
Abstract
Three bacterial strains, XENO-2T, XENO-7T, and XENO-10T, isolated from Steinernema entomopathogenic nematodes, were found to represent novel Xenorhabdus species. In this study, we describe these new species by whole-genome and whole-proteome phylogenomic reconstructions, by calculating sequence identity scores using core genome sequences, and by phenotypic characterization. Phylogenomic reconstructions using ribosomal and house-keeping genes, and whole-genome and whole-proteome sequences show that XENO-2T and XENO-10T are closely related to Xenorhabdus japonica DSM 16522T and that XENO-7T is closely related to Xenorhabdus bovienii subsp. africana XENO-1T and to X. bovienii subsp. bovienii T228T. The dDDH values between XENO-2T and XENO-10T and between XENO-2T and X. japonica DSM 16522T are 56.4 and 51.8%, respectively. The dDDH value between XENO-10T and X. japonica DSM 16522T is 53.4%. The dDDH values between XENO-7T and X. bovienii subsp. africana XENO-1T and between XENO-7T and X. bovienii subsp. bovienii T228T are 63.6 and 69.4%, respectively. These dDDH values are below the 70% divergence threshold for prokaryotic species delineation. The newly described species are highly pathogenic to G. mellonella larvae, grow at pH between 5 and 9 (optimum 5–7), at salt concentrations of 1–3% (optimum 1–2%), and temperatures between 20 and 37 °C (optimum 28–30 °C). Biochemical tests such as lysine decarboxylase, ornithine decarboxylase, urease, gelatinase, citrate utilization, indole and acetoin production, and cytochrome oxidase tests allow to differentiate the novel species from their more closely related species. Considering these genetic and phenotypic divergencies, we propose the following new species: Xenorhabdus aichiensis sp. nov. with XENO-7T (= CCM 9233T = CCOS 2024T) as the type strain, Xenorhabdus anantnagensis sp. nov., with XENO-2T (= CCM 9237T = CCOS 2023T) as the type strain, and Xenorhabdus yunnanensis sp. nov., with XENO-10T (= CCM 9322T = CCOS 2071T) as the type strain. Our study contributes to a better understanding of the biodiversity and phylogenetic relationships of entomopathogenic bacteria associated with insect parasitic nematodes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Species of the bacterial genus Xenorhabdus establish a symbiotic association with Steinernema entomopathogenic nematodes (EPNs) [1]. Steinernema EPNs are soil-living organisms which parasitize and reproduce inside insects and certain other small arthropods [1]. These nematodes carry Xenorhabdus bacteria inside a specialized vesicle, which is located at the anterior part of the intestine of infective juveniles, and release them immediately after colonizing a host. Xenorhabdus bacteria produce different digestive enzymes, toxins, immunosuppressors, and antibiotics, which serve to kill and pre-digest the infected host [2]. Then, nematodes and bacteria proliferate in the cadaver [1]. Upon resource depletion, nematodes and bacteria re-establish their symbiosis and abandon the cadaver in search of a new host. Due to their remarkable ability to search for and kill insects, including crop pests, and due to the enormous biochemical capabilities of Xenorhabdus bacteria, these organisms are of high agricultural, biotechnological, and medical relevance [3].
The genus Xenorhabdus was created by Thomas and Poinar to accommodate large, gram-negative, rod-shaped, facultatively anaerobic, entomopathogenic bacteria which establish an obligated symbiosis with entomogenous nematodes [4]. Initially, it contained species of bacteria associated with Steinernema and with Heterorhabditis nematodes. Later, the creation of the genus Photorhabdus was proposed, and the symbiotic bacteria associated with Heterorhabditis nematodes, were transferred to this new genus [5, 6]. At that time, the taxonomic decisions were made mainly based on wet-lab DNA-DNA hybridization techniques and biochemical tests [7,8,9,10,11]. A few years later, the use 16S rRNA gene sequences was introduced [12] and successfully used to describe several novel species of the genus Xenorhabdus [13,14,15]. Shortly after, the use of multiple house-keeping genes for taxonomic purposes was introduced for the first time [16], which significantly improved the robustness of the phylogenetic reconstructions, and again led to the description of several novel species [17,18,19]. Boosted by the advances in genome sequencing techniques, the use of core genome sequences and in silico DNA-DNA hybridization techniques are the gold standard nowadays [20,21,22]. After decades of efforts to understand the biodiversity of this agriculturally and biotechnologically relevant group of bacteria, several species have been described. Currently, this genus contains 28 taxa with validly published names: 27 species and 1 subspecies [4, 10, 11, 13,14,15,16,17,18,19,20,21,22,23,24,25,26]. The complete list of species is found under: https://lpsn.dsmz.de/genus/xenorhabdus [26].
In this study, we phenotypically and molecularly characterized several bacterial strains, isolated from different species of Steinernema entomopathogenic nematodes, which represent new species in the genus Xenorhabdus for which we propose the following names: Xenorhabdus aichiensis sp. nov., Xenorhabdus anantnagensis sp. nov., and Xenorhabdus yunnanensis sp. nov. Our study contributes to a better understanding of the biodiversity of a bacterial group of biotechnological and agricultural relevance and thereby further advances our efforts toward developing more biocontrol tools for sustainable and environmentally friendly agriculture.
Materials and Methods
Strain Isolation and Biogeography
The three bacterial strains characterized in this study, XENO-2T, XENO-7T, and XENO-10T, were isolated from Steinernema entomopathogenic nematodes. XENO-2T was isolated from a novel Steinernema nematode species, which will be described somewhere else [27]. These nematodes were isolated from soil samples collected in Anantnag (Jammu and Kashmir, India) (GPS coordinates: 33.828914, 75.100091) using Corcyra cephalonica Stainton (Lepidoptera: Pyralidae) larvae as baits. XENO-7T was isolated from S. litorale nematodes. These nematodes were isolated from soil samples collected from a coastal pine forest (Cape Irago-zaki, Atsumi Peninsula, Aichi Prefecture, Honshu, Japan) using the Galleria baiting technique [28]. XENO-10T was isolated from S. akhursti nematodes. These nematodes were isolated from soil samples collected from a grassland in the Cigu village (Deqen, Diqing Tibetan Autonomous Prefecture, Yunnan province, China) using G. mellonella larvae as baits [29]. Nematode identification was carried out as described below. To isolate the bacterial strains, G. mellonella larvae were infested with the different nematode strains. Three to four days later, several insect cadavers were surface-sterilized and dissected with a sterile surgical blade. Insect internal organs were spread onto Lysogeny Broth (LB) agar plates (Sigma-Aldrich, Switzerland) and incubated at 28 °C for 48-96 h. Xenorhabdus-like colonies were subcultured until monocultures were obtained [30]. Different procedures, such as the characterization of colony and cell morphology, and 16S rRNA gene sequencing were carried out to determine the culture purity.
