (2022) Risk of disease relapse following COVID-19 vaccination in patients with AQP4-IgG-positive NMOSD and MOGAD. Mult Scler Relat Disord 58:103424
Dong GY, Meng YH, Xiao XJ (2022) A meta-analysis on efficacy and safety of rituximab for neuromyelitis optica spectrum disorders. Medicine (Baltimore) 101:e30347
Dörner T, Schulze-Koops H, Burmester G-R, Iking-Konert C, Schmalzing M, Engel A, Kästner P, Kellner H, Kurthen R, Krüger K, Rubbert-Roth A, Schwenke H, Peters MA, Tony H-P (2019) Early and late responses in patients with rheumatoid arthritis who were conventional synthetic disease-modifying anti-rheumatic drug inadequate responders and were treated with tocilizumab or switched to rituximab: an open-label phase 3 trial (MIRAI). Clin Exp Rheumatol 37:937–945
Du C, Zeng P, Han JR, Zhang TX, Jia D, Shi FD, Zhang C (2021) Early initiation of tocilizumab treatment against moderate-to-severe myelitis in neuromyelitis optica spectrum disorder. Front Immunol 12:660230
Duan T, Tradtrantip L, Phuan PW, Bennett JL, Verkman AS (2020) Affinity-matured “aquaporumab” anti-aquaporin-4 antibody for therapy of seropositive neuromyelitis optica spectrum disorders. Neuropharmacology 162:107827
Elsone L, Kitley J, Luppe S, Lythgoe D, Mutch K, Jacob S, Brown R, Moss K, McNeillis B, Goh YY, Leite MI, Robertson N, Palace J, Jacob A (2014) Long-term efficacy, tolerability and retention rate of azathioprine in 103 aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder patients: a multicentre retrospective observational study from the UK. Mult Scler (Houndmills, Basingstoke, Engl) 20:1533–1540
Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, Lennon VA (2016) Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol 73:1297–1307
Flanagan EP, Aksamit AJ, Kumar N, Morparia NP, Keegan BM, Weinshenker BG (2013) Simultaneous PML-IRIS and myelitis in a patient with neuromyelitis optica spectrum disorder. Neurol Clin Pract 3:448–451
Flanagan EP, Levy M, Katz E, Cimbora D, Drappa J, Mealy MA, She D, Cree BAC (2022) Inebilizumab for treatment of neuromyelitis optica spectrum disorder in patients with prior rituximab use from the N-MOmentum Study. Mult Scler Relat Disord 57:103352
Food and Drug Administration US (2019) SOLIRIS Product information. In https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125166s431lbledt.pdf
Fujihara K, Bennett JL, de Seze J, Haramura M, Kleiter I, Weinshenker BG, Kang D, Mughal T, Yamamura T (2020) Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol Neuroimmunol Neuroinflamm 7:e841
Gable KL, Guptill JT (2019) Antagonism of the neonatal fc receptor as an emerging treatment for myasthenia gravis. Front Immunol 10:3052
Galati A, McElrath T, Bove R (2022) Use of B cell-depleting therapy in women of childbearing potential with multiple sclerosis and neuromyelitis optica spectrum disorder. Neurol Clin Pract 12:154–163
Giglhuber K, Berthele A (2022) Adverse events in NMOSD therapy. Int J Mol Sci 23:4154
Giovannelli J, Ciron J, Cohen M, Kim HJ, Kim SH, Stellmann JP, Kleiter I, McCreary M, Greenberg BM, Deschamps R, Audoin B, Maillart E, Papeix C, Collongues N, Bourre B, Laplaud D, Ayrignac X, Durand-Dubief F, Ruet A, Vukusic S, Marignier R, Dauchet L, Zephir H, Nemos NOi (2021) A meta-analysis comparing first-line immunosuppressants in neuromyelitis optica. Ann Clin Transl Neurol 8:2025–2037
Greco R, Bondanza A, Oliveira MC, Badoglio M, Burman J, Piehl F, Hagglund H, Krasulova E, Simoes BP, Carlson K, Pohlreich D, Labopin M, Saccardi R, Comi G, Mancardi GL, Bacigalupo A, Ciceri F, Farge D (2015) Autologous hematopoietic stem cell transplantation in neuromyelitis optica: a registry study of the EBMT Autoimmune Diseases Working Party. Mult Scler (Houndmills, Basingstoke, Engl) 21:189–197
Greenberg BM, Graves D, Remington G, Hardeman P, Mann M, Karandikar N, Stuve O, Monson N, Frohman E (2012) Rituximab dosing and monitoring strategies in neuromyelitis optica patients: creating strategies for therapeutic success. Mult Scler (Houndmills, Basingstoke, Engl) 18:1022–1026
Hamid SHM, Whittam D, Mutch K, Linaker S, Solomon T, Das K, Bhojak M, Jacob A (2017) What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol 264:2088–2094
Hatayama Y, Hashimoto Y, Motokura T (2020) Frequent co-reactivation of Epstein-Barr virus in patients with cytomegalovirus viremia under immunosuppressive therapy and/or chemotherapy. J Int Med Res 48:300060520972880
Heigl F, Hettich R, Fassbender C, Klingel R, Mauch E, Durner J, Kern R, Kleiter I (2023) Immunoadsorption as maintenance therapy for refractory neuromyelitis optica spectrum disorder. Ther Adv Neurol Disord 16:17562864221150314
Hoeltzenbein M, Beck E, Rajwanshi R, Gøtestam Skorpen C, Berber E, Schaefer C, Østensen M (2016) Tocilizumab use in pregnancy: analysis of a global safety database including data from clinical trials and post-marketing data. Semin Arthritis Rheum 46:238–245
Howard JF Jr, Bril V, Vu T, Karam C, Peric S, Margania T, Murai H, Bilinska M, Shakarishvili R, Smilowski M, Guglietta A, Ulrichts P, Vangeneugden T, Utsugisawa K, Verschuuren J, Mantegazza R (2021) Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol 20:526–536
Howard JF, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I, Jacob S, Vissing J, Burns TM, Kissel JT, Muppidi S, Nowak RJ, O'Brien F, Wang J-J, Mantegazza R, Mazia CG, Wilken M, Ortea C, Saba J, Rugiero M, Bettini M, Vidal G, Garcia AD, Lamont P, Leong W-K, Boterhoven H, Fyfe B, Roberts L, Jasinarachchi M, Willlems N, Wanschitz J, Löscher W, De Bleecker J, Van den Abeele G, de Koning K, De Mey K, Mercelis R, Wagemaekers L, Mahieu D, Van Damme P, Smetcoren C, Stevens O, Verjans S, D'Hondt A, Tilkin P, Alves de Siqueira Carvalho A, Hasan R, Dias Brockhausen I, Feder D, Ambrosio D, Melo AP, Rocha R, Rosa B, Veiga T, Augusto da Silva L, Gonçalves Geraldo J, da Penha Morita Ananias M, Nogueira Coelho E, Paiva G, Pozo M, Prando N, Dada Martineli Torres D, Fernanda Butinhao C, Coelho E, Renata Cubas Volpe L, Duran G, Gomes da Silva TC, Otavio Maia Gonçalves L, Pazetto LE, Souza Duca L, Suriane Fialho TA, Gheller Friedrich MA, Guerreiro A, Mohr H, Pereira Martins M, da Cruz Pacheco D, Macagnan AP, de Cassia Santos A, Bulle Oliveira AS, Amaral de Andrade AC, Annes M, Cavalcante Lino V, Pinto W, Miranda C, Carrara F, Souza I, Genge A, Massie R, Campbell N, Bril V, Katzberg H, Soltani M, Ng E, Siddiqi Z, Phan C, Blackmore D, Vohanka S, Bednarik J, Chmelikova M, Cierny M, Toncrova S, Junkerova J, Kurkova B, Reguliova K, Zapletalova O, Pitha J, Novakova I, Tyblova M, Wolfova M, Jurajdova I, Andersen H, Harbo T, Vinge L, Mogensen A, Højgaard J, Witting N, Autzen AM, Pedersen J, Färkkilä M, Atula S, Nyrhinen A, Erälinna J-P, Laaksonen M, Oksaranta O, Eriksson J, Harrison T, Desnuelle C, Sacconi S, Soriani M-H, Decressac S, Moutarde J, Lahaut P, Solé G, Le Masson G, Wielanek-Bachelet A-C, Gaboreau M, Moreau C, Wilson A, Vial C, Bouhour F, Gervais-Bernard H, Merle H, Hourquin C, Lacour A, Outteryck O, Vermersch P, Zephir H, Millois E, Deneve M, Deruelle F, Schoser B, Wenninger S, Stangel M, Alvermann S, Gingele S, Skripuletz T, Suehs K-W, Trebst C, Fricke K, Papagiannopoulos S, Bostantzopoulou S, Vlaikidis N, Zampaki M, Papadopoulou N, Mitsikostas D-D, Kasioti E, Mitropoulou E, Charalambous D, Rozsa C, Horvath M, Lovas G, Matolcsi J, Szabo G, Szabadosne B, Vecsei L, Dezsi L, Varga E, Konyane M, Gross B, Azrilin O, Greenbereg N, Bali Kuperman H, Antonini G, Garibaldi M, Morino S, Troili F, Di Pasquale A, Filla A, Costabile T, Marano E, Sacca F, Marsil A, Puorro G, Maestri Tassoni M, De Rosa A, Bonanno S, Antozzi C, Maggi L, Campanella A, Angelini C, Cudia P, Pegoraro V, Pinzan E, Bevilacqua F, Orrico D, Bonifati DM, Evoli A, Alboini PE, D'Amato V, Iorio R, Inghilleri M, Fionda L, Frasca V, Giacomelli E, Gori M, Lopergolo D, Onesti E, Gabriele M, Patti F, Salvatore Caramma A, Messina S, Reggio E, Caserta C, Uzawa A, Kanai T, Mori M, Kaneko Y, Kanzaki A, Kobayashi E, Masaki K, Matsuse D, Matsushita T, Uehara T, Shimpo M, Jingu M, Kikutake K, Nakamura Y, Sano Y, Nagane Y, Kamegamori I, Fujii Y, Futono K, Tsuda T, Saito Y, Suzuki H, Morikawa M, Samukawa M, Kamakura S, Shiraishi H, Mitazaki T, Motomura M, Mukaino A, Yoshimura S, Asada S, Kobashikawa T, Koga M, Maeda Y, Takada K, Takada MT, Yamashita Y, Yoshida S, Suzuki Y, Akiyama T, Narikawa K, Tsukita K, Meguro F, Fukuda Y, Sato M, Matsuo H, Fukudome T, Gondo Y, Maeda Y, Nagaishi A, Nakane S, Okubo Y, Okumura M, Funaka S, Kawamura T, Makamori M, Takahashi M, Hasuike T, Higuchi E, Kobayashi H, Osakada K, Taichi N, Tsuda E, Hayashi T, Hisahara S, Imai T, Kawamata J, Murahara T, Saitoh M, Shimohama S, Suzuki S, Yamamoto D, Konno S, Imamura T, Inoue M, Murata M, Nakazora H, Nakayama R, Ikeda Y, Ogawa M, Shirane M, Kanda T, Kawai M, Koga M, Ogasawara J, Omoto M, Sano Y, Arima H, Fukui S, Shimose S, Shinozaki H, Watanabe M, Yoshikawa C, van der Kooi A, de Visser M, Gibson T, Maessen J, de Baets M, Faber C, Keijzers MJ, Miesen M, Kostera-Pruszczyk A, Kaminska A, Kim B-J, Lee CN, Koo YS, Seok HY, Kang HN, Ra H, Kim BJ, Cho EB, Lee H, Min J-H, Seok J, Koh DY, Kwon J, Lee J, Park S, Hong Y-H, Lim J-S, Kim M, Kim SM, Kim Y-H, Lee HS, Shin HY, Hwang EB, Shin M, Sazonov D, Yarmoschuk A, Babenko L, Malkova N, Melnikova A, Korobko D, Kosykh E, Pokhabov D, Nesterova Y, Abramov V, Balyazin V, Casasnovas Pons