
Abstract
In our westernised society, the level of physical activity is low. Interventions that increase energy expenditure are generally associated with an improvement in metabolic health. Exercise and exercise training increase energy metabolism and are considered to be among the best strategies for prevention of type 2 diabetes mellitus. More recently, cold exposure has been suggested to have a therapeutic value in type 2 diabetes. At a cellular level, there is evidence that increasing the turnover of cellular substrates such as fatty acids is associated with preventive effects against lipid-induced insulin resistance. Cellular energy sensors may underlie the effects linking energy turnover with metabolic health effects. Here we review data supporting the hypothesis that increasing energy and substrate turnover has beneficial effects on insulin sensitivity and should be considered a target for the prevention and treatment of type 2 diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obesity is the major risk factor for the development of type 2 diabetes mellitus. It is not only characterised by an increased storage of fat in subcutaneous white adipose tissue but is also associated with increased storage of fat in non-adipose tissues, such as muscle and liver. This so-called ectopic fat accumulation is thought to lead to the development of insulin resistance, an early hallmark in the development of diabetes [1, 2]. Therefore, weight loss is the first-choice preventive and therapeutic intervention for type 2 diabetes; indeed, a reduction in body weight leads to improvement in insulin sensitivity and weight reduction is a good strategy for preventing the development of diabetes. However, long-term maintenance of a healthy weight is difficult.
Although whole-body energy balance is determined by both energy intake and energy expenditure, the window of opportunity for achieving a negative energy balance (and thus weight loss) is much larger when energy intake is challenged. A diet very low in energy reduces energy intake by up to 70–80%, whereas a similar percentage increase in energy expenditure would require a very substantial amount of physical activity, such as that achieved by participating in competitive sports. Indeed, exercise training programmes do not lead to major weight loss; for example, it has been shown that jogging the equivalent of 20 miles per week or walking 12 miles per week only resulted in a weight loss of 3.5 kg and 1.1 kg, respectively, after an 8 month training period [3]. Consequently, over recent decades most of the research in the obesity field has focused on reducing energy intake to combat obesity and type 2 diabetes, and the role of energy expenditure in metabolic health has been underappreciated and understudied. However, it is becoming increasingly evident that enhancing energy metabolism per se can counterbalance the metabolic consequences of obesity and that increasing energy turnover is an important target in the prevention of obesity-related metabolic disturbances, such as insulin resistance. For example, even though exercise has only minor effect on body weight, it is known to have a beneficial effect on many metabolism-related diseases such as type 2 diabetes [4], cardiovascular disease [5], the metabolic syndrome [6] and even cancer [7]. This notion is underscored further by the recent evidence that breaking sedentary time can have major effects on insulin sensitivity and metabolic profile [8–12], suggesting that even small increases in physical activity levels can have a major impact on health. Similarly, other environmental factors such as cold exposure can have substantial boosting effects on energy metabolism and are associated with metabolic health effects (see below). Here we review the evidence, focusing on human intervention studies, suggesting that increasing energy turnover by exercise and/or cold exposure can offset obesity-related insulin resistance and be a preventive strategy for type 2 diabetes mellitus.
