
Abstract
Since the first report in 2009, at least ten additional viruses have been identified and assigned to the proposed virus family Alternaviridae. Here we report two new mycoviruses tentatively assigned to this family, both identified as members of the fungal family Nectriaceae, which were isolated from surface-disinfected apple roots (Malus x domestica, Borkh.) affected by apple replant disease (ARD). ARD is a highly complex, worldwide-occurring disease resulting from plant reactions to a disturbed (micro)-biome and leads to high economic losses every year. The first alternavirus characterized in this study was identified in a Dactylonectria torresensis isolate. The virus was tentatively named dactylonectria torresensis alternavirus 1 (DtAV1) as the first member of the proposed new species Alternavirus dactylonectriae. The second virus was identified in an isolate of Ilyonectria robusta and was tentatively named ilyonectria robusta alternavirus 1 (IrAV1) as the first member of the proposed new species Alternavirus ilyonectriae. Full genomic sequences of the viruses were determined and are presented. Further, we found hints for putative components of a methyl transferase machinery using in silico approaches. This putative protein domain is encoded by segment 2. However, this result only establishes the basis for subsequent studies in which the function must be confirmed experimentally in vitro. Thus, this is the first study where a function is predicted to all three genomic segments within the group of the alternaviruses. These findings provide further insights into the virome of ARD-associated fungi and are therefore another brick in the wall of understanding the complexity of the disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mycoviruses are the subject of research in the virology field since their first description in 1962 (Hollings 1962). In recent years, especially since HTS techniques have become affordable for many research institutions worldwide, an increasing number of new viruses are continuously being assigned to the individual mycoviral taxa. Meanwhile, viral infections have been detected in all major fungal taxa (Ghabrial et al. 2015; Pearson et al. 2009).
Most mycoviruses possess a dsRNA or + ssRNA genome. Just a few -ssRNA or DNA viruses are known to infect fungi (Khalifa and MacDiarmid 2021; Kotta-Loizou 2021; Yu et al. 2010). For the vast majority of mycoviruses, no direct impact on their host could be shown so far. However, when observing viral infections of (phyto)-pathogenic fungi, some viruses were found to have either a hypo- or hypervirulent impact on their host (Kotta-Loizou 2021; Deng et al. 2003; Nuss 2005; Olivé and Campo 2021; Özkan and Coutts 2015; Polashock and Hillman 1994).
Here, we report the identification of two new mycoviruses, assigned to the proposed family Alternaviridae. Both were identified in dsRNA extracts from fungi assigned to the family Nectriaceae. The first virus belongs to the virome of a Dactylonectria torresensis isolate and was tentatively named dactylonectria torresensis alternavirus 1 (DtAV1), as a member of the proposed new virus species Alternavirus dactylonectriae. Secondly, we report a virus identified in an Ilyonectria robusta isolate, which was tentatively named ilyonectria robusta alternavirus 1 (IrAV1) as a member of the proposed new species Alternavirus ilyonectriae.
Up to now, eleven viruses have been assigned to the proposed family Alternaviridae. The eponymous alternaria alternata virus 1 (AaV-1) was identified in 2009 and the previously found aspergillus mycovirus 341 (AMV) was assigned to the family of which nine more viruses were discovered all around the world in the following years (Aoki et al. 2009; Gilbert et al. 2019; Hammond et al. 2008; He et al. 2018; Kozlakidis et al. 2013; Osaki et al. 2016; Wen et al. 2021; Zhang et al. 2019, 2022; Lutz et al. 2022). One of the most prominent features of this family is the ADD sequence triplet (motif VI) in the RdRp amino acid (aa) sequence. This motif is highly conserved among all RNA viruses and usually consists of a GDD (or less frequent SDD) triplet (Koonin 1991; Kamer and Argos 1984).
Both viruses characterized in this work were identified in members of the fungal family Nectriaceae. The fungi were in turn isolated and identified as part of the endophytic microbiome of apple plants (Malus x domestica, Borkh.) affected by apple replant disease (ARD). ARD is a worldwide problem, occurring when apple plants are replanted repeatedly in the same site and are thought to be caused by plant reactions due to a disturbed (micro)-biome (Winkelmann et al. 2019). The disease can lead to losses of up to 50% of the profitability of apple orchards by reduction of yield and a delay of the plants in bearing fruits (Schoor et al. 2009). Up to now, the disease etiology is still unclear. However, fungi of the family Nectriaceae were found repetitively at high abundance in suffering apple plants and seem to influence the disease expression (Manici et al. 2018; Popp et al. 2019).
Since there is still no effective and sustainable control strategy against ARD, all involved biotic factors like nematodes, bacteria, oomycetes, and fungi have to be investigated in detail. As part of the virome of associated endophytic fungi, the mycoviruses described in this work may play a role in the occurrence of the disease. This study is thus the basis for further analyses to evaluate the targeted deployment of mycoviruses in control strategies to counteract the ARD.
Materials and methods
Fungal material
Both fungal isolates investigated in this study were obtained among others in a central experiment in 2017 from apple roots, grown in ARD soil (Mahnkopp et al. 2018). The D. torresensis isolate O6-1-A and the I. robusta isolate O16-2-D were isolated from plants grown in soil from the experimental sites Ellerhoop, (53.714361, 9.770139; Schleswig–Holstein, Germany) and Heidgraben (53.699199, 9.683171; Schleswig–Holstein, Germany). Plant roots were surface disinfected and subsequently plated on water agar with rifampicin (25 µg mL−1), penicillin (50 µg mL−1), and pimaricin (25 µg mL−1). Outgrown fungi were picked and cultured on malt extract agar (MEA, 2%).
