
Abstract
Acute kidney injury (AKI) is now recognized as a heterogeneous syndrome that not only affects acute morbidity and mortality, but also a patient’s long-term prognosis. In this narrative review, an update on various aspects of AKI in critically ill patients will be provided. Focus will be on prediction and early detection of AKI (e.g., the role of biomarkers to identify high-risk patients and the use of machine learning to predict AKI), aspects of pathophysiology and progress in the recognition of different phenotypes of AKI, as well as an update on nephrotoxicity and organ cross-talk. In addition, prevention of AKI (focusing on fluid management, kidney perfusion pressure, and the choice of vasopressor) and supportive treatment of AKI is discussed. Finally, post-AKI risk of long-term sequelae including incident or progression of chronic kidney disease, cardiovascular events and mortality, will be addressed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Acute kidney injury (AKI) is recognized as an heterogeneous syndrome affecting short- and long-term morbidity and mortality. Progress on prediction and early detection, clinical phenotypes, pathophysiology, nephrotoxicity, organ cross-talk, prevention and supportive treatment of AKI as well as long-term sequelae are addressed in this review paper |
Introduction
The availability of a consensus definition of acute kidney injury (AKI) [1] has been an important step in establishing AKI epidemiology. AKI affects 30–60% of critically ill patients and is associated with acute morbidity and mortality [2]. Evidence is also accumulating that the burden of AKI extends beyond the acute phase with progression to chronic kidney disease (CKD), increased risk of cardiovascular complications, recurrent episodes of AKI and long-term mortality [3]. Prevention of development and/or progression is currently limited to hemodynamic and fluid status optimization and avoidance of nephrotoxins. Search for a specific pharmacologic treatment is hampered by the late diagnosis and the complex and incompletely elucidated pathophysiology. Progress in the management of AKI is to be expected from the recognition that AKI is a very heterogeneous syndrome with variable etiology, pathophysiology and clinical presentation [4].
In this narrative review, we discuss methods for early diagnosis of AKI, clinical phenotypes, pathophysiology, nephrotoxicity, optimal supportive management, as well as the importance of recovery and long-term follow-up. A discussion of renal replacement therapy (RRT) is outside the scope of this review.
Towards an improved AKI diagnosis?
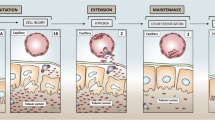
Although the kidney has many functions, AKI is mostly defined as a decrease of glomerular filtration rate (GFR). The KDIGO workgroup proposed a consensus definition and staging system for clinical practice (the KDIGO definition) that relies on the increase of serum creatinine (Scr) within 7 days and/or the presence of oliguria (Fig. 1), both surrogate markers of GFR [1]. This definition has enabled to streamline research in the field. However, both Scr and urine output may also be influenced by non-renal and non-GFR-related factors and are, therefore, imperfect markers of reduced GFR [5]. Consequently, the diagnosis of AKI by KDIGO criteria should be interpreted in the clinical context. Controversy on how to determine baseline kidney function is another drawback of the consensus definition. Despite the concerns about oliguria as a marker of kidney function, there is evidence that oliguria identifies patients with worse outcomes [6, 7]. In addition, because of the long half-life of creatinine and the presence of renal functional reserve (renal reserve capacity that can be recruited before basal GFR starts to decline [8]), Scr requires time to accurately reflect GFR resulting in delayed recognition of kidney dysfunction. Potential solutions for a more timely detection of a reduced GFR could be measurement of creatinine clearance over 2 or 4 h, kinetic eGFR calculated from two serial creatinine measurements [9], or utilizing the plasma disappearance of an injected compound like iohexol that is dependent on renal clearance. These methods have not been extensively investigated or used in the ICU setting. Bedside real-time GFR measurements using injection of a dye and fluorescent probes and enabling earlier diagnosis of kidney dysfunction are under investigation, but do not yet have regulatory approval [10]. Cystatin-C, another marker of glomerular filtration, might be useful in settings of muscle wasting, but is also affected by comorbidity.

Different phases of AKI development and progression and associated diagnostic tests. AKI acute kidney injury, TIMP tissue inhibitor of metalloproteinases, IGFBP insulin-like growth factor-binding protein, NGAL neutrophil gelatinase-associated lipocalin, UO urine output
Late AKI diagnosis has been implicated for the lack of efficacy success in drug trials. This explains the interest in biomarkers to predict KDIGO AKI (with 1700 publications over the past 5 years). Ideally, increased biomarker levels indicate kidney injury before the KDIGO criteria for AKI are met (so-called “subclinical AKI”), and thus might trigger early diagnostic and preventive measures [11]. The most widely investigated markers are neutrophil gelatinase-associated lipocalin (NGAL), kidney injury molecule -1 (KIM-1), liver fatty acid binding protein (LFABP) and the product of and tissue inhibitor of metalloproteinase 2 and insulin-like growth factor-binding protein 7 (TIMP-2*IGFBP7; Nephrocheck™) (Fig. 1) (Table 1). Only NGAL and TIMP-2*IGFBP7 are available for clinical use. TIMP-2*IGFBP7 is a urinary marker of cell cycle arrest, reflecting cellular stress that precedes tissue damage (Fig. 1). It has FDA and EMEA approval for the prediction of AKI stage 2 and 3 within 12 h in critically ill patients with cardiac and respiratory failure [12]. High sensitivity (> 0.3) and high specificity (> 2.0) cutoffs have been defined, allowing risk stratification [13]. Two recent meta-analyses showed an AUROC 0.83 for the prediction of AKI within 24 h in cardiac surgery [14] and 0.74 for the prediction of stage 2 and 3 within 12 h in the critically ill [15]. A meta-analysis on the accuracy of urinary NGAL showed an AUROC of 0.75 for severe AKI with cut-offs of 12 ng/ml for 95% sensitivity and 580 ng/ml for 95% specificity [16]. Significant heterogeneity is, however, reported related to the study population (pre-test probability), timing of sampling, prediction window and severity of predicted AKI. It should also be acknowledged that the gold standard definition (KDIGO criteria) refers to kidney function, not damage. “False positives” may reflect “subclinical” injury and false negatives may reflect “hemodynamic” (previously called prerenal) AKI. Combining damage biomarkers with the functional KDIGO criteria may allow better characterization of AKI phenotypes and improve the diagnostic accuracy [11]. Incorporation of biomarkers in the AKI definition has, therefore, been suggested [11].
Despite extensive research, guidance on how AKI biomarkers should be used in clinical practice is lacking. Currently, these biomarkers have been successful in identifying high-risk patients for clinical trials investigating early prevention strategies [17, 18]. Demonstration of utility/effectiveness in a real-world setting would, however, require a comparison of two strategies where clinicians have or do not have access to the biomarker result. Costs, availability, and the paucity of therapeutic options are other barriers to widespread clinical use.
Another recently proposed AKI predictor is loss of renal functional reserve (measured with a high oral protein load), that has shown to predict postoperative AKI in cardiac surgery [19] and might be a marker of incomplete recovery [20]. However, this clinical utilization of this parameter also remains to be evaluated.
Electronic alerts (e-alerts) have been suggested as solutions for early diagnosis of AKI [21]. They are based on prevalent KDIGO criteria and their main advantage is to be expected in the less monitored non-ICU setting and only when linked to an order set and/or action [22]. The evidence for benefit in the ICU is limited [23].
More promising are prediction models either based on logistic regression or machine-learning methodologies taking advantage of the large amount of data available in electronic health records (EHR). The past years have seen a proliferation of AKI prediction models based on artificial intelligence [24, 25]. Only a few have focused on ICU patients (Table 2). Most models have been developed retrospectively in a preprocessed dataset (research dataset or electronic health records) and only use the creatinine criteria. They show fair to excellent accuracy (AUC 0.75–0.90) and good calibration. Accuracy is generally higher with a shorter prediction window (lead time) and more severe AKI. These models either provide snap-shot scores [26], moving windows [27] or continuous AKI prediction [28]. Only one of these ICU models has undergone external validation confirming good accuracy in a multicenter independent dataset [29]. The last step in the validation process of these prediction models, that still has to be taken, is translation into a bedside real-time prediction tool (using the “uncleaned” EHR) providing continuously updated AKI probability with uncertainty levels. This model should then be evaluated in RCT’s testing the impact of its use on patient-centered outcomes when integrated in the real-world clinical workflow, eventually linked to a clinical decision support system (“turning predictions into action”). Such a trial is currently ongoing (NCT03590028) (Table 3).