Bacterial 16S rRNA Gene Phylogeny and Sequence Comparisons
To determine the taxonomic identities of XENO-2T, XENO-7T, and XENO-10T, genomic DNA was first extracted and purified from bacterial monocultures using the GenElute Bacterial Genomic DNA Kit (Sigma–Aldrich, Switzerland) following the manufacturer’s instructions. The resulting DNA was used to obtain 16S rRNA gene sequences by polymerase chain reaction (PCR) using the following universal primers: 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1525R (5′-AAGGAGGTGWTCCARCC-3′) and the following cycling conditions: 1 cycle at 94 °C for 10 min followed by 40 cycles at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 60 s, followed by a final extension at 72 °C for 5 min as described previously [31,32,33]. PCR products were separated by electrophoresis in a 1% TAE-agarose gel stained with GelRed nucleic acid gel stain (Biotium), gel-purified (QIAquick Gel Purification Kit, Qiagen), and sequenced by Sanger sequencing (Microsynth AG, Balgach, Switzerland). The resulting 16S rRNA raw sequences were manually curated using Bioedit 7.2.5 [34]. In addition, 16S rRNA gene sequences were obtained directly from whole-genome sequences using the bacterial ribosomal RNA predictor Barrnap 0.7 using the following parameters: reject length threshold = 0.5; length cutoff = 0.8; and e-value = 0.00001 [35]. The obtained sequences were identical to those obtained by Sanger sequencing. 16S rRNA gene-based phylogenetic relationships were reconstructed using the Maximum Likelihood method based on the Kimura 2-parameter model in MEGA7 [36,37,38]. To this end, sequences were aligned with MUSCLE (v3.8.31) [39]. The tree with the highest log likelihood was obtained. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were calculated automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Graphical representation and edition of the phylogenetic trees were performed with Interactive Tree of Life (v3.5.1) [40, 41].
Recombinase A Gene-Based Phylogenetic Reconstructions
Recombinase A (recA) gene sequences were obtained directly from the whole-genome sequences using BLAST. The obtained sequences were used to reconstruct phylogenetic relationships using the maximum likelihood method based on the Kimura 2-parameter model in MEGA7 as described above [36,37,38]. Graphical representation and edition of the phylogenetic trees were performed with Interactive Tree of Life (v3.5.1) [40, 41].
Genome Sequencing and Genomic Features
The genomes of the following strains were obtained in this study: X. aichiensis sp. nov. XENO-7 T, X. anantnagensis sp. nov. XENO-2T, X. griffiniae ID10T, X. khoisanae SF87T, X. magdalenensis IMI 397775T, X. romanii PR06-AT, and X. yunnanensis sp. nov. XENO-10T. Genomes sequences were obtained as described previously [42, 43]. Briefly, genomic DNA was extracted and purified using the GenElute Bacterial Genomic DNA Kit (Sigma–Aldrich, Switzerland) following the manufacturer’s instructions. The resulting DNA was used for library preparation using the TruSeq DNA PCR–Free LT Library Prep (FC–121–3003) kit. Indexed libraries were then pooled at equimolar concentrations and sequenced (2 × 150 bp) on an Illumina HiSeq 3000 instrument. Genomes were assembled using the Bactopia pipeline [44]. To this end, the raw Illumina reads were quality trimmed using Trimmomatic 0.39 (options: slidingwindow:4: 8, minlen:127) [45]. The resulting reads were assembled with SPAdes 3.14.1 (-careful, -mismatch-correction, k–mer sizes of 31, 51, 71, 91, and 111 bp) [46]. Scaffolds with a mean read–depth smaller than 20% of the median read–depth of the longer scaffolds (≥ 5000 bp) as well as scaffolds that were shorter than 200 bp were removed. Minor assembly errors were corrected using Pilon 1.22 with default parameters [47]. Completeness and contamination of the assembled genomes were assessed using checkM v1.1.6 with default parameters [48].
Core Genome- and Core Proteome-Based Phylogenetic Reconstructions and Sequence Comparisons
To reconstruct whole-genome-based phylogenetic relationships, genomes were first aligned using Roary 3.13.0. Genes to be considered core should be presented in 85% of the genomes with an 85% protein identity, and a coverage higher than 90%. Obtained alignments were used to build phylogenomic trees using FastTree 2.1.10 based on the Generalized Time Reversible Model (GTR). Branch support was assessed using the Shimodaira-Hasegawa-like procedure based on 100 replicates. To reconstruct whole-proteome-based phylogenetic relationships, first, all ORFs from all genomes were extracted using Prodigal [49]. Then, homologous genes (80% or higher similarity and a coverage higher than 90%) were clustered using MMSEQS2 (e-value: 0.001, sensitivity: 7.5, and cover: 0.5) and MCL (Inflation = 2) [50,51,52]. Orthologous genes were then translated and aligned using MAFFT [53]. Orthologous genes were considered core genes if they were present in more than 80% of the genomes analyzed. Lastly, a maximum likelihood-based phylogenetic tree was reconstructed based on the inferred core-proteome alignment using RAxML [54, 55]. Branch support was assessed using the rapid bootstrap method based on 100 replicates [56]. Graphical representation and edition of the phylogenetic trees were performed with Interactive Tree of Life (v3.5.1) [40, 41]. Whole-genome sequence similarities were calculated by the GBPD (Genome Blast Distance Phylogeny) method using the Genome-to-Genome Distance Calculator 2.1 and formula 2 of the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) web service (http://ggdc.dsmz.de) using default parameters [57,58,59,60].
Physiological, Biochemical, and Morphological Characterization
To physiologically, biochemically and morphologically characterize the newly isolated bacterial strains, bacterial cultures from single primary form colonies were used. Bacterial primary forms were determined by examining their ability to absorb dye from NBTA culturing plates (LB agar plates supplemented with 25 mg L−1 bromothymol blue and 4 mg L−1 triphenyl-2,3,5-tetrazolium chloride) or by their ability to produce yellow, orange, and reddish pigments. The selected primary form colonies were further subcultured and maintained on Lysogeny Broth (LB) agar plates at 28–30 °C. Cell morphology was observed under a Leica DM4B optical microscope at ×1000 magnification, with cells grown for 16 h at 28 °C on LB. Light microscopy photographs were captured using a Leica DM4 B optical microscope equipped with a Leica DFC 7000T Camera (Leica, Wetzlar, Germany). The optimum temperature for bacterial growth was evaluated on regular LB agar medium (pH 7, 1% NaCl) at 20 °C, 24 °C, 28 °C, 30 °C, 37 °C, and 42 °C. Growth on medium containing different salt concentrations was evaluated in 3 mL of LB medium (pH 7) in 15 mL Falcon tubes. Three NaCl concentrations were used: 1% (Regular LB medium), 2%, and 3%. Growth at different pH was evaluated in 3 mL of LB medium (1% NaCl) in 15 mL Falcon tubes. Five different pH were used: 3, 5, 7 (Regular LB medium), 8, and 9. Each tube was inoculated with 0.1 mL of an overnight bacterial culture, then incubated for 24 h at 28 °C and 180 rpm. Three tubes per treatment were considered. Antibiotic resistance was evaluated on regular LB medium (pH 7, 1% NaCl) containing either tetracycline, vancomycin, or gentamicin at concentrations of 30 mg/L. Cytochrome oxidase production was tested on disks containing N,N-dimethyl-p-phenylenediamine oxalate and α-naphthol (Sigma-Aldrich, Switzerland). Catalase activity was determined by adding a drop of 10% (v/v) H2O2 into 50 µL of a 16 h-old liquid LB-bacterial culture. The ability to absorb dye was tested by growing the cells on NBTA agar. Biochemical characterization was carried out using the API20E system (bioMérieux, Inc. Durham, NC) according to the manufacturer’s instructions. To this end, bacteria were grown for 16 h at 28 °C in LB agar Petri plates. Then, one single colony was re-suspended in 5 mL of 0.85% NaCl. The resulting bacterial solution was used to inoculate the different microtubes containing the biochemical tests. Samples were incubated at 28 °C. Results were evaluated after 24 h. Gram staining was carried out using the Gram-Color modified (phenol-free) staining kit following the manufacturer’s instructions (Sigma–Aldrich, Switzerland). Cell morphology, optimum temperature for bacterial growth, growth on medium containing different salt concentrations and pH, and Gram reaction were evaluated only in the novel species described in this study. Biochemical tests such as cytochrome oxidase production, catalase activity, and the API20E tests were evaluated, in parallel, two independent times, in all the eight strains as listed in Table 1.
