
Abstract
Secondary immunodeficiencies (SIDs) are acquired conditions that may occur as sequelae of immune therapy. In recent years a number of disease-modifying therapies (DMTs) has been approved for multiple sclerosis and related disorders such as neuromyelitis optica spectrum disorders, some of which are frequently also used in- or off-label to treat conditions such as chronic inflammatory demyelinating polyneuropathy (CIDP), myasthenia gravis, myositis, and encephalitis. In this review, we focus on currently available immune therapeutics in neurology to explore their specific modes of action that might contribute to SID, with particular emphasis on their potential to induce secondary antibody deficiency. Considering evidence from clinical trials as well as long-term observational studies related to the patients’ immune status and risks of severe infections, we delineate long-term anti-CD20 therapy, with the greatest data availability for rituximab, as a major risk factor for the development of SID, particularly through secondary antibody deficiency. Alemtuzumab and cladribine have relevant effects on circulating B-cell counts; however, evidence for SID mediated by antibody deficiency appears limited and urgently warrants further systematic evaluation. To date, there has been no evidence suggesting that treatment with fingolimod, dimethyl fumarate, or natalizumab leads to antibody deficiency. Risk factors predisposing to development of SID include duration of therapy, increasing age, and pre-existing low immunoglobulin (Ig) levels. Prevention strategies of SID comprise awareness of risk factors, individualized treatment protocols, and vaccination concepts. Immune supplementation employing Ig replacement therapy might reduce morbidity and mortality associated with SIDs in neurological conditions. In light of the broad range of existing and emerging therapies, the potential for SID warrants urgent consideration among neurologists and other healthcare professionals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
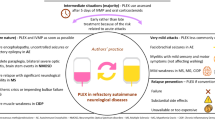
In patients with neuroimmunological disorders, secondary immunodeficiency (SID) is a complication on the rise and awareness needs to be raised not only among neurologists to improve diagnosis and treatment. |
Some risk factors predisposing to the development of SID have been identified. These include the duration of therapy, increasing age and pre-existing low Ig levels before therapy initiation. Compared to other DMTs, long-term anti-CD20 therapy is associated with a particularly high risk for development of SID and contraction of infections. |
A prior assessment and, if possible, elimination of risk factors by more personalized treatment approaches may substantially reduce morbidity and mortality associated with SID. |
1 Introduction
Immunodeficiencies comprise a heterogeneous group of disorders that are defined according to their etiology as primary or secondary. Primary immunodeficiencies (PIDs) are hereditary disorders and may result from either a single mutation or can be multifactorial. The overall estimated prevalence of PID is 1:1200 individuals [1, 2].
Secondary immunodeficiencies (SIDs) are acquired conditions that may arise from a variety of causes. For example, SIDs may occur as sequelae of immunotherapies; they may occur as a consequence of various diseases and age (immunosenescence), or malnutrition [3,4,5]. Although detailed epidemiological studies on SID are not available to date, the available evidence suggests that SIDs are more common than PIDs. Furthermore, it is thought that the steadily expanding clinical use of immunosuppressive and immunomodulatory drugs may be increasing the occurrence of SID [6].
In neurology, autoimmune diseases such as multiple sclerosis (MS), chronic inflammatory demyelinating polyneuropathy (CIDP), myasthenia gravis, myositis, and encephalitis can be treated with drugs that may cause SID, and the frequency of their use has increased in recent years. The risk of serious infections is known to be increased in MS patients [7, 8] and may further be influenced by factors such as age, comorbidities, and the individual’s immune status. This is complicated by the fact that therapeutic outcomes greatly rely on early treatment initiation and potentially lifelong treatment maintenance, resulting in cumulative risks of developing SID.
Over the past decade, a handful of novel immune therapies has been approved in neurology. The majority of these drugs are disease-modifying therapies (DMTs) to treat MS, including relapsing and progressive forms. Immune therapies differentially target various aspects of cellular and humoral immunity (see Fig. 1). While some compounds cause a broad depletion of immune cell subsets and can, therefore, be considered potently immunosuppressive, others specifically modulate more subtle processes such as polarization or cell trafficking [9]. Therefore, the risks of developing SID likely differ depending on the mode of action. Secondary antibody deficiency is one type of SID and can be quantified with blood analysis. Other potential surrogate markers of SID may be peripheral lymphocyte counts and their subtypes; however, the absolute counts may have different meaning depending on the mode of action of the compound used, or the timing of the blood analysis, complicating its clinical use.

Therapeutic targets of immune therapy in neurology. Rituximab, ocrelizumab, and ofatumumab are directed against different epitopes on the CD20-antigen, leading to a depletion of B cells. Alemtuzumab targets CD52, causing a broad depletion of lymphocytes. Cladribine is a purine analogue that requires incorporation into the DNA of lymphocytes, ultimately initiating apoptosis. Dimethyl fumarate exerts pleiotropic actions, including activation of Nrf2-regulated genes such the anti-inflammatory factor heme oxygenase-1 (HO-1). Fingolimod causes internalization of the S1P1 receptor, sequestering lymphocytes in lymphoid organs. Natalizumab blocks the transmigration of lymphocytes through the blood-brain-barrier (BBB) by inhibiting VLA-4 (α-4 integrin)
In this review, we focus on currently available immune therapeutics in neurology to explore the specific modes of action that might contribute to SID, with particular emphasis on their potential to induce secondary antibody deficiency. Findings from clinical trials as well as long-term observational studies related to the patients’ immune and antibody status and risks of severe infections are taken into consideration, and therapeutic strategies for SID are discussed.
2 Immunotherapies in Neurology
2.1 B-Cell Depletion
2.1.1 Clinical Use of B-Cell-Depleting Therapies in Neurological Disorders
Multiple lines of evidence support a central role of B cells in the pathology of various neurological autoimmune conditions such as myasthenia gravis, autoimmune encephalitis, CIDP, or MS [10,11,12,13,14]. B-cell-depleting therapies have only been approved in MS to date [15]. However, there has been an extensive off-label use of the chimeric human/murine anti-CD20 rituximab (Table 1) in other inflammatory neurological conditions including CIDP, myasthenia gravis, and encephalitis. In relapsing-remitting multiple sclerosis (RRMS), the efficacy of B-cell-depleting therapies was first demonstrated in a phase 2 trial employing rituximab [16]. In 2017, the fully humanized anti-CD20 Ab ocrelizumab (Table 1), which targets an overlapping epitope with rituximab, was approved for RRMS and also became the first available treatment option for primary progressive multiple sclerosis (PPMS) patients [17,18,19]. Ofatumumab, a fully humanized monoclonal antibody targeting CD20 (Table 1), was recently approved in MS by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) [20]. Ublituximab, a monoclonal glyco-engineered anti-CD20 antibody, is currently under investigation in phase 3 clinical trials (ULTIMATE-I + II) in patients with relapsing multiple sclerosis [21].
2.1.2 Change in Circulating Immune-Cell Populations with B-Cell-Depleting Therapies
Unlike CD19, which is expressed across the entire B-cell lineage, CD20 is expressed in most developmental B cell stages except for early pro-B cells and plasma cells. As autoantibody-secreting cells rarely express CD20, the efficacy of anti-B cell therapies has been ascribed to the substantial role of B cells in antigen-presentation and cytokine release [22], thereby potentially aggravating T-cell responses. However, long-lasting depletion of B cells is also likely to impact the number and replenishment of plasma blasts [18] and antibody levels.
The data on B-cell numbers from the phase 2 clinical trial (HERMES) to assess the efficacy of rituximab in RRMS suggest a nearly complete depletion of CD19+ peripheral B cells (> 95% reduction compared to baseline) 14 days after treatment; 30.7% of patients had returned to baseline after 48 weeks [16]. Nearly identical findings were reported in the phase 2 trial of rituximab in PPMS (OLYMPUS) [23]. Moreover, rituximab has been reported to reduce the number of B cells (as well as T cells) in cerebrospinal fluid [24].
In the phase 3 clinical trials of ocrelizumab, similarly effective depletion of CD19+ B cells was observed after 14 days and B-cell numbers remained negligible until the study end at 96 weeks for RRMS (OPERA I+II) and 120 weeks for PPMS (ORATORIO) [25, 26]. As treatment was administered every 24 weeks in both trials, data on B-cell recovery are not available, demonstrating that near-complete depletion persists for at least 5 months. There was no overall impact on CD16+/CD56+ cell count; however, a slight initial reduction by 4–6% from baseline of CD3+ and CD8+ T cells was observed after 2 weeks, with an additional reduction of CD8+ cells by 2% until week 96 in the OPERA trials [25, 26].
Regarding rituximab therapy, a study in rheumatoid arthritis (RA) patients has indicated that B-cell reconstitution may require a mean of 8 months [27]. From clinical experience, this is a highly inter-individual variable and also depends on the number of treatment cycles. In addition to its effect on B cells, rituximab has also been shown to exert profound effects on T-cell activation and proliferation. While these effects are mostly mediated by the absence of B cells as antigen-presenting cells, accumulating evidence suggests direct effects of rituximab on T-cell subpopulations expressing varying levels of CD20 and possibly even independent of CD20 expression [28, 29]. Indeed, CD20+ T cells with proinflammatory Th1/Tc1 phenotype have recently been shown to contribute to disease severity in RRMS patients [30]. As such, the efficacy of B-cell depletion therapy may partly also be related to depletion of T-cell subpopulations.