Clinical phenotypes
Over the past decade, it is increasingly recognized that AKI is a heterogeneous syndrome not only with regard to exposure (low cardiac output, sepsis, major surgery, toxicity, etc.) and pathophysiology (hypoperfusion, inflammation, etc.) but also with regard to the clinical presentation (severity and evolution). A potential approach to distinguishing clinical phenotypes is the application of latent class analysis to a set of clinical and biological variables to define subgroups with different outcomes and treatment responses [30, 31]. The prognostic importance of AKI duration and recovery pattern has been demonstrated in several analyses. A recent ADQI conference defined transient AKI and persistent AKI based on duration of more or less than 48 h [32]. Predicting the course of AKI could enable to define different phenotypes requiring different management. Traditional urinary biochemistry [33] and renal resistive index [34] perform poorly in this regard, especially in sepsis. Biomarkers may be helpful [35, 36] although results are not uniformly positive [37, 38]. A new biomarker, urinary C–C motif chemokine ligand-14 (CCL14) has recently been identified as a very accurate predictor (outperforming all other biomarkers) of persistent AKI stage 3 in ICU patients with severe AKI [39]. Limited data suggest that kinetic eGFR better predicts AKI progression better than some biomarkers [40] and the same applies to the furosemide stress test (FST) that performed better than biochemical biomarkers in predicting the progression to AKI stage 3 [41, 42]. Future developments in measuring real-time GFR or using real-time AKI prediction models will certainly contribute to this field.
Acute kidney disease: a recent entity
Acute kidney disease (AKD) defined as an AKI episode that lasts longer than 7 days but less than 90 days has recently been proposed as a concept (Fig. 1) [32]. It aims at closing the gap between AKI and CKD (which requires 3 months to be diagnosed). AKD uses the creatinine criteria of the KDIGO definition. It is important to note that the diagnosis of AKD (severity) or apparent recovery may be affected by the decrease of Scr related to muscle mass loss associated with chronic critical illness [5]. The relationship between persistent AKI, AKD and CKD as well as interventions that may interfere with this evolution require further study (Fig. 2).

Following the development of AKI, several scenarios are possible that may lead to recovery of renal function or to more prolonged dysfunction. Acute kidney disease (AKD) is assessed between 7 and 90 days after AKI. In patients that do not improve, chronic kidney disease (CKD) is established after day 90. Biomarkers of renal injury and function may be able to refine the prediction of rapid recovery (i.e., transient AKI) or transition to more persistent impairment of renal function and several therapeutic interventions may be able to modulate the progression of the disease course
Pathophysiology of a heterogeneous syndrome
The pathophysiology of AKI has been insufficiently elucidated not in the least because kidneys are complex and rather inaccessible organs. Animal models poorly reflect human pathophysiology (where comorbidities play an important role) and the AKI syndrome is heterogenous [43], illustrated by recent studies demonstrating different genomic responses in volume depletion, ischemic and septic animal models of AKI [44, 45]. In clinical practice, it is likely that there are distinct, but overlapping pathophysiological paradigms of AKI that may require individualized treatments [4], explaining in part the failure of many interventions in clinical trials. With the exception of specific intrinsic renal disease, AKI pathology can range from decreased GFR, mediated purely by systemic or local hemodynamic alterations through reversible tubular stress/injury to frank tubular necrosis. Histologic changes in AKI of critical illness are generally focal and modest [46]. Within this complex pathophysiology, a number of common themes emerge with patterns of inflammatory, ischemic and nephrotoxic kidney injury that can occur sequentially and concomitantly and may be differently influenced by underlying diseases (Fig. 3).

Simplified overview of AKI pathophysiology illustrating the heterogeneity in etiology, presentation, pathology, progression and outcomes and how investigations may help us understand underlying AKI phenotypes at various stages in illness. Green indicates functional/reversible processes; red indicates acute and chronic tissue injury. Yellow boxes indicate etiological factors in AKI pathogenesis, blue boxes diagnostic tests indicative of underlying pathophysiological processes
Identification of clinical phenotypes with different pathophysiology and outcome is essential to identify new therapeutic targets [4]. The importance of clinical context is illustrated in cardiorenal syndrome due to acute decompensation of chronic heart failure, where renal congestion is the predominant driver of worsening kidney function. Although successful resolution of fluid overload with diuretics or ultrafiltration may induce an increase of Scr, it is, however, associated with improved longer term kidney function [47], even in patients with elevated renal injury markers [48]. This suggests that the benefit of decongestion outweigh the modest increase of Scr and, for instance, that NT-proBNP might be a more useful prognostic biomarker than markers of kidney damage in this specific setting [49]. It is important to note, however, that often numerous AKI risk factors and clinical settings co-exist or follow sequentially so that clear clinical inferences are difficult to draw, which emphasizes the importance of further research to identify underlying dominant AKI phenotypes to guide therapeutic intervention.
The interplay of different pathophysiological pathways is most pronounced in sepsis, the most common cause of AKI in the critically ill [2]. Generally, the ischemic component does not appear to result from reduced global kidney blood flow. Instead, periglomerular shunting may reduce glomerular blood flow and inflammation-induced endothelial dysfunction induces microvascular disturbances and microthrombi formation [50, 51]. Afferent vasoconstriction due to tubulo-glomerular feedback is considered a consequence rather than a cause of tubular dysfunction [51]. The inflammatory component results from damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) that are present in the peritubular capillaries and undergo glomerular filtration and subsequently interact with Toll-like receptors located on the brush border membrane of epithelial cells in the proximal tubule [50, 51]. Recruitment of immune cells further contributes to an immuno-pathophysiological response and immune-mediated damage [52]. Besides inflammatory damage, recent experimental data have suggested that cell cycle arrest, deficient autophagy, ferroptosis, mitochondrial dysfunction and metabolic reprogramming are mechanisms that contribute to tubular dysfunction in septic AKI [50].
Some of these pathways have already resulted in exploration of potential interventions in patients. The kidney being a highly metabolically active organ, one of the pathways that recently received significant attention is mitochondrial dysfunction and impaired energy metabolism due to deficiency of nicotinamide adenine dinucleotide (NAD+) and metabolic reprogramming [50, 51]. Experimental AKI is characterized by deficiency of PPARgamma-coactivator 1a (PGC-1a), a critical regulator of mitochondrial biogenesis. Deficiency of PGC-1a is linked to impaired synthesis of NAD+, an essential player in cellular energy metabolism (mainly fatty acid oxidation and glycolysis). Markers of decreased NAD+ have been demonstrated in patients developing AKI after cardiac surgery. In addition, nicotinamide, a precursor of NAD+ reduced postoperative creatinine levels in a phase I RCT [53]. A larger RCT is currently ongoing (NCT04342975). Metabolic reprogramming refers to a switch from oxidative phosphorylation to less efficient energy production through glycolysis in response to decreased oxygen and substrate supply. It decreases the production of reactive oxygen species (ROS) and is a survival mechanism with reduction of non-vital functions paying the price for maintaining cellular integrity, which could explain the dissociation between structure and function that characterizes AKI. However, switchback to oxidative phosphorylation (mediated by AMPK) appears necessary for survival but requires restoration of functional mitochondria by mitophagy and mitochondrial biogenesis [50, 51].
Biomarkers have contributed to unravelling the pathophysiology of AKI. Besides metabolic reprogramming, cell cycle arrest is another protective mechanism that prevents cells with DNA damage from dividing and downregulates energy expenditure. However, as with metabolic reprogramming, it appears to be a double edge sword because with persistent cell cycle arrest repair becomes maladaptive resulting in AKI to CKD transition [54]. Chemokine C–C motif ligand-14 (CCL14) was recently described as a marker of AKI persistence. The hypothesized mechanism (and potential therapeutic target) is CCL14 release by tubular cells in response to inflammatory mediators, triggering infiltration and differentiation of monocytes and T cell-mediating fibrosis and incomplete kidney recovery [39]. Soluble urokinase plasminogen activator receptor (suPAR), a marker of chronic inflammation and immune activation is increased in a number of exposures including increasing age, diabetes, cardiovascular disease, infection and smoking. It also predicts incident CKD and CKD progression. Admission levels independently predicted development of AKI within the first 7 day after various procedures [55]. Importantly, targeting suPAR oxidative pathways appears to attenuate damage in experimental AKI [55] suggesting potential as therapeutic target. Similarly, urinary Dickkopf-3 (DKK3) is a pro-fibrotic glycoprotein secreted by renal tubular cells that modulates the Wnt/b pathway involved in tubulo-interstitial fibrosis and predicts GFR loss and renal fibrosis in CKD. It has recently been described as a strong pre-operative predictor of AKI post-cardiac surgery and, importantly, worsening of long-term renal function after AKI [56]. These findings emphasize the interconnected pathophysiology of AKI episodes and CKD progression. The previously mentioned study using latent class analysis for detection of clinical phenotypes found two sub-phenotypes (AKI-SP1 and AKI-SP2) with different biomarker profiles (markers of endothelial dysfunction such as tumor necrosis factor receptor-1, angiopoietin-1 and 2), kidney and patient outcome and response to treatment with vasopressin [57], apparently genetically determined [58]. These are only the first steps in the search for pathophysiological pathways that could lead to precision medicine and clinical benefit.
What is new in nephrotoxicity?
There are drugs that are directly nephrotoxic, drugs that are not nephrotoxic but interfere with intrarenal hemodynamics (i.e., ACE-inhibitors, NSAIDs) and drugs that are not nephrotoxic, but accumulate in renal failure and should, therefore, also be prescribed with caution. The most commonly incriminated drugs are contrast agents and antibiotics but multidrug toxicity maybe the predominant problem. When prescribing drugs, clinicians should consider how they affect kidney function and whether their clearance is affected by the presence of AKI. Prescription of nephrotoxic medications should be minimized in terms of frequency and duration, however, they should not be withheld in life-threatening situations owing to concern for AKI [59]. Drugs with potentially renoprotective effects should be continued even if they are associated with a mild rise in serum creatinine (i.e., ACE-inhibitors in diabetic nephropathy).