C, Alberti Aguilo M, Homedes-Pedret C, Palacios NJ, Lazaro A, Diez Tejedor E, Fernandez-Fournier M, Lopez Ruiz P, Rodriguez de Rivera FJ, Salvado Figueras M, Gamez J, Salvado M, Cortes Vicente E, Diaz-Manera J, Querol Gutierrez L, Rojas Garcia R, Vidal N, Arribas-Ibar E, Piehl F, Hietala A, Bjarbo L, Lindberg C, Jons D, Andersson B, Sengun I, Ozcelik P, Tuga C, Ugur M, Boz C, Altiparmak D, Gazioglu S, Ozen Aydin C, Erdem-Ozdamar S, Bekircan-Kurt CE, Yilmaz E, Acar NP, Caliskan Y, Efendi H, Aydinlik S, Cavus H, Semiz C, Tun O, Terzi M, Dogan B, Onar MK, Sen S, Cavdar TK, Norwood F, Dimitriou A, Gollogly J, Mahdi-Rogers M, Seddigh A, Maier G, Sohail F, Sathasivam S, Arndt H, Davies D, Watling D, Rivner M, Hartmann JE, Quarles B, Smalley N, Amato A, Cochrane T, Salajegheh M, Roe K, Amato K, Toska S, Wolfe G, Silvestri N, Patrick K, Zakalik K, Katz J, Miller R, Engel M, Bravver E, Brooks B, Plevka S, Burdette M, Sanjak M, Kramer M, Nemeth J, Schommer C, Juel V, Guptill J, Hobson-Webb L, Beck K, Carnes D, Loor J, Anderson A, Lange D, Agopian E, Goldstein J, Manning E, Kaplan L, Holzberg S, Kassebaum N, Pascuzzi R, Bodkin C, Kincaid J, Snook R, Guinrich S, Micheels A, Chaudhry V, Corse A, Mosmiller B, Ho D, Srinivasan J, Vytopil M, Ventura N, Scala S, Carter C, Donahue C, Herbert C, Weiner E, McKinnon J, Haar L, McKinnon N, Alcon K, Daniels K, Sattar N, Jeffery D, McKenna K, Guidon A, David W, Dheel C, Levine-Weinberg M, Nigro C, Simpson E, Appel SH, Lai E, Lay JL, Pleitez M, Halton S, Faigle C, Thompson L, Sivak M, Shin S, Bratton J, Jacobs D, Brown G, Bandukwala I, Brown M, Kane J, Blount I, Freimer M, Hoyle JC, Agriesti J, Khoury J, Marburger T, Kaur H, Dimitrova D, Mellion M, Sachs G, Crabtree B, Keo R, Perez EK, Taber S, Gilchrist J, Andoin A, Darnell T, Goyal N, Sakamuri S, So YT, Welsh LW, Bhavaraju-Sanka R, Tobon Gonzalez A, Jones F, Saklad A, Nations S, Trivedi J, Hopkins S, Kazamel M, Alsharabati M, Lu L, Mumfrey-Thomas S, Woodall A, Richman D, Butters J, Lindsay M, Mozaffar T, Cash T, Goyal N, Roy G, Mathew V, Maqsood F, Minton B, Jones HJ, Rosenfeld J, Garcia R, Garcia S, Echevarria L, Pulley M, Aranke S, Berger AR, Shah J, Shabbir Y, Smith L, Varghese M, Gutmann L, Gutmann L, Swenson A, Olalde H, Hafer-Macko C, Kwan J, Zilliox L, Callison K, DiSanzo B, Naunton K, Bilsker M, Sharma K, Reyes E, Cooley A, Michon S-C, Steele J, Karam CK, Chopra M, Bird S, Kaufman J, Gallatti N, Vu T, Katzin L, McClain T, Harvey B, Hart A, Huynh K, Beydoun S, Chilingaryan A, Droker B, Lin F, Shah A, Tran A, Akhter S, Malekniazi A, Tandan R, Hehir M, Waheed W, Lucy S, Weiss M, Distad J, Downing S, Strom S, Lisak R, Bernitsas E, Khan O, Kumar Sriwastava S, Tselis A, Jia K, Bertorini T, Arnold T, Henderson K, Pillai R, Liu Y, Wheeler L, Hewlett J, Vanderhook M, Dicapua D, Keung B, Kumar A, Patwa H, Robeson K, Nye J, Vu H (2017) Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. The Lancet Neurology 16:976–986
Huang W, Wang L, Zhang B, Zhou L, Zhang T, Quan C (2019) Effectiveness and tolerability of immunosuppressants and monoclonal antibodies in preventive treatment of neuromyelitis optica spectrum disorders: a systematic review and network meta-analysis. Mult Scler Relat Disord 35:246–252
Huang X, Wu J, Xiao Y, Zhang Y (2021) Timing of plasma exchange for neuromyelitis optica spectrum disorders: a meta-analysis. Mult Scler Relat Disord 48:102709
Hümmert MW, Butow F, Tkachenko D, Ayzenberg I, Pakeerathan T, Hellwig K, Klotz L, Haussler V, Stellmann JP, Warnke C, Goereci Y, Etgen T, Luessi F, Bronzlik P, Gingele S, Lauenstein AS, Kleiter I, Rommer PS, Paul F, Bellmann-Strobl J, Duchow A, Then Bergh F, Pul R, Walter A, Pellkofer H, Kumpfel T, Pompsch M, Kraemer M, Albrecht P, Aktas O, Ringelstein M, Senel M, Giglhuber K, Berthele A, Jarius S, Wildemann B, Trebst C, Neuromyelitis Optica Study G (2023) Effects of the COVID-19 pandemic on patients with NMO spectrum disorders and MOG-antibody-associated diseases: COPANMO(G)-study. Neurol Neuroimmunol Neuroinflamm 10:e200082
Ismail II, Salama S (2022) A systematic review of cases of CNS demyelination following COVID-19 vaccination. J Neuroimmunol 362:577765
Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, Lang W, Reindl M, Vincent A, Kristoferitsch W (2008) Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 131:3072–3080
Jarius S, Aktas O, Ayzenberg I, Bellmann-Strobl J, Berthele A, Giglhuber K, Haussler V, Havla J, Hellwig K, Hummert MW, Kleiter I, Klotz L, Krumbholz M, Kumpfel T, Paul F, Ringelstein M, Ruprecht K, Senel M, Stellmann JP, Bergh FT, Tumani H, Wildemann B, Trebst C, Neuromyelitis Optica Study G (2023) Update on the diagnosis and treatment of neuromyelits optica spectrum disorders (NMOSD) - revised recommendations of the Neuromyelitis Optica Study Group (NEMOS). Part I: diagnosis and differential diagnosis. J Neurol 270:3341–3368
Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, Franciotta D, Fujihara K, Jacob A, Kim HJ, Kleiter I, Kumpfel T, Levy M, Palace J, Ruprecht K, Saiz A, Trebst C, Weinshenker BG, Wildemann B (2018) MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflamm 15:134
Jarius S, Paul F, Weinshenker BG, Levy M, Kim HJ, Wildemann B (2020) Neuromyelitis optica. Nat Rev Dis Primers 6:85
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, Beume LA, Hummert MW, Trebst C, Ringelstein M, Aktas O, Winkelmann A, Buttmann M, Schwarz A, Zimmermann H, Brandt AU, Franciotta D, Capobianco M, Kuchling J, Haas J, Korporal-Kuhnke M, Lillevang ST, Fechner K, Schanda K, Paul F, Wildemann B, Reindl M, in cooperation with the Neuromyelitis Optica Study G (2016) MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflamm 13:279
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, Kleiter I, Kleinschnitz C, Berthele A, Brettschneider J, Hellwig K, Hemmer B, Linker RA, Lauda F, Mayer CA, Tumani H, Melms A, Trebst C, Stangel M, Marziniak M, Hoffmann F, Schippling S, Faiss JH, Neuhaus O, Ettrich B, Zentner C, Guthke K, Hofstadt-van Oy U, Reuss R, Pellkofer H, Ziemann U, Kern P, Wandinger KP, Bergh FT, Boettcher T, Langel S, Liebetrau M, Rommer PS, Niehaus S, Munch C, Winkelmann A, Zettl UU, Metz I, Veauthier C, Sieb JP, Wilke C, Hartung HP, Aktas O, Paul F (2012) Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflamm 9:14
Jarius S, Wandinger KP, Borowski K, Stoecker W, Wildemann B (2012) Antibodies to CV2/CRMP5 in neuromyelitis optica-like disease: case report and review of the literature. Clin Neurol Neurosurg 114:331–335
Jarius S, Wildemann B (2013) Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol 23:661–683
Javadian N, Magrouni H, Ghaffarpour M, Ranji-Burachaloo S (2021) Severe relapses of neuromyelitis optica spectrum disorder during treatment with dimethyl fumarate. Clin Neuropharmacol 44:21–22
Jiao Y, Fryer JP, Lennon VA, Jenkins SM, Quek AM, Smith CY, McKeon A, Costanzi C, Iorio R, Weinshenker BG, Wingerchuk DM, Shuster EA, Lucchinetti CF, Pittock SJ (2013) Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology 81:1197–1204
Kastrati K, Aletaha D, Burmester GR, Chwala E, Dejaco C, Dougados M, McInnes IB, Ravelli A, Sattar N, Stamm TA, Takeuchi T, Trauner M, van der Heijde D, Voshaar MJH, Winthrop K, Smolen JS, Kerschbaumer A (2022) A systematic literature review informing the consensus statement on efficacy and safety of pharmacological treatment with interleukin-6 pathway inhibition with biological DMARDs in immune-mediated inflammatory diseases. RMD Open 8:e002359
Katz Sand I, Fabian MT, Telford R, Kraus TA, Chehade M, Masilamani M, Moran T, Farrell C, Ebel S, Cook LJ, Rose J, Lublin FD (2018) Open-label, add-on trial of cetirizine for neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 5:e441
Kelly RJ, Hochsmann B, Szer J, Kulasekararaj A, de Guibert S, Roth A, Weitz IC, Armstrong E, Risitano AM, Patriquin CJ, Terriou L, Muus P, Hill A, Turner MP, Schrezenmeier H, Peffault de Latour R (2015) Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 373:1032–1039
Kessler RA, Mealy MA, Levy M (2016) Early indicators of relapses vs pseudorelapses in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 3:e269
Kim H, Lee EJ, Kim S, Choi LK, Kim HJ, Kim HW, Chung K, Seo D, Moon S, Kim KK, Lim YM (2022) Longitudinal follow-up of serum biomarkers in patients with neuromyelitis optica spectrum disorder. Mult Scler (Houndmills, Basingstoke, Engl) 28:512–521
Kim SH, Huh SY, Jang H, Park NY, Kim Y, Jung JY, Lee MY, Hyun JW, Kim HJ (2020) Outcome of pregnancies after onset of the neuromyelitis optica spectrum disorder. Eur J Neurol 27:1546–1555
Kim SH, Hyun JW, Kim HJ (2019) Individualized B cell-targeting therapy for neuromyelitis optica spectrum disorder. Neurochem Int 130:104347
Kim SH, Jang H, Park NY, Kim Y, Kim SY, Lee MY, Hyun JW, Kim HJ (2021) Discontinuation of immunosuppressive therapy in patients with neuromyelitis optica spectrum disorder with aquaporin-4 antibodies. Neurol Neuroimmunol Neuroinflamm 8:e947
Kim SH, Jeong IH, Hyun JW, Joung A, Jo HJ, Hwang SH, Yun S, Joo J, Kim HJ (2015) Treatment outcomes with rituximab in 100 patients with neuromyelitis optica: influence of FCGR3A polymorphisms on the therapeutic response to rituximab. JAMA Neurol 72:989–995
Kim SH, Kim W, Park MS, Sohn EH, Li XF, Kim HJ (2011) Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol 68:473–479