Fungi are usually identified by ITS-PCR and BLAST. Since this method is insufficient for the accurate identification of members of the Nectriaceae family, multi-locus analyses were performed (Lawrence et al. 2019; Cabral et al. 2012). O6-1-A was identified as D. torresensis by amplification of the translation elongation factor 1-α (TEF) and histone 3 (HIS) genes. TEF was amplified with the primer pair CylEF-1 (Groenewald, J. Z., unpublished) and CylEF-R2 (Crous et al. 2004). For the identification by HIS, the primers CYLH3F and CYLH3R were used (Crous et al. 2004). O16-2-D was identified as I. robusta by amplification of three loci. In addition to TEF and HIS, the β-tubulin gene (TUB) was analyzed with the primers T1 and Bt2b (O'Donnell and Cigelnik 1997; Glass and Donaldson 1995). All primers used for fungal identification are summed up in Table 1. PCR-fragments were purified, sent to Microsynth SeqLab (Goettingen, Germany) for Sanger sequencing, and the sequences were analyzed by NCBI BLASTn, subsequently (Popp et al. 2019; Sanger et al. 1977).
Nucleic acid extraction
To obtain enough material for nucleic acid extraction, fungi were grown in 2% malt extract broth for two weeks before the mycelium was filtered and ground in liquid nitrogen. DsRNA was extracted from 20 g ground mycelium with a modified protocol, based on the method published by Morris and Dodds in 1979 as described in Lesker et al. 2013 except for the use of a different cellulose (Merck; Darmstadt, Germany; product nr. 22,184) (Morris and Dodds 1979; Lesker et al. 2013). 20 U RNase T1 and 40 U DNase I (both: Roche; Basel, Switzerland) were used to digest 20 mL eluate at 37 °C for 30 min each. The dsRNA extracts were purified and concentrated in a 3 mL storage volume. 500 µL eluate were precipitated with ethanol and solved in 20 µL water for subsequent analyses. The purified dsRNA extracts were used for the identification of viral segments (S1–S3) by agarose gel electrophoresis and subsequent Illumina sequencing at the Leibniz Institute DSMZ (Brunswick, Germany).
RT-PCRs for virus detection, end determination by poly (A) tail hybridization, and 5′-RACEs (rapid amplification of cDNA ends) were performed with whole nucleic acid extractions. Those extractions were done with the same ground fungal material as for dsRNA extraction. The methodology for the whole nucleic acid extractions and purification was obtained from a simpler protocol as described in (Menzel et al. 2002).
High-throughput sequencing and bioinformatics analysis
The isolated dsRNA was used as input for cDNA synthesis with random octamer primers and the Maxima H Minus Reverse Transcriptase (Thermo Fisher Scientific, Waltham, USA), followed by second-strand synthesis and library preparation according to the Nextera XT DNA Library Preparation Kit (Illumina; San Diego, USA). Sequencing of the libraries was performed at the Leibniz-Institute DSMZ on a MiSeq instrument as paired-end reads (2 × 301 bp). Raw reads were trimmed and de novo assembled with the Geneious Prime® software (Biomatters, Auckland, New Zealand) using an in-house established workflow, and putative virus contigs were identified by BLASTn/BLASTp alignment against a custom database of NCBI nuclear-core reference sequences. The contigs were subsequently ordered and trimmed according to reference sequences. The resulting sequence information was used to design primers for validation and determination of full-length genome sequences by RT-PCR and RACE reactions.
RT-PCR and RACE
To detect the viral segments in fungal subcultures, to confirm the HTS results, and assemble full-length genomes, RT-PCRs with specific primers were developed for each virus. A cDNA synthesis mix was set up with 3 µL whole nucleic acid extract, 1 µL antisense identification primer for the respective viral segment (primer no.: 8, 10, 12, 23, 25, 27, Table 2), 0.5 µL dNTPs (10 mM each; Carl Roth; Karlsruhe, Germany), 2 µL RT buffer, and 20 U RevertAid reverse transcriptase (both: Thermo Fisher Scientific, Waltham, USA) in a total volume of 10 µL. cDNA synthesis was performed at 42 °C for 1 h. Subsequent PCR set-up with 5 µL Phusion Flash High-Fidelity PCR Master Mix (Thermo Fisher Scientific, Waltham, USA), 0.5 µL of each forward and reverse identification primer for the respective viral segment (primer no.: 7–12, 22–27; Table 2) and 1 µL cDNA from the previous step, in a final volume of 10 µL. The PCR program consisted of a denaturation step of 15 s at 98 °C, followed by 34 cycles with a 15 s denaturation at 98 °C, 5 s primer hybridization at the respective annealing temperature (TA), and 20 s elongation at 72 °C. A final elongation was performed at 72 °C for 5 min.
The 5′ ends of the six viral segments were determined with a modified RACE protocol, based on the method described by Frohman et al. (Frohman et al. 1988). The cDNA used for the RACE was synthesized as described for the RT-PCRs apart from using different primers (primer no.: 13–15; 28–30; Table 2). After purifying with SureClean Plus (Bioline; London, UK), 3 µL cDNA was tailed in two reactions with 1 µL of either dGTP or dCTP (100 mM; Thermo Fisher Scientific; Waltham, USA) in a mix with 1 µL terminal deoxynucleotidyl transferase (TdT; 20 U mL−1), 4 µL 5 × TdT Reaction Buffer (both: Thermo Fisher Scientific; Waltham, USA) and 11 µL water at 37 °C for 30 min, followed by an inactivation step for 10 min at 70 °C. The subsequent PCR setup was identical to the RT-PCR described above, aside from using the respective nested primers (primer no.: 16–18; 31–33; Table 2) and the corresponding poly (G) or poly (C) primer (primer no.: 37, 38; Table 2).
Determination of 3′-ends was done by detecting the poly (A) tail that all segments have in common. A detectable linker was hybridized to each poly (A) tail by cDNA synthesis with the RACE-BOE1 primer (primer no. 39; Table 2) in a mixture as described above. The following PCR was set up with the corresponding RACE-BOE2 primer (no. 40; Table 2) and the specific 3′ primer (no.: 19–21; 34–36; Table 2) in a mixture as described for the detection PCR.