Historically, contrast agents have been considered an important cause of AKI. However, recent observational studies with propensity score-adjusted models found no relevant difference in AKI incidence between those exposed or not exposed to modern contrast agents, even in septic and ICU patients [60,61,62], suggesting that the risk of contrast-induced AKI (causal association) is much lower than previously thought and that contrast-associated AKI (temporal association) frequently has other causes. The absence of a post-contrast increase of biomarkers of kidney injury [63] supports this concept. Hence, contrast-enhanced CT scan should not be postponed if required for the diagnosis of a life-threatening condition. In all other situations, the uncertain risk of contrast-induced AKI should be balanced against the risk of missing an important diagnosis, taking into account the possibility of alternative imaging procedures [64]. Recent guidelines suggest using moderate contrast doses and prophylactic isotonic hydration in patients at risk [65] although even the latter has been questioned [66]. It is evident that in critically ill patients hydration should account for the volume status of the individual patient, balancing the risk of kidney hypoperfusion against the risk of fluid overload. A large RCT confirmed that acetylcysteine has no benefit and that bicarbonate is not better than saline in preventing contrast nephropathy [67].
With regard to dosing of antibiotics, recent literature mainly stresses the danger of subtherapeutic concentrations in the treatment of multidrug-resistant (MDR) microorganisms, which makes dosing challenging for antibiotics with narrow therapeutic index such as vancomycin, aminoglycosides and polymyxins. Trials are ongoing to investigate optimal administration and duration of antibiotics. (https://warwick.ac.uk/fac/sci/med/research/ctu/trials/adaptsepsis and ClinicalTrials.gov Identifier: NCT03213990).
The nephrotoxicity of vancomycin has been debated over many years and is probably lower than previously suggested [68]. A recent meta-analysis showed that the incidence of AKI increases with higher trough concentrations and is significantly higher for trough concentrations ≥ 20 μg/mL. It also suggested that AUC/MIC monitoring strategy (aiming at a target of 400) could result in less nephrotoxicity [69]. In patients at high risk or with early signs of kidney dysfunction, a switch to a less toxic alternative should be considered. Several observational trials showed increased nephrotoxicity when vancomycin was combined with piperacillin/tazobactam in comparison with vancomycin alone or in combination with other beta-lactams [70]. Others suggested that the Scr increase does not reflect true AKI (a decrease of GFR), but an inhibition of tubular secretion of creatinine by piperacillin/tazobactam but this may be valid only for lower stages of AKI [71]. Future RCTs with non-creatinine markers of GFR should clarify the issue. The emergence of difficult-to-treat MDR gram-negative bacteria has resulted in renewed interest in polymyxins and aminoglycosides, mainly amikacin. Most recent trials on amikacin focus on optimal dosing with regard to efficacy, whereas nephrotoxicity has been poorly studied in ICU patients. A small propensity-based study suggested no toxicity with short (< 3 days) amikacin treatment, which might be sufficient to bridge the waiting time for bacteriological results [72]. Polymyxins are nephrotoxic [73] with colistin being more harmful than polymyxin B [74]. However, since they are mostly used as a last resort, kidney dysfunction is often an unavoidable side effect. A combination of injury biomarkers and therapeutic drug monitoring could help to reduce nephrotoxicity, although evidence from clinical studies is lacking.
Organ cross-talk
During the development and presence of AKI in the critically ill, virtually all non-renal organs are affected. This generalized systemic process could be representing the impact of the underlying disease (shock, systemic inflammation) on multiple organ systems. However, an alternative explanation suggests mutual impact of failing organs, known as organ cross-talk could also play a role. AKI can indeed result from failure of other organs, most well-known examples being cardiorenal syndrome, from a failing heart, and hepatorenal syndrome, from a failing liver. There are also indications that ARDS and (the modality of) mechanical ventilation may affect kidney function [75]. However, the concept of cross-talk also works in the other direction and considers AKI as a systemic disease with impact on other organs such as the heart [76], lung [75], liver and brain [77, 78]. Instead of being an innocent bystander in the process of multiple organ failure, the kidneys may indeed initiate metabolic or humoral pathways affecting distant organ function. Potential mechanisms are the consequences of decreased kidney function, leading to accumulation of uremic toxins, fluid overload, electrolyte disturbances and acid–base dysbalance. Alternatively, an inflammatory mechanism involving neutrophil migration and inflammatory mediators originating in the kidney or side effects of supportive treatment with renal replacement therapy (RRT) may play a role. Cross-talk also exists between the kidney and the immune system with inflammation as being an important pathophysiological mechanism of AKI on the one hand and AKI-induced immunosuppression resulting in a higher susceptibility to develop secondary infections on the other hand. The uremic toxin resistin appears to be an important mediator of impaired cellular immunity [79]. The clinical relevance of AKI-induced immune paralysis is illustrated by the observation that renal dysfunction increases the chances of developing serious infections following heart surgery [80] and approximately half of the patients with AKI who do not survive, die of sepsis [81, 82].
Direct evidence for an impact of AKI on other organs mostly results from animal experiments (summarized in [77, 78]) since the impact of a common etiology and organ cross-talk is difficult to discern in the clinical situation. For instance, a galectin-3-dependent pathway has been shown to be involved in the reno-cardiac syndrome [83]. Recent animal studies also suggest that acute renal ischemia may induce both functional and transcriptional changes in the lung, independent of uremia, but related to leukocyte trafficking [84].
Clearly, cross-talk between the kidney and other organs may be an important contributor to the increased morbidity and mortality that is associated with AKI [85] and may explain why, as compared to matched controls, patients with AKI are more likely to die from sepsis, bleeding, delirium and respiratory failure [75, 76, 86].
Prevention of AKI
General principles
General preventive measures should be applied to all patients admitted to the ICU, including correction of hypovolemia and hypotension, discontinuation and avoidance of nephrotoxic agents and correction of hyperglycemia [64].
Fluid management
The aim of fluid administration is to correct intravascular hypovolemia without causing fluid overload and associated complications, including new development and progression of AKI [87]. An association between elevated central venous pressure, renal venous congestion and development of AKI, mainly reported in congestive heart failure, has also been found in other ICU patient cohorts [88, 89]. A randomized trial in ARDS patients found fluid restrictive strategies to be safe [90]. In contrast, peri-operative restrictive fluid management increased the risk of AKI in patients undergoing major elective abdominal surgery [91]. In established AKI, the role of fluid restriction remains uncertain and likely depends on pre-existing intravascular volume status. A pilot study in critically ill patients with AKI showed that restricting fluid intake with the aim to prevent fluid overload was associated a lower incidence of adverse effects and less need for RRT [92]. Similarly, a strategy of active fluid restriction after initial fluid resuscitation in patients with septic shock was associated with less AKI progression [93] but this finding could not be reproduced in two subsequent trials using a similar approach [94, 95]. The results of ongoing RCTs examining the effectiveness and safety of fluid restriction and the role of active de-resuscitation in high-risk patients are awaited [96].
The type of crystalloid fluid for resuscitation has also been evaluated in recent large RCTs in non-critically ill and critically ill patients. The SMART study, which compared saline with buffered crystalloids, showed a lower incidence of major adverse kidney events (MAKE) in those receiving buffered crystalloids, but there was no significant difference in maximum stage of AKI, need for RRT or proportion of patients with at least a doubling of Scr. There was also no difference in median volume of fluid between both groups. Among patients with sepsis, the use of buffered crystalloids was associated with a lower 30-day in-hospital mortality compared with use of saline [97].
Kidney perfusion pressure
Conditions with non-fluid-responsive impaired cardiac output (CO) may require inotropes. Interestingly, the intervention arm of the PREV-AKI study, showing a beneficial effect of a care bundle, used more dobutamine [17]. With regards to early AKI, observational evidence suggests a higher CO and oxygen delivery (DO2) may be beneficial to prevent its progression [98], although early goal-direct therapy in general does not impact AKI [99]. It is important to note that AKI may also occur in situations of normal or increased kidney perfusion pressure due to development of intrarenal shunting and microcirculatory disturbances [50]. Every effort should be made to avoid severe hypotension, a definite cause for AKI, especially in situations of disturbed autoregulation. The ideal mean arterial pressure (MAP) to avoid AKI remains to be determined and might need to be tailored to patients’ characteristics [100]. In 2463 sepsis patients aged > 65 years, “permissive hypotension” (MAP 60–65 mm Hg) was not associated with need for RRT or an increased 90-day mortality compared to usual care; less severe AKI was not assessed though [101]. On the other hand, in a RCT in septic shock patients, a lower MAP target was related to doubling of creatinine or need for RRT in the subgroup of patients with chronic hypertension [102]. Similarly, maintaining systolic BP in the operating room within 10% of resting systolic BP resulted in a significantly lower incidence of postoperative AKI compared to a fixed (80 mmHg) target in high-risk adults undergoing major surgery [103]. A retrospective study of septic shock patients, stratified according to the difference between pre-morbid and post-resuscitation MAP, showed that the incidence of AKI was lowest among those whose post-resuscitation MAP was closest to or higher than their pre-morbid MAP [104]. Therefore, it appears plausible that a more personalized approach, mainly based on pre-existing values, may be the optimal way to manage blood pressure. In addition, more attention should be given to importance of renal perfusion pressure (MAP-CVP) [105, 106].