Kim SH, Park NY, Kim KH, Hyun JW, Kim HJ (2022) Rituximab-induced hypogammaglobulinemia and risk of infection in neuromyelitis optica spectrum disorders: a 14-year real-life experience. Neurol Neuroimmunol Neuroinflamm 9:e1179
Kitley J, Leite MI, Nakashima I, Waters P, McNeillis B, Brown R, Takai Y, Takahashi T, Misu T, Elsone L, Woodhall M, George J, Boggild M, Vincent A, Jacob A, Fujihara K, Palace J (2012) Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 135:1834–1849
Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, Palace J, Vincent A (2012) Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 79:1273–1277
Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Hellwig K, Pache F, Ruprecht K, Havla J, Kumpfel T, Aktas O, Hartung HP, Ringelstein M, Geis C, Kleinschnitz C, Berthele A, Hemmer B, Angstwurm K, Stellmann JP, Schuster S, Stangel M, Lauda F, Tumani H, Mayer C, Krumbholz M, Zeltner L, Ziemann U, Linker R, Schwab M, Marziniak M, Then Bergh F, Hofstadt-van Oy U, Neuhaus O, Zettl UK, Faiss J, Wildemann B, Paul F, Jarius S, Trebst C, Nemos (2018) Apheresis therapies for NMOSD attacks: a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm 5:e504
Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Wegner B, Hellwig K, Pache F, Ruprecht K, Havla J, Krumbholz M, Kumpfel T, Aktas O, Hartung HP, Ringelstein M, Geis C, Kleinschnitz C, Berthele A, Hemmer B, Angstwurm K, Stellmann JP, Schuster S, Stangel M, Lauda F, Tumani H, Mayer C, Zeltner L, Ziemann U, Linker R, Schwab M, Marziniak M, Then Bergh F, Hofstadt-van Oy U, Neuhaus O, Winkelmann A, Marouf W, Faiss J, Wildemann B, Paul F, Jarius S, Trebst C (2016) Neuromyelitis optica: evaluation of 871 attacks and 1153 treatment courses. Ann Neurol 79:206–216
Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung HP, Paul F, Aktas O (2012) Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol 69:239–245
Kleiter I, Traboulsee A, Palace J, Yamamura T, Fujihara K, Saiz A, Javed A, Mayes D, Büdingen HCV, Klingelschmitt G, Stokmaier D, Bennett JL (2022) Long-term efficacy of satralizumab in AQP4-IgG–seropositive neuromyelitis optica spectrum disorder from SAkuraSky and SAkuraStar. Neurol Neuroimmunol Neuroinflamm 10:e200071
Kojima M, Oji S, Tanaka S, Izaki S, Hashimoto B, Fukaura H, Nomura K (2019) Tacrolimus is effective for neuromyelitis optica spectrum disorders with or without anti-AQP4 antibody. Mult Scler Relat Disord 39:101907
Konen FF, Schwenkenbecher P, Jendretzky KF, Gingele S, Grote-Levi L, Mohn N, Suhs KW, Eiz-Vesper B, Maecker-Kolhoff B, Trebst C, Skripuletz T, Hummert MW (2022) Stem cell therapy in neuroimmunological diseases and its potential neuroimmunological complications. Cells 11:2165
Kowarik MC, Hoshi M, Hemmer B, Berthele A (2016) Failure of alemtuzumab as a rescue in a NMOSD patient treated with rituximab. Neurol Neuroimmunol Neuroinflamm 3:e208
Kremer S, Renard F, Achard S, Lana-Peixoto MA, Palace J, Asgari N, Klawiter EC, Tenembaum SN, Banwell B, Greenberg BM, Bennett JL, Levy M, Villoslada P, Saiz A, Fujihara K, Chan KH, Schippling S, Paul F, Kim HJ, de Seze J, Wuerfel JT, Cabre P, Marignier R, Tedder T, van Pelt D, Broadley S, Chitnis T, Wingerchuk D, Pandit L, Leite MI, Apiwattanakul M, Kleiter I, Prayoonwiwat N, Han M, Hellwig K, van Herle K, John G, Hooper DC, Nakashima I, Sato D, Yeaman MR, Waubant E, Zamvil S, Stüve O, Aktas O, Smith TJ, Jacob A, O’Connor K (2015) Use of advanced magnetic resonance imaging techniques in neuromyelitis optica spectrum disorder. JAMA Neurol 72:815–822
Krysko KM, Bove R, Dobson R, Jokubaitis V, Hellwig K (2021) Treatment of women with multiple sclerosis planning pregnancy. Curr Treat Options Neurol 23:11
Krysko KM, LaHue SC, Anderson A, Rutatangwa A, Rowles W, Schubert RD, Marcus J, Riley CS, Bevan C, Hale TW, Bove R (2019) Minimal breast milk transfer of rituximab, a monoclonal antibody used in neurological conditions. Neurol Neuroimmunol Neuroinflamm 7:e637
Kümpfel T, Thiel S, Meinl I, Ciplea AI, Bayas A, Hoffmann F, Hofstadt-van Oy U, Hoshi M, Kluge J, Ringelstein M, Aktas O, Stoppe M, Walter A, Weber MS, Ayzenberg I, Hellwig K (2020) Anti-CD20 therapies and pregnancy in neuroimmunologic disorders: a cohort study from Germany. Neurol Neuroimmunol Neuroinflamm 8:e913
LaHue SC, Anderson A, Krysko KM, Rutatangwa A, Dorsey MJ, Hale T, Mahadevan U, Rogers EE, Rosenstein MG, Bove R (2020) Transfer of monoclonal antibodies into breastmilk in neurologic and non-neurologic diseases. Neurol Neuroimmunol Neuroinflamm 7:e769
Lebrun C, Rocher F (2018) Cancer risk in patients with multiple sclerosis: potential impact of disease-modifying drugs. CNS Drugs 32:939–949
Lee WJ, Lee ST, Moon J, Sunwoo JS, Byun JI, Lim JA, Kim TJ, Shin YW, Lee KJ, Jun JS, Lee HS, Kim S, Park KI, Jung KH, Jung KY, Kim M, Lee SK, Chu K (2016) Tocilizumab in autoimmune encephalitis refractory to rituximab: an institutional cohort study. Neurotherapeutics 13:824–832
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 202:473–477
Lerch M, Schanda K, Lafon E, Wurzner R, Mariotto S, Dinoto A, Wendel EM, Lechner C, Hegen H, Rostasy K, Berger T, Wilflingseder D, Hoftberger R, Reindl M (2023) More efficient complement activation by anti-aquaporin-4 compared with anti-myelin oligodendrocyte glycoprotein antibodies. Neurol Neuroimmunol Neuroinflamm 10:e200059
Levy M, Haycox AR, Becker U, Costantino C, Damonte E, Klingelschmitt G, von Büdingen HC, Wallenstein G, Maio DD, Szczechowski L (2022) Quantifying the relationship between disability progression and quality of life in patients treated for NMOSD: insights from the SAkura studies. Mult Scler Relat Disord 57:103332
Levy M, Mealy MA (2014) Purified human C1-esterase inhibitor is safe in acute relapses of neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 1:e5
Li J, Bazzi SA, Schmitz F, Tanno H, McDaniel JR, Lee CH, Joshi C, Kim JE, Monson N, Greenberg BM, Hedfalk K, Melamed E, Ippolito GC (2021) Molecular level characterization of circulating aquaporin-4 antibodies in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 8:e1034
Li R, Li C, Huang Q, Liu Z, Chen J, Zhang B, Liu C, Shu Y, Wang Y, Kermode AG, Qiu W (2022) Immunosuppressant and neuromyelitis optica spectrum disorder: optimal treatment duration and risk of discontinuation. Eur J Neurol 29:2792–2800
Lim YM, Kim H, Lee EJ, Kim HW, Kim HJ, Kim KK (2020) Beneficial effects of intravenous immunoglobulin as an add-on therapy to azathioprine for NMO-IgG-seropositive neuromyelitis optica spectrum disorders. Mult Scler Relat Disord 42:102109
Lim YM, Pyun SY, Kang BH, Kim J, Kim KK (2013) Factors associated with the effectiveness of plasma exchange for the treatment of NMO-IgG-positive neuromyelitis optica spectrum disorders. Mult Scler (Houndmills, Basingstoke, Engl) 19:1216–1218
Lin J, Xue B, Zhu R, Pan J, Li J, Lin Y, Li X, Xia J (2021) Intravenous immunoglobulin as the rescue treatment in NMOSD patients. Neurol Sci 42:3857–3863
Liu J, Tan G, Li B, Zhang J, Gao Y, Cao Y, Jia Z, Sugimoto K (2021) Serum aquaporin 4-immunoglobulin G titer and neuromyelitis optica spectrum disorder activity and severity: a systematic review and meta-analysis. Front Neurol 12:746959
Liu YP, Xu HQ, Li M, Yang X, Yu S, Fu WL, Huang Q (2015) Association between thiopurine S-methyltransferase polymorphisms and azathioprine-induced adverse drug reactions in patients with autoimmune diseases: a meta-analysis. PLoS ONE 10:e0144234
Longoni G, Banwell B, Filippi M, Yeh EA (2014) Rituximab as a first-line preventive treatment in pediatric NMOSDs: preliminary results in 5 children. Neurol Neuroimmunol Neuroinflamm 1:e46
Lotan I, Charlson RW, Ryerson LZ, Levy M, Kister I (2019) Effectiveness of subcutaneous tocilizumab in neuromyelitis optica spectrum disorders. Mult Scler Relat Disord 39:101920
Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, Trebst C, Weinshenker B, Wingerchuk D, Parisi JE, Lassmann H (2002) A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 125:1450–1461
Mader S, Kümpfel T, Meinl E (2020) Novel insights into pathophysiology and therapeutic possibilities reveal further differences between AQP4-IgG- and MOG-IgG-associated diseases. Curr Opin Neurol 33:362–371
Majed M, Sanchez CV, Bennett JL, Fryer J, Mulligan MD, Redenbaugh V, McKeon A, Mills JR, Wingerchuk DM, Lennon VA, Weinshenker B, Chen JJ, Flanagan EP, Pittock SJ, Kunchok A (2023) Alterations in aquaporin-4-IgG serostatus in 986 patients: a laboratory-based longitudinal analysis. Ann Neurol. https://doi.org/10.1002/ana.26722. (Online ahead of print)
Mao-Draayer Y, Thiel S, Mills EA, Chitnis T, Fabian M, Katz Sand I, Leite MI, Jarius S, Hellwig K (2020) Neuromyelitis optica spectrum disorders and pregnancy: therapeutic considerations. Nat Rev Neurol 16:154–170
Marcinno A, Marnetto F, Valentino P, Martire S, Balbo A, Drago A, Leto M, Capobianco M, Panzica G, Bertolotto A (2018) Rituximab-induced hypogammaglobulinemia in patients with neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm 5:e498
Marignier R, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk D, Fujihara K, Paul F, Cutter GR, Green AJ, Aktas O, Hartung HP, Lublin FD, Williams IM, Drappa J, She D, Cimbora D, Rees W, Smith M, Ratchford JN, Katz E, Cree BAC, Investigators NMS (2021) Disability outcomes in the N-MOmentum trial of inebilizumab in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 8:e978