2.1.3 Immunoglobulin (Ig) Levels and Clinical Risk of Infection with B-Cell-Depleting Therapies
Due to the short observation periods, the pivotal phase 2 and 3 clinical trials on rituximab, ocrelizumab, and ofatumumab only report a moderate proportion of patients with decreases in IgM, and even fewer to none with regard to IgA and IgG [16, 20, 23, 25, 26]. Nevertheless, several long-term studies on anti-CD20 therapy point towards the risk of hypogammaglobulinemia (i.e., reduction of IgG levels) (see Table 2), which is associated with an increased risk of infection [8, 31,32,33,34,35].
Most of these studies relate to the use of rituximab. A study assessing 3,194 rheumatoid arthritis (RA) patients who had received up to 17 rituximab courses over a period of 9.5 years [31] found that 22.4% of patients developed IgM levels below the lower limit of normal (LLN) and 3.5% of patients had IgG levels below the LLN. The occurrence of serious infection events was significantly higher in patients with low IgG compared with patients who had never developed IgG levels below the LLN. There was no such association between low IgM levels and serious infection events. Patients developing low IgG associated with serious infection events were characterized by an older age, longer disease duration, and lower mean IgG levels at baseline. However, hypogammaglobulinemia in these patients did not appear to result from a pre-existing condition, as low Ig levels were an exclusion criterion for trial entry.
A French cohort study aimed to determine risk factors for severe infection in 1303 rituximab-treated RA patients, with a mean follow-up period of 1.2 years after the first or any subsequent rituximab cycles [32]. Pre-existing low IgG levels (< 6 g/L) or general hypogammaglobulinemia before treatment initiation, higher doses of concomitant corticosteroids, and an older age (mean 64.7 years) were identified as significant risk factors for severe infections. There was no correlation between low IgM (< 0.5 g/L) before rituximab infusion and development of serious infection events. Thus, in contrast to IgG, low baseline IgM at treatment initiation or subsequent development of hypo-IgM do not appear to be associated with elevated risks of infection in RA patients.
Concerning neurological diseases, a recent case-series study investigated the long-term effects of rituximab in 15 neuromyelitis optica spectrum disorder (NMOSD) patients [33]. The median follow-up was 70 months. The authors observed a significant reduction in IgG, IgA, and IgM levels, as well as a strong correlation between the isotype classes. Across the group, 73% of patients were identified as hypo-IgG (IgG < 7.0 g/L), 40% as hypo-IgA (IgA < 0.7 g/L), and 60% as hypo-IgM (IgM < 0.4 g/L). Rituximab significantly reduced total IgG by 0.42 g/L per year (follow-up of 70 months). Moreover, the assessment of specific anti-pathogen IgGs revealed reduced levels of anti-varicella-zoster virus (VZV), anti-Epstein-Barr virus (EBV) and anti-tetanus IgGs, perhaps indicative of insufficient protection thresholds. In support of these findings, another case series study indicated development of hypogammaglobulinemia in 32 of 50 NMOSD patients (64%) following treatment with rituximab. Of these, five patients contracted serious infections associated with development of antibody deficiency [34].
A Swedish retrospective, observational study of the safety and effectiveness of rituximab in 822 patients with MS (557 RRMS, 198 SPMS, 67 PPMS) indicated a decrease of IgG below the LLN in 3% of patients. The remaining patients only showed slight decreases in IgG levels. Severe infections occurred in 1.7% of patients; however, any potential association between IgG levels and risk of infection were not evaluated in this study [36].
Still, there is evidence that by presence of long-lived plasma cells or the persistence of B cells in protective niches a protective antibody pattern is restored, despite B-cell-depletion therapy. This was supported by an observational study that assessed levels of autoantibodies as well as antimicrobial antibodies following B-cell-depletion therapy using rituximab in 16 patients with systemic lupus erythematosus. While anti-nucleosome and anti-dsDNA-autoantibodies were significantly decreased by 6–8 months of rituximab treatment, there was no impact on antimicrobial antibodies against tetanus toxoid or pneumococcal capsular polysaccharide [37].
With regard to ocrelizumab, long-term follow-up data are clearly limited at this point. Nevertheless, preliminary data from the pivotal phase 3 trials on ocrelizumab and their open-label extensions have been reported [35]. After 264 weeks of ocrelizumab treatment, there was a mean reduction from baseline of 17% for IgG, 21% for IgA, and 58% (RRMS) and 56% (PPMS) for IgM. The proportion of patients who reached Ig levels below the LLN were 5.7% for IgG, 5.4% for IgA, and 29.2% for IgM. Within the first 6 years, treatment with ocrelizumab reduced serum IgG concentration at an average rate of 0.32 g/L per year (3.0% per year). The reduction of Ig levels below LLN was associated with an increased occurrence of serious infection events. The strongest association was observed for hypo-IgG; however, no clear association between serious infection events and distinct pathogens, organ affected, disease severity, or infection duration could be determined for the different Ig classes.
The clinical trials—most likely due to the short-term observation periods—indicated low evidence for severe Ig decrease or an overall increase in serious infections. The HERMES trial reported that 22.4% of rituximab-treated patients displayed IgM levels below the LLN compared with 8.6% in the placebo arm over an observation period of 48 weeks [16]. In the OLYMPUS trial, IgM levels were below the LLN in 31.7% of rituximab-treated compared with 5.9% of placebo-treated patients over an observation period of 96 weeks [23]. For the OPERA and ORATORIO trials (ocrelizumab), 16.5% and 15.5% of treated patients, respectively, showed IgM levels below the LLN. A small proportion of patients showed reduced levels of IgA and IgG (2.4 and 1.5% below the LLN) in the OPERA trial. There is no mention of reduced IgA and IgG levels in the ORATORIO trial [25, 26].
No definitive data are available regarding the effects of ofatumumab on Ig levels, although this monoclonal B-cell-targeting antibody did undergo phase 3 trials in MS. Initial indications of minor effects on Ig levels have been shown [38], analogous to the effects seen following use of ofatumumab in oncological indications [39, 40]. Interestingly, ofatumumab may show greater effectivity in targeting B cells in lymphoid organs, but could also be associated with faster repletion of B cells than rituximab therapy. This may hold implications with regard to infection risks associated with B-cell depletion [41].
The increased risk of infections following anti-CD20 MS therapy in a real-world setting was recently confirmed in a Swedish register-based cohort study that compared rituximab, fingolimod, natalizumab, and interferon beta/glatiramer acetate [8]. Of these disease-modifying therapies, rituximab was associated with the highest rate of serious infections. Interestingly, antibiotic usage was highest with rituximab, while the prescription of herpes antiviral drugs was approximately 70% higher with fingolimod and natalizumab than rituximab, suggesting that patients who received anti-CD20 therapy might be more prone to bacterial than viral infections [8]. A key difference between this real-world data and the phase 3 studies mentioned above is the longer observation period.
In summary, there is a considerable risk of developing SID in terms of hypogammaglobulinemia with anti-CD20 therapy. SID appears to be correlated with treatment duration and leads to increased risk of bacterial infections.
2.2 Alemtuzumab
2.2.1 Clinical Use of Alemtuzumab in Neurological Disorders
Alemtuzumab is a humanized monoclonal anti-CD52 antibody initially approved in 2001 for the treatment of B-cell chronic lymphocytic leukemia (B-CLL), and has since been used in various hematological malignancies. In 2013, alemtuzumab was approved for RRMS patients with active disease, and following recent recommendations should be reserved for patients with first-line treatment-refractory RRMS [42].
2.2.2 Change in Circulating Immune-Cell Populations with Alemtuzumab
CD52 is highly expressed on T and B lymphocytes and to a lesser degree on monocytes and dendritic and natural killer (NK) cells, whereas expression in plasma cells as well as bone-marrow stem cells is negligible. Therefore, alemtuzumab causes a massive depletion of circulating lymphocytes; however, it induces a less profound impact on cells resident in lymphoid organs [42]. Before its approval for RRMS, the therapeutic efficacy of alemtuzumab in MS patients had been explored since the early 1990s. Long-term follow-up data from 37 treatment-naïve patients (median age of 39 years) with progressive MS who had received a single course of alemtuzumab between 1991 and 1997 provide valuable insight into the duration of lymphopenia and time to lymphocyte reconstitution, respectively [43]. The geometric mean recovery time for total lymphocyte count to reach the LLN was 12.7 months, for CD19+ B lymphocytes recovery to LLN was 7.1 months, and median recovery time for CD8+ and CD4+ lymphocytes was 20 and 35 months, respectively. Of note, only 30% and 21% of patients, respectively, recovered CD8+ and CD4+ lymphocytes to baseline values, with follow-up lymphocyte counts available for a median of 12 years. Baseline values for B lymphocytes were not available in this study. These findings suggest long-lasting lymphopenia after only one single course of alemtuzumab, even in relatively young patients without previous immunomodulatory therapy.