Genomic Comparative Analyses
Genomic comparative analyses to annotate and determine the presence/absence of genes that are involved in antibiotic resistance or in the production of specialized metabolites were carried out by aligning draft genome assemblies or predicted proteomes against The Comprehensive Antibiotic Resistance Database (“CARD”) [61,62,63,64,65,66] and against the antibiotics and secondary metabolite analysis shell (antiSMASH) database [67, 68]. Genes that passed the threshold values (antibiotic resistance: ≥ 70% nucleotide identity and ≥ 50% coverage; and antiSMASH: ≥ 50% nucleotide identity) were considered as present in the genome. Below these thresholds, genes were considered absent or non-functional.
Ecological Characterization
Xenorhabdus bacteria are characterized by their ability to produce different insecticidal molecules [3]. To evaluate the entomopathogenic potential of the novel species, bacteria were cultured overnight in LB liquid medium. Then, the bacterial cultures were collected and their optical densities at 600 nm (OD600) were measured. All cultures were then diluted to reach an OD600 = 1. The resulting cultures were further diluted to obtain four bacterial solutions with OD600= 0.1, 0.25, 0.5, or 1. Then, 10 µL of the resulting bacterial solutions were injected into third-instar G. mellonella larvae. Ten larvae per bacterial strain and dilution were injected (n = 10). Mortality was evaluated at 48 and 72 h after injections. Results were statistically assessed by repeated-measures ANOVA with bacterial strain and time as factors. Normality and equality of variance were verified using Shapiro–Wilk and Levene's tests, respectively. Holm–Sidak post hoc tests were used for multiple comparisons. All statistical analyses were conducted using Sigma Plot 14.5 (Systat Software Inc., San Jose, CA, USA).
Nematode Molecular Characterization and Phylogenetic Analyses
The nematodes that host the novel bacterial species described in this study were molecularly identified based on rRNA gene sequences [69]. To extract genomic DNA from single virgin females, several individuals were collected and washed with Ringer’s solution, then with PBS buffer, and then transferred individually into sterile PCR tubes (0.2 mL), each containing 20 μL extraction buffer (17.6 μL nuclease-free dH2O, 2 μL 5X PCR buffer, 0.2 μL 1% Tween, and 0.2 μL proteinase K). Samples were then frozen at − 20 °C for 60 min and then incubated in a water bath at 65 °C for 1 h, and at 95 °C for 10 min. The lysates were incubated on ice for 20 min and centrifuged at 6500 g for 2 min. No further DNA purification was carried out and the supernatants were directly used for PCRs. To amplify the internal transcribed spacer regions (ITS1-5.8S-ITS2), the following primers were used: 18S: (5′-TTGATTACGTCCCTGCCCTTT-3′) (forward), and 28S: (5′-TTTCACTCGCCGTTACTAAGG-3′) (reverse) [70]. The 25 µL PCRs consisted of 12.5 µL of Dream Taq Green PCR Master Mix (Thermo Scientific, USA), 0.5 µL of each forward and reverse primer at 10 µM, 2 µL of DNA extract, and 9.5 µL of nuclease-free distilled water. The PCR was performed using a thermocycler with the following settings: 1 cycle of 5 min at 94 °C followed by 37 cycles of 30 s at 94 °C, 30 s at 50 °C, 1 min 30 s at 72 °C, and by a single final elongation step at 72 °C for 10 min. PCR products were separated by electrophoresis (40 min, 130 V) in a 1% TBA (Tris–boric acid–EDTA)-buffered agarose gel stained with SYBR Safe DNA Gel Stain (Invitrogen, Carlsbad, California, USA). PCR products were purified using QIAquick PCR Purification Kit (Qiagen, Valencia, CA) and sequenced using reverse and forward primers by Sanger sequencing (Bioserve Ltd., Hyderabad, India). The obtained sequences were manually curated and trimmed using BioEdit, and deposited in the NCBI. To obtain sequences of closely related nematodes, we searched the database of NCBI by the Basic Local Alignment Search Tool (BLAST) [71]. The resulting sequences were used to reconstruct phylogenetic relationships by the maximum likelihood method based on Hasegawa-Kishino-Yano model (HKY+G) nucleotide substitution model as described above. To select the best substitution model, best–fit nucleotide substitution model analyses were conducted in MEGA 7 [38, 72].
Results and Discussion
Bacterial 16S rRNA Gene-Based Phylogeny and Sequence Comparisons
Phylogenetic reconstructions based on 16S rRNA gene sequences show that X. anantnagensis sp. nov. XENO-2T and X. yunnanensis sp. nov. XENO-10T form a unique clade together with X. japonica DSM 16522T and X. vietnamensis VN01T (Fig. S1, Table S1). In turn, X. aichiensis sp. nov. XENO-7T forms a unique clade together with X. bovienii subsp. africana XENO-1T and X. bovienii subsp. bovienii T228T (Fig. S1). 16S rRNA gene sequence similarity scores between X. anantnagensis sp. nov. XENO-2T and X. vietnamensis VN01T, and between X. anantnagensis sp. nov. XENO-2T and X. japonica DSM 16522T are 98.9% in both cases (Fig. S2). 16S rRNA gene sequence similarity scores between X. yunnanensis sp. nov. XENO-10T and X. vietnamensis VN01T, and between X. yunnanensis sp. nov. XENO-10T and X. japonica DSM 16522T are 98.3% in both cases. The 16S rRNA gene sequence similarity score between X. anantnagensis sp. nov. XENO-2T and X. yunnanensis sp. nov. XENO-10T is 98.7%. Lastly, 16S rRNA gene sequence similarity scores between X. aichiensis sp. nov. XENO-7T and X. bovienii subsp. africana XENO-1T, and between X. aichiensis sp. nov. XENO-7T and X. bovienii subsp. bovienii T228T are 99.2 and 99.4%, respectively. Lower sequence similarities were observed when the sequences of X. anantnagensis sp. nov. XENO-2T, X. aichiensis sp. nov. XENO-7T, and X. yunnanensis sp. nov. XENO-10T were compared to the sequences of the type strains of all the species of the genus Xenorhabdus with validly published names (Fig. S2). 16S rRNA gene sequences are deposited in the National Center for Biotechnology Information (NCBI) databank under the accession numbers OQ439938, OQ439939, and OQ439940, respectively.