Mbaeyi SA, Bozio CH, Duffy J, Rubin LG, Hariri S, Stephens DS, MacNeil JR (2020) Meningococcal vaccination: recommendations of the Advisory Committee on Immunization Practices, United States, 2020. MMWR Recomm Rep 69:1–41
McNamara LA, Topaz N, Wang X, Hariri S, Fox LA, MacNeil JR (2017) High risk for invasive meningococcal disease among patients receiving eculizumab (soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep 66(27):734–737
Mealy MA, Cook LJ, Pache F, Velez DL, Borisow N, Becker D, Arango JAJ, Paul F, Levy M (2018) Vaccines and the association with relapses in patients with neuromyelitis optica spectrum disorder. Mult Scler Relat Disord 23:78–82
Mealy MA, Kim SH, Schmidt F, López R, Jimenez Arango JA, Paul F, Wingerchuk DM, Greenberg BM, Kim HJ, Levy M (2018) Aquaporin-4 serostatus does not predict response to immunotherapy in neuromyelitis optica spectrum disorders. Mult Scler (Houndmills, Basingstoke, Engl) 24:1737–1742
Mealy MA, Levy M (2019) A pilot safety study of ublituximab, a monoclonal antibody against CD20, in acute relapses of neuromyelitis optica spectrum disorder. Medicine (Baltimore) 98:e15944
Mealy MA, Mossburg SE, Kim SH, Messina S, Borisow N, Lopez-Gonzalez R, Ospina JP, Scheel M, Yeshokumar AK, Awad A, Leite MI, Arango JJ, Paul F, Palace J, Kim HJ, Levy M (2019) Long-term disability in neuromyelitis optica spectrum disorder with a history of myelitis is associated with age at onset, delay in diagnosis/preventive treatment, MRI lesion length and presence of symptomatic brain lesions. Mult Scler Relat Disord 28:64–68
Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M (2014) Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol 71:324–330
Meyer A, Drouin J, Weill A, Carbonnel F, Dray-Spira R (2021) Comparative study of pregnancy outcomes in women with inflammatory bowel disease treated with thiopurines and/or anti-TNF: a French nationwide study 2010–2018. Aliment Pharmacol Ther 54:302–311
Mills EA, Mao-Draayer Y (2018) Aging and lymphocyte changes by immunomodulatory therapies impact PML risk in multiple sclerosis patients. Mult Scler (Houndmills, Basingstoke, Engl) 24:1014–1022
Min JH, Kim BJ, Lee KH (2012) Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler (Houndmills, Basingstoke, Engl) 18:113–115
Miyamoto K, Kusunoki S (2009) Intermittent plasmapheresis prevents recurrence in neuromyelitis optica. Ther Apher Dial 13:505–508
Molazadeh N, Filippatou AG, Vasileiou ES, Levy M, Sotirchos ES (2021) Evidence for and against subclinical disease activity and progressive disease in MOG antibody disease and neuromyelitis optica spectrum disorder. J Neuroimmunol 360:577702
Molloy ES, Calabrese LH (2009) Progressive multifocal leukoencephalopathy: a national estimate of frequency in systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum 60:3761–3765
Montcuquet A, Collongues N, Papeix C, Zephir H, Audoin B, Laplaud D, Bourre B, Brochet B, Camdessanche JP, Labauge P, Moreau T, Brassat D, Stankoff B, de Seze J, Vukusic S, Marignier R, group Ns, the Observatoire Francais de la Sclerose en P (2017) Effectiveness of mycophenolate mofetil as first-line therapy in AQP4-IgG, MOG-IgG, and seronegative neuromyelitis optica spectrum disorders. Mult Scler (Houndmills, Basingstoke, Engl) 23:1377–1384
Muller YD, Aubert JD, Vionnet J, Rotman S, Sadallah S, Aubert V, Pascual M (2018) Acute antibody-mediated rejection 1 week after lung transplantation successfully treated with eculizumab, intravenous immunoglobulins, and rituximab. Transplantation 102:e301–e303
Nabizadeh F, Masrouri S, Sharifkazemi H, Azami M, Nikfarjam M, Moghadasi AN (2022) Autologous hematopoietic stem cell transplantation in neuromyelitis optica spectrum disorder: a systematic review and meta-analysis. J Clin Neurosci 105:37–44
Niederberger M, Spranger J (2020) Delphi technique in health sciences: a map. Front Public Health 8:457
Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F (2017) Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol 264:2003–2009
Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, Noji H, Kitamura K, Eto T, Takahashi T, Masuko M, Matsumoto T, Wano Y, Shichishima T, Shibayama H, Hase M, Li L, Johnson K, Lazarowski A, Tamburini P, Inazawa J, Kinoshita T, Kanakura Y (2014) Genetic variants in C5 and poor response to eculizumab. N Engl J Med 370:632–639
Oertel FC, Havla J, Roca-Fernández A, Lizak N, Zimmermann H, Motamedi S, Borisow N, White OB, Bellmann-Strobl J, Albrecht P, Ruprecht K, Jarius S, Palace J, Leite MI, Kuempfel T, Paul F, Brandt AU (2018) Retinal ganglion cell loss in neuromyelitis optica: a longitudinal study. J Neurol Neurosurg Psychiatry 89:1259–1265
Oertel FC, Kuchling J, Zimmermann H, Chien C, Schmidt F, Knier B, Bellmann-Strobl J, Korn T, Scheel M, Klistorner A, Ruprecht K, Paul F, Brandt AU (2017) Microstructural visual system changes in AQP4-antibody-seropositive NMOSD. Neurol Neuroimmunol Neuroinflamm 4:e334
Oertel FC, Specovius S, Zimmermann HG, Chien C, Motamedi S, Bereuter C, Cook L, Lana Peixoto MA, Fontanelle MA, Kim HJ, Hyun JW, Palace J, Roca-Fernandez A, Leite MI, Sharma S, Ashtari F, Kafieh R, Dehghani A, Pourazizi M, Pandit L, D’Cunha A, Aktas O, Ringelstein M, Albrecht P, May E, Tongco C, Leocani L, Pisa M, Radaelli M, Martinez-Lapiscina EH, Stiebel-Kalish H, Siritho S, de Seze J, Senger T, Havla J, Marignier R, Cobo-Calvo A, Bichuetti D, Tavares IM, Asgari N, Soelberg K, Altintas A, Yildirim R, Tanriverdi U, Jacob A, Huda S, Rimler Z, Reid A, Mao-Draayer Y, Soto de Castillo I, Petzold A, Green AJ, Yeaman MR, Smith T, Brandt AU, Paul F (2021) Retinal optical coherence tomography in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 8:e1068
Oomen I, Nassar-Sheikh Rashid A, Bouts AHM, Gouw SC, Kuijpers TW, Rispens T, de Vries A, Wolbink G, van den Berg JM, Schonenberg-Meinema D (2022) Anti-rituximab antibodies affect pharmacokinetics and pharmacodynamics of rituximab in children with immune-mediated diseases. Clin Exp Rheumatol 40:183–190
Oray M, Abu Samra K, Ebrahimiadib N, Meese H, Foster CS (2016) Long-term side effects of glucocorticoids. Expert Opin Drug Saf 15:457–465
Oren S, Mandelboim M, Braun-Moscovici Y, Paran D, Ablin J, Litinsky I, Comaneshter D, Levartovsky D, Mendelson E, Azar R, Wigler I, Balbir-Gurman A, Caspi D, Elkayam O (2008) Vaccination against influenza in patients with rheumatoid arthritis: the effect of rituximab on the humoral response. Ann Rheum Dis 67:937–941
Ortiz S, Pittock SJ, Berthele A, Levy M, Nakashima I, Oreja-Guevara C, Kim HJ (2023) Pharmacokinetics and pharmacodynamics of ravulizumab in adults with anti-aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder during the phase 3 CHAMPION-NMOSD trial. AAN Meeting S5:004
Pache F, Zimmermann H, Finke C, Lacheta A, Papazoglou S, Kuchling J, Wuerfel J, Hamm B, Ruprecht K, Paul F, Brandt AU, Scheel M (2016) Brain parenchymal damage in neuromyelitis optica spectrum disorder—a multimodal MRI study. Eur Radiol 26:4413–4422
Padmanabhan A, Connelly-Smith L, Aqui N, Balogun RA, Klingel R, Meyer E, Pham HP, Schneiderman J, Witt V, Wu Y, Zantek ND, Dunbar NM, Schwartz GEJ (2019) Guidelines on the use of therapeutic apheresis in clinical practice—evidence-based approach from the writing committee of the American Society for Apheresis: the eighth special issue. J Clin Apher 34:171–354
Palace J, Lin DY, Zeng D, Majed M, Elsone L, Hamid S, Messina S, Misu T, Sagen J, Whittam D, Takai Y, Leite MI, Weinshenker B, Cabre P, Jacob A, Nakashima I, Fujihara K, Pittock SJ (2019) Outcome prediction models in AQP4-IgG positive neuromyelitis optica spectrum disorders. Brain 142:1310–1323
Palace J, Wingerchuk DM, Fujihara K, Berthele A, Oreja-Guevara C, Kim HJ, Nakashima I, Levy M, Terzi M, Totolyan N, Viswanathan S, Wang KC, Pace A, Yountz M, Miller L, Armstrong R, Pittock S, Group PS (2021) Benefits of eculizumab in AQP4+ neuromyelitis optica spectrum disorder: subgroup analyses of the randomized controlled phase 3 PREVENT trial. Mult Scler Relat Disord 47:102641
Papadopoulos MC, Bennett JL, Verkman AS (2014) Treatment of neuromyelitis optica: state-of-the-art and emerging therapies. Nat Rev Neurol 10:493–506
Papeix C, Vidal J-S, Seze Jd, Pierrot-Deseilligny C, Tourbah A, Stankoff B, Lebrun C, Moreau T, Vermersch P, Fontaine B, Lyon-Caen O, Gout O (2007) Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler (Houndmills, Basingstoke, Engl) 13:256–259
Park SH, Park CY, Shin YJ, Jeong KS, Kim NH (2020) Low contrast visual acuity might help to detect previous optic neuritis. Front Neurol 11:602193
Paul F, Marignier R, Palace J, Arrambide G, Asgari N, Bennett JL, Cree BAC, De Seze J, Fujihara K, Kim HJ, Hornby R, Huda S, Kissani N, Kleiter I, Kuwabara S, Lana-Peixoto M, Law L, Leite MI, Pandit L, Pittock SJ, Quan C, Ramanathan S, Rotstein D, Saiz A, Sato DK, Vaknin-Dembinsky A (2023) International delphi consensus on the management of AQP4-IgG+ NMOSD: recommendations for eculizumab, inebilizumab, and satralizumab. Neurol Neuroimmunol Neuroinflamm 10:e200124
Paul S, Dickstein A, Saxena A, Terrin N, Viveiros K, Balk EM, Wong JB (2017) Role of surface antibody in hepatitis B reactivation in patients with resolved infection and hematologic malignancy: a meta-analysis. Hepatology 66:379–388
Pittock SJ, Barnett M, Bennett JL, Berthele A, de Seze J, Levy M, Nakashima I, Oreja-Guevara C, Palace J, Paul F, Pozzilli C, Yountz M, Allen K, Mashhoon Y, Kim HJ (2023) Ravulizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. Ann Neurol 93:1053–1068
Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, Nakashima I, Terzi M, Totolyan N, Viswanathan S, Wang KC, Pace A, Fujita KP, Armstrong R, Wingerchuk DM (2019) Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med 381:614–625
Pittock SJ, Fujihara K, Palace J, Berthele A, Kim HJ, Guevara CO, Nakashima I, Levy M, Shang S, Yountz M, Miller L, Armstrong R, Wingerchuk DM (2022) Eculizumab monotherapy for NMOSD: Data from PREVENT and its open-label extension. Mult Scler (Houndmills, Basingstoke, Engl) 28:480–486
Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, O’Toole O, Wingerchuk DM (2013) Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol 12:554–562
Plate A, Havla J, Kumpfel T (2014) Late-onset neutropenia during long-term rituximab therapy in neuromyelitis optica. Mult Scler Relat Disord 3:269–272
Poupart J, Giovannelli J, Deschamps R, Audoin B, Ciron J, Maillart E, Papeix C, Collongues N, Bourre B, Cohen M, Wiertlewski S, Outteryck O, Laplaud D, Vukusic S, Marignier R, Zephir H, group Ns (2020) Evaluation of efficacy and tolerability of first-line therapies in NMOSD. Neurology 94:e1645–e1656
Qin C, Tian DS, Zhou LQ, Shang K, Huang L, Dong MH, You YF, Xiao J, Xiong Y, Wang W, Pang H, Guo JJ, Cai SB, Wang D, Li CR, Zhang M, Bu BT, Wang W (2023) Anti-BCMA CAR T cell therapy CT103A in relapsed or refractory AQP4-IgG seropositive neuromyelitis optica spectrum disorders: phase 1 trial interim results. Signal Transduct Target Ther 8:5
Qiu W, Kermode AG, Li R, Dai Y, Wang Y, Wang J, Zhong X, Li C, Lu Z, Hu X (2015) Azathioprine plus corticosteroid treatment in Chinese patients with neuromyelitis optica. J Clin Neurosci 22:1178–1182
Ramanathan RS, Malhotra K, Scott T (2014) Treatment of neuromyelitis optica/neuromyelitis optica spectrum disorders with methotrexate. BMC Neurol 14:1–5
Ramanathan S, Reddel SW, Henderson A, Parratt JD, Barnett M, Gatt PN, Merheb V, Kumaran RY, Pathmanandavel K, Sinmaz N, Ghadiri M, Yiannikas C, Vucic S, Stewart G, Bleasel AF, Booth D, Fung VS, Dale RC, Brilot F (2014) Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm 1:e40
Rensel M, Zabeti A, Mealy MA, Cimbora D, She D, Drappa J, Katz E (2022) Long-term efficacy and safety of inebilizumab in neuromyelitis optica spectrum disorder: analysis of aquaporin-4–immunoglobulin G–seropositive participants taking inebilizumab for ⩾4 years in the N-MOmentum trial. Mult Scler (Houndmills, Basingstoke, Engl) 28:925–932
Reuss R, Rommer P, Brueck W, Jarius S, Bolz M, Zettl U (2010) Anti-AQP4 ab might be relevant in pregnancy. BMJ cited electronic resource, available from: http://www.bmj.com/rapid-response/2011/2011/2002/anti-aqp2014-ab-might-be-relevant-pregnancy
Reuss R, Rommer PS, Brück W, Paul F, Bolz M, Jarius S, Boettcher T, Grossmann A, Bock A, Zipp F, Benecke R, Zettl UK (2009) A woman with acute myelopathy in pregnancy: case outcome. BMJ 339:b4026
Ribes D, Belliere J, Piedrafita A, Faguer S (2019) Glucocorticoid-free induction regimen in severe ANCA-associated vasculitis using a combination of rituximab and eculizumab. Rheumatology (Oxford) 58:2335–2337
Rigal J, Ciron J, Lépine Z, Biotti D (2020) Late-onset neutropenia after RITUXIMAB therapy for multiple sclerosis, neuromyelitis optica spectrum disorders and MOG-antibody-associated diseases. Mult Scler Relat Disord 41:102019
Ringelstein M (2022) ECTRIMS 2022—ePoster. Mult Scler J 28:692–945
Ringelstein M, Ayzenberg I, Harmel J, Lauenstein AS, Lensch E, Stogbauer F, Hellwig K, Ellrichmann G, Stettner M, Chan A, Hartung HP, Kieseier B, Gold R, Aktas O, Kleiter I (2015) Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol 72:756–763
Ringelstein M, Ayzenberg I, Lindenblatt G, Fischer K, Gahlen A, Novi G, Hayward-Konnecke H, Schippling S, Rommer PS, Kornek B, Zrzavy T, Biotti D, Ciron J, Audoin B, Berthele A, Giglhuber K, Zephir H, Kumpfel T, Berger R, Rother J, Haussler V, Stellmann JP, Whittam D, Jacob A, Kraemer M, Gueguen A, Deschamps R, Bayas A, Hummert MW, Trebst C, Haarmann A, Jarius S, Wildemann B, Grothe M, Siebert N, Ruprecht K, Paul F, Collongues N, Marignier R, Levy M, Karenfort M, Deppe M, Albrecht P, Hellwig K, Gold R, Hartung HP, Meuth SG, Kleiter I, Aktas O, Neuromyelitis Optica Study G (2022) Interleukin-6 receptor blockade in treatment-refractory MOG-IgG-associated disease and neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm 9:e1100
Ringelstein M, Harmel J, Distelmaier F, Ingwersen J, Menge T, Hellwig K, Kieseier B, Mayatepek E, Hartung HP, Kuempfel T, Aktas O (2013) Neuromyelitis optica and pregnancy during therapeutic B cell depletion: infant exposure to anti-AQP4 antibody and prevention of rebound relapses with low-dose rituximab postpartum. Mult Scler (Houndmills, Basingstoke, Engl) 19:1544–1547
Ringelstein M, Harmel J, Zimmermann H, Brandt AU, Paul F, Haarmann A, Buttmann M, Hümmert MW, Trebst C, Schroeder C, Ayzenberg I, Kleiter I, Hellwig K, Havla J, Kümpfel T, Jarius S, Wildemann B, Rommer P, Weber MS, Pellkofer H, Röpke L, Geis C, Retzlaff N, Zettl U, Deppe M, Klotz L, Young K, Stellmann JP, Kaste M, Kermer P, Marouf W, Lauda F, Tumani H, Graf J, Klistorner A, Hartung HP, Aktas O, Albrecht P (2020) Longitudinal optic neuritis-unrelated visual evoked potential changes in NMO spectrum disorders. Neurology 94:e407–e418
Roberts DM, Jones RB, Smith RM, Alberici F, Kumaratne DS, Burns S, Jayne DR (2015) Rituximab-associated hypogammaglobulinemia: incidence, predictors and outcomes in patients with multi-system autoimmune disease. J Autoimmun 57:60–65
Rojas JI, Pappolla A, Patrucco L, Cristiano E, Miguez J, Liwacki S, Tkachuk V, Balbuena ME, Vrech C, Deri N, Correale J, Marrodan M, Ysrraelit MC, Fiol M, Leguizamon F, Luetic G, Menichini ML, Lopez PA, Pettinicchi JP, Criniti J, Caride A, Tavolini D, Mainella C, Zanga G, Burgos M, Hryb J, Barboza A, Lazaro L, Alonso R, Silva B, Fernández Liguori N, Nadur D, Chercoff A, Martinez A, Steinberg J, Garcea O, Carrá A, Alonso Serena M, Carnero Contentti E (2022) Disability outcomes in NMOSD and MOGAD patients: data from a nationwide registry in Argentina. Neurol Sci 44:281–286
Rondeau E, Ardissino G, Caby-Tosi MP, Al-Dakkak I, Fakhouri F, Miller B, Scully M (2022) Pregnancy in women with atypical hemolytic uremic syndrome. Nephron 146:1–10
Saadoun S, Waters P, Leite MI, Bennett JL, Vincent A, Papadopoulos MC (2013) Neuromyelitis optica IgG causes placental inflammation and fetal death. J Immunol 191:2999–3005
Schindler P, Aktas O, Ringelstein M, Wildemann B, Jarius S, Paul F, Ruprecht K (2023) Glial fibrillary acidic protein as a biomarker in neuromyelitis optica spectrum disorder: a current review. Expert Rev Clin Immunol 19:71–91
Schmetzer O, Lakin E, Roediger B, Duchow A, Asseyer S, Paul F, Siebert N (2021) Anti-aquaporin 4 IgG is not associated with any clinical disease characteristics in neuromyelitis optica spectrum disorder. Front Neurol 12:635419
Schweitzer F, Laurent S, Fink GR, Barnett MH, Reddel S, Hartung HP, Warnke C (2019) Age and the risks of high-efficacy disease modifying drugs in multiple sclerosis. Curr Opin Neurol 32:305–312
Sechi E, Zarbo R, Biancu MA, Chessa P, Idda ML, Orrù V, Lai S, Leoni S, Solla P (2021) Prolonged B cell depletion after rituximab in AQP4-IgG-positive neuromyelitis optica spectrum disorder. J Neuroimmunol 358:577666
Sepúlveda M, Armangué T, Sola-Valls N, Arrambide G, Meca-Lallana JE, Oreja-Guevara C, Mendibe M, Alvarez de Arcaya A, Aladro Y, Casanova B, Olascoaga J, Jiménez-Huete A, Fernández-Fournier M, Ramió-Torrentà L, Cobo-Calvo A, Viñals M, de Andrés C, Meca-Lallana V, Cervelló A, Calles C, Rubio MB, Ramo-Tello C, Caminero A, Munteis E, Antigüedad AR, Blanco Y, Villoslada P, Montalban X, Graus F, Saiz A (2016) Neuromyelitis optica spectrum disorders: comparison according to the phenotype and serostatus. Neurol Neuroimmunol Neuroinflamm 3:e225
Shi B, Zhao M, Qiao L, Huang F, Zhou S, Wei Y, Wang J, Wang N (2021) Relapses shortly after rituximab treatment in neuromyelitis optica spectrum disorder. Mult Scler Relat Disord 54:103143
Shosha E, Pittock SJ, Flanagan E, Weinshenker BG (2017) Neuromyelitis optica spectrum disorders and pregnancy: interactions and management. MSJ 23:1808–1817
Siebert N, Duchow A, Paul F, Infante-Duarte C, Bellmann-Strobl J (2021) Inebilizumab in AQP4-Ab-positive neuromyelitis optica spectrum disorder. Drugs Today (Barc) 57:321–336
Siritho S, Nopsopon T, Pongpirul K (2021) Therapeutic plasma exchange vs conventional treatment with intravenous high dose steroid for neuromyelitis optica spectrum disorders (NMOSD): a systematic review and meta-analysis. J Neurol 268:4549–4562
Stastna D, Menkyova I, Drahota J, Hrnciarova T, Kubala Havrdova E, Vachova M, Andelova M, Kleinova P, Kovarova I, Krasulova E, Preiningerova JL, Novakova I, Novotna K, Novotna M, Nytrova P, Pavlickova J, Srpova B, Storey K, Ticha V, Tyblova M, Uher T, Vodehnalova K, Horakova D (2022) To be or not to be vaccinated: the risk of MS or NMOSD relapse after COVID-19 vaccination and infection. Mult Scler Relat Disord 65:104014
Steinman L, Bar-Or A, Behne JM, Benitez-Ribas D, Chin PS, Clare-Salzler M, Healey D, Kim JI, Kranz DM, Lutterotti A, Martin R, Schippling S, Villoslada P, Wei CH, Weiner HL, Zamvil SS, Yeaman MR, Smith TJ (2016) Restoring immune tolerance in neuromyelitis optica: part I. Neurol Neuroimmunol Neuroinflamm 3:e276