Defining components of human energy metabolism
Heat production
In the (human) body, almost all energy is ultimately converted into heat [13], and heat production is the gold standard for the measurement of energy expenditure under resting conditions. Apart from the performance of external work, energy in the body is used in the form of ATP for all metabolic processes that have heat production as a final end-product. The resting metabolic rate (RMR) is largely determined by the sum and the efficiency of these processes; for example, the continuous cycle of protein synthesis and breakdown is an energy-requiring process, and an increase in protein turnover results in the extra production of heat and thus elevated energy expenditure. The basal metabolic rate (BMR) is defined as the fasting resting energy expenditure in the morning in thermoneutral conditions; under less strict conditions this is the RMR [14]. The metabolic rate during sleep is slightly lower than the BMR, as being awake requires energy. As well as the BMR there is diet-induced thermogenesis (DIT), also known as thermogenic effect of food, which is the amount of extra heat related to the digestion, absorption and intermediary processing of food. DIT is in the order of magnitude of 5–10% of the total energy intake under energy balance conditions. Physical activity (exercise) also increases heat production, due to the increased energy demand necessary to perform the external work, particularly since only 20–25% of the produced energy can be used for external work (i.e. low mechanical energy efficiency with 75–80% of produced energy lost as heat) [15]. Although physical activity energy expenditure can be large—up to 4–5 times the BMR—it is highly variable both within and between individuals. Only low levels of physical activity can be sustained for long periods and in the general population intensive exercise only tends to consist of short bouts. As a result, physical activity energy expenditure typically forms about 30% of the 24 h energy expenditure in free-living individuals [16].
Heat regulation
Heat produced by the above processes is lost via respiration (evaporation) and via the skin (conduction, convection, radiation and evaporation). Under resting, thermoneutral conditions there is heat balance at a core body temperature of around 37°C, so no extra heat production is needed. However, if the environmental temperature is above or below the thermoneutral zone heat production increases [17]. This increased production of heat can be achieved by increased turnover of metabolic processes and induction of futile cycles, by muscle contraction (shivering) and also by so-called mitochondrial uncoupling. Thus in the cell and mitochondria, energy substrates are broken down in processes such as β-oxidation, glycolysis and the tricarboxylic acid cycle, ultimately leading to the build-up of a proton gradient over the inner mitochondrial membrane and ATP generation [18]. Mitochondrial uncoupling lowers the proton gradient without ATP formation, thereby reducing energy efficiency and indirectly stimulating heat production. Shivering can increase energy expenditure to up to four times the BMR [19], but cannot be maintained for prolonged periods as it is uncomfortable, decreases coordination and results in muscle fatigue. Non-shivering thermogenesis (NST), on the other hand, among other processes occurring via regulated mitochondrial uncoupling, can be sustained, is not uncomfortable (it is insensible) and does not affect coordination. The maximal reported NST is 40% of the RMR [20] but varies between individuals; in healthy lean individuals it ranges from 0% to 30% of the RMR [21–23].
Unless physical activity levels are raised to those seen in competitive sports, whole-body 24 h energy expenditure can typically only be sustainably elevated by ∼10–20% in humans. Nevertheless, many of the interventions that increase energy expenditure have marked metabolic health effects. Given the relatively minor effects on whole-body 24 h energy expenditure, which is often also compensated for by increased energy intake, the beneficial effects of interventions such as exercise and cold exposure cannot be attributed to weight loss. It is worth noting that this does not imply that energy expenditure has no role in body-weight regulation since a small increase or decrease in the BMR of 5% could theoretically lead to a reduction in body weight of ∼5–10 kg in a year, if not compensated by other means [24]. Indeed, a low RMR has been shown to be a risk factor for the development of obesity [25]. Nevertheless, most intervention studies in which energy expenditure is elevated do show beneficial metabolic health effects without changes in body weight.