The impact of different vasoactive or inotropic medications on kidney function varies and may depend on the underlying condition. The most frequently used vasopressor to maintain renal perfusion pressure is norepinephrine [107]. The effects of phenylephrine, a pure α-1 agonist, on renal function are poorly investigated, but without evidence for benefit [108]. Catecholamines may have side effects at higher doses. Vasopressin, an endogenous non-catecholamine vasopressor, has the ability to preferentially constrict efferent glomerular arterioles, thus increasing glomerular perfusion pressure and urine generation. While in the Vasopressin vs Norepinephrine as Initial Therapy in Septic Shock (VANISH) trial [109], the number of kidney failure-free days was similar in patients who received noradrenaline or vasopressin, patients in the vasopressin group had lower Scr levels and higher urinary output in the first 7 days, leading to lower use of RRT (25.4% vs. 35.3%). A meta-analysis including 4 RCTs concluded that vasopressin reduced the requirement for RRT (RR 0.86, 95% CI 0.74–0.99), but this finding was not robust to sensitivity analyses [110]. More recently, the Vasopressin versus Norepinephrine in Patients with Vasoplegic Shock after Cardiac Surgery (VANCS) trial demonstrated a significantly better primary composite outcome in patients randomized to vasopressin versus norepinephrine, an effect primarily driven by a lower rate of AKI [111]. Angiotensin II infusion has recently been investigated in patients with shock demonstrating equal hemodynamic stabilization as achieved by norepinephrine (ATHOS-3, [112]). A post hoc analysis of this trial showed that in a subgroup of patients on RRT, duration of RRT was shorter and survival higher in patients who received angiotensin-II compared to norepinephrine [113], a finding that warrants confirmation. Naturally, potential impact on kidney function should always be weighed against potential adverse effects.
Biomarkers to guide management
Several single-center studies in patients undergoing major surgery suggested that initiation of a KDIGO prevention bundle in high-risk patients identified by biomarkers could reduce the incidence and progression of AKI however, without beneficial effect on patient-centered outcome [17, 18]. Biomarker-guided management of nephrotoxic drugs has also been advocated as a form of nephrotoxin stewardship [114]. The strength of evidence currently precludes the routine use of biomarkers to guide decision-making on when to initiate RRT [115]. New biomarkers of kidney non-recovery have been discovered, i.e., CCL14 [39]. Their potential role in guiding RRT initiation needs to be determined in future studies. A study in 162 patients with AKI showed that the furosemide stress test (FST) had excellent predictive ability for subsequent use of RRT [116]. However, there was no difference in outcome between early versus standard initiation of RRT in FST non-responders.
New drugs
To date, there are no specific drugs or therapies that prevent or treat AKI. A recent multicenter RCT demonstrated that human recombinant alkaline phosphatase (AP), an enzyme that dephosphorylates endotoxin and ATP, was not associated with a significant improvement of endogenous creatinine clearance during the first 7 days. However, creatinine clearance up to day 28 was better and all-cause mortality was lower [117]. Other promising agents include novel compounds, repurposed drugs and cell-based therapies targeting a variety of pathways, including mitochondrial stress, cell metabolism, inflammation, antioxidant effects, apoptosis, repair mechanisms and systemic hemodynamics [118] (Supplemental Table 1). Some of these compounds are progressing through early-phase clinical trials.
Initiation of RRT
Four out of five recent RCTs [119,120,121,122,123,124] (Supplemental Table 2) failed to demonstrate a survival benefit of early RRT initiation, in patients without evident urgent indications. Concern exists about possible harm of early initiation (hypotension, hypophosphataemia, prolonged dialysis dependence) and a “watch and wait” strategy appears to be safe up to a certain point [125]. The recent AKIKI 2 trial compared ‘delayed’ with ‘very delayed’ initiation of RRT and demonstrated no difference in the number of RRT-free days between both groups [124] but 60-day mortality was higher in the ‘very delayed’ arm. Robust evidence for biomarker-guided RRT initiation is lacking [114]. The role of furosemide stress test is still to be established. In some patients, a more individualized decision may be appropriate [59].
Long-term outcome following AKI
Several retrospective studies, mostly based on administrative databases, have shown that AKI, even after apparent full recovery, is associated with unfavorable long-term outcomes, including an increased risk of dying, occurrence of cardiovascular events and development of (or progression to) CKD [2, 3]. These findings were recently confirmed in a prospective cohort (ASSESS study) including hospital survivors at 3 months with or without AKI during hospitalization with a median of 4.7-year follow-up [126]. The increased risk of death appears to prevail over the risk of new CKD. Main causes of mortality are cardiovascular events and cancer [3]. Besides the expected risk factors (e.g., age, comorbidities, AKI severity), the recovery pattern of AKI also appears to be associated with long-term mortality [127, 128].
Whether the association of AKI with long-term cardiovascular events is related to shared risk factors (diabetes, hypertension, heart failure, pre-existing CKD), the increased incidence of CKD (a known risk factor for cardiovascular disease [76]) or to a causal relationship has not been clearly established but evidence is accumulating that AKI itself may accelerate cardiovascular disease [3, 129], likely related to AKI-induced remote organ injury and systemic inflammation (organ cross-talk). The pathophysiology is likely multifactorial. Galectin-3, a substance that induces cardiac inflammation, cardiac fibrosis and cardiac dysfunction may contribute [83]. Activation of the RAAS after AKI has also been identified as a mediator of cardiovascular damage after AKI as Angiotensin II induces macrophage infiltration, cardiac inflammation and myocardial fibrosis ultimately leading to cardiac dysfunction and heart failure [130]. The risk of incident and progressive CKD has been shown in retrospective and prospective studies [3, 126] and can contribute to poor long-term outcomes. AKI is a risk factor for subsequent proteinuria [131], an independent predictor of adverse outcomes [126]. Other risk factors for the AKI–CKD transition include duration and severity of kidney injury, older age and chronic health conditions, including baseline kidney function. The ASSESS study showed that patients with AKI lasting more than 72 h had a higher risk of CKD than those with resolving AKI and that proteinuria at 3 months was a strong predictor of further deterioration of kidney function [132]. The mechanisms underlying the AKI–CKD transition are incompletely elucidated but thought to include maladaptive tubular repair, persistent microvascular damage and inflammation leading to fibrosis [133].
The need for better follow-up and long-term care of patients recovering from AKI or AKD is increasingly being recognized. Several institutions and health care systems have developed AKI follow-up clinics [134]. However, there is also a need for specific quality indicators and quality measures and better quantification of the impact of these organization on patient-centered outcomes [135]. Strategies to prevent long-term complications of AKI have emerged recently. First, prevention of new episodes of AKI (e.g., with avoidance of nephrotoxins) is important [64, 136]. Recognition and treatment of hypertension, diabetes, or obesity are potential key factors to improve long-term outcomes. In this line, an observational study showed that follow-up by a nephrologist was associated with lower long-term all-cause mortality following AKI [137], but implementation in clinical practice would impose a substantial burden on the nephrology community. This calls for risk-stratified follow-up [138], driven by proteinuria [129] and estimated GFR at discharge, potentially aided by biomarker levels [11, 56, 139].
Several cardio- and renoprotective pharmacological strategies hold promise to improve post-AKI outcomes. Reducing the consequences of RAAS activation after AKI appears to improve long-term mortality in patients recovering from AKI [140]. Treatments with selective mineralocorticoid receptor antagonists or sodium-glucose co-transporter-2 inhibitors have recently been shown to improve clinical outcomes (mortality or progression of CKD and cardiovascular events) in CKD patients [141, 142]. Whether these results translate into better post-AKI outcomes needs to be explored.
Conclusion
AKI is a heterogenous syndrome that may present in different phenotypes, not reflected in the current criteria that define AKI. Novel developments including biomarkers and machine learning hold promise to be more sensitive and predictive of the development of AKI. In addition, biomarker kinetics may reveal more insight into the pathogenesis as observed in the subclinical AKI groups identified. Nevertheless, clinical trials are needed to assess their real usefulness. If so, a more personalized approach, taking into account the underlying pathophysiology, may change the way AKI is viewed and managed in the future. In those patients who may need RRT, more clinical data regarding timing of initiation are now available illustrating that a somewhat reserved approach is acceptable. In addition, it is becoming clear that, apart from short-term consequences of AKI, long-term sequelae may occur in AKI survivors and follow-up of this specific patient group warrants further attention.