Stellmann JP, Krumbholz M, Friede T, Gahlen A, Borisow N, Fischer K, Hellwig K, Pache F, Ruprecht K, Havla J, Kumpfel T, Aktas O, Hartung HP, Ringelstein M, Geis C, Kleinschnitz C, Berthele A, Hemmer B, Angstwurm K, Young KL, Schuster S, Stangel M, Lauda F, Tumani H, Mayer C, Zeltner L, Ziemann U, Linker RA, Schwab M, Marziniak M, Then Bergh F, Hofstadt-van Oy U, Neuhaus O, Zettl U, Faiss J, Wildemann B, Paul F, Jarius S, Trebst C, Kleiter I (2017) Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J Neurol Neurosurg Psychiatry 88:639–647
Stiebel-Kalish H, Hellmann MA, Mimouni M, Paul F, Bialer O, Bach M, Lotan I (2019) Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol Neuroimmunol Neuroinflamm 6:e572
Szepanowski F, Warnke C, Meyer-Zu-Hörste G, Mausberg AK, Hartung HP, Kleinschnitz C, Stettner M (2021) Secondary immunodeficiency and risk of infection following immune therapies in neurology. CNS Drugs 35:1173–1188
Tahara M, Oeda T, Okada K, Kiriyama T, Ochi K, Maruyama H, Fukaura H, Nomura K, Shimizu Y, Mori M, Nakashima I, Misu T, Umemura A, Yamamoto K, Sawada H (2020) Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 19:298–306
Tahara M, Oeda T, Okada K, Ochi K, Maruyama H, Fukaura H, Nomura K, Shimizu Y, Nakashima I, Misu T, Umemura A, Yamamoto K, Sawada H (2022) Compassionate open-label use of rituximab following a randomised clinical trial against neuromyelitis optica (RIN-2 study): B cell monitoring-based administration. Mult Scler Relat Disord 60:103730
Takai Y, Kuroda H, Misu T, Akaishi T, Nakashima I, Takahashi T, Nishiyama S, Fujihara K, Aoki M (2021) Optimal management of neuromyelitis optica spectrum disorder with aquaporin-4 antibody by oral prednisolone maintenance therapy. Mult Scler Relat Disord 49:102750
Takeshita Y, Fujikawa S, Serizawa K, Fujisawa M, Matsuo K, Nemoto J, Shimizu F, Sano Y, Tomizawa-Shinohara H, Miyake S, Ransohoff RM, Kanda T (2021) New BBB model reveals that IL-6 blockade suppressed the BBB disorder, preventing onset of NMOSD. Neurol Neuroimmunol Neuroinflamm 8:e1076
Tenembaum S, Yeh EA (2020) Pediatric NMOSD: a review and position statement on approach to work-up and diagnosis. Front Pediatr 8:339
Thai TN, Sarayani A, Wang X, Albogami Y, Rasmussen SA, Winterstein AG (2020) Risk of pregnancy loss in patients exposed to mycophenolate compared to azathioprine: a retrospective cohort study. Pharmacoepidemiol Drug Saf 29:716–724
Traboulsee A, Fujihara K, Kim HJ, Marignier R, O’Connor K, Sergott R, Wiendl H, Wuerfel J, Zamvil S, Pittock S, Anania V, Buffels R, Künzel T, Lennon-Chrimes S, Bennett J (2022) SAkuraBONSAI: a prospective, open-label study of satralizumab investigating novel imaging, biomarker, and clinical outcomes in patients with aquaporin-4-IgG-seropositive (AQP4-IgG+) neuromyelitis optica spectrum disorder (NMOSD). AAN Meet P15:002
Traboulsee A, Greenberg BM, Bennett JL, Szczechowski L, Fox E, Shkrobot S, Yamamura T, Terada Y, Kawata Y, Wright P, Gianella-Borradori A, Garren H, Weinshenker BG (2020) Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol 19:402–412
Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, Bennett JL, Verkman AS (2012) Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol 71:314–322
Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, Borisow N, Kleiter I, Aktas O, Kumpfel T, Neuromyelitis Optica Study G (2014) Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol 261:1–16
Venero JL, Vizuete ML, Machado A, Cano J (2001) Aquaporins in the central nervous system. Prog Neurobiol 63:321–336
Viswanathan S, Wong AH, Quek AM, Yuki N (2015) Intravenous immunoglobulin may reduce relapse frequency in neuromyelitis optica. J Neuroimmunol 282:92–96
Vollmer BL, Wallach AI, Corboy JR, Dubovskaya K, Alvarez E, Kister I (2020) Serious safety events in rituximab-treated multiple sclerosis and related disorders. Ann Clin Transl Neurol 7:1477–1487
Vu T, Ortiz S, Katsuno M, Annane D, Mantegazza R, Beasley KN, Aguzzi R, Howard JF Jr (2023) Ravulizumab pharmacokinetics and pharmacodynamics in patients with generalized myasthenia gravis. J Neurol 270:3129–3137
Vukusic S, Marignier R, Ciron J, Bourre B, Cohen M, Deschamps R, Guillaume M, Kremer L, Pique J, Carra-Dalliere C, Michel L, Leray E, Guennoc AM, Laplaud D, Androdias G, Bensa C, Bigaut K, Biotti D, Branger P, Casez O, Daval E, Donze C, Dubessy AL, Dulau C, Durand-Dubief F, Hebant B, Kwiatkowski A, Lannoy J, Maarouf A, Manchon E, Mathey G, Moisset X, Montcuquet A, Roux T, Maillart E, Lebrun-Frenay C (2023) Pregnancy and neuromyelitis optica spectrum disorders: 2022 recommendations from the French Multiple Sclerosis Society. Mult Scler (Houndmills, Basingstoke, Engl) 29:37–51
Wang J, Johnson T, Sahin L, Tassinari MS, Anderson PO, Baker TE, Bucci-Rechtweg C, Burckart GJ, Chambers CD, Hale TW, Johnson-Lyles D, Nelson RM, Nguyen C, Pica-Branco D, Ren Z, Sachs H, Sauberan J, Zajicek A, Ito S, Yao LP (2017) Evaluation of the safety of drugs and biological products used during lactation: workshop summary. Clin Pharmacol Ther 101:736–744
Wang L, Du L, Li Q, Li F, Wang B, Zhao Y, Meng Q, Li W, Pan J, Xia J, Wu S, Yang J, Li H, Ma J, ZhangBao J, Huang W, Chang X, Tan H, Yu J, Zhou L, Lu C, Wang M, Dong Q, Lu J, Zhao C, Quan C (2022) Neuromyelitis optica spectrum disorder with anti-aquaporin-4 antibody: outcome prediction models. Front Immunol 13:873576
Wang L, Liu K, Tan X, Zhou L, Zhang Y, Liu X, Fu Y, Qiu W, Yang H (2021) Remedial effect of intravenous cyclophosphamide in corticosteroid-refractory patients in the acute phase of neuromyelitis optica spectrum disorder-related optic neuritis. Front Neurol 11:612097
Wang L, Su M, Zhou Z, Zhou L, ZhangBao J, Tan H, Huang W, Chang X, Lu C, Yu J, Wang M, Lu J, Zhao C, Zhang T, Quan C (2022) Analysis of pregnancy-related attacks in neuromyelitis optica spectrum disorder: a systematic review and meta-analysis. JAMA Netw Open 5:e2225438
Wang Y, Ma J, Chang H, Zhang X, Yin L (2021) Efficacy of mycophenolate mofetil in the treatment of neuromyelitis optica spectrum disorders: an update systematic review and meta-analysis. Mult Scler Relat Disord 55:103181
Wang Y, Zhong X, Wang H, Peng Y, Shi F, Jia D, Yang H, Zeng Q, Quan C, ZhangBao J, Lee M, Qi J, Chen X, Qiu W, Batoclimab NSG (2022) Batoclimab as an add-on therapy in neuromyelitis optica spectrum disorder patients with acute attacks. Eur J Neurol 30:195–203
Watanabe S, Misu T, Miyazawa I, Nakashima I, Shiga Y, Fujihara K, Itoyama Y (2007) Low-dose corticosteroids reduce relapses in neuromyelitis optica: a retrospective analysis. Mult Scler (Houndmills, Basingstoke, Engl) 13:968–974
Weber-Schoendorfer C, Schaefer C (2016) Pregnancy outcome after tocilizumab therapy in early pregnancy-a case series from the German Embryotox Pharmacovigilance Center. Reprod Toxicol 60:29–32
Weinfurtner K, Graves J, Ness J, Krupp L, Milazzo M, Waubant E (2015) Prolonged remission in neuromyelitis optica following cessation of rituximab treatment. J Child Neurol 30:1366–1370
Weinshenker BG, Barron G, Behne JM, Bennett JL, Chin PS, Cree BA, de Seze J, Flor A, Fujihara K, Greenberg B, Higashi S, Holt W, Khan O, Knappertz V, Levy M, Melia AT, Palace J, Smith TJ, Sormani MP, Van Herle K, VanMeter S, Villoslada P, Walton MK, Wasiewski W, Wingerchuk DM, Yeaman MR (2015) Challenges and opportunities in designing clinical trials for neuromyelitis optica. Neurology 84:1805–1815
Weinshenker BG, O’Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW, Pineda AA, Stevens LN, Rodriguez M (1999) A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol 46:878–886
Weinshenker BG, Wingerchuk DM, Vukusic S, Linbo L, Pittock SJ, Lucchinetti CF, Lennon VA (2006) Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 59:566–569
Wijnsma KL, Ter Heine R, Moes D, Langemeijer S, Schols SEM, Volokhina EB, van den Heuvel LP, Wetzels JFM, van de Kar N, Bruggemann RJ (2019) Pharmacology, pharmacokinetics and pharmacodynamics of eculizumab, and possibilities for an individualized approach to eculizumab. Clin Pharmacokinet 58:859–874
Wilson R, Makuch M, Kienzler AK, Varley J, Taylor J, Woodhall M, Palace J, Leite MI, Waters P, Irani SR (2018) Condition-dependent generation of aquaporin-4 antibodies from circulating B cells in neuromyelitis optica. Brain 141:1063–1074
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, de Seze J, Fujihara K, Greenberg B, Jacob A, Jarius S, Lana-Peixoto M, Levy M, Simon JH, Tenembaum S, Traboulsee AL, Waters P, Wellik KE, Weinshenker BG (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85:177–189
Wingerchuk DM, Fujihara K, Palace J, Berthele A, Levy M, Kim HJ, Nakashima I, Oreja-Guevara C, Wang KC, Miller L, Shang S, Sabatella G, Yountz M, Pittock SJ, Group PS (2021) Long-term safety and efficacy of eculizumab in aquaporin-4 IgG-positive NMOSD. Ann Neurol 89:1088–1098
Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG (2007) A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 68:603–605
Wingerchuk DM, Zhang I, Kielhorn A, Royston M, Levy M, Fujihara K, Nakashima I, Tanvir I, Paul F, Pittock SJ (2022) Network meta-analysis of food and drug administration-approved treatment options for adults with aquaporin-4 immunoglobulin G-positive neuromyelitis optica spectrum disorder. Neurol Ther 11:123–135