2.2.3 Ig Levels and Clinical Risk of Infection with Alemtuzumab
Humoral responses to recall antigens as well as T-cell-dependent and -independent responses to vaccination have been reported to be largely normal following alemtuzumab treatment in a small pilot case-control study [44]. Serum antibodies against common viruses remained detectable after treatment [44], probably owing to negligible expression of CD52 on plasma cells. Nevertheless, a recent investigation of 38 RRMS patients demonstrated significant reductions in serum Ig levels (IgG, IgA, and IgM) at 12 and 24 months following two courses of alemtuzumab [45]. Some patients who required an additional third course due to persistent disease activity exhibited a further decline in IgG, but not IgA or IgM. Those patients with IgG below the LLN were shown to be at increased risk of developing pneumonia, sinusitis, and otitis [45].
Pooled 6-year data from the pivotal phase 2 (CAMMS233) and phase 3 (CARE-MS I + CARE-MS II) trials comparing two courses of 12 mg alemtuzumab versus 44 µg subcutaneous interferon beta in RRMS indicate that infections occurred more frequently with alemtuzumab. Severity of infections was graded as mild to moderate in approximately 95% of cases and predominantly included upper respiratory tract, urinary tract, and herpetic infections. The rate of serious infections was 1.0–1.9% per year. Moreover, lymphocyte counts were not predictive of infection risks [46].
Given the rapid and long-lasting induction of lymphopenia following alemtuzumab treatment, the reported occurrence of infections has been surprisingly infrequent and of rather mild to moderate severity [47]. However, a recent review of post-marketing surveillance data and several single case reports concluded that patients who received alemtuzumab may be at increased risk of serious opportunistic infections such as Listeria monocytogenes, Cytomegalovirus, and Pneumocystis jiroveci [47].
In summary, owing to the broad and long-lasting immune-cell-depleting effects of alemtuzumab, patients are at considerable risk of developing both cellular and humoral SID. Antibody deficiency may play a role after long-term treatment. However, the exact incidence of serious infections associated with low Ig levels in patients treated with alemtuzumab requires further systematic evaluation.
2.3 Natalizumab
2.3.1 Clinical Use of Natalizumab in Neurological Disorders
Natalizumab is a humanized monoclonal antibody targeting the cell-adhesion molecule α-4 integrin (VLA-4), which is required for the vascular transmigration of leukocytes. Natalizumab was approved for RRMS treatment and prevents immune-cell infiltration of the CNS across the blood-brain barrier [48].
2.3.2 Change in Circulating Immune-Cell Populations with Natalizumab
In contrast to other immune-suppressive drugs, natalizumab treatment is not generally associated with an increased risk of lymphopenia, but conversely leads to increases of circulating T and B lymphocytes as well as lymphoid progenitor cells [48] while blocking their egress to the brain and cerebrospinal fluid [49]. Also, natalizumab does not seem to affect cytokine responses in T cells derived from RRMS patients [50].
2.3.3 Ig Levels and Clinical Risk of Infection with Natalizumab
Due to its mode of action, a relevant reduction in serum Ig level with natalizumab would not intuitively be expected. However, two cross-sectional cohort studies revealed a significant reduction in serum IgM and IgG. A longitudinal cohort study confirmed IgM and IgG reductions during natalizumab treatment, although the reduction in IgG was less pronounced and appeared rather inconsistent [51]. One possible mechanism underlying diminished serum Ig levels during natalizumab treatment might be an impairment of B-lymphocyte homing, preventing follicle formation and finally leading to reduced generation of plasma cells [51,52,53]. However, vaccination responses to both recall (tetanus toxoid) and neoantigen (keyhole limpet hemocyanin) are largely normal in natalizumab-treated patients [54]. As such, the clinical relevance of any natalizumab-induced reduction in serum IgM, and possibly IgG, with regard to infection risk remains unclear and warrants further investigation.
Regarding Ig levels in the cerebrospinal fluid (CSF), a recent longitudinal, observational study demonstrated an impaired intrathecal production of antiviral antibodies after 1 year of natalizumab treatment [55]. The authors reported a significant decline in CSF levels of IgG, IgM, and IgA, whereas in serum only IgM was found to be reduced. Intrathecal polyoma JC virus (JCV) and BK-virus (BKV) specific antibody production was identified in 20% of treatment-naïve RRMS patients, but became undetectable under natalizumab, while peripheral JCV and BKV antibody production persisted. Interestingly, intrathecal antibody production against measles, rubella, and zoster antigens remained mostly stable, suggesting that natalizumab may predominantly affect intrathecal humoral immune responses against JC rather than other neurotropic viruses [55].
The pivotal phase 3 trials on natalizumab in RRMS, AFFIRM (natalizumab vs. placebo) and SENTINEL (natalizumab vs. interferon beta), did not identify any increased risk for infections or serious infections [56, 57]. However, in the SENTINEL trial, two cases of progressive multifocal leukoencephalopathy (PML) were reported [56]. As a consequence of impaired CNS immunosurveillance, a subgroup of patients receiving natalizumab has an increased risk of developing serious opportunistic brain infections, most notably symptomatic JCV infection causing PML. The overall natalizumab-associated PML incidence has been indicated as 4.14 per 1000 patients [58]. Three main risk factors for the development of PML with natalizumab have been identified: positivity for anti-JCV antibodies (approximately 55% of patients), duration of natalizumab treatment with the highest risk level occurring beyond 2 years of continuous therapy, and preceding use of immunosuppressive drugs [59]. Additionally, a review of post-marketing adverse events indicated a potential for increased risk of CNS herpetic infections, predominantly herpes simplex virus (HSV) and VZV, although these may occur less frequently than JCV reactivation and development of PML, respectively [60].
In summary, patients treated with natalizumab do not appear at a relevant risk of developing SID in terms of either hypogammaglobulinemia or decreased cellular immunity.
2.4 Fingolimod
2.4.1 Clinical Use of Fingolimod in Neurological Disorders
Fingolimod (FTY720) is a first-in-class sphingosine-1-phosphate (S1P) receptor agonist approved for the treatment of RRMS since 2010. As a sphingosine analogue, fingolimod becomes phosphorylated by sphingosine kinases and modulates four of the five known S1P receptors, except for S1P2. Structurally a S1P receptor agonist, its immunomodulatory effect is presumably based on its “functionally antagonistic” effect on the S1P1 receptor expressed on lymphocytes, causing receptor internalization and abrogation of S1P1 signaling. It thereby interferes with the egress of predominately CCR7+ lymphocytes from lymphoid tissues and prevents infiltration of the CNS [61]. Recently, more selective S1P receptor modulators with a comparable mode of action such as siponimod, ozanimod, and ponesimod have been approved or are in clinical development [62].
2.4.2 Change in Circulating Immune-Cell Populations with Fingolimod
As a consequence of lymphocyte sequestration, fingolimod causes a reversible decrease in circulating lymphocytes of 70–80% within 2 weeks of treatment initiation, resulting in mean lymphocyte counts of approximately 0.5 × 109/L that remain relatively stable over several months and years on therapy [63]. Since the induction and maintenance of a “therapeutic lymphopenia” is considered integral to the efficacy of fingolimod, lymphocyte counts below 0.2 × 109/L have been defined as the criterion for treatment discontinuation. An increased risk of developing severe lymphopenia has been identified in patients with baseline lymphocyte counts below 1.6 × 109/L and in female patients with low body mass index (BMI; < 18.5 kg/m2) [64]. In the majority of patients, lymphocyte reconstitution begins 2 weeks following cessation of therapy and lymphocyte counts usually reach ~ 80% of baseline values within 2–3 months. However, patients with markedly delayed lymphocyte reconstitution have been described and only partial recovery may persist years post-cessation [65, 66]. However, the degree and heterogeneity of lymphocyte reconstitution do not seem to be related to age, treatment duration, or lymphocyte count pre-treatment [65].
2.4.3 Ig Levels and Clinical Risk of Infection with Fingolimod
A recent retrospective, cohort study of 327 RRMS patients comprising six patients receiving fingolimod found significantly reduced IgG levels in fingolimod-treated patients compared with untreated patients, but two of them had IgG levels below LLN [67]. In line with this, a recent case series study investigated IgG titers in response to varicella-zoster virus (VZV) vaccination in 11 fingolimod-treated patients. Following initiation of fingolimod, there was a general decrease in antibody titers, and a disappearance occurred in 7/11 patients. After cessation of fingolimod therapy, antibody titers reappeared [68]. However, previous studies have indicated that patients treated with fingolimod are still able to mount sufficient adaptive immune responses to influenza vaccines [69], although the avidity of anti-influenza IgG may fail to increase in fingolimod-treated patients compared with interferon beta-treated patients or healthy controls [70]. Similarly, a randomized, multicenter, placebo-controlled study of 136 RRMS patients demonstrated that fingolimod-treated patients can indeed mount sufficient immune responses to vaccination, but seroconversion might be impaired compared to placebo controls [71].
In contrast to its effects on peripheral immune cells, fingolimod has a less profound impact on T and B cells in cerebrospinal fluid. Consistently, fingolimod shows little effect on intrathecal Ig synthesis [72].
Regarding risk of infections, all three trials (FREEDOMS I+II and TRANSFORMS) have reported similar overall infection risks between fingolimod and placebo or interferon-beta treatment, respectively. However, the risk of specific infections might be elevated with fingolimod. In the TRANSFORMS trial, a slightly increased incidence of upper and lower respiratory tract infections as well as herpes virus infections was found with fingolimod versus interferon beta. Of note, two fatal herpetic infections were reported with the 1.25 mg dose—disseminated primary varicella zoster and herpes simplex encephalitis [73].