References
Disease K (2012) Improving global outcomes (KDIGO) acute kidney injury work group (2012) KDIGO clinical practice guidelines AKI: AKI definition. Kidney Int Supplements 2(1):19–36
Hoste EAJ, Kellum JA, Selby NM, Zarbock A, Palevsky PM, Bagshaw SM et al (2018) Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrol 14(10):607–625
James MT, Bhatt M, Pannu N, Tonelli M (2020) Long-term outcomes of acute kidney injury and strategies for improved care. Nat Rev Nephrol 16(4):193–205
Kellum JA, Prowle JR (2018) Paradigms of acute kidney injury in the intensive care setting. Nat Rev Nephrol 14(4):217–230
Schetz M, Schortgen F (2017) Ten shortcomings of the current definition of AKI. Intensive Care Med 43(6):911–913
Priyanka P, Zarbock A, Izawa J, Gleason TG, Renfurm RW, Kellum JA (2020) The impact of acute kidney injury by serum creatinine or urine output criteria on major adverse kidney events in cardiac surgery patients. J Thorac Cardiovasc Surg. https://doi.org/10.1016/j.jtcvs.2019.11.137
Kellum JA, Sileanu FE, Murugan R, Lucko N, Shaw AD, Clermont G (2015) Classifying AKI by urine output versus serum creatinine level. J Am Soc Nephrol 26(9):2231–2238
Ronco C, Bellomo R, Kellum J (2017) Understanding renal functional reserve. Intensive Care Med 43(6):917–920
Chen S (2013) Retooling the creatinine clearance equation to estimate kinetic GFR when the plasma creatinine is changing acutely. J Am Soc Nephrol 24(6):877–888
Schneider AG, Molitoris BA (2020) Real-time glomerular filtration rate: improving sensitivity, accuracy and prognostic value in acute kidney injury. Curr Opin Crit Care 26(6):549–555
Ostermann M, Zarbock A, Goldstein S, Kashani K, Macedo E, Murugan R, et al. Recommendations on acute kidney injury biomarkers from the acute disease quality initiative consensus conference: a consensus statement. JAMA Netw Open. 2020;3(10):e2019209.
Kashani K, Al-Khafaji A, Ardiles T, Artigas A, Bagshaw SM, Bell M et al (2013) Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Critical care (London, England) 17(1):R25
Hoste EA, McCullough PA, Kashani K, Chawla LS, Joannidis M, Shaw AD et al (2014) Derivation and validation of cutoffs for clinical use of cell cycle arrest biomarkers. Nephrol Dial Transpl 29(11):2054–2061
Su LJ, Li YM, Kellum JA, Peng ZY (2018) Predictive value of cell cycle arrest biomarkers for cardiac surgery-associated acute kidney injury: a meta-analysis. Br J Anaesth 121(2):350–357
Zhang D, Yuan Y, Guo L, Wang Q (2019) Comparison of urinary TIMP-2 and IGFBP7 cut-offs to predict acute kidney injury in critically ill patients: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 98(26):e16232
Albert C, Zapf A, Haase M, Rover C, Pickering JW, Albert A, et al. (2020) Neutrophil gelatinase-associated lipocalin measured on clinical laboratory platforms for the prediction of acute kidney injury and the associated need for dialysis therapy: a systematic review and meta-analysis. Am J Kidney Dis 76(6):826–41 e1.
Meersch M, Schmidt C, Hoffmeier A, Van Aken H, Wempe C, Gerss J et al (2017) Prevention of cardiac surgery-associated AKI by implementing the KDIGO guidelines in high risk patients identified by biomarkers: the PrevAKI randomized controlled trial. Intensive Care Med 43(11):1551–1561
Göcze I, Jauch D, Götz M, Kennedy P, Jung B, Zeman F et al (2018) Biomarker-guided Intervention to prevent acute kidney injury after major surgery: the prospective randomized BigpAK study. Ann Surg 267(6):1013–1020
Husain-Syed F, Ferrari F, Sharma A, Danesi TH, Bezerra P, Lopez-Giacoman S et al (2018) Preoperative renal functional reserve predicts risk of acute kidney injury after cardiac operation. Ann Thorac Surg 105(4):1094–1101
Husain-Syed F, Ferrari F, Sharma A, Hinna Danesi T, Bezerra P, Lopez-Giacoman S et al (2019) Persistent decrease of renal functional reserve in patients after cardiac surgery-associated acute kidney injury despite clinical recovery. Nephrol Dialysis Transpl 34(2):308–317
Hoste EA, Kashani K, Gibney N, Wilson FP, Ronco C, Goldstein SL et al (2016) Impact of electronic-alerting of acute kidney injury: workgroup statements from the 15(th) ADQI Consensus Conference. Can J Kidney Health Dis 3:10
Al-Jaghbeer M, Dealmeida D, Bilderback A, Ambrosino R, Kellum JA (2018) Clinical decision support for in-hospital AKI. J Am Soc Nephrol 29(2):654–660
Colpaert K, Hoste EA, Steurbaut K, Benoit D, Van Hoecke S, De Turck F et al (2012) Impact of real-time electronic alerting of acute kidney injury on therapeutic intervention and progression of RIFLE class. Crit Care Med 40(4):1164–1170
De Vlieger G, Kashani K, Meyfroidt G (2020) Artificial intelligence to guide management of acute kidney injury in the ICU: a narrative review. Curr Opin Crit Care 26(6):563–573
Gameiro J, Branco T, Lopes JA (2020) Artificial intelligence in acute kidney injury risk prediction. J Clin Med. 9(3):678. https://doi.org/10.3390/jcm9030678
Flechet M, Guiza F, Schetz M, Wouters P, Vanhorebeek I, Derese I et al (2017) AKIpredictor, an online prognostic calculator for acute kidney injury in adult critically ill patients: development, validation and comparison to serum neutrophil gelatinase-associated lipocalin. Intensive Care Med 43(6):764–773
Koyner JL, Carey KA, Edelson DP, Churpek MM (2018) The development of a machine learning inpatient acute kidney injury prediction model. Crit Care Med 46(7):1070–1077
Chiofolo C, Chbat N, Ghosh E, Eshelman L, Kashani K (2019) Automated continuous acute kidney injury prediction and surveillance: a random forest model. Mayo Clin Proc 94(5):783–792
Churpek MM, Carey KA, Edelson DP, Singh T, Astor BC, Gilbert ER et al (2020) Internal and external validation of a machine learning risk score for acute kidney injury. JAMA Netw Open 3(8):e2012892
Chaudhary K, Vaid A, Duffy A, Paranjpe I, Jaladanki S, Paranjpe M et al (2020) Utilization of deep learning for subphenotype identification in sepsis-associated acute kidney injury. Clin J Am Soc Nephrol 15(11):1557–1565
Wiersema R, Jukarainen S, Vaara ST, Poukkanen M, Lakkisto P, Wong H et al (2020) Two subphenotypes of septic acute kidney injury are associated with different 90-day mortality and renal recovery. Critical care (London, England) 24(1):150
Chawla LS, Bellomo R, Bihorac A, Goldstein SL, Siew ED, Bagshaw SM et al (2017) Acute kidney disease and renal recovery: consensus report of the acute disease quality initiative (ADQI) 16 Workgroup. Nat Rev Nephrol 13(4):241–257
Pons B, Lautrette A, Oziel J, Dellamonica J, Vermesch R, Ezingeard E et al (2013) Diagnostic accuracy of early urinary index changes in differentiating transient from persistent acute kidney injury in critically ill patients: multicenter cohort study. Critical care (London, England) 17(2):R56
Darmon M, Bourmaud A, Reynaud M, Rouleau S, Meziani F, Boivin A et al (2018) Performance of Doppler-based resistive index and semi-quantitative renal perfusion in predicting persistent AKI: results of a prospective multicenter study. Intensive Care Med 44(11):1904–1913
Coca SG, Nadkarni GN, Garg AX, Koyner J, Thiessen-Philbrook H, McArthur E et al (2016) First post-operative urinary kidney injury biomarkers and association with the duration of AKI in the TRIBE-AKI Cohort. PLoS ONE 11(8):e0161098
Meersch M, Schmidt C, Van Aken H, Martens S, Rossaint J, Singbartl K et al (2014) Urinary TIMP-2 and IGFBP7 as early biomarkers of acute kidney injury and renal recovery following cardiac surgery. PLoS ONE 9(3):e93460
Legrand M, Jacquemod A, Gayat E, Collet C, Giraudeaux V, Launay JM et al (2015) Failure of renal biomarkers to predict worsening renal function in high-risk patients presenting with oliguria. Intensive Care Med 41(1):68–76