Yamamura T, Araki M, Fujihara K, Okuno T, Misu T, Guo YC, Hemingway C, Matsushima J, Sugaya N, Yamashita M, von Büdingen HC, Miyamoto K (2022) Exploring steroid tapering in patients with neuromyelitis optica spectrum disorder treated with satralizumab in SAkuraSky: a case series. Mult Scler Relat Disord 61:103772
Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, Patti F, Tsai CP, Saiz A, Yamazaki H, Kawata Y, Wright P, De Seze J (2019) Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med 381:2114–2124
Yamamura T, Weinshenker B, Yeaman MR, De Seze J, Patti F, Lobo P, von Budingen HC, Kou X, Weber K, Greenberg B (2022) Long-term safety of satralizumab in neuromyelitis optica spectrum disorder (NMOSD) from SAkuraSky and SAkuraStar. Mult Scler Relat Disord 66:104025
Yang S, Zhang C, Zhang TX, Feng B, Jia D, Han S, Li T, Shen Y, Yan G, Zhang C (2022) A real-world study of interleukin-6 receptor blockade in patients with neuromyelitis optica spectrum disorder. J Neurol 270:348–356
Zéphir H, Bernard-Valnet R, Lebrun C, Outteryck O, Audoin B, Bourre B, Pittion S, Wiertlewski S, Ouallet JC, Neau JP, Ciron J, Clavelou P, Marignier R, Brassat D (2015) Rituximab as first-line therapy in neuromyelitis optica: efficiency and tolerability. J Neurol 262:2329–2335
Zhang C, Tian DC, Yang CS, Han B, Wang J, Yang L, Shi FD (2017) Safety and efficacy of bortezomib in patients with highly relapsing neuromyelitis optica spectrum disorder. JAMA Neurol 74:1010–1012
Zhang C, Zhang M, Qiu W, Ma H, Zhang X, Zhu Z, Yang C-S, Jia D, Zhang T-X, Yuan M, Feng Y, Yang L, Lu W, Yu C, Bennett JL, Shi F-D (2020) Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, phase 2 trial. Lancet Neurol 19:391–401
Zhang P, Liu B (2020) Effect of autologous hematopoietic stem cell transplantation on multiple sclerosis and neuromyelitis optica spectrum disorder: a PRISMA-compliant meta-analysis. Bone Marrow Transplant 55:1928–1934
Zhang Y, Li D, Guo H, Wang W, Li X, Shen S (2021) Association between thiopurines use and pregnancy outcomes in female patients with inflammatory bowel disease: a meta-analysis. Curr Pharm Des 27:2317–2324
Zhong X, Zhou Y, Lu T, Wang Z, Fang L, Peng L, Kermode AG, Qiu W (2018) Infections in neuromyelitis optica spectrum disorder. J Clin Neurosci 47:14–19
Zubizarreta I, Florez-Grau G, Vila G, Cabezon R, Espana C, Andorra M, Saiz A, Llufriu S, Sepulveda M, Sola-Valls N, Martinez-Lapiscina EH, Pulido-Valdeolivas I, Casanova B, Martinez Gines M, Tellez N, Oreja-Guevara C, Espanol M, Trias E, Cid J, Juan M, Lozano M, Blanco Y, Steinman L, Benitez-Ribas D, Villoslada P (2019) Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc Natl Acad Sci U S A 116:8463–8470
Acknowledgements
The authors are grateful to Kathleen Ingenhoven for continuous excellent organizational support. We thank Mr. Ilya Demchenko from Insight Editing London for help in scientific language editing.
Members of the Neuromyelitis Optica Study Group (NEMOS) in alphabetical order: Orhan Aktas, Heinrich Heine University Düsseldorf, Düsseldorf, Germany; Philipp Albrecht, Heinrich Heine University Düsseldorf, Germany; Klemens Angstwurm, University Hospital, Regensburg, Germany; Susanna Asseyer, Charité – Universitätsmedizin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Ana Beatriz Ayroza Galvao Ribeiro Gomes, Universitätsspital Basel; Ilya Ayzenberg, St. Josef Hospital, Ruhr University Bochum, Bochum, Germany; Antonios Bayas, University Hospital, Augsburg, Germany; Stefanie Behnke, Knappschaftsklinikum Saar GmbH, Sulzbach, Germany; Judith Bellmann-Strobl, Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany, and Experimental and Clinical Research Center, Berlin, Germany, and Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Berlin, Germany; Achim Berthele, Klinikum rechts der Isar, School of Medicine, Technical University Munich, Munich, Germany; Stefan Bittner, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; Franziska Buetow, Hannover Medical School, Germany; Mathias Buttmann, Caritas Krankenhaus Bad Mergentheim, Germany; Ankelien Duchow, Charité -Universitätsmedizin Berlin, Germany; Daniel Engels, Ludwig-Maximilians University, Munich, Germany; Thorleif Etgen, Kliniken Südostbayern-Klinikum Traunstein, Germany; Katinka Fischer, MSc, Heinrich Heine University, Düsseldorf, Germany; Benedikt Frank, University Hospital, Essen, Germany; Anna Gahlen, MVZ für Neurologie, Psychiatrie und Physiotherapie, Herne, Germany; Achim Gass, University Hospital, Mannheim, Germany; Johannes Gehring; University Hospital, Frankfurt, Germany; Christian Geis, Department of Neurology, Jena University Hospital, Germany; Katrin Giglhuber, Klinikum rechts der Isar, School of Medicine, Technical University Munich, Munich, Germany; Ralf Gold, Ruhr University of Bochum, Germany; Yasemin Göreci, Faculty of Medicine and University Hospital Cologne, Germany; Jonas Graf, Heinrich Heine University Düsseldorf, Düsseldorf, Germany; Sergiu Groppa, University Medical Center of the Johannes Gutenberg University Mainz, Germany; Matthias Grothe, University Hospital, Greifswald, Germany; Julia Gutbrod, University Hospital, Augsburg, Germany; Kersten Guthke, Klinikum Görlitz, Germany; Axel Haarmann, University of Würzburg, Germany; Vivien Häußler, University Medical Center Hamburg-Eppendorf, Hamburg, Germany; Maria Hastermann—- Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Joachim Havla, Ludwig-Maximilians-Universität München, Munich, Germany; Kerstin Hellwig, St. Josef Hospital, Ruhr University Bochum, Germany; Bernhard Hemmer, Klinikum rechts der Isar, School of Medicine, Technical University Munich, Germany; Mariella Herfurth, University of Leipzig, Leipzig, Germany; Marina Herwerth, Universitätsspital Zürich, Schweiz; Frank Hoffmann, Krankenhaus Martha-Maria, Halle, Germany; Olaf Hoffmann, St. Josefs-Krankenhaus, Potsdam, Germany; Ulrich Hofstadt-van Oy, Klinikum Westfalen, Dortmund, Germany; Martin W. Hümmert, Hannover Medical School, Hanover, Germany; Leila Husseini, University Hospital, Göttingen, Germany; Sven Jarius, University of Heidelberg, Heidelberg, Germany; Jutta Junghans, Krankenhaus Martha-Maria, Halle, Germany; Matthias Kaste, Nordwest Hospital Sanderbusch, Sande, Germany; Peter Kern, Asklepios Klinik, Teupitz, Germany; Karsten Kern, Knappschaftsklinikum Saar GmbH, Sulzbach, Germany; Pawel Kermer, Nordwest Hospital Sanderbusch, Sande, Germany; Christoph Kleinschnitz, University Hospital, Essen, Germany; Ingo Kleiter, Marianne-Strauß-Klinik, Berg, Germany; Luisa Klotz, University of Münster, Münster, Germany; Wolfgang Köhler, University Hospital Leipzig, Germany; Kimberly Körbel, University Hospital, Frankfurt, Germany; Markus Kowarik, University Hospital, Tübingen, Germany; Markus Kraemer, Alfried-Krupp-Krankenhaus Essen, Germany; Julian Kretschmer, Hannover Medical School, Hanover, Germany; Markus Krumbholz, Brandenburg Medical School, Rüdersdorf bei Berlin, Germany; Tania Kümpfel, LMU Hospital, Ludwig- Maximilians-Universität München, Munich, Germany; Natalia Kurka, University Hospital, Frankfurt, Germany; Theodoros Ladopoulus, St. Josef Hospital, Ruhr University Bochum, Bochum, Germany; Ann-Sophie Lauenstein, German Diagnostic Clinic, DKD Helios Clinic Wiesbaden, Germany; Sarah Laurent, Faculty of Medicine and University Hospital Cologne, Germany; De-Hyung Lee, University Hospital, Regensburg, Germany; Dominik Lehrieder; Department of Neurology, University Hospital of Würzburg, Germany; Frank Leypoldt, University Medical Center Schleswig Holstein – Kiel, Germany; Martin Liebetrau, St. Josefs-Hospital, Wiesbaden, Germany; Ralf Linker, University Hospital, Regensburg, Germany; Gero Lindenblatt Heinrich Heine University Düsseldorf, Düsseldorf, Germany; Lisa Lohmann, University of Münster, Münster, Germany; Felix Lüssi, University Medical Center of the Johannes Gutenberg University Mainz, Germany; Peter Luedemann; Agaplesion ev. Bathildiskrankenhaus Bad Pyrmont, Germany; Michelle Maiworm, University Hospital, Frankfurt, Germany; Martin Marziniak, Isar-Amper Klinik Ost, Munich, Germany; Christoph Mayer, Neurologischen Gemeinschaftspraxis am Kaiserplatz, Frankfurt, Germany; Stefanie Meister, University Hospital, Rostock, Germany; Mathias von Mering, Klinikum Bremen Nord, Germany; Imke Metz, University Hospital, Göttingen, Germany; Sven Meuth, Heinrich Heine University,Düsseldorf, Germany; Jasmin Naumann, Knappschaftsklinikum Saar GmbH, Sulzbach, Germany; Oliver Neuhaus, SRH Krankenhaus Sigmaringen, Germany; Tradite Neziraj, Universitätsspital Basel, Schweiz; Moritz Niederschweiberer, Charité Universitätsmedizin, Berlin, Germany; Sabine Niehaus, Klinikum Dortmund, Germany; Carolin Otto, Charité Universitätsmedizin, Berlin, Germany; Florence Pache, Charité Universitätsmedizin, Berlin, Germany; Thivya Pakeerathan, St. Josef Hospital, Ruhr University Bochum, Bochum, Germany; Sarah Passoke Hannover Medical School, Hanover, Germany; Friedemann Paul, Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany, and Experimental and Clinical Research Center, a cooperation between the Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC), Berlin, Germany; Marc Pawlitzki, Heinrich Heine University Düsseldorf, Düsseldorf, Germany; Hannah Pellkofer, Ludwig-Maximilians University, Munich, Germany; Mosche Pompsch, Alfried-Krupp-Krankenhaus Essen, Germany; Roxanne Alice Pretzsch, Universitätsspital