In FREEDOM I, the incidence of lower respiratory tract infections including pneumonia was increased with fingolimod versus placebo. While herpes virus infections were found at similar proportions between fingolimod and placebo, two cases in the fingolimod groups were classified as serious—genital herpes and herpes simplex labialis [74]. FREEDOM II pointed to a greater occurrence of lower respiratory, influenza, and herpes zoster infections in the fingolimod groups [75].
An analysis of pooled data from the controlled phase 2 and 3 trials as well as post-marketing surveillance data further indicated a heightened susceptibility to herpes zoster infections among patients treated with fingolimod. Interestingly, no risk accumulation associated with long-term exposure to fingolimod could be identified [76].
Recently, the LONGTERMS study provided insight into the safety and efficacy of fingolimod for up to 14 years of continuous treatment [77]. Overall, 51.9% of patients reported adverse events related to infection, of which 19.9% were suggested to be related to fingolimod treatment. Notably, 17.3% of patients experienced viral upper respiratory tract infections, and 2.4% of patients were diagnosed with herpes zoster. Furthermore, cryptococcal meningitis has been reported as a relevant risk in the post-marketing setting, occurring after 2–3 years of fingolimod therapy [77]. Moreover, there is a low risk of developing progressive multifocal leukoencephalopathy (PML) for patients treated with fingolimod, estimated at 0.13 per 1000 patients, with 37 confirmed cases so far [78].
Interestingly, infection risks with fingolimod appear to be unrelated to the degree of lymphopenia. Furthermore, development of severe lymphopenia, as defined above, has not been confirmed as a risk factor [63, 64, 77, 79].
In summary, while the development of lymphopenia is integral to the efficacy, fingolimod leads to an increased risk of mostly viral infections. Antibody deficiency is not known to play a relevant role. However, humoral responses to vaccination may be impaired in patients treated with fingolimod.
2.5 Dimethyl Fumarate
2.5.1 Clinical Use of Dimethyl Fumarate in Neurological Disorders
Dimethyl fumarate (DMF) is an established immunomodulatory agent for the treatment of MS and psoriasis. In 2013, DMF received approval as the second orally available treatment for RRMS. While the mode of action underlying its efficacy in autoimmune diseases has still not been fully elucidated, DMF has been demonstrated to function as an activator of the NFE2-related factor 2 (Nrf2) transcription factor, regulating the expression of antioxidant response element (ARE) genes, including a wide range of anti-oxidative and anti-inflammatory factors [80].
2.5.2 Change in Circulating Immune-Cell Populations with Dimethyl Fumarate
The clinical efficacy of DMF is not solely related to a depletion of immune cells or the induction of lymphopenia. Nevertheless, the two pivotal phase 3 clinical trials in RRMS (DEFINE and CONFIRM) indicated that patients receiving DMF experienced a mean decline in absolute lymphocyte counts by approximately 30% during the first year of treatment [81, 82]. Mild to moderate lymphopenia was observed in 30% of patients. About 2% of patients developed severe lymphopenia (≤ 0.5 × 109/L) that persisted for more than 6 months [83], associated with pleiotropic changes in intracellular calcium homeostasis [84]. Development of lymphopenia was mainly correlated with a decline in T-cell subsets; thereby, DMF appeared to have greater impact on CD8+ rather than CD4+ T cells by inducing apoptosis. However, a reduction in most immune cell types with DMF has been described, including B cells, NK-cells, dendritic cells, and monocytes [85,86,87].
2.5.3 Ig Levels and Clinical Risk of Infection with Dimethyl Fumarate
A recent prospective, open-label phase 3b study (PROCLAIM) of patients with RRMS (218 patients enrolled and 158 completed) assessed Ig levels after 48 and 96 weeks of DMF treatment. Total IgA, IgM, IgG, as well as IgG1-4 subclass levels remained stable during DMF treatment and during follow-up assessments. Interestingly, 16 patients exhibited baseline IgA, IgM, and IgG levels below the LLN. Of these, seven reached normal Ig values over the course of DMF treatment [88]. Regarding immune responses to vaccination, an open-label, multicenter study compared patients receiving DMF and interferon beta. T-cell-dependent recall antigen and T-cell-independent humoral as well as T-cell-dependent neoantigen responses were comparable between the DMF- and interferon-treated patients [89].
In the two pivotal, phase 3 clinical trials, similar rates of infection were reported between the DMF and placebo groups. There was a tendency towards an increased incidence of nasopharyngitis, urinary tract infection, upper respiratory tract infection, bronchitis, sinusitis, and gastroenteritis [81, 82]. Nevertheless, DMF treatment has been recognized to confer an increased risk of developing PML, with 11 cases reported to date [90]. Of note, this appears to be independent of the development of severe lymphopenia [91, 92]. The overall risk of infections, including serious and opportunistic, appears to be generally low, and there is a lack of clear correlation to absolute lymphocyte counts [88].
In summary, antibody deficiency does not appear to be a frequent complication leading to SID with DMF treatment.
2.6 Cladribine
2.6.1 Clinical Use of Cladribine in Neurological Disorders
Cladribine was originally approved for the treatment of hematological malignancies, including hairy cell leukemia, due to its potent antiproliferative properties in hematopoietic cells. In 2017, cladribine also received approval for RRMS in the EU and, since 2019, it is approved for both RRMS and active secondary-progressive multiple sclerosis (SPMS) in the USA, recommended after inadequate response to an alternative DMT. Cladribine is a chlorinated deoxyadenosine analogue that requires intracellular phosphorylation by deoxycytidine kinase (DCK) to be incorporated into DNA. This causes DNA strand brakes and subsequently induction of apoptosis. Therefore, cladribine preferentially targets rapidly dividing cells such as lymphocytes expressing high levels of DCK [93].
2.6.2 Change in Circulating Immune-Cell Populations with Cladribine
Oral cladribine is administered as a cumulative dose in two 4-week cycles over the course of 2 years. Following the first treatment cycle, absolute lymphocyte counts reached a nadir at 1.0 × 109 cells/L after 2 months. Following the second cycle, a nadir of 0.81 × 109 cells/L was reached at week 55 of treatment [94]. A quarter of patients (25%) experienced severe lymphopenia grade 3 (< 0.5 × 109/L) and < 1% of patients experienced lymphopenia grade 4 (< 0.2 × 109/L) [95, 96]. Overall decline in absolute lymphocyte counts (ALCs) was closely correlated with a decrease in CD19+ B lymphocytes as well as CD4+ and CD8+ T lymphocytes, albeit the decrease in CD8+ cells was less pronounced [94]. Furthermore, cladribine may target memory B lymphocytes to a greater extent due to increased expression of DCK in these cells [97].
2.6.3 Ig Levels and Clinical Risk of Infection with Cladribine
To date, no systematic investigation of Ig levels in patients treated with cladribine has been published. One observational study reported a loss of oligoclonal bands in 55% of patients observed over the course of 10 years, suggesting that cladribine may impact intrathecal humoral immune responses [98].
A combined analysis of the safety data for oral cladribine monotherapy across the three pivotal phase 3 clinical trials (CLARITY; CLARITY EXTENSION, and ORACLE-MS) and the PREMIERE registry (a prospective, observational registry for MS patients who have participated in the clinical trials) has been performed [99]. An increased incidence of herpetic infections, in particular herpes zoster followed by oral herpes and herpes simplex, was found following cladribine treatment. During periods of grade 3 or 4 lymphopenia, a greater frequency of herpes zoster and upper respiratory tract infections was observed [99].
In summary, the decrease of CD19+ B-lymphocytes in patients treated with cladribine points to the potential to cause antibody deficiency in long-term-treated patients. However, further studies will show whether impaired humoral immune response leads to SID in cladribine-treated patients.
3 Risk of Developing Secondary Immunodeficiency (SID) Under Immunotherapy in Neurological Diseases
Overall, in contrast to non-neurological conditions, immunotherapy in neurology often requires long-term maintenance therapeutic regimens from a young age, further increasing the likelihood of developing SID. All the drugs discussed above have the potential to contribute to an increased risk of infections. For some of these drugs, the immune mechanism underlying an increased infection rate is not clearly known, but secondary antibody deficiency is particularly relevant in the occurrence of SID. It has been estimated that treatment-associated secondary antibody deficiency might be 30 times more frequent than primary antibody deficiency [4], and the incidence of treatment-associated secondary antibody deficiency seems likely to increase further in the coming years, especially considering new therapies targeting B lymphocytes in neurology alone.
The identification of and screening for risk factors that may predispose patients to develop SID is critical to avoid the contraction of serious, potentially life-threatening infections. Impairment of the humoral immune compartment as a cause for SID has been best characterized for anti-CD20 therapy, but is likely to also apply to other potent immunosuppressive drugs such as alemtuzumab or fingolimod. Further studies are required to better define and understand this relevant aspect.
Regarding the example of rituximab, the risk for serious infections has been found to be elevated in individuals with low baseline IgG levels at treatment initiation, long treatment duration, increasing age, structural lung disease, cardiovascular diseases, and use of previous or concomitant immunosuppressive agents [31, 32]. These factors should be considered before starting any immunomodulatory therapy in patients with neurological conditions (Table 3).