Evidence that enhancing energy turnover improves insulin sensitivity: mitochondrial uncoupling
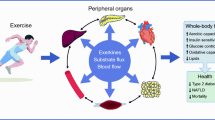
If enhancing energy turnover does not lead to major weight loss, the question arises as to what mechanism can explain the beneficial health effects of enhanced energy turnover (Fig. 1). A variety of mechanisms, such as muscle remodelling, sympathetic nervous system activation, hormonal changes and mitochondrial biogenesis, have been suggested to underlie the metabolic health effects of specific interventions like exercise and exposure to cold, and it is difficult to prove that these effects are direct. Nevertheless, there is clear evidence that boosting energy turnover may have a direct beneficial health effect as it is underscored by studies in which energy turnover is increased by inducing mitochondrial uncoupling. Overexpression of the mitochondrial uncoupling proteins UCP1 or UCP3 in skeletal muscle increases energy expenditure and improves insulin sensitivity [26–28]. Mitochondrial uncoupling can, apart from exercise training or cold exposure, also be increased by chemical agents such as 2,4-dinitrophenol (DNP), although the use of DNP in humans was banned in the 1930s after several cases of lethal hyperthermia. Recently, however, it was shown that targeting DNP towards the liver reduced hypertriacylglycerolaemia, fatty liver and whole-body insulin resistance in high-fat-fed rats and decreased hyperglycaemia in a rat model of type 2 diabetes [29]. Similarly, it was recently shown that niclosamide ethanolamine salt (NEN) induced mitochondrial uncoupling in mice, increased energy expenditure and lipid metabolism and was efficacious in preventing and treating hepatic steatosis and insulin resistance induced by a high-fat diet [30]. Moreover, NEN improved glycaemic control and delayed disease progression in db/db mice [30]. So far, drugs that induce mitochondrial uncoupling specifically in certain tissues are not available for use in humans. Nevertheless, it has been shown that insulin-sensitive endurance-trained athletes display elevated mitochondrial uncoupling and enhanced substrate turnover [31]. Recently, we showed that endurance-trained athletes have an increased sensitivity to fatty acid-induced uncoupling, together with elevated levels of the mitochondrial protein adenine nucleotide translocase 1 (ANT1), which is thought to be involved in facilitating fatty acid-induced uncoupling [32, 33]. Interestingly, the level of fatty acid-induced uncoupling was related to the level of insulin sensitivity and reducing ANT1 levels in C2C12 muscle cells reduced fatty acid-induced uncoupling and insulin-stimulated glucose uptake [34]. In addition, thyroid hormone has been shown to induce mitochondrial uncoupling in skeletal muscle and to increase thermogenesis and RMR in humans [35]. Together, these results suggest that mitochondrial uncoupling, likely to lead to enhanced energy turnover, improves glucose homeostasis in rodents and humans.

Schematic overview of how interventions that target energy turnover, such as exercise and cold exposure, may affect metabolic health. Although the exact mechanisms are unknown, cold exposure and exercise have been shown to increase or improve the following factors (indicated by green arrows): turnover of fat in ectopic sites (liver, skeletal muscle); mitochondrial function and mitochondrial uncoupling (in liver, BAT and skeletal muscle); energy expenditure (in all tissues); and fatty acid turnover (in liver and skeletal muscle). These effects may lead to alterations in energy balance and body weight but may also lead to improvement in insulin sensitivity independent of change in body weight
Link between energy turnover, ectopic fat and insulin sensitivity
Athlete’s paradox
As outlined above, one of the major determinants of obesity-associated metabolic complications is the location of the storage of excess fat, with fat accumulation in metabolically active tissues such as muscle and liver leading to insulin resistance of these tissues [1, 2]. One could thus speculate that enhancing mitochondrial uncoupling, thereby increasing cellular energy expenditure, would lead to the burning-off of intracellular fat and reduction of ectopic fat stores. Although this may be true for the studies mentioned above, improvement in metabolic health via energy metabolism-enhancing interventions do not necessarily require a reduction in ectopic fat. The best known example of this is the so-called athletes paradox—even though accumulation of fat in skeletal muscle is associated with insulin resistance, highly insulin-sensitive endurance-trained athletes also have very high levels of intramyocellular lipid [36]. This discrepancy has often been explained by the notion that it is fatty acid intermediates (such as ceramides and diacylglycerol) and not intramyocellular triacylglycerol (the major form in which fat is accumulated in lipid droplets) that