Titeca-Beauport D, Daubin D, Van Vong L, Belliard G, Bruel C, Alaya S et al (2020) Urine cell cycle arrest biomarkers distinguish poorly between transient and persistent AKI in early septic shock: a prospective, multicenter study. Critical care (London, England) 24(1):280
Hoste E, Bihorac A, Al-Khafaji A, Ortega LM, Ostermann M, Haase M et al (2020) Identification and validation of biomarkers of persistent acute kidney injury: the RUBY study. Intensive Care Med 46(5):943–953
Dewitte A, Joannes-Boyau O, Sidobre C, Fleureau C, Bats ML, Derache P et al (2015) Kinetic eGFR and novel AKI biomarkers to predict renal recovery. Clin J Am Soc Nephrol 10(11):1900–1910
Koyner JL, Davison DL, Brasha-Mitchell E, Chalikonda DM, Arthur JM, Shaw AD et al (2015) Furosemide stress test and biomarkers for the prediction of AKI severity. J Am Soc Nephrol 26(8):2023–2031
Chen JJ, Chang CH, Huang YT, Kuo G (2020) Furosemide stress test as a predictive marker of acute kidney injury progression or renal replacement therapy: a systemic review and meta-analysis. Critical care (London, England) 24(1):202
Barasch J, Zager R, Bonventre JV (2017) Acute kidney injury: a problem of definition. Lancet 389(10071):779–781
Xu K, Rosenstiel P, Paragas N, Hinze C, Gao X, Huai Shen T et al (2017) Unique transcriptional programs identify subtypes of AKI. J Am Soc Nephrol 28(6):1729–1740
Mar D, Gharib SA, Zager RA, Johnson A, Denisenko O, Bomsztyk K (2015) Heterogeneity of epigenetic changes at ischemia/reperfusion- and endotoxin-induced acute kidney injury genes. Kidney Int 88(4):734–744
Garofalo AM, Lorente-Ros M, Goncalvez G, Carriedo D, Ballén-Barragán A, Villar-Fernández A et al (2019) Histopathological changes of organ dysfunction in sepsis. Intensive Care Med Exp 7(Suppl 1):45
Ahmad T, Jackson K, Rao VS, Tang WHW, Brisco-Bacik MA, Chen HH et al (2018) Worsening renal function in patients with acute heart failure undergoing aggressive diuresis is not associated with tubular injury. Circulation 137(19):2016–2028
Rao VS, Ahmad T, Brisco-Bacik MA, Bonventre JV, Wilson FP, Siew ED et al (2019) Renal effects of intensive volume removal in heart failure patients with preexisting worsening renal function. Circ Heart Fail 2(6):e005552
Yoshioka K, Matsue Y, Okumura T, Kida K, Oishi S, Akiyama E et al (2020) Impact of brain natriuretic peptide reduction on the worsening renal function in patients with acute heart failure. PLoS ONE 15(6):e0235493
Peerapornratana S, Manrique-Caballero CL, Gómez H, Kellum JA (2019) Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int 96(5):1083–1099
Gomez H, Ince C, De Backer D, Pickkers P, Payen D, Hotchkiss J et al (2014) A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock (Augusta, Ga) 41(1):3–11
Radi ZA (2018) Immunopathogenesis of Acute Kidney Injury. Toxicol Pathol 46(8):930–943
Poyan Mehr A, Tran MT, Ralto KM, Leaf DE, Washco V, Messmer J et al (2018) De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat Med 24(9):1351–1359
Kellum JA, Chawla LS (2016) Cell-cycle arrest and acute kidney injury: the light and the dark sides. Nephrol Dial Transplant 31(1):16–22
Hayek SS, Leaf DE, Samman Tahhan A, Raad M, Sharma S, Waikar SS et al (2020) Soluble urokinase receptor and acute kidney injury. N Engl J Med 382(5):416–426
Schunk SJ, Zarbock A, Meersch M, Kullmar M, Kellum JA, Schmit D et al (2019) Association between urinary dickkopf-3, acute kidney injury, and subsequent loss of kidney function in patients undergoing cardiac surgery: an observational cohort study. Lancet (London, England) 394(10197):488–496
Bhatraju PK, Zelnick LR, Herting J, Katz R, Mikacenic C, Kosamo S et al (2019) Identification of acute kidney injury subphenotypes with differing molecular signatures and responses to vasopressin therapy. Am J Respir Crit Care Med 199(7):863–872
Bhatraju PK, Cohen M, Nagao RJ, Morrell ED, Kosamo S, Chai XY et al (2020) Genetic variation implicates plasma angiopoietin-2 in the development of acute kidney injury sub-phenotypes. BMC Nephrol 21(1):284
Ostermann M, Bellomo R, Burdmann EA, Doi K, Endre ZH, Goldstein SL, et al. Controversies in acute kidney injury: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Conference. Kidney international. 2020;98(2):294–309.
McDonald JS, McDonald RJ, Williamson EE, Kallmes DF, Kashani K (2017) Post-contrast acute kidney injury in intensive care unit patients: a propensity score-adjusted study. Intensive Care Med 43(6):774–784
Hinson JS, Al Jalbout N, Ehmann MR, Klein EY (2019) Acute kidney injury following contrast media administration in the septic patient: a retrospective propensity-matched analysis. J Crit Care 51:111–116
Miyamoto Y, Iwagami M, Aso S, Yasunaga H, Matsui H, Fushimi K et al (2019) Association between intravenous contrast media exposure and non-recovery from dialysis-requiring septic acute kidney injury: a nationwide observational study. Intensive Care Med 45(11):1570–1579
Rouve E, Lakhal K, Salmon Gandonnière C, Jouan Y, Bodet-Contentin L, Ehrmann S (2018) Lack of impact of iodinated contrast media on kidney cell-cycle arrest biomarkers in critically ill patients. BMC Nephrol 19(1):308
Joannidis M, Druml W, Forni LG, Groeneveld ABJ, Honore PM, Hoste E et al (2017) Prevention of acute kidney injury and protection of renal function in the intensive care unit: update 2017: expert opinion of the working group on prevention, AKI section, European society of intensive care medicine. Intensive Care Med 43(6):730–749
Davenport MS, Perazella MA, Yee J, Dillman JR, Fine D, McDonald RJ et al (2020) Use of intravenous iodinated contrast media in patients with kidney disease: consensus statements from the american college of radiology and the national kidney foundation. Radiology 294(3):660–668
Timal RJ, Kooiman J, Sijpkens YWJ, de Vries JPM, Verberk-Jonkers I, Brulez HFH et al (2020) Effect of no prehydration vs sodium bicarbonate prehydration prior to contrast-enhanced computed tomography in the prevention of postcontrast acute kidney injury in adults with chronic kidney disease: the kompas randomized clinical trial. JAMA Intern Med 180(4):533–541
Weisbord SD, Gallagher M, Jneid H, Garcia S, Cass A, Thwin SS et al (2018) Outcomes after angiography with sodium bicarbonate and acetylcysteine. N Engl J Med 378(7):603–614
Arnaud FCS, Libório AB (2020) Attributable nephrotoxicity of vancomycin in critically ill patients: a marginal structural model study. J Antimicrob Chemother 75(4):1031–1037
Tsutsuura M, Moriyama H, Kojima N, Mizukami Y, Tashiro S, Osa S et al (2021) The monitoring of vancomycin: a systematic review and meta-analyses of area under the concentration-time curve-guided dosing and trough-guided dosing. BMC Infect Dis 21(1):153
Covert KL, Knoetze D, Cole M, Lewis P (2020) Vancomycin plus piperacillin/tazobactam and acute kidney injury risk: a review of the literature. J Clin Pharm Ther 45(6):1253–1263
Selby AR, Hall RG, 2nd. Utilizing the Patient Care Process to Minimize the Risk of Vancomycin-Associated Nephrotoxicity. J Clin Med. 2019;8(6).
Picard W, Bazin F, Clouzeau B, Bui HN, Soulat M, Guilhon E et al (2014) Propensity-based study of aminoglycoside nephrotoxicity in patients with severe sepsis or septic shock. Antimicrob Agents Chemother 58(12):7468–7474
Wagenlehner F, Lucenteforte E, Pea F, Soriano A, Tavoschi L, Steele VR, et al. Systematic review on estimated rates of nephrotoxicity and neurotoxicity in patients treated with polymyxins. Clin Microbiol Infect. 2021.