Basel, Schweiz; Anne-Katrin Pröbstel, Universitätsspital Basel, Schweiz; Refik Pul, University of Essen, Germany; Sebastian Rauer, University Medical Center Freiburg, Germany; Nele Retzlaff, University Hospital, Rostock, Germany; Arne Riedlinger, Asklepios Klinik, Teupitz, Germany; Marius Ringelstein, Heinrich Heine University Düsseldorf, Düsseldorf, Germany; Paulus Rommer, Medical University of Vienna, Austria; Veith Rothhammer Department of Neurology, University Hospital Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen-Nürnberg, Germany; Kevin Rostásy, Vestische Caritas-Kliniken GmbH, Datteln, Germany; Klemens Ruprecht, Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Rebekka Rust—Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany Christoph Ruschil, University Hospital, Tübingen, Germany; Matthias Schwab, Jena University Hospital, Jena, Germany; Makbule Senel, University of Ulm, Ulm, Germany; Maria Seipelt, University Hospital Marburg, Marburg, Germany; Jörn Peter Sieb, Helios Hanseklinikum, Stralsund, Germany; Patrick Schindler, Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Carolin Schwake, St. Josef Hospital, Ruhr University Bochum, Bochum, Germany; Patricia Schwarz, University Hospital, Tübingen, Germany; Gilberto Solorza Buenrostro—Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Claudia Sommer, University Hospital, Würzburg, Germany; Alexander Stefanou, Klinikum der Landeshauptstadt Stuttgart gKAöR Katharinenhospital, Stuttgart, Germany; Till Sprenger, German Diagnostic Clinic, DKD Helios Clinic Wiesbaden, Germany; Andreas Steinbrecher, Helios Klinikum, Erfurt, Germany; Heike Stephanik, University Hospital, Magdeburg, Germany; Jan-Patrick Stellmann, CEMEREM, Marseille, France, and Aix Marseille Univ,Marseille, France; Muriel Stoppe, University Hospital, Leipzig, Germany; Klarissa Stürner, University Medical Center Schleswig Holstein – Kiel, Germany; Marie Süße, University Hospital, Greifswald, Germany; Athanasios Tarampanis, Heinrich Heine University Düsseldorf, Düsseldorf, Germany; Simone Tauber Department of Neurology, RWTH Aachen University, Germany Daria Tkachenko, Hannover Medical School, Germany; Florian Then Bergh, University of Leipzig, Leipzig, Germany; Corinna Trebst, Hannover Medical School, Hanover, Germany; Hayrettin Tumani, University of Ulm, Ulm, Germany; Annette Walter, Herford Hospital, Germany; Clemens Warnke, Faculty of Medicine and University Hospital Cologne, Germany; Klaus-Peter Wandinger, University Medical Center Schleswig-Holstein Campus, Lübeck, Germany; Anna Walz, Hannover Medical School, Hanover, Germany; Martin Weber, University Hospital, Göttingen, Germany; Jens Weise, Helios Vogtland-Klinikum Plauen, Germany; Jonathan Wickel, Department of Neurology, Hospital, Germany; Heinz Wiendl, University Hospital, Münster, Germany; Brigitte Wildemann, University of Heidelberg, Heidelberg, Germany; Alexander Winkelmann, University Hospital, Rostock, Germany; Yavor Yalachkov, University Hospital, Frankfurt, Germany; Uwe Zettl, University Hospital, Rostock, Germany; Ulf Ziemann, University Hospital, Tübingen, Germany; Frauke Zipp, University Medical Center of the Johannes Gutenberg University, Mainz, Germany.
Funding
Open Access funding enabled and organized by Projekt DEAL. There was no specific funding for these recommendations.
Author information
Authors and Affiliations
Consortia
Contributions
The initial version of this manuscript was prepared by TK, KG, and AB with contributions from all authors. All authors were involved in critical revision of the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest
TK has received speaker honoraria and/or personal fees for advisory boards from Roche Pharma, Alexion/Astra Zeneca, Horizon, Merck, Chugai and Biogen. The Institution she works for has received grant support for her research from Bayer-Schering AG, Novartis and Chugai Pharma in the past. None resulted in a conflict of interest. KG has received travel support from UCB. OA has received speaking honoraria and travel grants from Alexion, Almirall, Biogen, Celgene, Merck, Novartis, Roche, and VielaBio. IA has received speaking honoraria and scientific advisory board compensation from Alexion, Roche, Merck Serono, Santhera, and Sanofi Genzyme and research support from Chugai and Diamed. JBS has received speaking honoraria and travel grants from Bayer Healthcare, sanofiaventis/Genzyme, and Biogen and compensation for serving on a scientific advisory board from Roche. VH reports no conflicts of interest. JH reports a grant for OCT research from the Friedrich-Baur-Stiftung, personal fees and nonfinancial support from Merck, Alexion, Novartis, Roche, Santhera, Biogen, Heidelberg Engineering, and Sanofi Genzyme, and non-financial support from the Guthy-Jackson Charitable Foundation; JH is partially funded by the German Federal Ministry of Education and Research [grant numbers 01ZZ1603[A-D] and 01ZZ1804[A-H] (DIFUTURE)]. KH reports no conflicts of interest. MH reports no conflicts of interest. SJ reports no conflicts of interest. IK has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities from Alexion, Almirall, Bayer, Biogen, Hexal, Horizon, Merck, Neuraxpharm, Sanofi, and Roche/Chugai. LK has received compensation for serving on scientific advisory boards from Alexion, Biogen, BMS, Celgene GmbH, Genzyme, Horizon, Janssen, Merck Serono, Novartis, and Roche. She has received speaker honoraria and travel support from Bayer, Biogen, Celgene GmbH, Genzyme, Grifols, Merck Serono, Novartis, Roche, Santhera, and Teva. She receives research support from the German Research Foundation, the IZKF Münster, IMF Münster, Biogen, Immunic AG, Novartis, and Merck Serono. MK has received travel funding, speaker honoraria, and research support from Bristol Myers Squibb, Merck, Novartis, and Roche. FP has received grants from the Guthy Jackson Charitable Foundation, German Research Foundation, German Federal Ministry of Education and Research, Novartis, Bayer, Novartis, Biogen Idec, Teva, Alexion, Roche, Horizon, Sanofi-Aventis/Genzyme, Merck Serono, Chugai, German Research Council, Werth Stiftung of the City of Cologne, Arthur Arnstein Stiftung Berlin, EU FP7 Framework Program, National Multiple Sclerosis (USA), MedImmune, Shire, and Alexion, and has a patent on Foveal Morphometry pending to Nocturne GmbH; he is Academic Editor at PLoS ONE and Associate Editor of Neurology® Neuroimmunology & Neuroinflammation. MR has received speaker honoraria from Novartis, Bayer Vital GmbH, Roche, Alexion, and Ipsen and travel reimbursement from Bayer Schering, Biogen Idec, Merz, Genzyme, Teva, Roche, and Merck. KR received research support from Novartis Pharma, Merck Serono, German Ministry of Education and Research, European Union (821283–2), Stiftung Charité (BIH Clinical Fellow Program) and Arthur Arnstein Foundation, and received speaker honoraria from Bayer and travel grants from Guthy Jackson Charitable Foundation. MS has received consulting and/or speaker honoraria from Alexion, Bayer, Biogen, Bristol-Myers-Squibb, Merck, Roche, and Sanofi Genzyme. She has received research funding from the Hertha-Nathorff-Program. PS reports no conflict of interest. FTB has received personal compensation for advisory board participation or speaking, and through his institution has received research support for investigator-initiated studies or for attending scientific meetings, from Actelion, Alexion, Bayer-Schering, Biogen, Merck, Novartis, Roche, Sanofi-Genzyme, Teva, UCB, the German Research Foundation (DFG), and the German Federal Ministry of Education and Research (BMBF). CT has received honoraria for consultation and expert testimony from Alexion Pharma Germany GmbH, Chugai Pharma Germany GmbH, and Roche Pharma GmbH HT received research institutional support and/or consulting/speaker honoraria from Alexion, Bayer, Biogen, Bristol-Myers Squibb, Celgene, Diamed, Fresenius, Fujirebio, GlaxoSmithKline, Horizon, Janssen-Cilag, Merck, Novartis, Roche, Sanofi Genzyme, TEVA. His research is also funded by Ministry of Education and Research (BMBF), Ministry of Science, Research and Arts Baden Württemberg (MWK-BW), German Society of Multiple Sclerosis (DMSG), DMS-Stiftung, AMSEL-Stiftung, Bayern-DMSG and Chemische Fabrik Karl Bucher GmbH BW has received grants from the German Ministry of Education and Research, Deutsche Forschungsgemeinschaft, Dietmar Hopp Stiftung, Klaus Tschira Stiftung, and Merck, and personal fees from Alexion, Bayer, Biogen, Roche. CW has received institutional support from Novartis, Alexion, Sanofi Genzyme, Biogen, Merck, and Roche AB has received consulting and/or speaking fees from Alexion, Bayer Healthcare, Biogen, Celgene, Horizon and Roche. His institution has received compensations for clinical trials from Alexion, Biogen, Merck, Novartis, Roche, and Sanofi.
Ethical statement
The manuscript does not contain patient or animal data.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kümpfel, T., Giglhuber, K., Aktas, O. et al. Update on the diagnosis and treatment of neuromyelitis optica spectrum disorders (NMOSD) – revised recommendations of the Neuromyelitis Optica Study Group (NEMOS). Part II: Attack therapy and long-term management. J Neurol 271, 141–176 (2024). https://doi.org/10.1007/s00415-023-11910-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-11910-z