Low Ig levels resulting from anti-CD20 therapy predispose to heightened risks of infections [34, 100]. A retrospective cohort study of 4,479 patients found that the majority of patients (3,824) receiving rituximab had not been examined for hypogammaglobulinemia prior to treatment initiation [101]. However, 47.8% of those patients with determined Ig levels displayed hypogammaglobulinemia at treatment start and worsening of this condition was noted following rituximab administration. There was an association between increased mortality and severe infections within 6 months before and after the first rituximab infusion [101].
A link between low baseline Ig levels and risk of SID also seems likely for other B-cell-targeting therapies, as interim reports indicated an association between hypogammaglobulinemia and serious infections with ocrelizumab [35].
Therefore, the assessment of pre-existing Ig levels as well as of the other risk factors should be standard procedure before therapy initiation of immune therapies.
Aside from the assessment of serum Igs, the determination of B-cell reconstitution might prove useful to achieve a more personalized treatment approach, at least in patients treated with B-cell-targeting therapies. In studies involving NMSOD patients, maintenance therapy was only initiated after a certain threshold of re-emerging CD27+ B memory cells was surpassed following the first rituximab cycle. This allowed for a cumulative dose reduction while sustaining clinical efficacy of rituximab [102, 103]. Such treatment strategies may not only reduce drug exposure and costs, but also increase safety with regard to infection risk and overall adverse events (Table 4).
4 Prevention and Treatment of SID in Patients with Neurological Conditions
Although the induction of immunodeficiency is not an intended consequence, it is, to some extent, integral to the clinical efficacy of immunotherapies. Therefore, in patients at high risk of serious infectious complications, “immune supplementation” without compromising treatment efficacy may be desirable. Currently, there are no consensus guidelines available for the treatment of patients with neurological conditions developing SID. Nevertheless, there is long-standing experience regarding the management of primary immunodeficiencies, immunodeficiencies in hematological malignancies, and consecutive antibody deficiency that may indicate a potential for using Ig replacement therapy (IGRT). Firstly, the ability of intravenous Igs (IVIgs) to reduce bacterial infections was demonstrated three decades ago in patients with chronic lymphocytic leukemia or non-Hodgkin lymphoma who developed hypogammaglobulinemia or had a history of recurrent infections [104, 105]. The efficacy of IGRT to reduce bacterial infections is dose dependent [106]. Several studies have strengthened the evidence base for IGRT in these conditions; however, clear recommendations concerning dosing and timing are still required [107, 108]. Recently, the European Medicines Agency (EMA) recommended IVIg to those patients “who suffer from severe or recurrent infections, ineffective antimicrobial treatment and either proven specific antibody failure (PSAF) or serum IgG level of < 4 g/L” (with PSAF defined as failure to mount at least a twofold rise in IgG antibody titer to pneumococcal polysaccharide and polypeptide antigen vaccines) [109]. The recommended dose for IGRT is 0.2–0.4 g/kg body weight every 3–4 weeks; however, dosing and timing can be adjusted to the patient’s individual requirements. In clinical practice, IGRT can be administered intravenously (IVIg) or subcutaneously (SCIg). While both routes of administration produce favorable outcomes, SCIg may have advantages over IVIg, especially with regard to convenient self-administration and improved pharmacokinetic properties, i.e., near steady-state IgG serum concentrations and a reduced incidence of systemic adverse events [107, 110, 111].
Studies report that patients with B-cell-targeting therapy-associated hypogammaglobulinemia who received Ig replacement therapy had a lower risk of serious infectious complications, indicating that excess mortality and morbidity might be avoided by identifying patients requiring Ig replacement therapy [101]. While several reports clearly indicate that IGRT significantly reduces the risk of infections, the routine use of prophylactic IVIg for this purpose is debated. A Cochrane database review does not support routine primary prophylactic IVIg treatment in patients with hematopoietic stem cell transplant, while IVIg secondary prophylaxis may be considered in patients with hematological malignancies for reducing infection frequency [112]. While the prophylactic administration of antibiotics is common practice in patients with malignancies and primary immunodeficiencies, this is not a usual procedure in patients treated for neuroinflammatory conditions. However, given the risk of the emergence of multi-resistant bacterial strains with antibiotic prophylaxis, the use of IGRT should be considered as an alternative even in patients with mild to moderate antibody deficiency [1, 107, 108].
Moreover, concomitant corticosteroid therapy should be avoided if possible, as the extent of hypogammaglobulinemia has been shown to correlate with corticosteroid dosage in asthmatic patients receiving daily prednisolone [113].
There are no consensus guidelines regarding vaccination strategies particularly for patients with MS or other neurological disorders under various immunotherapies [114]. Prophylactic vaccination employing live and attenuated vaccines is generally not recommended for patients with SID; however, despite an impaired humoral immune compartment, a certain degree of protection can be achieved using inactivated vaccines [4, 108]. Before initiating immunomodulatory/-suppressive therapy, vaccination status should be checked and, if necessary, vaccinations should be carried out according to national recommendations. Particular attention should be paid to vaccination against VZV before initiation of immunotherapy [114]. Even for VZV-seropositive patients, vaccination might be considered to prevent zoster reactivation [114]. In this regard, a recombinant VZV vaccine is available for immunocompromised patients [115]. Furthermore, aside from quantitative serum Ig measurements, patients at high risk of developing antibody deficiency might benefit from an assessment of vaccination responses, preferably using conjugate polysaccharide vaccines [4, 108].
5 Covid-19 and Immunotherapy in Neurology
Since the occurrence of infections with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and identification of the coronavirus disease 2019 (Covid-19) in China in late 2019 [116, 117], more than 200,000,000 confirmed cases have been verified [118]. While up to 60% of Covid-19 patients may show neurological symptoms [119], the question arises as to whether Covid-19 severity is worsened in patients treated with immunosuppressive/-modulatory agents and whether neuroinflammatory conditions are exacerbated by SARS-CoV-2 infection. Although availability of reliable data sets regarding the risks of SARS-CoV-2 infection is still limited for most neuroimmunological conditions, a handful of studies have analyzed Covid-19 disease severity in mostly MS patients with and without DMT.
A survey conducted shortly after the beginning of the Covid-19 pandemic found no increased risk of contracting SARS-CoV-2 infection in MS and NMOSD patients, irrespective of whether patients had received DMTs [120].
Similarly, a French observational study encompassing 347 MS patients found that Covid-19 symptom profiles are consistent with that of the general population [121]. While a correlation between neurological disability as assessed by EDSS and Covid-19 severity was identified, there was no elevated risk for increased Covid-19 severity in patients receiving DMTs. Interestingly, there was even a lower risk for hospitalization under DMTs [121]. This is in line with case reports, i.e. relating to the use milder immunomodulatory therapies such as dimethyl fumarate [122, 123], indicating potentially protective effects against overshooting immune responses triggered by SARS-CoV-2 infection, possibly depending on the respective mode of action of the DMT.
As for more potent immunosuppressive therapies, one study observed an increased risk for severe Covid-19 in MS patients treated with anti-CD20 agents compared to other DMTs [124]. Another recent study found no overall heightened risk of infection or severity of Covid-19 with DMTs, but observed a tendency towards an increased risk of infection with alemtuzumab and cladribine [125]. An Austrian investigation of 126 MS patients with Covid-19 concluded that severity is not affected by DMTs when accounting for already known risk factors such as age, obesity, comorbidities, and degree of physical disability.
Likewise, with regard to neuromuscular disorders including myasthenia gravis, inflammatory myositis and chronic inflammatory neuropathies, the risk of infection does not seem to be increased, albeit the risk for hospitalization might be increased in patents receiving immunomodulatory therapies. Nevertheless, the majority of patients appear likely to experience mild to moderate Covid-19 [126].
Collectively, based on the evidence available so far, neither the risk of SARS-CoV-2 infection nor the development of severe Covid-19 symptoms appears to be significantly increased in patients with MS and other neuroimmunological disorders, with or without DMTs. With regard to Covid-19 vaccination, humoral responses are partly impaired in patients receiving certain DMTs such as ocrelizumab or fingolimod [118, 127]; however, expert consensus states that discontinuation or modification of DMTs should not be considered, as the risk of disease reactivation or progression outweighs the potential benefit [128, 129].
6 Conclusion
SID in neurology is only partly understood, and further research is needed. However, in recent years progress has been made in the identification of risk factors that predispose patients to the development of SID. These include the duration of therapy, increasing age, and pre-existing low Ig levels before therapy initiation. A better risk stratification, more individualized treatment approaches, including the assessment of immune cell reconstitution, and adjustment of treatment courses, as well as immune supplementation employing IGRT might substantially reduce morbidity and mortality associated with SID in neurological conditions. In light of the broad range of existing and emerging therapies, the potential for SID warrants urgent consideration among neurologists and other healthcare professionals to improve diagnosis and treatment. Treatment strategy studies considering SID as potential adverse effect are needed, and data available from routine clinical practice, such as anonymized data from electronic records, should be analyzed in a global “big data” approach to shed more light on this topic that is only partly elucidated so far.
References
McCusker C, Upton J, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol. 2018;14(Suppl 2):61.
Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27(5):497–502.