Alignments of UTRs and amino acid motifs
5′ ends of the three segments of both DtAV1 and IrAV1 were compared to identify conserved regions. Alignments were performed in MEGA X using the MUSCLE algorithm (Edgar 2004; Kumar et al. 2018). The GeneDoc software (National Resource for Biomedical Supercomputing, Pittsburgh, USA) was used to visualize the RNA alignments. To calculate aa alignments and identify conserved domains within the RdRps and methyltransferases (MTase), the Clustal Omega algorithm was used (Sievers et al. 2011). Conservation indices were calculated and annotated according to the Gonnet PAM250 matrix (Gonnet et al. 1992).
Phylogenetic analyses
To perform comparative phylogenetic analyses, all available RdRp and CP amino acid sequences of members assigned to the proposed family Alternaviridae were compiled. Since the CP was not yet annotated for most of the viruses within this group, comparative BLASTp searches were performed and the respective viral segments were assigned to their function as major CP according to the protein determination of (Wu et al. 2021). Penicillium chrysogenum virus (PcV) was chosen as an outgroup for the phylogenetic trees since it has been shown that chrysoviruses are closely related to alternaviruses (Aoki et al. 2009; Castón et al. 2003). The set of RdRp sequences was composed of 14 different proteins. The set of CP sequences comprised 13 sequences since segment 1 (S1) is the only available sequence of AMV. Respective sequence sets were aligned using the MUSCLE algorithm with default settings (gap opening: −2.9, gap extension: 0) in MEGA X (Edgar 2004; Kumar et al. 2018). Best-performing substitution models were calculated for both alignments. For the RdRp-based tree, the Le_Gascuel_2008 was determined as the best model with gamma-distributed rates and empirical base frequencies (LG + G + F) (Le and Gascuel 2008). The calculations for the CP-tree are based on the empirical model by Whelan and Goldman with gamma-distributed rates and base frequencies (WAG + G + F) (Whelan and Goldman 2001). Finally, Maximum Likelihood trees were calculated using the bootstrap method with 1000 replicates and all sites from the data subsets. Moreover, pairwise sequence identity matrices we calculated comparing the RdRP and CP sequences of all available alternaviruses using the EMBOSS/Needle tool with default settings (Madeira et al. 2019).
Protein folding modeling and function prediction
To get hints on the function of the protein encoded by dsRNA2, the protein folding was predicted by using the ColabFold code, which is available for free as a GoogleColab worksheet (Mirdita et al. 2022). With this worksheet, protein structures are predicted by using AlphaFold2 and the Alphafold2-multimer (Jumper et al. 2021; Evans et al. 2021). Hints on protein functions were subsequently calculated by using the structure-based function tool DeepFRI (Gligorijević et al. 2021). After the identification of high-scoring functional domains, respective regions of the amino acid sequence were analyzed using the homology modeling service of the SWISS-MODEL server to confirm the domains by comparison with PDB-deposited templates and specify their putative function (Waterhouse et al. 2018).
Results and discussion
Discovery of two new alternaviruses
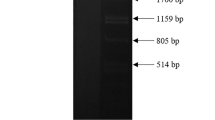
Gel electrophoresis of the dsRNA extracts of Dactylonectria torresensis O6-1-A and Ilyonectria robusta O16-2-D revealed four and two fragments, respectively. For D. torresensis, the sizes of the fragments were estimated to be 3600 bp, 2700 bp, 2500 bp, and 1300 bp, and for I. robusta about 3600 bp and 2500 bp. Both dsRNA extractions are shown in Fig. 1b, together with two additional D. torresensis isolates (22-1-B-D, 20-1-B). Both of these isolates (22–1–B–D and 20–1–B) were not part of this study and are therefore negligible.

Agarose gel electrophoreses: a Detection RT-PCR of DtAV1 (left) and IrAV1 (right). Marker (M): PstI digested Lambda-Phage DNA, Polymerase: PhusionFlash (ThermoScientific™), Primers according to Table 2: DtAV1_RNA1: 7 + 8; DtAV1_RNA2: 9 + 10; DtAV1_RNA3: 11 + 12; IrAV1_RNA1: 22 + 23; IrAV1_RNA2: 24 + 25; IrAV1_RNA3: 26 + 27; Primer concentration: 10 µM. b dsRNA-extractions of 22–1–B–D, 20–1–B (D. torresensis isolates irrelevant to this study), O16–2–D (I. robusta) and O6-1-A (D. torresensis), Marker (M): PstI digested Lambda-Phage DNA
Illumina sequencing of the libraries generated 408.650 and 473.416 reads from samples DtO6-1-A and IrO16-2-D, respectively, and the bioinformatic analyses revealed a number of contigs which were assigned to putative alternaviruses (Online Resource Table ESM1). To validate the results, all of the sequences were subsequently confirmed by RT-PCRs in the respective hosts, and 5′- and 3′-ends were determined by RACE to assemble the full-length genomic sequences.
The three virus genome segments assembled from D. torresensis have a length of 3578 bp, 2668 bp, and 2467 bp, which correlated to the size of the three larger fragments visible on the gel (Fig. 1b). These segments were assigned to a new member of the proposed family Alternaviridae, tentatively named dactylonectria torresensis alternavirus 1 (DtAV1) being the first member of the proposed new species Alternavirus dactylonectriae. A fourth fragment which was visualized by electrophoresis and putatively assigned to a 4th RNA of DtAV1 was not identified in the bioinformatics analysis by homology search against the segment 4 (S4) of known alternaviruses. Further, this putative S4 appeared to be lost by serial subcultures, supporting that it is not strictly necessary for viral replication. Such loss of S4 during serial fungal passage without significant impact on the host morphology and growth was described before for other alternaviruses (Wen et al. 2021). Moreover, many alternaviruses lack an S4 completely (Table 3).
The segments discovered in the dsRNA of I. robusta have a total length of 3529 bp, 2464 bp, and 2469 bp, corresponding to the estimated size of the bands visible in the agarose gel (Fig. 1b). These segments were assigned to a putative new member of the proposed family Alternaviridae, which was tentatively named ilyonectria robusta alternavirus 1 (IrAV1), as the first member of the proposed new species Alternavirus ilyonectriae.