Bacterial Recombinase A Gene-Based Phylogeny
Phylogenetic reconstructions based on the recombinase A (recA) gene sequences show that X. anantnagensis sp. nov. XENO-2T and X. yunnanensis sp. nov. XENO-10T form a unique clade together with X. japonica DSM 16522T and X. vietnamensis VN01T (Fig. S3). In turn, X. aichiensis sp. nov. XENO-7T forms a unique clade together with X. bovienii subsp. africana XENO-1T and X. bovienii subsp. bovienii T228T (Fig. S3).
Genome Sequencing and Genomic Features
The main characteristics of the genomes obtained in this study are summarized in Tables S2, S3, and S4. The main characteristics of the genomes of the novel species are as follows. The genome of X. aichiensis sp. nov. XENO-7T contains 4,699,893 bp, a G+C content of 44.63%, and 4196 proteins (Tables S2, S3). The genome of X. anantnagensis sp. nov. XENO-2T contains 4,318,764 bp, a G+C content of 42.88%, and 3905 proteins (Tables S2, S3). The genome of X. yunnanensis sp. nov. XENO-10T contains 4,667,994 bp, a G+C content of 43.14%, and 4534 proteins (Tables S2, S3). These genomes are predicted to be more than 99% complete and contain less than 2% of contamination (Table S4). Whole-genome sequences of XENO-2T, XENO-7T, and XENO-10T are deposited in the National Center for Biotechnology Information (NCBI) databank under the accession numbers JAQRFN01, JAQRFO01, and JAQRFI01, respectively.
Core Genome- and Core Proteome-Based Phylogenetic Reconstructions and Sequence Comparisons
Core-genome and core-proteome phylogenies show that X. aichiensis sp. nov. XENO-7T forms a unique clade together with X. bovienii subsp. africana XENO-1T and X. bovienii subsp. bovienii T228T (Figs. 1, S4). In addition, X. anantnagensis sp. nov. XENO-2T and X. yunnanensis sp. nov. XENO-10T form a unique clade together with X. vietnamensis VN01T, X. japonica DSM 16522T, and X. poinarii G6T (Figs. 1, S4). The dDDH values between XENO-7T and X. bovienii subsp. africana XENO-1T, and between XENO-7T and X. bovienii subsp. bovienii T228T are 63.6 and 69.4%, respectively (Fig. 2). The dDDH values between XENO-2T and XENO-10T, and between XENO-2T and X. japonica DSM 16522T are 56.4 and 51.8%, respectively. The dDDH value between XENO-10T and X. japonica DSM 16522T is 53.4%. Lower sequence similarities were observed when the sequences of X. anantnagensis sp. nov. XENO-2T, X. aichiensis sp. nov. XENO-7T, and X. yunnanensis sp. nov. XENO-10T were compared to the sequences of the type strains of all the species of the genus Xenorhabdus with validly published names. Given that the observed dDDH values are below the 70% divergence threshold for prokaryotic species delineation, XENO-2T, XENO-7T, and XENO-10T represent novel species within the genus Xenorhabdus [73].

Phylogenetic reconstruction based on core genome sequences of Xenorhabdus bacterial strains. 1,485,300 nucleotide positions (1454 core genes) were used in the analysis. Numbers at the nodes represent SH-like branch supports. Bar represents 0.05 nucleotide substitutions per sequence position. The National Center for Biotechnology Information (NCBI) accession numbers of the sequences used for these analyses are shown in Table S1

Pairwise comparison of digital DNA-DNA Hybridization (dDDH) scores (%) of Xenorhabdus strains. NCBI accession numbers of gene sequences used are shown in Table S1
Physiological and Biochemical Characterization
Biochemical tests show that the three novel species exhibit biochemical capacities that are similar to the biochemical capacities of several members of the Xenorhabdus genus (Table 1). However, the three novel species also exhibit unique biochemical capacities that differ from the biochemical capacities of their most closely related species (Table 1). In particular, lysine decarboxylase, citrate utilization, and urease and gelatinase activity allow to differentiate X. aichiensis sp. nov. XENO-7T, X. bovienii subsp. africana XENO-1T, and X. bovienii subsp. bovienii T228T. In addition, lysine decarboxylase, ornithine decarboxylase, citrate utilization, urease activity, indole and acetoin production, gelatinase activity, cytochrome oxidase, and NO2 reduction to N2 gas allow to differentiate X. anantnagensis sp. nov. XENO-2T, X. yunnanensis sp. nov. XENO-10T, X. vietnamensis VN01T, X. japonica DSM 16522T, and X. poinarii G6T (Table 1).
Antibiotic Resistance
The genomes of X. aichiensis sp. nov. XENO-7T, X. anantnagensis sp. nov. XENO-2T, and X. yunnanensis sp. nov. XENO-10T and the genomes of their more closely related species contain several antibiotic resistance genes (Table 2). In vitro experiments confirm the predicted antibiotic resistance patterns of these strains (Table S5). In particular, X. aichiensis sp. nov. XENO-7T, X. anantnagensis sp. nov. XENO-2T, X. yunnanensis sp. nov. XENO-10T, X. bovienii subsp. africana XENO-1T, and X. bovienii subsp. bovienii T228T are susceptible to tetracycline, and X. japonica DSM 16522T, X. poinarii G6T, and X. vietnamensis VN01T are resistant to this antibiotic at the dose tested (Table S5). All these strains resist gentamicin, and only X. bovienii subsp. africana XENO-1T was found vancomycin susceptible (Table S5).
Genomic Comparative Analyses
At the functional level, according to the antibiotics and secondary metabolite analysis shell (antiSMASH) database, the genomes of X. aichiensis sp. nov. XENO-7T, X. anantnagensis sp. nov. XENO-2T, and X. yunnanensis sp. nov. XENO-10T, and the genome of their more closely relates species contain biosynthetic gene clusters dedicated to the production of several phenazines, aryl polyenes, polyketides, and non-ribosomal peptides such as 1,6-phenazinedimethanol, acinetobactin, ambactin, bicornutin A1-A2, cycloserine, fabclavine, glidopeptin, indigoidine, kleboxymycin, luminmide, nematophin, puromycin, putrebactin, pyrrolizixenamide A, ralsolamycin, rhizomide A-C, syringomycin, syringopeptin 25A, taxlllaid A, thiomarinol, turnerbactin, xefoampeptides AG, xenematide, xeneprotides A-C, xenoamicin A-B, xenocoumacin I-II, and xenortide A-D (Table 3). The production of several of these compounds occurs in a species-specific manner as not all the species analyzed contain the gene cluster necessary for their biosynthesis (Table 3). In particular, only X. aichiensis sp. nov. XENO-7T contains the genes to produce kleboxymycin, Syringopeptin 25A, and Xeneprotides A-C; only X. bovienii subsp. africana XENO-1T contains the genes to produce closerine and syringomycin; only X. bovienii subsp. bovienii T228T contains the genes to produce glidopeptin and puromicin; and only X. yunnanensis sp. nov. XENO-10T contains the genes to produce ralsolamycin (Table 3).