Sisay M, Hagos B, Edessa D, Tadiwos Y, Mekuria AN (2021) Polymyxin-induced nephrotoxicity and its predictors: a systematic review and meta-analysis of studies conducted using RIFLE criteria of acute kidney injury. Pharmacol Res 163:105328
Joannidis M, Forni LG, Klein SJ, Honore PM, Kashani K, Ostermann M et al (2020) Lung-kidney interactions in critically ill patients: consensus report of the acute disease quality initiative (ADQI) 21 workgroup. Intensive Care Med 46(4):654–672
Rangaswami J, Bhalla V, Blair JEA, Chang TI, Costa S, Lentine KL et al (2019) Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American heart association. Circulation 139(16):e840–e878
Depret F, Prud’homme M, Legrand M (2017) A role of remote organs effect in acute kidney injury outcome. Nephron 137(4):273–276
Lee SA, Cozzi M, Bush EL, Rabb H (2018) Distant organ dysfunction in acute kidney injury: a review. Am J Kidney Dis 72(6):846–856
Singbartl K, Formeck CL, Kellum JA (2019) Kidney-immune system crosstalk in AKI. Semin Nephrol 39(1):96–106
Thakar CV, Yared JP, Worley S, Cotman K, Paganini EP (2003) Renal dysfunction and serious infections after open-heart surgery. Kidney Int 64(1):239–246
Liaño F, Junco E, Pascual J, Madero R, Verde E (1998) The spectrum of acute renal failure in the intensive care unit compared with that seen in other settings. The madrid acute renal failure study group. Kidney Int Suppl 66:S16-24
Woodrow G, Turney JH (1992) Cause of death in acute renal failure. Nephrol Dial Transpl 7(3):230–234
Prud’homme M, Coutrot M, Michel T, Boutin L, Genest M, Poirier F et al (2019) Acute kidney injury induces remote cardiac damage and dysfunction through the galectin-3 pathway. JACC Basic Transl Sci 4(6):717–732
Hassoun HT, Lie ML, Grigoryev DN, Liu M, Tuder RM, Rabb H (2009) Kidney ischemia-reperfusion injury induces caspase-dependent pulmonary apoptosis. Am J Physiol Renal Physiol 297(1):F125–F137
Girling BJ, Channon SW, Haines RW, Prowle JR (2020) Acute kidney injury and adverse outcomes of critical illness: correlation or causation? Clin Kidney J 13(2):133–141
Tanaka S, Okusa MD (2020) Crosstalk between the nervous system and the kidney. Kidney Int 97(3):466–476
Ostermann M, Liu K, Kashani K (2019) Fluid management in acute kidney injury. Chest 156(3):594–603
Gambardella I, Gaudino M, Ronco C, Lau C, Ivascu N, Girardi LN (2016) Congestive kidney failure in cardiac surgery: the relationship between central venous pressure and acute kidney injury. Interact Cardiovasc Thorac Surg 23(5):800–805
Legrand M, Dupuis C, Simon C, Gayat E, Mateo J, Lukaszewicz AC et al (2013) Association between systemic hemodynamics and septic acute kidney injury in critically ill patients: a retrospective observational study. Crit Care 17(6):R278
Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B et al (2006) Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 354(24):2564–2575
Myles PS, Bellomo R, Corcoran T, Forbes A, Peyton P, Story D et al (2018) Restrictive versus liberal fluid therapy for major abdominal surgery. N Engl J Med 378(24):2263–2274
Vaara ST, Ostermann M, Bitker L, Schneider A, Poli E, Hoste E, et al. Restrictive fluid management versus usual care in acute kidney injury (REVERSE-AKI): a pilot randomized controlled feasibility trial. Intensive Care Med. 2021.
Hjortrup PB, Haase N, Bundgaard H, Thomsen SL, Winding R, Pettilä V et al (2016) Restricting volumes of resuscitation fluid in adults with septic shock after initial management: the CLASSIC randomised, parallel-group, multicentre feasibility trial. Intensive Care Med 42(11):1695–1705
Macdonald SPJ, Keijzers G, Taylor DM, Kinnear F, Arendts G, Fatovich DM et al (2018) Restricted fluid resuscitation in suspected sepsis associated hypotension (REFRESH): a pilot randomised controlled trial. Intensive Care Med 44(12):2070–2078
Corl KA, Prodromou M, Merchant RC, Gareen I, Marks S, Banerjee D et al (2019) The restrictive IV fluid trial in severe sepsis and septic shock (RIFTS): a randomized pilot study. Crit Care Med 47(7):951–959
Agrinier N, Monnier A, Argaud L, Bemer M, Virion JM, Alleyrat C et al (2019) Effect of fluid balance control in critically ill patients: Design of the stepped wedge trial POINCARE-2. Contemp Clin Trials 83:109–116
Brown RM, Wang L, Coston TD, Krishnan NI, Casey JD, Wanderer JP et al (2019) Balanced crystalloids versus saline in sepsis A secondary analysis of the SMART clinical trial. Am J Respir Crit Care Med 200(12):1487–1495
Raimundo M, Crichton S, Syed Y, Martin JR, Beale R, Treacher D et al (2015) Low systemic oxygen delivery and bp and risk of progression of early AKI. Clin J Am Soc Nephrol 10(8):1340–1349
Kellum JA, Chawla LS, Keener C, Singbartl K, Palevsky PM, Pike FL et al (2016) The effects of alternative resuscitation strategies on acute kidney injury in patients with septic shock. Am J Respir Crit Care Med 193(3):281–287
Asfar P, Radermacher P, Ostermann M (2018) MAP of 65: target of the past? Intensive Care Med 44(9):1551–1552
Lamontagne F, Richards-Belle A, Thomas K, Harrison DA, Sadique MZ, Grieve RD et al (2020) Effect of reduced exposure to vasopressors on 90-day mortality in older critically Ill patients with vasodilatory hypotension: a randomized clinical trial. JAMA 323(10):938–949
Asfar P, Meziani F, Hamel JF, Grelon F, Megarbane B, Anguel N et al (2014) High versus low blood-pressure target in patients with septic shock. N Engl J Med 370(17):1583–1593
Futier E, Lefrant JY, Guinot PG, Godet T, Lorne E, Cuvillon P et al (2017) Effect of individualized vs standard blood pressure management strategies on postoperative organ dysfunction among high-risk patients undergoing major surgery: a randomized clinical trial. JAMA 318(14):1346–1357
Moman RN, Ostby SA, Akhoundi A, Kashyap R, Kashani K (2018) Impact of individualized target mean arterial pressure for septic shock resuscitation on the incidence of acute kidney injury: a retrospective cohort study. Ann Intensive Care 8(1):124
Ostermann M, Hall A, Crichton S (2017) Low mean perfusion pressure is a risk factor for progression of acute kidney injury in critically ill patients—a retrospective analysis. BMC Nephrol 18(1):151
Wong BT, Chan MJ, Glassford NJ, Martensson J, Bion V, Chai SY et al (2015) Mean arterial pressure and mean perfusion pressure deficit in septic acute kidney injury. J Crit Care 30(5):975–981
Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R et al (2017) Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med 43(3):304–377
Morelli A, Ertmer C, Rehberg S, Lange M, Orecchioni A, Laderchi A et al (2008) Phenylephrine versus norepinephrine for initial hemodynamic support of patients with septic shock: a randomized, controlled trial. Crit Care 12(6):R143
Gordon AC, Mason AJ, Thirunavukkarasu N, Perkins GD, Cecconi M, Cepkova M et al (2016) Effect of early vasopressin vs norepinephrine on kidney failure in patients with septic shock: the VANISH randomized clinical trial. JAMA 316(5):509–518
Nagendran M, Russell JA, Walley KR, Brett SJ, Perkins GD, Hajjar L et al (2019) Vasopressin in septic shock: an individual patient data meta-analysis of randomised controlled trials. Intensive Care Med 45(6):844–855
Hajjar LA, Vincent JL, Barbosa Gomes Galas FR, Rhodes A, Landoni G, Osawa EA et al (2017) Vasopressin versus norepinephrine in patients with vasoplegic shock after cardiac surgery: the VANCS randomized controlled trial. Anesthesiology 126(1):85–93
Khanna A, English SW, Wang XS, Ham K, Tumlin J, Szerlip H et al (2017) Angiotensin II for the treatment of vasodilatory shock. N Engl J Med 377(5):419–430
Tumlin JA, Murugan R, Deane AM, Ostermann M, Busse LW, Ham KR et al (2018) Outcomes in patients with vasodilatory shock and renal replacement therapy treated with intravenous angiotensin II. Crit Care Med 46(6):949–957
Kane-Gill SL, Meersch M, Bell M (2020) Biomarker-guided management of acute kidney injury. Curr Opin Crit Care 26(6):556–562
Klein SJ, Brandtner AK, Lehner GF, Ulmer H, Bagshaw SM, Wiedermann CJ et al (2018) Biomarkers for prediction of renal replacement therapy in acute kidney injury: a systematic review and meta-analysis. Intensive Care Med 44(3):323–336
Lumlertgul N, Peerapornratana S, Trakarnvanich T, Pongsittisak W, Surasit K, Chuasuwan A et al (2018) Early versus standard initiation of renal replacement therapy in furosemide stress test non-responsive acute kidney injury patients (the FST trial). Critical care (London, England) 22(1):101
Pickkers P, Mehta RL, Murray PT, Joannidis M, Molitoris BA, Kellum JA et al (2018) Effect of human recombinant alkaline phosphatase on 7-day creatinine clearance in patients with sepsis-associated acute kidney injury: a randomized clinical trial. JAMA 320(19):1998–2009
Côté JM, Murray PT, Rosner MH (2020) New drugs for acute kidney injury. Curr Opin Crit Care 26(6):525–535
Zarbock A, Kellum JA, Schmidt C, Van Aken H, Wempe C, Pavenstädt H et al (2016) Effect of early vs delayed initiation of renal replacement therapy on mortality in critically ill patients with acute kidney injury: the ELAIN randomized clinical trial. JAMA 315(20):2190–2199