RT-PCR assays were developed to amplify the genomic fragments of the two new alternaviruses, with amplicons of expected fragment sizes of 846 bp, 548 bp, and 561 bp for DtAV1 and 943 bp, 622 bp, and 625 bp for IrAV1 (Fig. 1a). Sequencing of all amplicons confirmed the presence of DtAV1 and IrAV1, respectively, underlining the specificity of the primers (data not shown). Viral infection could be verified in the fungal isolates grown in culture over multiple generations.
Genome organization
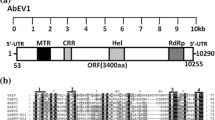
Both viruses DtAV1 and IrAV1 possess a genome composed of three dsRNA segments with a single ORF each and poly (A) tails at the 3′ end. DtAV1 has a total genome size of 8713 bp with a 3578 bp segment 1 (S1) encoding a 126.6 kDa RdRp on a 3375 bp open reading frame (ORF), flanked by a 62 bp 5′ UTR and a 141 bp 3′ UTR. A conserved RdRp_4 (pfam02123) site was found on aa position 641–754 with a confidence E-value of 0.002 calculated with NCBI-CDD search. Segment 2 (S2) has a length of 2668 bp with a 66 bp-long 5′ UTR and a 334 bp-long 3′ UTR. ORF 2 encodes an 81.7 kDa protein with a so far unknown function. The third fragment has a total length of 2467 bp with a 75 bp and a 178 bp UTR at the 5′- and 3′-end, respectively. The ORF of this segment encodes the 79.7 kDa major coat protein, which was identified, based on the study by (Wu et al. 2021).
IrAV1 has a total genome size of 8462 bp. S1 is 3529 bp long and encodes a 126.3 kDa RdRp on a 3375 bp ORF, flanked by an 80 bp 5′ UTR and a 74 bp 3′ UTR. A conserved RdRp_4 (pfam02123) site was found on aa position 587–753 with an E-value of 0.046. S2 has a total length of 2464 bp with a 77 bp-long 5′ UTR and a 116 bp-long 3′ UTR. The ORF encodes an 83.7 kDa protein with a so far unknown function. The third fragment has a total length of 2469 bp with a 77 bp and a 160 bp UTR at the 5′- and 3′-end, respectively. The ORF of this segment encodes the 81.6 kDa major coat protein. A scaled genome map of both viruses is shown in Fig. 2.

Scaled genome map of the identified viruses dactylonectria torresensis alternavirus 1 (DtAV1, light red) and ilyonectria robusta alternavirus 1 (IrAV1, light green). Double lines:genomic dsRNA. Digits: nucleotide positions of 5′-ends, ORFs and 3′-ends. Boxes: ORFs with the respective encoding protein and protein size in brackets. Grey boxes: Conserved RdRp domains. A(n): Poly(A)-tail. Scale bar: 500 bp. “RdRP” = RNA-dependent RNA-polymerase; “put. MTase” = putative Methyltransferase; “CP” = Coat protein
The analyses of conserved 5′ end lead to diverse results when comparing DtAV1 and IrAV1. For IrAV1, the first 16 nucleotides (5′-GGCUCUCUCUUUAGUU-3′) are conserved in all three fragments. When aligning the 5′ ends of DtAV1 the segments are far more variable with 8 common nucleotides (5′-GCUYUUAM-3′) and an additional G as the first nucleotides of segment 1. The 5′ UTR alignments are shown in Fig. 3.

Alignments of 5 ‘ UTRs of a Dactylonectria torresensis alternavirus 1 (DtAV1) and b Ilyonectria robusta alternavirus 1 (IrAV1). Consensus sequences are shown above the alignment. Digits: Position of last shown nucleotide. Conservation indices: Black = 100%; grey ≥ 66.67%. Displayed with the GeneDoc software
Since the 5′ RACE of DtAV1 provided clear results, the 5′ terminal nucleotides of the other alternaviruses were aligned to compare the results. These alignments ranged from high conservation (e.g., CcAV1, FoAV1, IrAV1) to high diversity (e.g., FpAV1, SlV, AheAV1). Therefore, we conclude that conserved 5′ ends are no common feature of alternaviruses. Nevertheless, the conserved RdRp motifs I–VIII were identified by alignments according to Gilbert et al. 2019 for all available alternaviruses and the results are summarized in Fig. 4 (Gilbert et al. 2019).

Alignment of conserved RdRp motifs I–VIII of available alternaviruses according to Gilbert et al. 2019, calculated with Clustal Omega (Gilbert et al. 2019; Sievers et al. 2011). Digits in brackets: Unlisted amino acids. Indices: Asterisk (black): Full conservation; colon (dark grey): Conservation of aa with strongly similar properties (> 0.5 [Gonnet PAM250]); period (light grey): Conservation of aa with weakly similar properties (> 0, = < 0.5 [Gonnet PAM250]) (Gonnet et al. 1992)
The crucial metal ion binding ADD triplet in motif VI is rather unusual, as most of the RNA viruses share a GDD or SDD at this position (Venkataraman et al. 2018). DtAV1 and IrAV1 both match the conserved areas with the other alternaviruses in the alignment.
Determination of putative protein functions
After the identification of the major coat protein on segment 3 (S3), the functions of two of the three segments are decrypted for DtAV1 and IrAV1 (Wu et al. 2021). With this information and BLASTp alignments, the CP was identified for all available alternaviruses on S3 regardless of whether the respective virus is tri- or quadripartite. A full overview of the current status of the genome organization of all alternaviruses is shown in Table 3.