Ecological Characterization
When injected into the hemocoel of G. mellonella larvae, all the strains evaluated in this study rapidly killed the insects in a bacterial density-dependent manner (Fig. S5). At the lowest bacterial density tested (OD600 = 0.1), all the bacterial strains killed between 50 and 80% of the larvae in less than 48 h after injections, being X. aichiensis sp. nov. XENO-7T and X. anantnagensis sp. nov. XENO-2T the less pathogenic strains. More insects were killed when the bacterial densities injected were increased, and in some cases, even 100% of the insects were killed within 48 h. 72 h after injections, almost all insects were killed, especially when the bacterial densities injected were high (Fig. S5). Overall, X. anantnagensis sp. nov. XENO-2T was apparently the less pathogenic strain, but they still killed more than 70% of the insects at the lowest bacterial density tested (Fig. S5).
Nematode and Bacteria Co-phylogenies
To infer about potential co-evolutionary processes between the newly described bacterial species and their nematode hosts, we reconstructed phylogenetic relationships of these two groups of organisms and of closely related species and compared the resulting phylogenetic trees (Fig. S6). We observed very interesting patterns. Specifically, the nematode hosts of the following four closely related strains: X. anantnagensis sp. nov. XENO-2T, X. yunnanensis sp. nov. XENO-10T, X. japonica DSM 16522T, and X. vietnamensis VN01T are S. akhursti, Steinernema sp., S. kushidai, and S. sangi, respectively. These nematode species are also closely related (Fig. S6). Similarly, the nematodes that host the following three closely related bacterial strains: X. aichiensis sp. nov. XENO-7T, X. bovienii subsp. africana XENO-1T, and X. bovienii subsp. bovienii T228T are S. litorale, S. africanum, and S. feltiae, respectively. These nematode species are also closely related (Fig. S6). The observed phylogenetic congruence points toward potential co-evolution between these two groups of organisms, which has been already suggested in the past [74,75,76]. Further studies with deeper sampling sizes will be required to conclusively test this hypothesis.
Taxonomic Conclusions and Final Considerations
There is a strong scientific consensus around the use of dDDH values to delimit bacterial species and subspecies [57, 58, 77]. When dDDH values between two strains are below 70%, the two bacterial strains belong to different species. If their dDDH values are between 70 and 79%, the two bacterial species belong to the same species but to different subspecies. When their dDDH values are above 79%, the two bacterial strains belong to the same species and subspecies. However, when dDDH values between two bacterial strains are very close to 70% or to 79%, the taxonomic decisions are not that straightforward, and additional factors should be considered. In this study, for instance, we observed that the dDDH value between XENO-7T and X. bovienii subsp. bovienii T228T is 69.4%, which is very close to the species threshold, motivating the question whether XENO-7T should actually be considered to represent a subspecies within the species X. bovienii, or a different species. The fact that dDDH value between XENO-7T and X. bovienii subsp. africana XENO-1T is 63.6% which indicates that XENO-7T does belong to a different species, in spite that dDDH value between XENO-7T and X. bovienii subsp. bovienii T228T is close to the species threshold. Considering the above and based on the results of this polyphasic taxonomic study, we recommend the use of whole-genome sequences and sequence comparison methods such as the GBPD (Genome Blast Distance Phylogeny) for future description of novel Xenorhabdus species. Hence, the following novel species are proposed: Xenorhabdus aichiensis sp. nov. with XENO-7T (= CCM 9233T = CCOS 2024T) as the type strain, Xenorhabdus anantnagensis sp. nov. with XENO-2T (= CCM 9237T = CCOS 2023T) as the type strain, and Xenorhabdus yunnanensis sp. nov. with XENO-10T (= CCM 9322T = CCOS 2071T) as the type strain.
Description of Xenorhabdus aichiensis sp. nov.
(ai.chi.en’sis. N.L. fem. adj. aichiensis, pertaining to the Aichi, Japan, the Japanese prefecture where the nematodes hosting the bacterial type strain were collected). Cells are rod-shaped, approx. 1.1–1.8 µm wide and 3.6–5.3 µm long (Fig. 3). Cells are highly pathogenic when injected in G. mellonella larvae. Colonies are light or dark yellow and of about 4–5 mm after 48 h on LB agar plates. When cultured in LB agar plates (pH 7, 1% NaCl), growth is observed at temperatures between 20 and 37 °C, but not at 42 °C, and optimal growth occurs at 28–30 °C. When cultured in LB (28 °C, 1% NaCl), bacterial growth occurs at pH between 5 and 9 (optimum 5–7) and not at pH 3. Bacterial growth occurs in LB medium (28 °C, pH 7.0) containing NaCl between 1 and 3% (optimum 1–2%), negative for β-galactosidase, lysine decarboxylase, ornithine decarboxylase, and tryptophan deaminase, positive for arginine dihydrolase and gelatinase, oxidase positive, catalase negative, positive for citrate utilization, does not produce hydrogen sulfide or indole, produces acetoin, oxidizes glucose but does not oxidize mannitol, inositol sorbitol, rhamnose, sucrose, melibiose, amygdalin, or arabinose. The type strain was isolated from Steinernema litorale nematodes. These nematodes were isolated from soil samples collected from a coastal pine forest (Cape Irago-zaki, Atsumi Peninsula, Aichi Prefecture, Honshu, Japan). The type strain of the species is XENO-7T (= CCM 9233T = CCOS 2024T). Whole-genome sequences of XENO-7T are deposited in the NCBI databank under the accession number JAQRFO01 and 16S rRNA gene sequences under the accession number OQ439939. The genome assembled contains 4,699,893 bp, a G+C content of 44.63, and 4196 proteins.

Light microscopy microphotographs of the newly described bacterial species. a Xenorhabdus aichiensis sp. nov. XENO-7T. b Xenorhabdus anantnagensis sp. nov. XENO-2T. c Xenorhabdus yunnanensis sp. nov. XENO-10T. Bars correspond to 20 µm
Description of Xenorhabdus anantnagensis sp. nov.