Bagshaw SM, Wald R, Adhikari NKJ, Bellomo R, da Costa BR, Dreyfuss D et al (2020) Timing of initiation of renal-replacement therapy in acute kidney injury. N Engl J Med 383(3):240–251
Gaudry S, Hajage D, Benichou N, Chaïbi K, Barbar S, Zarbock A et al (2020) Delayed versus early initiation of renal replacement therapy for severe acute kidney injury: a systematic review and individual patient data meta-analysis of randomised clinical trials. Lancet 395(10235):1506–1515
Gaudry S, Hajage D, Schortgen F, Martin-Lefevre L, Pons B, Boulet E et al (2016) Initiation strategies for renal-replacement therapy in the intensive care unit. N Engl J Med 375(2):122–133
Barbar SD, Clere-Jehl R, Bourredjem A, Hernu R, Montini F, Bruyere R et al (2018) Timing of renal-replacement therapy in patients with acute kidney injury and sepsis. N Engl J Med 379(15):1431–1442
Gaudry S, Hajage D, Martin-Lefevre L, Lebbah S, Louis G, Moschietto S et al (2021) Comparison of two delayed strategies for renal replacement therapy initiation for severe acute kidney injury (AKIKI 2): a multicentre, open-label, randomised, controlled trial. Lancet (London, England) 397(10281):1293–1300
Ostermann M, Lumlertgul N (2021) Wait and see for acute dialysis: but for how long? Lancet (London, England) 397(10281):1241–1243
Ikizler TA, Parikh CR, Himmelfarb J, Chinchilli VM, Liu KD, Coca SG et al (2021) A prospective cohort study of acute kidney injury and kidney outcomes, cardiovascular events, and death. Kidney Int 99(2):456–465
Bhatraju PK, Zelnick LR, Chinchilli VM, Moledina DG, Coca SG, Parikh CR et al (2020) Association between early recovery of kidney function after acute kidney injury and long-term clinical outcomes. JAMA Netw Open 3(4):e202682
Fiorentino M, Tohme FA, Wang S, Murugan R, Angus DC, Kellum JA (2018) Long-term survival in patients with septic acute kidney injury is strongly influenced by renal recovery. PLoS ONE 13(6):e0198269
Bansal N, Matheny ME, Greevy RA Jr, Eden SK, Perkins AM, Parr SK et al (2018) Acute kidney injury and risk of incident heart failure among US veterans. Am J Kidney Dis 71(2):236–245
Mehta RL, Rabb H, Shaw AD, Singbartl K, Ronco C, McCullough PA et al (2013) Cardiorenal syndrome type 5: clinical presentation, pathophysiology and management strategies from the eleventh consensus conference of the Acute dialysis quality initiative (ADQI). Contrib Nephrol 182:174–194
Parr SK, Matheny ME, Abdel-Kader K, Greevy RA Jr, Bian A, Fly J et al (2018) Acute kidney injury is a risk factor for subsequent proteinuria. Kidney Int 93(2):460–469
Hsu CY, Chinchilli VM, Coca S, Devarajan P, Ghahramani N, Go AS et al (2020) Post-acute kidney injury proteinuria and subsequent kidney disease progression: the assessment, serial evaluation, and subsequent sequelae in acute kidney injury (ASSESS-AKI) study. JAMA Intern Med 180(3):402–410
Ferenbach DA, Bonventre JV (2015) Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11(5):264–276
Silver SA, Goldstein SL, Harel Z, Harvey A, Rompies EJ, Adhikari NK et al (2015) Ambulatory care after acute kidney injury: an opportunity to improve patient outcomes. Can J Kidney Health Dis 2:36
Liu KD, Forni LG, Heung M, Wu VC, Kellum JA, Mehta RL et al (2020) Quality of care for acute kidney disease: current knowledge gaps and future directions. Kidney Int Rep 5(10):1634–1642
Kashani K, Rosner MH, Haase M, Lewington AJP, O’Donoghue DJ, Wilson FP et al (2019) Quality improvement goals for acute kidney injury. Clin J Am Soc Nephrol 14(6):941–953
Harel Z, Wald R, Bargman JM, Mamdani M, Etchells E, Garg AX et al (2013) Nephrologist follow-up improves all-cause mortality of severe acute kidney injury survivors. Kidney Int 83(5):901–908
James MT, Pannu N, Hemmelgarn BR, Austin PC, Tan Z, McArthur E et al (2017) Derivation and external validation of prediction models for advanced chronic kidney disease following acute kidney injury. JAMA 318(18):1787–1797
Puthumana J, Thiessen-Philbrook H, Xu L, Coca SG, Garg AX, Himmelfarb J, et al. (2021) Biomarkers of inflammation and repair in kidney disease progression. J Clin Invest 131(3).
Gayat E, Hollinger A, Cariou A, Deye N, Vieillard-Baron A, Jaber S et al (2018) Impact of angiotensin-converting enzyme inhibitors or receptor blockers on post-ICU discharge outcome in patients with acute kidney injury. Intensive Care Med 44(5):598–605
Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF et al (2020) Dapagliflozin in patients with chronic kidney disease. N Engl J Med 383(15):1436–1446
Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P et al (2020) Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med 383(23):2219–2229
Dai X, Zeng Z, Fu C, Zhang S, Cai Y, Chen Z (2015) Diagnostic value of neutrophil gelatinase-associated lipocalin, cystatin C, and soluble triggering receptor expressed on myeloid cells-1 in critically ill patients with sepsis-associated acute kidney injury. Critical care (London, England) 19:223
Delanaye P, Cavalier E, Morel J, Mehdi M, Maillard N, Claisse G et al (2014) Detection of decreased glomerular filtration rate in intensive care units: serum cystatin C versus serum creatinine. BMC Nephrol 15:9
Herget-Rosenthal S, Marggraf G, Husing J, Goring F, Pietruck F, Janssen O et al (2004) Early detection of acute renal failure by serum cystatin C. Kidney Int 66(3):1115–1122
Khorashadi M, Beunders R, Pickkers P, Legrand M (2020) Proenkephalin: a new biomarker for glomerular filtration rate and acute kidney injury. Nephron 144(12):655–661
Martensson J, Bellomo R (2014) The rise and fall of NGAL in acute kidney injury. Blood Purif 37(4):304–310
Glassford NJ, Schneider AG, Xu S, Eastwood GM, Young H, Peck L et al (2013) The nature and discriminatory value of urinary neutrophil gelatinase-associated lipocalin in critically ill patients at risk of acute kidney injury. Intensive Care Med 39(10):1714–1724
Geng J, Qiu Y, Qin Z, Su B (2021) The value of kidney injury molecule 1 in predicting acute kidney injury in adult patients: a systematic review and Bayesian meta-analysis. J Transl Med 19(1):105
Koyner JL, Garg AX, Shlipak MG, Patel UD, Sint K, Hong K et al (2013) Urinary cystatin C and acute kidney injury after cardiac surgery. Am J Kidney Dis 61(5):730–738
Faubel S (2020) SuPAR: a potential predictive biomarker for acute kidney injury. Nat Rev Nephrol 16(7):375–376
Lin X, Yuan J, Zhao Y, Zha Y (2015) Urine interleukin-18 in prediction of acute kidney injury: a systemic review and meta-analysis. J Nephrol 28(1):7–16
Xu Y, Xie Y, Shao X, Ni Z, Mou S (2015) L-FABP: a novel biomarker of kidney disease. Clin Chim Acta 445:85–90
De Loor J, Herck I, Francois K, Van Wesemael A, Nuytinck L, Meyer E et al (2017) Diagnosis of cardiac surgery-associated acute kidney injury: differential roles of creatinine, chitinase 3-like protein 1 and neutrophil gelatinase-associated lipocalin: a prospective cohort study. Ann Intensive Care 7(1):24
Hoste EA, Vaara ST, De Loor J, Haapio M, Nuytinck L, Demeyere K et al (2020) Urinary cell cycle arrest biomarkers and chitinase 3-like protein 1 (CHI3L1) to detect acute kidney injury in the critically ill: a post hoc laboratory analysis on the FINNAKI cohort. Critical care (London, England) 24(1):144
Zarbock A, Kellum JA, Schmidt C, Van Aken H, Wempe C, Pavenstadt H et al (2016) Effect of early vs delayed initiation of renal replacement therapy on mortality in critically ill patients with acute kidney injury: the elain randomized clinical trial. JAMA 315(20):2190–2199
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
PP declares having received speaker fees from Baxter and consultancy fees from AM-Pharma. MD declares having received speaker fees from Astelas, Gilead-Kite and MSD along with a grant from MSD outside the scope of this manuscript. EH declares speaker fees from Alexion and Sopachem (paid to the university), travel grant from AM-Pharma. MJ has received honoraria and research support from Baxter Healthcare Corp, AM-Pharma, CLS Behring, Fresenius, and Astute Medical. ML declares no conflict of interest. MO declares speaker fees from Fresenius and Biomerieux, research funding from Baxter, Biomerieux and LaJolla Pharma and consultancy fee from NxStage. JRP has received grant and/or research support from bioMérieux, consulting fees from Medibeacon Inc., Beckton Dickinson Inc and Jafron Biomedical Co Ltd and speaker’s honoraria from Nikkiso Europe GmbH, Baxter, Braun Medical Ltd, Fresenius Medical Care and Fresenius-Kabi UK. AS has received a grant from the Leenaards foundation, speaker honoraria from Fresenius Medical Care and consulting honoraria from B Braun Melsungen AG. MS has no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Pickkers, P., Darmon, M., Hoste, E. et al. Acute kidney injury in the critically ill: an updated review on pathophysiology and management. Intensive Care Med 47, 835–850 (2021). https://doi.org/10.1007/s00134-021-06454-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-021-06454-7