To assign a putative function of the protein encoded by S2, further bioinformatic analyses were performed. Since Wu et al. 2021 described a methylated cap at the 5′ ends of the alternaviruses analyzed in that study, it is plausible that other alternaviruses possess capped RNAs as well (Wu et al. 2021). Terminal structures like m7G caps and polyadenylated 3′-ends were reported several times for dsRNA viruses (Aoki et al. 2009; Furuichi and Miura 1975; Wei and Moss 1975). Nevertheless, the simultaneous occurrence of both structures is very rare (Wu et al. 2021). Caps and polyadenylation of mRNAs usually serve to recruit RNA by translation factors and increase translation efficiency. In addition, the terminal structures protect against degradation by exonucleases (Gallie 1991). As for cellular mRNA, this also applies to viruses that take evolutionary advantage of these terminal structures (Wu et al. 2021; Furuichi and Miura 1975; Wei and Moss 1975; Schneider and Mohr 2003). So far, three mechanisms for capping viral RNAs are known. In the first case, mRNA capping is usually dependent on the host RNA polymerase II (Schneider and Mohr 2003). However, this is rather unlikely for the alternaviruses, since they are transcribed by their own RdRp, and RNA pol II is DNA dependent. The second scenario is cap-snatching, where viral RNAs take over the 5' caps of cellular mRNAs, by the activity of viral endonucleases (Wu et al. 2021; Schneider and Mohr 2003; Fujimura and Esteban 2011). As we were not able to identify conserved domains or motifs of known viral endonucleases in the genomes of DtAV1 or IrAV1, neither in the ORF of S2 nor in segments 1 and 3, we conclude that it is improbable that alternaviruses are able to reutilize mRNA caps from host transcripts. This fact suggests that it is likely that alternaviruses would use the third capping option and encode their own capping proteins. In the case of an m7G cap, as shown by Wu et al. 2021, a viral MTase would be required (Wu et al. 2021; Schneider and Mohr 2003).
By evaluating the output of the function predictions for the protein with so far unknown function with ColabFold, followed by DeepFRI of DtAV1 and IrAV1, we received evidence of a possible RNA-binding domain for both viruses (Mirdita et al. 2022; Gligorijević et al. 2021). The highest-scoring molecular functions are heterocyclic compound binding (GO:1901363), organic cyclic compound binding (GO:0097159), nucleotide binding (GO:0000166), and nucleoside phosphate binding (GO:1901265). The template modeling scores (TM-scores), which evaluate how trustworthy the results are, range from 0.79 to 0.89 for IrAV1 and 0.94 for DtAV1. When evaluating the DeepFRI algorithm, Gligorijević et al. found TM-scores > 0.5 to be significant and scores > 0.73 to be very specific (Gligorijević et al. 2021). Since these results are giving reliable hints for a possible MTase activity of a protein encoded by S2, the amino acids sequences were screened for possible MTase domains by searching for templates with the SWISS-MODEL server (Waterhouse et al. 2018). The best matches of IrAV1 S2 were found within aa 233 to aa 286. Analyses of this region revealed a high identity (34.78%) with a heterocyclic toxin methyltransferase (SWISS-MODEL Template Library ID (SMTL ID): 7bgg.1.A). For DtAV1 S2, the best matches were found in aa position 233–285. Searching for templates within the section resulted in the best matches with the same MTase (7bgg.1.A) and an identity of 36.36%. These results suggest that (i) DtAV1 and IrAV1 may have a methyltransferase function in vivo and (ii) components of the necessary machinery are possibly encoded by S2. By considering these results, it was possible to identify a putative MTase domain in the ORF of every available S2 within the group of alternaviruses. To determine conserved motifs of the putative MTase domains, an alignment was computed (Fig. 5). Since the domains of SlV and AaV-1 stand out in comparison to the other viruses, they were considered separately and not included for the determination of conserved regions. The special position of these two viruses can also be observed in the phylogenetic results. However, possible methyltransferase domains were also found for these viruses. These showed the best matches with a cobalt-precorrin-6Y C(15)-methyltransferase (2yxd.1.A).

Alignment of conserved MTase (methyltransferase)-motifs on dsRNA2 of available alternaviruses, calculated with Clustal Omega (Sievers et al. 2011). Digits in brackets: unlisted amino acids. Indices: Asterisk (black): full conservation; colon (dark grey): conservation of aa with strongly similar properties (> 0.5 [Gonnet PAM250]); period (light grey): conservation of aa with weakly similar properties (> 0, = < 0.5 [Gonnet PAM250]).”SlV” and “AaV-1” were not taken into account in the calculation of the indices
According to the sequence of DtAV1, the regions from aa 258 to aa 268 (GDXPG[T/S][L/F][G/A/S]RXL) and aa 275 to aa 282 (V[V/T]GXDP[K/R]N) stand out with high conservation. Performing a blastp search using aa 259 to aa 283 (GDHPGTLARALVSRGIDVVGVDPRN) of IrAV1 S2, containing the conserved sequences indicated above, delivered expected hits for ORF2 of alternaviruses and for more than 40 classI SAM-dependent metyhltransferases of bacteria, further strengthening the hypothesis for a methyltransferase function of the ORF of dsRNA2. However, since all of these results were generated via in silico calculations, it is crucial that future studies confirm the methyltransferase function with in vitro assays before it can be unequivocally assumed that alternaviruses indeed encode such a function on dsRNA2. Other possible methods to confirm the protein function would be docking with ligands or molecular dynamics studies (Kar et al. 2017). Thus, by providing a strong hypothesis, this study provides the basis for targeted protein function analyses in follow-up studies.
Phylogenetics
To confirm DtAV1 and IrAV1 as members of the proposed family Alternaviridae, different phylogenetic analyses were performed in the course of this study. Both the RdRp and CP amino acid sequences of all available alternaviruses were analyzed and maximum likelihood trees, as well as pairwise sequence identity matrices, were calculated.