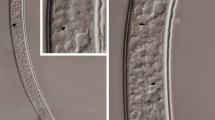
(anan.tnag.en’sis. N.L. fem. adj. anantnagensis, pertaining to Anantnag, India, the Indian District where the nematodes hosting the bacterial type strain were collected). Cells are rod-shaped, approx. 1.2–2.1 µm wide and 4.1–6.3 µm long (Fig. 3). Cells are highly pathogenic when injected in G. mellonella larvae. Colonies are light or dark yellow and of about 4–5 mm after 48 h on LB agar plates. When cultured in LB agar plates (pH 7, 1% NaCl), growth is observed at temperatures between 20 and 37 °C, but not at 42 °C, and optimal growth occurs at 28–30 °C. When cultured in LB (28 °C, 1% NaCl), bacterial growth occurs at pH between 5 and 9 (optimum 5–7) and not at pH 3. Bacterial growth occurs in LB medium (28 °C, pH 7.0) containing NaCl between 1 and 3% (optimum 1–2%), negative for β-galactosidase and tryptophan deaminase, positive for arginine dihydrolase, lysine decarboxylase, ornithine decarboxylase, urease, and gelatinase activity, utilizes citrate, oxidase positive, catalase negative, produces indole, does not produce acetoin or hydrogen sulfide, oxidizes glucose but does not oxidize mannitol, inositol sorbitol, rhamnose, sucrose, melibiose, amygdalin, or arabinose. The type strain was isolated from a novel Steinernema species, which will be formally described elsewhere (Fig. 4). These nematodes were isolated from soil samples collected in Anantnag (Jammu and Kashmir, India) (GPS coordinates: 33.828914, 75.100091) using Corcyra cephalonica Stainton (Lepidoptera: Pyralidae) larvae as baits. The type strain of the species is XENO-2T (= CCM 9237T = CCOS 2023T). Whole-genome sequences of XENO-2T are deposited in the NCBI databank under the accession number JAQRFN01 and 16S rRNA gene sequences under the accession number OQ439938. The genome assembled contains 4,318,764 bp, a G+C content of 42.88%, and 3905 proteins.

Light microscopy microphotographs of Xenorhabdus anantnagensis sp. nov. XENO-2T within its Steinernema nematode host. Arrows point to the bacterial sac
Description of Xenorhabdus yunnanensis sp. nov.
(yun.nan.en'sis. N.L. fem. adj. yunnanensis pertaining to Yunnan, a province of south-west China where the nematodes hosting the bacterial type strain were collected). Cells are rod-shaped, approx. 1.5–2.0 µm wide and 3.1–6.6 µm long. Cells are highly pathogenic when injected in G. mellonella larvae. Colonies are light or dark yellow and of about 4-5 mm after 48 h on LB agar plates. When cultured in LB agar plates (pH 7, 1% NaCl), growth is observed at temperatures between 20 and 37 °C, but not at 42 °C, and optimal growth occurs at 28–30 °C. When cultured in LB (28 °C, 1% NaCl), bacterial growth occurs at pH between 5 and 9 (optimum 5–7) and not at pH 3. Bacterial growth occurs in LB medium (28 °C, pH 7.0) containing NaCl between 1 and 3% (optimum 1–2%), negative for β-galactosidase, lysine decarboxylase, ornithine decarboxylase, tryptophan deaminase, and urease activity, positive for arginine dihydrolase and gelatinase activity, negative for citrate utilization, oxidase and catalase negative, produces indole and acetoin, does not produce hydrogen sulfide, oxidizes glucose but does not oxidize mannitol, inositol sorbitol, rhamnose, sucrose, melibiose, amygdalin, or arabinose. The type strain was isolated from Steinernema akhursti nematodes. These nematodes were isolated from soil samples collected from a grassland in the Cigu village (Deqen, Diqing Tibetan Autonomous Prefecture, Yunnan province, China). The type strain of the species is XENO-10T (= CCM 9322T = CCOS 2071T). Whole-genome sequences of XENO-10T are deposited in the NCBI databank under the accession number JAQRFI01 and 16S rRNA gene sequences under the accession number OQ439939. The genome assembled contains 4,667,994 bp, a G+C content of 43.14%, and 4534 proteins.
References
Kaya HK, Gaugler R (1993) Entomopathogenic nematodes. Annu Rev Entomol 38:181–206. https://doi.org/10.1146/annurev.en.38.010193.001145
Caldas C, Cherqui A, Pereira A et al (2002) Purification and characterization of an extracellular protease from Xenorhabdus nematophila involved in insect immunosuppression. Appl Environ Microbiol 68:1297–1304. https://doi.org/10.1128/AEM.68.3.1297-1304.2002
Cimen H, Touray M, Gulsen SH et al (2022) Natural products from Photorhabdus and Xenorhabdus: mechanisms and impacts. Appl Microbiol Biotechnol 106:4387–4399. https://doi.org/10.1007/s00253-022-12023-9
Thomas GM, Poinar GO (1979) Xenorhabdus gen. nov., a genus of entomopathogenic, nematophilic bacteria of the family Enterobacteriaceae. Int J Syst Bacteriol 29:352–360. https://doi.org/10.1099/00207713-29-4-352
Akhurst RJ, Mourant RG, Baud L et al (1996) Phenotypic and DNA relatedness between nematode symbionts and clinical strains of the genus Photorhabdus (Enterobacteriaceae). Int J Syst Bacteriol 46:1034–1041. https://doi.org/10.1099/00207713-46-4-1034
Boemare NE, Akhurst RJ, Mourant RG (1993) DNA relatedness between Xenorhabdus spp. (Enterobacteriaceae), symbiotic bacteria of entomopathogenic nematodes, and a proposal to transfer Xenorhabdus luminescens to a new genus, Photorhabdus gen. nov. Int J Syst Bacteriol 43:249–255. https://doi.org/10.1099/00207713-43-2-249
Grimont PAD, Steigerwalt AG, Boemare N et al (1984) Deoxyribonucleic acid relatedness and phenotypic study of the genus Xenorhabdus. Int J Syst Evol Microbiol 34:378–388. https://doi.org/10.1099/00207713-34-4-378
Farmer JJ, Jorgensen JH, Grimont PA et al (1989) Xenorhabdus luminescens (DNA hybridization group 5) from human clinical specimens. J Clin Microbiol 27:1594–1600. https://doi.org/10.1128/jcm.27.7.1594-1600.1989
Suzuki T, Yamanaka S, Nishimura Y (1990) Chemotaxonomic study of Xenorhabdus species-cellular fatty acids, ubiquinone and DNA-DNA hybridization. J Gen Appl Microbiol 36:393–401. https://doi.org/10.2323/jgam.36.393
Nishimura Y, Hagiwara A, Suzuki T et al (1994) Xenorhabdus japonicus sp. nov. associated with the nematode Steinernema kushidai. World J Microbiol Biotechnol 10:207–210. https://doi.org/10.1007/BF00360889
Akhurst RJ, Boemare NE (1988) A numerical taxonomic study of the genus Xenorhabdus (Enterobacteriaceae) and proposed elevation of the subspecies of X. nematophilus to species. Microbiology 134:1835–1845. https://doi.org/10.1099/00221287-132-7-1917
Rainey FA, Ehlers R-U, Stackebrandt E (1995) Inability of the polyphasic approach to systematics to determine the relatedness of the genera Xenorhabdus and Photorhabdus. Int J Syst Evol Microbiol 45:379–381. https://doi.org/10.1099/00207713-45-2-379