Aiello A, Farzaneh F, Candore G, Caruso C, Davinelli S, Gambino CM, et al. Immunosenescence and Its hallmarks: how to oppose aging strategically? A review of potential options for therapeutic intervention. Front Immunol. 2019;10:2247.
Patel SY, Carbone J, Jolles S. The expanding field of secondary antibody deficiency: causes, diagnosis, and management. Front Immunol. 2019;10:33.
Friman V, Winqvist O, Blimark C, Langerbeins P, Chapel H, Dhalla F. Secondary immunodeficiency in lymphoproliferative malignancies. Hematol Oncol. 2016;34(3):121–32.
Axelrod H, Adams M. Biologic agents and secondary immune deficiency. Pediatr Clin N Am. 2019;66(5):1007–20.
Wijnands JM, Kingwell E, Zhu F, Zhao Y, Fisk JD, Evans C, et al. Infection-related health care utilization among people with and without multiple sclerosis. Mult Scler. 2017;23(11):1506–16.
Luna G, Alping P, Burman J, Fink K, Fogdell-Hahn A, Gunnarsson M, et al. Infection risks among patients with multiple sclerosis treated with fingolimod, natalizumab, rituximab, and injectable therapies. JAMA Neurol. 2020;77(2):184–91.
Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron. 2018;97(4):742–68.
Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nat Clin Pract Neurol. 2008;4(10):557–67.
Sabatino JJ Jr, Probstel AK, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci. 2019;20(12):728–45.
Rajabally YA, Stettner M, Kieseier BC, Hartung HP, Malik RA. CIDP and other inflammatory neuropathies in diabetes-diagnosis and management. Nat Rev Neurol. 2017;13(10):599–611.
Graf J, Mares J, Barnett M, Aktas O, Albrecht P, Zamvil SS, et al. Targeting B cells to modify MS, NMOSD, and MOGAD: part 2. Neurol Neuroimmunol Neuroinflamm. 2020;16;8(1):e919.
Comi G, Bar-Or A, Lassmann H, Uccelli A, Hartung HP, Montalban X, et al. Role of b cells in multiple sclerosis and related disorders. Ann Neurol. 2021;89(1):13–23.
Greenfield AL, Hauser SL. B-cell therapy for multiple sclerosis: entering an era. Ann Neurol. 2018;83(1):13–26.
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–88.
Mulero P, Midaglia L, Montalban X. Ocrelizumab: a new milestone in multiple sclerosis therapy. Ther Adv Neurol Disord. 2018;11:1756286418773025.
Hauser SL. The Charcot Lecture | beating MS: a story of B cells, with twists and turns. Mult Scler. 2015;21(1):8–21.
Hoffmann F, Meinl E. B cells in multiple sclerosis: good or bad guys?: an article for 28 May 2014-World MS Day 2014. Eur J Immunol. 2014;44(5):1247–50.
Hauser SL, Bar-Or A, Cohen JA, Comi G, Correale J, Coyle PK, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546–57.
Steinman L, Fox E, Hartung H-P, Alvarez E, Qian P, Wray S, et al. Efficacy and safety of ublituximab versus teriflunomide in relapsing multiple sclerosis: results of the Phase 3 ULTIMATE I and II trials (4494). Neurology. 2021;96(15 Supplement):4494.
Crawford A, Macleod M, Schumacher T, Corlett L, Gray D. Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol. 2006;176(6):3498–506.
Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460–71.
Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol. 2006;180(1–2):63–70.
Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376(3):221–34.
Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209–20.
Leandro MJ, Cambridge G, Ehrenstein MR, Edwards JC. Reconstitution of peripheral blood B cells after depletion with rituximab in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(2):613–20.
Avivi I, Stroopinsky D, Katz T. Anti-CD20 monoclonal antibodies: beyond B-cells. Blood Rev. 2013;27(5):217–23.
Chen Q, Yuan S, Sun H, Peng L. CD3(+)CD20(+) T cells and their roles in human diseases. Hum Immunol. 2019;80(3):191–4.
von Essen MR, Ammitzboll C, Hansen RH, Petersen ERS, McWilliam O, Marquart HV, et al. Proinflammatory CD20+ T cells in the pathogenesis of multiple sclerosis. Brain. 2019;142(1):120–32.
van Vollenhoven RF, Emery P, Bingham CO 3rd, Keystone EC, Fleischmann RM, Furst DE, et al. Long-term safety of rituximab in rheumatoid arthritis: .5-year follow-up of the global clinical trial programme with a focus on adverse events of interest in RA patients. Ann Rheum Dis. 2013;72(9):1496–502.
Gottenberg JE, Ravaud P, Bardin T, Cacoub P, Cantagrel A, Combe B, et al. Risk factors for severe infections in patients with rheumatoid arthritis treated with rituximab in the autoimmunity and rituximab registry. Arthritis Rheum. 2010;62(9):2625–32.
Marcinno A, Marnetto F, Valentino P, Martire S, Balbo A, Drago A, et al. Rituximab-induced hypogammaglobulinemia in patients with neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm. 2018;5(6):e498.
Tallantyre EC, Whittam DH, Jolles S, Paling D, Constantinesecu C, Robertson NP, et al. Secondary antibody deficiency: a complication of anti-CD20 therapy for neuroinflammation. J Neurol. 2018;265(5):1115–22.
Derfuss T, Weber MS, Hughes R, Wang Q, Sauter A, Koendgen H, et al. Serum immunoglobulin levels and risk of serious infections in the pivotal Phase III trials of ocrelizumab in multiple sclerosis and their open-label extensions. ECTRIMS Online Library. 2019;279399:65.
Salzer J, Svenningsson R, Alping P, Novakova L, Bjorck A, Fink K, et al. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology. 2016;87(20):2074–81.
Cambridge G, Leandro MJ, Teodorescu M, Manson J, Rahman A, Isenberg DA, et al. B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 2006;54(11):3612–22.
de Seze J, Bar-Or AJC, Cross A-H, Kappos L, Selmaj K, et al. Effect of ofatumumab on serum immunoglobulin levels and infection risk in relapsing multiple sclerosis patients from the phase 3 ASCLEPIOS I and II Trials. Int J MS Care. 2020;22(S2):85.
Coiffier B, Lepretre S, Pedersen LM, Gadeberg O, Fredriksen H, van Oers MH, et al. Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: a phase 1–2 study. Blood. 2008;111(3):1094–100.
van Oers MH, Kuliczkowski K, Smolej L, Petrini M, Offner F, Grosicki S, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370–9.
Smith P, Huck C, Schmid C, Baumgartner R, Stuber N, Theil D, et al. Ofatumumab differs from rituximab by effectively targeting lymph node B cells and achieving faster post-treatment repletion (S24.03). Neurology. 2017;88(16 Supplement):S24.003.
Li Z, Richards S, Surks HK, Jacobs A, Panzara MA. Clinical pharmacology of alemtuzumab, an anti-CD52 immunomodulator, in multiple sclerosis. Clin Exp Immunol. 2018;194(3):295–314.
Hill-Cawthorne GA, Button T, Tuohy O, Jones JL, May K, Somerfield J, et al. Long term lymphocyte reconstitution after alemtuzumab treatment of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(3):298–304.
McCarthy CL, Tuohy O, Compston DA, Kumararatne DS, Coles AJ, Jones JL. Immune competence after alemtuzumab treatment of multiple sclerosis. Neurology. 2013;81(10):872–6.
Mohn N, Pfeuffer S, Ruck T, Gross CC, Skripuletz T, Klotz L, et al. Alemtuzumab therapy changes immunoglobulin levels in peripheral blood and CSF. Neurol Neuroimmunol Neuroinflamm. 2020;7(2):e654.
Wray S, Havrdova E, Snydman DR, Arnold DL, Cohen JA, Coles AJ, et al. Infection risk with alemtuzumab decreases over time: pooled analysis of 6-year data from the CAMMS223, CARE-MS I, and CARE-MS II studies and the CAMMS03409 extension study. Mult Scler. 2019;25(12):1605–17.
Buonomo AR, Zappulo E, Viceconte G, Scotto R, Borgia G, Gentile I. Risk of opportunistic infections in patients treated with alemtuzumab for multiple sclerosis. Expert Opin Drug Saf. 2018;17(7):709–17.
McCormack PL. Natalizumab: a review of its use in the management of relapsing-remitting multiple sclerosis. Drugs. 2013;73(13):1463–81.
Lohmann L, Janoschka C, Schulte-Mecklenbeck A, Klinsing S, Kirstein L, Hanning U, et al. Immune cell profiling during switching from natalizumab to fingolimod reveals differential effects on systemic immune-regulatory networks and on trafficking of non-T cell populations into the cerebrospinal fluid-results from the ToFingo successor study. Front Immunol. 2018;9:1560.
Bornsen L, Christensen JR, Ratzer R, Oturai AB, Sorensen PS, Sondergaard HB, et al. Effect of natalizumab on circulating CD4+ T-cells in multiple sclerosis. PLoS ONE. 2012;7(11):e47578.
Selter RC, Biberacher V, Grummel V, Buck D, Eienbroker C, Oertel WH, et al. Natalizumab treatment decreases serum IgM and IgG levels in multiple sclerosis patients. Mult Scler. 2013;19(11):1454–61.