Within the RdRp-based tree (Fig. 6a), four major clusters are formed. DtAV1 and IrAV1 are clustering together with FpAV1, FgAV1, and FiAV1. All these viruses are originally hosted by Nectriaceae fungi. The second cluster is composed of AheAV1, AMV, AfMV, and CcV1, originating from Aspergillus sp. or Cordyceps sp. Species, respectively. Two smaller groups are built from AaV-1 with SlV and FoAV1 with FsAV1. These results confirm those from other studies and extend the phylogenetic knowledge of the proposed family Alternaviridae (Zhang et al. 2019, 2022).

Phylogenetic trees and pairwise sequence identity matrix of available alternaviruses based on RdRp and CP amino acid sequences. Alignments were performed with the MUSCLE algorithm (Edgar 2004). Trees were constructed with the Maximum likelihood method and 1000 bootstraps in MEGA X (Kumar et al. 2018). All viruses are annotated with GenBank accession number. Colored dots indicate the viruses identified in this study. Scale bars representing substitutions per site. Numbers next to the branches are the percentage of trees, bootstrapped as shown. a: RdRp-based tree; substitution model: LG + G + F. b: CP-based tree; substitution model: WAG + G + F. c: Pairwise identity matrix of RdRp (red) and CP (blue) sequences, calculated with the EMBOSS/Needle tool with preset settings. Values are given in %. The scaling represents the color gradation of values from 0 to 100%
In comparison to that, the CP-based tree is composed of 3 major clusters. As in the RdRp-based tree, DtAV1 and IrAV1 are clustering with FpAV1, FgAV1, and FiAV1. In contrast to the RdRp tree, FoAV1 and FsAV1 are summed up in a bigger cluster together with CcAV1, AheAV1, and AfMV (Fig. 6b). After the calculation of percentage pairwise sequence identities of the amino acid sequences of both the RdRp and CP a comparative double matrix was generated (Fig. 6c), to show that DtAV1 and IrAV1 have the highest sequence identity with FgAV1 in both protein sequences.
The clustering observed in the CP-based phylogenetic tree shows up similarly in the matrices. IrAV1 builds a cluster together with FpAV1, FgAV1, FiAV1, and DtAV1, which is slightly outside the group. FsAV1 and FoAV1 have the highest identity but contrary to the phylogenetic trees, they do not build a distinct group and are rather combined in one cluster with AfMV, AheAV1, and CcAV1. SlV and AaV-1 are building an outstanding group as observed before.
In summary, the RdRp-based tree appears to be sharper resolved, however, the clustering of the CP-based tree and the sequence identity matrix are more similar. Since most dsRNA mycoviruses, such as chrysoviruses and megabirnaviruses are distinguished by the phylogenetic analysis of RdRp and previous studies on alternaviruses have used the same approach, we recommend to continue to follow this criterion in the future (Aoki et al. 2009; Gilbert et al. 2019; Kozlakidis et al. 2013; Osaki et al. 2016; Wen et al. 2021; Zhang et al. 2019, 2022; Kotta-Loizou et al. 2020; Sato et al. 2019).
Conclusions
In this study, two new mycoviruses were discovered and assigned to two new proposed species in the family Alternaviridae. We were further able to show that conserved 5′ ends do not exist for every virus and the RdRp motifs I–VIII are conserved with an active ADD triplet in motif VI for all alternaviruses. Further, as we showed that the ORF of S2 putatively encodes crucial motifs of a methyltransferase, it is possible that alternaviruses employ self-encoded proteins to cap their RNAs at the 5′ ends. Subsequent studies are crucial to confirm this hypothesis by specific work on segment S2 with techniques such as fusion-protein assays, protein–ligand docking, molecular dynamics or protein crystallization and X-ray analyses. The function of S3 could be inferred for all alternaviruses by BLAST alignments and the putative function of all confirmed ORFs were deciphered. After evaluation of the results of the phylogenetic analyses and the characteristics of the described viruses, we strongly support the assignment of the novel viruses to the proposed family Alternaviridae. The goal of this study was to get an insight into the virome of two ARD-associated endophytic fungi and to characterize the identified viruses in detail. To be of use for plant disease management, future studies need to focus on their biological influence by demonstrating whether there is a hyper- or hypovirulent effect of DtAV1 or IrAV1 on their hosts. These results will guide us to understand if the described alternaviruses could serve as possible biocontrol or mitigation agent to help counteract diseases like ARD.
Data availability
Sequences will be available under the following NCBI GenBank accession numbers: Dactylonectria torresensis alternavirus 1 (DtAV1): OM296437–OM296439, Ilyonectria robusta alternavirus 1 (IrAV1): ON721403–ON721405
References
Aoki N, Moriyama H, Kodama M et al (2009) A novel mycovirus associated with four double-stranded RNAs affects host fungal growth in Alternaria alternata. Virus Res 140:179–187. https://doi.org/10.1016/j.virusres.2008.12.003
Cabral A, Rego C, Nascimento T et al (2012) Multi-gene analysis and morphology reveal novel Ilyonectria species associated with black foot disease of grapevines. Fungal Biol 116:62–80. https://doi.org/10.1016/j.funbio.2011.09.010
Castón JR, Ghabrial SA, Jiang D et al (2003) Three-dimensional structure of Penicillium chrysogenum virus: a double-stranded RNA Virus with a Genuine T=1 Capsid. J Mol Biol 331:417–431. https://doi.org/10.1016/S0022-2836(03)00695-8
Crous PW, Groenewald JZ, Risède J-M et al (2004) Calonectria species and their Cylindrocladium anamorphs: species with sphaeropedunculate vesicles. Stud Mycol 50:415–430