Lengyel K, Lang E, Fodor A et al (2005) Description of four novel species of Xenorhabdus, family Enterobacteriaceae: Xenorhabdus budapestensis sp. nov., Xenorhabdus ehlersii sp. nov., Xenorhabdus innexi sp. nov., and Xenorhabdus szentirmaii sp. nov. Syst Appl Microbiol 28:115–122. https://doi.org/10.1016/j.syapm.2004.10.004
Tailliez P, Pages S, Ginibre N et al (2006) New insight into diversity in the genus Xenorhabdus, including the description of ten novel species. Int J Syst Evol Microbiol 56:2805–2818. https://doi.org/10.1099/ijs.0.64287-0
Somvanshi VS, Lang E, Ganguly S et al (2006) A novel species of Xenorhabdus, family Enterobacteriaceae: Xenorhabdus indica sp. nov., symbiotically associated with entomopathogenic nematode Steinernema thermophilum Ganguly and Singh, 2000. Syst Appl Microbiol 29:519–525. https://doi.org/10.1016/j.syapm.2006.01.004
Tailliez P, Laroui C, Ginibre N et al (2010) Phylogeny of Photorhabdus and Xenorhabdus based on universally conserved protein-coding sequences and implications for the taxonomy of these two genera. Proposal of new taxa: X. vietnamensis sp. nov., P. luminescens subsp. caribbeanensis subsp. nov., P. luminescens subsp. hainanensis subsp. nov., P. temperata subsp. khanii subsp. nov., P. temperata subsp. tasmaniensis subsp. nov., and the reclassification of P. luminescens subsp. thracensis as P. temperata subsp. thracensis comb. nov. Int J Syst Evol Microbiol 60:1921–1937. https://doi.org/10.1099/ijs.0.014308-0
Tailliez P, Pages S, Edgington S et al (2012) Description of Xenorhabdus magdalenensis sp. nov., the symbiotic bacterium associated with Steinernema australe. Int J Syst Evol Microbiol 62:1761–1765. https://doi.org/10.1099/ijs.0.034322-0
Kuwata R, Qiu L, Wang W et al (2013) Xenorhabdus ishibashii sp. nov., isolated from the entomopathogenic nematode Steinernema aciari. Int J Syst Evol Microbiol 63:1690–1695. https://doi.org/10.1099/ijs.0.041145-0
Ferreira T, van Reenen CA, Endo A et al (2013) Description of Xenorhabdus khoisanae sp. nov., the symbiont of the entomopathogenic nematode Steinernema khoisanae. Int J Syst Evol Microbiol 63:3220–3224. https://doi.org/10.1099/ijs.0.049049-0
Kämpfer P, Tobias NJ, Ke LP et al (2017) Xenorhabdus thuongxuanensis sp. nov. and Xenorhabdus eapokensis sp. nov., isolated from Steinernema species. Int J Syst Evol Microbiol 67:1107–1114. https://doi.org/10.1099/ijsem.0.001770
Castaneda-Alvarez C, Prodan S, Zamorano A et al (2021) Xenorhabdus lircayensis sp. Nov., the symbiotic bacterium associated with the entomopathogenic nematode Steinernema unicornum. Int J Syst Evol Microbiol 71:5151. https://doi.org/10.1099/ijsem.0.005151
Machado RAR, Bhat AH, Fallet P et al (2023) Xenorhabdus bovienii subsp. africana subsp. Nov., isolated from Steinernema africanum entomopathogenic nematodes. Int J Syst Evol Microbiol 73:5795. https://doi.org/10.1099/ijsem.0.005795
Akhurst RJ, Boemare NE (1986) A non-luminescent strain of Xenorhabdus luminescens (Enterobacteriaceae). Microbiology 132:1917–1922
Akhurst RJ (1983) Taxonomic study of Xenorhabdus, a genus of bacteria symbiotically associated with insect pathogenic nematodes. Int J Syst Evol Microbiol 33:38–45. https://doi.org/10.1099/00207713-33-1-38
Poinar GO, Thomas GM (1965) A new bacterium, Achromobacter nematophilus sp. nov. (Achromobacteriaceae: Eubacteriales) associated with a nematode. Int J Syst Evol Microbiol 15:249–252. https://doi.org/10.1099/00207713-15-4-249
Parte AC, Carbasse JS, Meier-Kolthoff JP et al (2020) List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int J Syst Evol Microbiol 70:5607. https://doi.org/10.1099/ijsem.0.004332
Bhat A, Machado R, Abolafia J et al. Multigene sequence based and phenotypic characterization reveals the occurrence of a novel entomopathogenic nematode species, Steinernema anantnagense n. sp. Aashaq Hussain Bhat1,2*, Ricardo A. R. Machado2, Joaquín Abolafia3, Vladimír Půža4, Alba Nazaret Ruiz-Cuenca3, Fuad Multigene sequence based and phenotypic characterization reveals the occurrence of a novel entomopathogenic nematode species, Steinernema anantnagense n. sp. J Nematol
Yoshida M (2004) Steinernema litorale n. sp. (Rhabditida: Steinernematidae), a new entomopathogenic nematode from Japan. Nematology 6:819–838. https://doi.org/10.1163/1568541044038650
Qiu L, Hu X, Zhou Y et al (2005) Steinernema akhursti sp. n. (Nematoda: Steinernematidae) from Yunnan, China. J Invertebr Pathol 90:151–160. https://doi.org/10.1016/j.jip.2005.09.004
Fukruksa C, Yimthin T, Suwannaroj M et al (2017) Isolation and identification of Xenorhabdus and Photorhabdus bacteria associated with entomopathogenic nematodes and their larvicidal activity against Aedes aegypti. Parasit Vectors 10:1–10. https://doi.org/10.1186/s13071-017-2383-2
Marchesi JR, Sato T, Weightman AJ et al (1998) Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl Environ Microbiol 64:795–799. https://doi.org/10.1128/AEM.64.2.795-799.1998
Hill V, Kuhnert P, Erb M et al (2020) Identification of Photorhabdus symbionts by MALDI-TOF MS. Microbiology. https://doi.org/10.1099/mic.0.000905
Lane D (1991) 16S/23S rRNA sequencing, 1st edn. Wiley, New York
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In: Nucleic Acids Symp, pp 95–98
Seemann T (2013) Barrnap 0.7: rapid ribosomal RNA prediction. https://github.com/tseemann/barrnap
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174. https://doi.org/10.1007/BF02101694
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, Oxford
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Chevenet F, Brun C, Bañuls A-L et al (2006) TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinform 7:439. https://doi.org/10.1186/1471-2105-7-439
Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. https://doi.org/10.1093/nar/gkw290
Machado RAR, Wüthrich D, Kuhnert P et al (2018) Whole-genome-based revisit of Photorhabdus phylogeny: proposal for the elevation of most Photorhabdus subspecies to the species level and description of one novel species Photorhabdus bodei sp. nov., and one novel subspecies Photorhabdus laumondii subsp. clarkei subsp. nov. Int J Syst Evol Microbiol 68:2664–2681. https://doi.org/10.1099/ijsem.0.002820
Machado RAR, Bhat AH, Abolafia J et al (2021) Multi-locus phylogenetic analyses uncover species boundaries and reveal the occurrence of two new entomopathogenic nematode species, Heterorhabditis ruandica n. sp. and Heterorhabditis zacatecana n. sp. J Nematol 53:1–42. https://doi.org/10.21307/jofnem-2021-089
Petit RA, Read TD (2020) Bactopia: a flexible pipeline for complete analysis of bacterial genomes. Msystems 5:e00190-20. https://doi.org/10.1128/mSystems.00190-20