Saure C, Warnke C, Zohren F, Schroeder T, Bruns I, Cadeddu RP, et al. Natalizumab and impedance of the homing of CD34+ hematopoietic progenitors. Arch Neurol. 2011;68(11):1428–31.
Planas R, Jelcic I, Schippling S, Martin R, Sospedra M. Natalizumab treatment perturbs memory- and marginal zone-like B-cell homing in secondary lymphoid organs in multiple sclerosis. Eur J Immunol. 2012;42(3):790–8.
Kaufman M, Pardo G, Rossman H, Sweetser MT, Forrestal F, Duda P. Natalizumab treatment shows no clinically meaningful effects on immunization responses in patients with relapsing-remitting multiple sclerosis. J Neurol Sci. 2014;341(1–2):22–7.
Largey F, Jelcic I, Sospedra M, Heesen C, Martin R, Jelcic I. Effects of natalizumab therapy on intrathecal antiviral antibody responses in MS. Neurol Neuroimmunol Neuroinflamm. 2019;6(6):e621.
Rudick RA, Stuart WH, Calabresi PA, Confavreux C, Galetta SL, Radue EW, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911–23.
Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910.
Giovannoni G, Kappos L, Berger J, Cutter G, Fox R, Wiendl H, et al. Updated incidence of natalizumab-associated progressive multifocal leukoencephalopathy (PML) and its relationship with natalizumab exposure over time (2815). Neurology. 2020;94(15 Supplement):2815.
Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870–80.
Fine AJ, Sorbello A, Kortepeter C, Scarazzini L. Central nervous system herpes simplex and varicella zoster virus infections in natalizumab-treated patients. Clin Infect Dis. 2013;57(6):849–52.
Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9(11):883–97.
Derfuss T, Mehling M, Papadopoulou A, Bar-Or A, Cohen JA, Kappos L. Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 2020;19(4):336–47.
Francis G, Kappos L, O’Connor P, Collins W, Tang D, Mercier F, et al. Temporal profile of lymphocyte counts and relationship with infections with fingolimod therapy. Mult Scler. 2014;20(4):471–80.
Warnke C, Dehmel T, Ramanujam R, Holmen C, Nordin N, Wolfram K, et al. Initial lymphocyte count and low BMI may affect fingolimod-induced lymphopenia. Neurology. 2014;83(23):2153–7.
Ghadiri M, Fitz-Gerald L, Rezk A, Li R, Nyirenda M, Haegert D, et al. Reconstitution of the peripheral immune repertoire following withdrawal of fingolimod. Mult Scler. 2017;23(9):1225–32.
Johnson TA, Shames I, Keezer M, Lapierre Y, Haegert DG, Bar-Or A, et al. Reconstitution of circulating lymphocyte counts in FTY720-treated MS patients. Clin Immunol. 2010;137(1):15–20.
Zoehner G, Miclea A, Salmen A, Kamber N, Diem L, Friedli C, et al. Reduced serum immunoglobulin G concentrations in multiple sclerosis: prevalence and association with disease-modifying therapy and disease course. Ther Adv Neurol Disord. 2019;12:1756286419878340.
Signoriello E, Bonavita S, Sinisi L, Russo CV, Maniscalco GT, Casertano S, et al. Is antibody titer useful to verify the immunization after VZV Vaccine in MS patients treated with Fingolimod? A case series. Mult Scler Relat Disord. 2020;40:101963.
Mehling M, Hilbert P, Fritz S, Durovic B, Eichin D, Gasser O, et al. Antigen-specific adaptive immune responses in fingolimod-treated multiple sclerosis patients. Ann Neurol. 2011;69(2):408–13.
Mehling M, Eichin D, Hafner P, Honger G, Kappos L, Hess C. Avidity of vaccine-induced influenza IgG fails to increase in fingolimod-treated patients with MS. Neurol Neuroimmunol Neuroinflamm. 2014;1(3):e28.
Kappos L, Mehling M, Arroyo R, Izquierdo G, Selmaj K, Curovic-Perisic V, et al. Randomized trial of vaccination in fingolimod-treated patients with multiple sclerosis. Neurology. 2015;84(9):872–9.
Kowarik MC, Pellkofer HL, Cepok S, Korn T, Kumpfel T, Buck D, et al. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology. 2011;76(14):1214–21.
Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–15.
Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401.
Calabresi PA, Radue EW, Goodin D, Jeffery D, Rammohan KW, Reder AT, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(6):545–56.
Arvin AM, Wolinsky JS, Kappos L, Morris MI, Reder AT, Tornatore C, et al. Varicella-zoster virus infections in patients treated with fingolimod: risk assessment and consensus recommendations for management. JAMA Neurol. 2015;72(1):31–9.
Cohen JA, Tenenbaum N, Bhatt A, Zhang Y, Kappos L. Extended treatment with fingolimod for relapsing multiple sclerosis: the 14-year LONGTERMS study results. Ther Adv Neurol Disord. 2019;12:1756286419878324.
Fox R CB, Greenberg B, Hemmer B, Ward BJ, Ontaneda D, Moore A, Zhang Y, Sullivan R, Girase P, Hach T, Berger JR. Update on the risk estimates of progressive multifocal leukoencephalopathy related to fingolimod. MSVirtual 2020. 2020; Abstract FC02.02.
Kappos L, Cohen J, Collins W, de Vera A, Zhang-Auberson L, Ritter S, et al. Fingolimod in relapsing multiple sclerosis: an integrated analysis of safety findings. Mult Scler Relat Disord. 2014;3(4):494–504.
Blair HA. Dimethyl fumarate: a review in relapsing-remitting MS. Drugs. 2019;79(18):1965–76.
Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–97.
Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–107.
Fox RJ, Chan A, Gold R, Phillips JT, Selmaj K, Chang I, et al. Characterizing absolute lymphocyte count profiles in dimethyl fumarate-treated patients with MS: Patient management considerations. Neurol Clin Pract. 2016;6(3):220–9.
Herrmann AK, Wullner V, Moos S, Graf J, Chen J, Kieseier B, et al. Dimethyl fumarate alters intracellular Ca(2+) handling in immune cells by redox-mediated pleiotropic effects. Free Radic Biol Med. 2019;141:338–47.
Mehta D, Miller C, Arnold DL, Bame E, Bar-Or A, Gold R, et al. Effect of dimethyl fumarate on lymphocytes in RRMS: Implications for clinical practice. Neurology. 2019;92(15):e1724–38.
Ghadiri M, Rezk A, Li R, Evans A, Luessi F, Zipp F, et al. Dimethyl fumarate-induced lymphopenia in MS due to differential T-cell subset apoptosis. Neurol Neuroimmunol Neuroinflamm. 2017;4(3):e340.
Longbrake EE, Ramsbottom MJ, Cantoni C, Ghezzi L, Cross AH, Piccio L. Dimethyl fumarate selectively reduces memory T cells in multiple sclerosis patients. Mult Scler. 2016;22(8):1061–70.
Longbrake EE, Mao-Draayer Y, Cascione M, Zielinski T, Bame E, Brassat D, et al. Dimethyl fumarate treatment shifts the immune environment toward an anti-inflammatory cell profile while maintaining protective humoral immunity. Mult Scler. 2020;27:1352458520937282.
von Hehn C, Howard J, Liu S, Meka V, Pultz J, Mehta D, et al. Immune response to vaccines is maintained in patients treated with dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm. 2018;5(1):e409.
Vola EA, Petracca M, Cocozza S, De Angelis M, Carotenuto A, Pontillo G, et al. Possible progressive multifocal leukoencephalopathy and active multiple sclerosis under dimethyl fumarate: the central role of MRI in informing therapeutic decisions. BMC Neurol. 2021;21(1):146.
Balak D, Hajdarbegovic E. PML in patients treated with dimethyl fumarate. N Engl J Med. 2015;373(6):582–3.
Lehmann-Horn K, Penkert H, Grein P, Leppmeier U, Teuber-Hanselmann S, Hemmer B, et al. PML during dimethyl fumarate treatment of multiple sclerosis: how does lymphopenia matter? Neurology. 2016;87(4):440–1.
Wiendl H. Cladribine—an old newcomer for pulsed immune reconstitution in MS. Nat Rev Neurol. 2017;13(10):573–4.
Comi G, Cook S, Giovannoni G, Rieckmann P, Sorensen PS, Vermersch P, et al. Effect of cladribine tablets on lymphocyte reduction and repopulation dynamics in patients with relapsing multiple sclerosis. Mult Scler Relat Disord. 2019;29:168–74.
Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Soelberg Sorensen P, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):416–26.
Giovannoni G, Soelberg Sorensen P, Cook S, Rammohan K, Rieckmann P, Comi G, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: Results from the randomized extension trial of the CLARITY study. Mult Scler. 2018;24(12):1594–604.
Ceronie B, Jacobs BM, Baker D, Dubuisson N, Mao Z, Ammoscato F, et al. Cladribine treatment of multiple sclerosis is associated with depletion of memory B cells. J Neurol. 2018;265(5):1199–209.
Rejdak K, Stelmasiak Z, Grieb P. Cladribine induces long lasting oligoclonal bands disappearance in relapsing multiple sclerosis patients: 10-year observational study. Mult Scler Relat Disord. 2019;27:117–20.