Deng F, Xu R, Boland GJ (2003) Hypovirulence-Associated Double-Stranded RNA from Sclerotinia homoeocarpa Is Conspecific with Ophiostoma novo-ulmi Mitovirus 3a-Ld. Phytopathology 93:1407–1414. https://doi.org/10.1094/PHYTO.2003.93.11.1407
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Evans R, O’Neill M, Pritzel A et al. (2021) Protein complex prediction with AlphaFold-Multimer
Frohman MA, Dush MK, Martin GR (1988) Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Nat Acad Sci USA 85:8998–9002. https://doi.org/10.1073/pnas.85.23.8998
Fujimura T, Esteban R (2011) Cap-snatching mechanism in yeast L-A double-stranded RNA virus. Proc Nati Acad Sci 108:17667–17671. https://doi.org/10.1073/pnas.1111900108
Furuichi Y, Miura K (1975) A blocked structure at the 5’ terminus of mRNA from cytoplasmic polyhedrosis virus. Nature 253:374–375. https://doi.org/10.1038/253374a0
Gallie DR (1991) The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev 5:2108–2116. https://doi.org/10.1101/gad.5.11.2108
Ghabrial SA, Castón JR, Jiang D et al (2015) 50-plus years of fungal viruses. Virology 479–480:356–368. https://doi.org/10.1016/j.virol.2015.02.034
Gilbert KB, Holcomb EE, Allscheid RL et al (2019) Hiding in plain sight: New virus genomes discovered via a systematic analysis of fungal public transcriptomes. PLoS ONE 14:e0219207. https://doi.org/10.1371/journal.pone.0219207
Glass NL, Donaldson GC (1995) Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl Environ Microbiol 61:1323–1330. https://doi.org/10.1128/aem.61.4.1323-1330.1995
Gligorijević V, Renfrew PD, Kosciolek T et al (2021) Structure-based protein function prediction using graph convolutional networks. Nat Commun 12:3168. https://doi.org/10.1038/s41467-021-23303-9
Gonnet GH, Cohen MA, Benner SA (1992) Exhaustive matching of the entire protein sequence database. Science 256:1443–1445. https://doi.org/10.1126/science.1604319
Hammond TM, Andrewski MD, Roossinck MJ et al (2008) Aspergillus mycoviruses are targets and suppressors of RNA silencing. Eukaryot Cell 7:350–357. https://doi.org/10.1128/EC.00356-07
He H, Chen X, Li P et al (2018) Complete Genome Sequence of a Fusarium graminearum Double-Stranded RNA Virus in a Newly Proposed Family, Alternaviridae. Genome Announc. https://doi.org/10.1128/genomeA.00064-18
Hollings M (1962) Viruses associated with a die-back disease of cultivated mushroom. Nature 196:962–965. https://doi.org/10.1038/196962a0
Jumper J, Evans R, Pritzel A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. https://doi.org/10.1038/s41586-021-03819-2
Kamer G, Argos P (1984) Primary structural comparison of RNA-dependent polymerases from plant, animal and bacterial viruses. Nucleic Acids Res 12:7269–7282. https://doi.org/10.1093/nar/12.18.7269
Kar RK, Kharerin H, Padinhateeri R et al (2017) multiple conformations of Gal3 protein drive the galactose-induced allosteric activation of the GAL genetic switch of saccharomyces cerevisiae. JMB 429:158–176. https://doi.org/10.1016/j.jmb.2016.11.005
Khalifa ME, MacDiarmid RM (2021) A Mechanically Transmitted DNA Mycovirus Is Targeted by the Defence Machinery of Its Host, Botrytis cinerea. Viruse. https://doi.org/10.3390/v13071315
Koonin EV (1991) The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J Gen Virol 72(Pt 9):2197–2206. https://doi.org/10.1099/0022-1317-72-9-2197
Kotta-Loizou I (2021) Mycoviruses and their role in fungal pathogenesis. Curr Opin Microbiol 63:10–18. https://doi.org/10.1016/j.mib.2021.05.007
Kotta-Loizou I, Castón JR, Coutts RHA et al (2020) ICTV virus taxonomy profile: chrysoviridae. J Gen Virol 101:143–144. https://doi.org/10.1099/jgv.0.001383
Kozlakidis Z, Herrero N, Ozkan S et al (2013) Sequence determination of a quadripartite dsRNA virus isolated from Aspergillus foetidus. Arch Virol 158:267–272. https://doi.org/10.1007/s00705-012-1362-3
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lawrence DP, Nouri MT, Trouillas FP (2019) Taxonomy and multi-locus phylogeny of cylindrocarpon-like species associated with diseased roots of grapevine and other fruit and nut crops in California. Fungal Syst Evol 4:59–75. https://doi.org/10.3114/fuse.2019.04.06
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320. https://doi.org/10.1093/molbev/msn067
Lesker T, Rabenstein F, Maiss E (2013) Molecular characterization of five betacryptoviruses infecting four clover species and dill. Arch Virol 158:1943–1952. https://doi.org/10.1007/s00705-013-1691-x
Lutz T, Japić E, Bien S et al (2022) Characterization of a novel alternavirus infecting the fungal pathogen Fusarium solani. Virus Res 317:198817. https://doi.org/10.1016/j.virusres.2022.198817
Madeira F, Park YM, Lee J et al (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47:W636–W641. https://doi.org/10.1093/nar/gkz268
Mahnkopp F, Simon M, Lehndorff E et al (2018) Induction and diagnosis of apple replant disease (ARD): a matter of heterogeneous soil properties? Sci Hortic 241:167–177. https://doi.org/10.1016/j.scienta.2018.06.076
Manici LM, Kelderer M, Caputo F et al (2018) Involvement of Dactylonectria and Ilyonectria spp. in tree decline affecting multi-generation apple orchards. Plant Soil 425:217–230. https://doi.org/10.1007/s11104-018-3571-3
Menzel W, Jelkmann W, Maiss E (2002) Detection of four apple viruses by multiplex RT-PCR assays with coamplification of plant mRNA as internal control. J Virol Methods 99:81–92. https://doi.org/10.1016/S0166-0934(01)00381-0
Mirdita M, Schütze K, Moriwaki Y et al (2022) ColabFold: making protein folding accessible to all. Nat Methods 19:679–682. https://doi.org/10.1038/s41592-022-01488-1
Morris TJ, Dodds JA (1979) Isolation and analysis of double-stranded RNA from virus-infected plant and fungal tissue. Phytopathology 69:854. https://doi.org/10.1094/Phyto-69-854