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bankevich A, Nurk S, Antipov D et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Walker BJ, Abeel T, Shea T et al (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. https://doi.org/10.1371/journal.pone.0112963
Parks DH, Imelfort M, Skennerton CT et al (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. https://doi.org/10.1101/gr.186072.114
Hyatt D, Chen G-L, LoCascio PF et al (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform 11:1–11. https://doi.org/10.1186/1471-2105-11-119
Steinegger M, Söding J (2017) MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 35:1026–1028. https://doi.org/10.1038/nbt.3988
van Dongen SM (2000) Graph clustering by flow simulation [Internet]. Ph. D Thesis, University of Utrecht, Utrecht University Repository
van Dongen S, Abreu-Goodger C (2012) Using MCL to extract clusters from networks. In: Bacterial molecular networks. Springer, New York, pp 281–295
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. https://doi.org/10.1093/bioinformatics/btl446
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57:758–771. https://doi.org/10.1080/10635150802429642
Meier-Kolthoff JP, Auch AF, Klenk H-P et al (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. https://doi.org/10.1186/1471-2105-14-60
Meier-Kolthoff JP, Hahnke RL, Petersen J et al (2014) Complete genome sequence of DSM 30083(T), the type strain (U5/41(T)) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genomic Sci 9:2. https://doi.org/10.1186/1944-3277-9-2
Auch AF, von Jan M, Klenk H-P et al (2010) Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci 2:117–134. https://doi.org/10.4056/sigs.531120
Auch AF, Klenk H-P, Göker M (2010) Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci 2:142–148. https://doi.org/10.4056/sigs.541628
Chen C-Y, Clark CG, Langner S et al (2020) Detection of antimicrobial resistance using proteomics and the comprehensive antibiotic resistance database: a case study. Proteomics Clin Appl 14:e1800182. https://doi.org/10.1002/prca.201800182
Alcock BP, Raphenya AR, Lau TTY et al (2020) CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res 48:D517–D525. https://doi.org/10.1093/nar/gkz935
Guitor AK, Raphenya AR, Klunk J et al (2019) Capturing the resistome: a targeted capture method to reveal antibiotic resistance determinants in metagenomes. Antimicrob Agents Chemother 64:e01324-19. https://doi.org/10.1128/AAC.01324-19
Tsang K, Speicher D, McArthur A (2019) Pathogen taxonomy updates at the comprehensive antibiotic resistance database: implications for molecular epidemiology. https://doi.org/10.20944/preprints201907.0222.v1
McArthur AG, Waglechner N, Nizam F et al (2013) The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57:3348–3357. https://doi.org/10.1128/AAC.00419-13
Jia B, Raphenya AR, Alcock B et al (2017) CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 45:D566–D573. https://doi.org/10.1093/nar/gkw1004
Blin K, Shaw S, Kloosterman AM et al (2021) antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res 49:W29–W35. https://doi.org/10.1093/nar/gkab335
Blin K, Shaw S, Steinke K et al (2019) antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res 47:W81–W87. https://doi.org/10.1093/nar/gkz310
Machado R, Bhat A, Abolafia J et al. (2022) Steinernema africanum n. sp. (Rhabditida, Steinernematidae), a new entomopathogenic nematode species isolated in the Republic of Rwanda. Int J Nematol
Vrain TC, Wakarchuk Da, Levesque AC et al (1992) Intraspecific rDNA restriction fragment length polymorphism in the Xiphinema americanum group. Fundam Appl Nematol 15:563–573
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Tamura K, Stecher G, Peterson D et al (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. https://doi.org/10.1093/molbev/mst197
Wayne LG, Brenner DJ, Colwell RR et al (1987) Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int J Syst Bacteriol 37:463–464. https://doi.org/10.1099/00207713-37-4-463
Lee M-M, Stock SP (2010) A multilocus approach to assessing co-evolutionary relationships between Steinernema spp. (Nematoda: Steinernematidae) and their bacterial symbionts Xenorhabdus spp. (γ-Proteobacteria: Enterobacteriaceae). Syst Parasitol 77:1–12. https://doi.org/10.1007/s11230-010-9256-9
Murfin KE, Lee M-M, Klassen JL et al (2015) Xenorhabdus bovienii strain diversity impacts coevolution and symbiotic maintenance with Steinernema spp. nematode hosts. MBio 6:e00076-e115. https://doi.org/10.1128/mBio.00076-15
Bhat AH, Chaubey AK, Půža V (2019) The first report of Xenorhabdus indica from Steinernema pakistanense: co-phylogenetic study suggests co-speciation between X. indica and its steinernematid nematodes. J Helminthol 93:81–90. https://doi.org/10.1017/S0022149X17001171
Chun J, Oren A, Ventosa A et al (2018) Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol 68:461–466. https://doi.org/10.1099/ijsem.0.002516
Acknowledgements
We thank the Swiss National Science Foundation and the Institute of Biology of the University of Neuchâtel (Switzerland) for their support. We thank Celine Terrettaz (Institute of Biology, University of Neuchâtel) for her technical support related to light microscopy.
Funding
Open access funding provided by University of Neuchâtel. The work of RARM and AHB is supported by the Swiss National Science Foundation (Grant 186094 to RARM). The work of AHB was supported by a Postdoctoral Swiss Government Excellence Scholarship (Grant Nr. 2021.0463 to AHB).
Author information
Authors and Affiliations
Contributions
RARM contributed to conceptualization, data curation, funding acquisition, investigation, project administration, supervision, validation, and writing – original draft; RARM and AHB contributed to formal analysis; RARM, AHB, CC-A, THA, VP, SP, and JA contributed to investigation; RARM and AHB contributed to methodology; RARM, THA, VP, and SP contributed to resources; RARM and JA contributed to visualization; RARM, AHB, CC-A, VP, and JA contributed to writing – review & editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest and that no humans or animals were used for experimental purposes.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Machado, R.A.R., Bhat, A.H., Castaneda-Alvarez, C. et al. Xenorhabdus aichiensis sp. nov., Xenorhabdus anantnagensis sp. nov., and Xenorhabdus yunnanensis sp. nov., Isolated from Steinernema Entomopathogenic Nematodes. Curr Microbiol 80, 300 (2023). https://doi.org/10.1007/s00284-023-03373-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-023-03373-2