Cook S, Leist T, Comi G, Montalban X, Giovannoni G, Nolting A, et al. Safety of cladribine tablets in the treatment of patients with multiple sclerosis: an integrated analysis. Mult Scler Relat Disord. 2019;29:157–67.
Tallantyre EC, Robertson NP, Jolles S. Secondary antibody deficiency in neurology. Curr Opin Allergy Clin Immunol. 2018;18(6):481–8.
Barmettler S, Ong MS, Farmer JR, Choi H, Walter J. Association of immunoglobulin levels, infectious risk, and mortality with rituximab and hypogammaglobulinemia. JAMA Netw Open. 2018;1(7):e184169.
Cohen M, Romero G, Bas J, Ticchioni M, Rosenthal M, Lacroix R, et al. Monitoring CD27+ memory B-cells in neuromyelitis optica spectrum disorders patients treated with rituximab: results from a bicentric study. J Neurol Sci. 2017;15(373):335–8.
Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70(9):1110–7.
Griffiths H, Brennan V, Lea J, Bunch C, Lee M, Chapel H. Crossover study of immunoglobulin replacement therapy in patients with low-grade B-cell tumors. Blood. 1989;73(2):366–8.
Gale RP, Chapel HM, Bunch C, Rai KR, Foon K, Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic L, et al. Intravenous immunoglobulin for the prevention of infection in chronic lymphocytic leukemia. A randomized, controlled clinical trial. N Engl J Med. 1988;319(14):902–7.
Chapel H, Dicato M, Gamm H, Brennan V, Ries F, Bunch C, et al. Immunoglobulin replacement in patients with chronic lymphocytic leukaemia: a comparison of two dose regimes. Br J Haematol. 1994;88(1):209–12.
Na IK, Buckland M, Agostini C, Edgar JDM, Friman V, Michallet M, et al. Current clinical practice and challenges in the management of secondary immunodeficiency in hematological malignancies. Eur J Haematol. 2019;102(6):447–56.
Agostini C, Blau IW, Kimby E, Plesner T. Prophylactic immunoglobulin therapy in secondary immune deficiency—an expert opinion. Expert Rev Clin Immunol. 2016;12(9):921–6.
European Medicines Agency: Core summary of product characteristics for human normal immunoglobulin for intravenous administration (IVIg). https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-core-smpc-human-normal-immunoglobulin-intravenous-administration-ivig-rev-5_en.pdf. 2019. Accessed 30 July 2021.
Jolles S, Orange JS, Gardulf A, Stein MR, Shapiro R, Borte M, et al. Current treatment options with immunoglobulin G for the individualization of care in patients with primary immunodeficiency disease. Clin Exp Immunol. 2015;179(2):146–60.
Abolhassani H, Sadaghiani MS, Aghamohammadi A, Ochs HD, Rezaei N. Home-based subcutaneous immunoglobulin versus hospital-based intravenous immunoglobulin in treatment of primary antibody deficiencies: systematic review and meta analysis. J Clin Immunol. 2012;32(6):1180–92.
Raanani P, Gafter-Gvili A, Paul M, Ben-Bassat I, Leibovici L, Shpilberg O. Immunoglobulin prophylaxis in hematological malignancies and hematopoietic stem cell transplantation. Cochrane Database Syst Rev. 2008;4:CD006501.
Kawano T, Matsuse H, Obase Y, Kondo Y, Machida I, Tomari S, et al. Hypogammaglobulinemia in steroid-dependent asthmatics correlates with the daily dose of oral prednisolone. Int Arch Allergy Immunol. 2002;128(3):240–3.
Zrzavy T, Kollaritsch H, Rommer PS, Boxberger N, Loebermann M, Wimmer I, et al. Vaccination in multiple sclerosis: friend or foe? Front Immunol. 2019;10:1883.
Heineman TC, Cunningham A, Levin M. Understanding the immunology of Shingrix, a recombinant glycoprotein E adjuvanted herpes zoster vaccine. Curr Opin Immunol. 2019;59:42–8.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–33.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3.
Achiron A, Mandel M, Dreyer-Alster S, Harari G, Magalashvili D, Sonis P, et al. Humoral immune response to COVID-19 mRNA vaccine in patients with multiple sclerosis treated with high-efficacy disease-modifying therapies. Ther Adv Neurol Disord. 2021;14:17562864211012836.
Fleischer M, Kohrmann M, Dolff S, Szepanowski F, Schmidt K, Herbstreit F, et al. Observational cohort study of neurological involvement among patients with SARS-CoV-2 infection. Ther Adv Neurol Disord. 2021;14:1756286421993701.
Fan M, Qiu W, Bu B, Xu Y, Yang H, Huang D, et al. Risk of COVID-19 infection in MS and neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm. 2020;4;7(5):e787.
Louapre C, Collongues N, Stankoff B, Giannesini C, Papeix C, Bensa C, et al. Clinical characteristics and outcomes in patients with coronavirus disease 2019 and multiple sclerosis. JAMA Neurol. 2020;77(9):1079–88.
Mantero V, Abate L, Basilico P, Balgera R, Salmaggi A, Nourbakhsh B, et al. COVID-19 in dimethyl fumarate-treated patients with multiple sclerosis. J Neurol. 2021;268(6):2023–5.
Capone F, Ferraro E, Motolese F, Di Lazzaro V. COVID-19 in multiple sclerosis patients treated with dimethyl fumarate. J Neurol. 2021;268(9):3132–4.
Sormani MP, De Rossi N, Schiavetti I, Carmisciano L, Cordioli C, Moiola L, et al. Disease-modifying therapies and coronavirus disease 2019 severity in multiple sclerosis. Ann Neurol. 2021;89(4):780–9.
Dalla Costa G, Leocani L, Montalban X, Guerrero AI, Sorensen PS, Magyari M, et al. Real-time assessment of COVID-19 prevalence among multiple sclerosis patients: a multicenter European study. Neurol Sci. 2020;41(7):1647–50.
Kovvuru S, Nalleballe K, Onteddu SR, Sharma R, Jasti M, Kapoor N, et al. Immunosuppression in chronic autoimmune neurological disorders during the COVID-19 pandemic. J Neurol Sci. 2021;15(420):117230.
Monschein T, Hartung HP, Zrzavy T, Barnett M, Boxberger N, Berger T, et al. Vaccination and multiple sclerosis in the era of the COVID-19 pandemic. J Neurol Neurosurg Psychiatry. 2021;92(10):1033–43.
Centonze D, Rocca MA, Gasperini C, Kappos L, Hartung HP, Magyari M, et al. Disease-modifying therapies and SARS-CoV-2 vaccination in multiple sclerosis: an expert consensus. J Neurol. 2021;1–8.
Korsukewitz C, Reddel SW, Bar-Or A, Wiendl H. Neurological immunotherapy in the era of COVID-19 - looking for consensus in the literature. Nat Rev Neurol. 2020;16(9):493–505.
Cotchett KR, Dittel BN, Obeidat AZ. Comparison of the efficacy and safety of anti-CD20 B cells depleting drugs in multiple sclerosis. Mult Scler Relat Disord. 2021;49:102787.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. Octapharma provided a grant for writing this review article. Octapharma did not have any influence on the contents, for which the authors are entirely responsible.
Conflicts of interests
F. Szepanowski has no conflicts of interest. C. Warnke has received institutional support from Novartis, Biogen, Alexion, Janssen, and Roche. G. Meyer zu Hörste served on scientific advisory boards for LFB and received speaker honoraria from Alexion. A. Mausberg has no conflicts of interest. H-P. Hartung received personal fees for consulting, serving on steering and data monitoring committees from Bayer Healthcare, Biogen, Celgene Receptos, CSL Behring, GeNeuro, MedImmune, Merck, Novartis, Octapharma, Roche, Sanofi Genzyme, Teva. C. Kleinschnitz has received remuneration for lectures and advisory boards from Alexion, Almirall, Amgen, Amicus, Bayer, Biogen, Biotronik, Boehringer Ingelheim, Bristol Myers-Squibb, Celgene, CSL Behring, Daiichi Sankyo, Desitin, Eisai, Ever Pharma, GE Healthcare, MedDay Pharmaceuticals, Merck Serono, Mylan/Viatris, Novartis, Pfizer, Roche, Sanofi-Genzyme, Siemens, STADA, Stago, Teva. C. Kleinschnitz owns shares: Sanofi < 30.000 €. Biontec < 30.000 €. M. Stettner served on the scientific advisory boards and/or received speaker honoraria, travel funding or honoraria for medical writing from UCB, Biogen Idec, Grifols, Genzyme, Roche, Merck, Novartis, Octapharma, CSL Behring, Sanofi-Aventis, TEVA, and Bayer.
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Availability of data and material
Not applicable
Code availability
Not applicable
Authors' contributions
All authors take responsibility for the integrity and accuracy of data interpretation and statements. Review concept and design: MS, FS. Drafting of the manuscript: MS, FS. Critical revision of the manuscript for intellectual content: MS, CW, GMH, H-PH, CK. All authors have read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Szepanowski, F., Warnke, C., Meyer zu Hörste, G. et al. Secondary Immunodeficiency and Risk of Infection Following Immune Therapies in Neurology. CNS Drugs 35, 1173–1188 (2021). https://doi.org/10.1007/s40263-021-00863-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-021-00863-4