Nuss DL (2005) Hypovirulence: mycoviruses at the fungal-plant interface. Nat Rev Microbiol 3:632–642. https://doi.org/10.1038/nrmicro1206
O’Donnell K, Cigelnik E (1997) Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Mol Phylogenet Evol 7:103–116. https://doi.org/10.1006/mpev.1996.0376
Olivé M, Campo S (2021) The dsRNA mycovirus ChNRV1 causes mild hypervirulence in the fungal phytopathogen Colletotrichum higginsianum. Arch Microbiol 203:241–249. https://doi.org/10.1007/s00203-020-02030-7
Osaki H, Sasaki A, Nomiyama K et al (2016) Multiple virus infection in a single strain of Fusarium poae shown by deep sequencing. Virus Genes 52:835–847. https://doi.org/10.1007/s11262-016-1379-x
Özkan S, Coutts RHA (2015) Aspergillus fumigatus mycovirus causes mild hypervirulent effect on pathogenicity when tested on Galleria mellonella. Fungal Genet Biol 76:20–26. https://doi.org/10.1016/j.fgb.2015.01.003
Pearson MN, Beever RE, Boine B et al (2009) Mycoviruses of filamentous fungi and their relevance to plant pathology. Mol Plant Pathol 10:115–128. https://doi.org/10.1111/j.1364-3703.2008.00503.x
Polashock JJ, Hillman BI (1994) A small mitochondrial double-stranded (ds) RNA element associated with a hypovirulent strain of the chestnut blight fungus and ancestrally related to yeast cytoplasmic T and W dsRNAs. Proc Nati Acad Sci 91:8680–8684. https://doi.org/10.1073/pnas.91.18.8680
Popp C, Grunewaldt-Stöcker G, Maiss E (2019) A soil-free method for assessing pathogenicity of fungal isolates from apple roots. J Plant Dis Prot 126:329–341. https://doi.org/10.1007/s41348-019-00236-6
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Nat Acad Sci USA 74:5463–5467. https://doi.org/10.1073/pnas.74.12.5463
Sato Y, Miyazaki N, Kanematsu S et al (2019) ICTV virus taxonomy profile: megabirnaviridae. J Gen Virol 100:1269–1270. https://doi.org/10.1099/jgv.0.001297
Schneider RJ, Mohr I (2003) Translation initiation and viral tricks. Trends Biochem Sci 28:130–136. https://doi.org/10.1016/S0968-0004(03)00029-X
Sievers F, Wilm A, Dineen D et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. https://doi.org/10.1038/msb.2011.75
van Schoor L, Denman S, Cook NC (2009) Characterisation of apple replant disease under South African conditions and potential biological management strategies. Sci Hortic 119:153–162. https://doi.org/10.1016/j.scienta.2008.07.032
Venkataraman S, Prasad BVLS, Selvarajan R (2018) RNA dependent RNA polymerases: insights from structure, function and evolution. Viruses. https://doi.org/10.3390/v10020076
Waterhouse A, Bertoni M, Bienert S et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. https://doi.org/10.1093/nar/gky427
Wei CM, Moss B (1975) Methylated nucleotides block 5’-terminus of vaccinia virus messenger RNA. Proc Nat Acad Sci USA 72:318–322. https://doi.org/10.1073/pnas.72.1.318
Wen C, Wan X, Zhang Y et al (2021) Molecular Characterization of the First Alternavirus Identified in Fusarium oxysporum. Viruses. https://doi.org/10.3390/v13102026
Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18:691–699. https://doi.org/10.1093/oxfordjournals.molbev.a003851
Winkelmann T, Smalla K, Amelung W et al (2019) Apple replant disease: causes and mitigation strategies. Curr Issues Mol Biol 30:89–106. https://doi.org/10.21775/cimb.030.089
Wu C-F, Aoki N, Takeshita N et al (2021) Unique terminal regions and specific deletions of the segmented double-stranded RNA Genome of Alternaria Alternata Virus 1, in the proposed family Alternaviridae. Front Microbiol 12:773062. https://doi.org/10.3389/fmicb.2021.773062
Yu X, Li B, Fu Y et al (2010) A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc Nati Acad Sci 107:8387–8392. https://doi.org/10.1073/pnas.0913535107
Zhang X, Xie Y, Zhang F et al (2019) Complete genome sequence of an alternavirus from the phytopathogenic fungus Fusarium incarnatum. Arch Virol 164:923–925. https://doi.org/10.1007/s00705-018-04128-2
Zhang Y, Shi N, Wang P et al (2022) Molecular characterization of a novel alternavirus infecting the entomopathogenic fungus Cordyceps chanhua. Arch Virol. https://doi.org/10.1007/s00705-022-05446-2
Acknowledgements
This work was funded by the Federal Ministry of Education and Research and is part of the ORDIAmur network (FKZ: 031B0025A, 031B0512), which is allocated to the BONARES program. We thank Mr. Aaron Balke for his assistance with dsRNA extractions.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was funded by the Federal Ministry of Education and Research and is part of the ORDIAmur network (FKZ: 031B0025A, 031B0512), which is allocated to the BONARES program.
Author information
Authors and Affiliations
Contributions
TPP and CP performed the experiments, analyzed data and wrote the main manuscript text. SF performed and evaluated experiments. PM and DK perfomed NGS analyses and evaluated NGS data. EM conceived and designed the experiments. All authors contributed to the writing and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no financial or proprietary interests in any material discussed in this article.
Research involving human participants and/or animals
The authors declare that no human and/or animal material, data, or cell lines were involved in this study.
Informed consent
The authors declare that no individual rights were infringed during this study. All involved individuals have been informed and gave their consent to publication.
Additional information
Communicated by Yusuf Akhter.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pielhop, T.P., Popp, C., Fricke, S. et al. Molecular characterization of two new alternaviruses identified in members of the fungal family Nectriaceae. Arch Microbiol 205, 129 (2023). https://doi.org/10.1007/s00203-023-03477-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-023-03477-0