lead to insulin resistance, and that these intermediates accumulate when the mitochondrial oxidative capacity of the muscle is low [37, 38]. However, endurance-trained athletes also have elevated levels of diacylglycerol [39], and muscle-specific overexpression of diacylglycerol acyltransferase (the enzyme converting diacylglycerol into triacylglycerol) leads to improved insulin sensitivity despite elevated diacylglycerol levels [40]. Furthermore, the concept that simply having more mitochondria could counterbalance lipid-induced skeletal muscle insulin resistance is not entirely correct, as it has been shown that overexpression of peroxisome proliferator-activated receptor γ coactivator 1 (PGC1) in mice leads to an improved mitochondrial function, yet such mice are not protected from high-fat-diet-induced insulin resistance [41]. However, when these mice are stimulated to become physically active, and thereby use their capacity for elevated energy and substrate turnover, they are protected from insulin resistance [42]. Interestingly in that context, PGC1 not only regulates mitochondrial function but is also involved in the transcriptional regulation of lipid droplet coating proteins, which contribute to the regulation of fatty acid delivery to the mitochondria [43]. This suggests that intracellular transcriptional programs exist that not only regulate cellular oxidative capacity but are also tightly involved in the regulation of substrate release and delivery, thereby laying the basis for efficient energy and substrate turnover. In accordance, overexpression of the lipid droplet coating proteins perilipin (PLIN) 2 or PLIN5, which are involved in the fine-tuning of fatty acid release for mitochondrial use, in skeletal muscle or liver also prevents lipid-induced insulin resistance, despite increased fat accumulation [44–46].
Energy turnover
Human studies also show that it is not the level of lipid or intermediates per se that leads to insulin resistance, but that it is the turnover of these substrates that determines whether fat accumulation leads to insulin resistance (Fig. 1). Thus, we have previously shown that in individuals with type 2 diabetes the capacity to convert fatty acids into inert triacylglycerols is reduced when compared with obese controls, suggesting that triacylglycerol turnover is reduced. In accordance, Listenberger et al [47] showed that the capacity for triacylglycerol accumulation is an important determinant of fatty-acid-induced insulin resistance in skeletal muscle cells and that impairing triacylglycerol synthesis induced lipotoxicity. These in vitro and ex vivo findings are in accordance with those of Perreault et al [48] who elegantly determined fractional synthesis rate and intramyocellular lipid concentration in obese volunteers with impaired glucose tolerance vs BMI-matched normoglycaemic controls. Interestingly, it was found that intramyocellular lipid concentration was higher but fractional synthesis rate was lower in the glucose-intolerant individuals. These findings were confirmed in a later report by the same authors, although the finding could only be verified in men and not in women [49]. Also in liver, hepatic fat is not detrimental per se as it is known that fat in the liver has the important physiological function of temporarily buffering circulating fatty acids and triacylglycerols. However, chronic oversupply of fat to the liver, such as in obesity, leads to hepatic steatosis, increased VLDL-triacylglycerol production, hepatic insulin resistance and ultimately hepatic failure [50, 51]. Taken together, these findings suggest that the link between cellular fat accumulation and insulin sensitivity is not straightforward, but depends on the tight balance between cellular fat storage capacity and mitochondrial oxidative capacity, and specifically the fine-tuned regulation of the turnover of fatty acids. It may be hypothesised that increasing energy turnover—by exercise, cold exposure, thyroid hormone, uncoupling or other interventions—may improve insulin sensitivity by enhancing the turnover of fat in ectopic fat stores, and thereby preventing deleterious effects of excessive ectopic fat storage (Fig. 1). However, this concept would need to be tested.
Cellular mechanisms linking energy turnover to metabolic adaptations
At the cellular level, turnover of energy and substrates is driven by energy demand, either because ATP is needed for cellular processes or because the efficiency of ATP formation is reduced by mitochondrial uncoupling (Fig. 2). In the cell, an increase in energy use can lead to alterations in the AMP/ATP and NAD+/NADH ratios resulting in the activation of among others AMPK (Fig. 2) [52–54] and sirtuin 1 (SIRT1) [55]. These energy sensors are strongly involved in the regulation of cellular energy metabolism, and activation of these factors is associated with metabolic health effects, as has been reviewed extensively elsewhere [54, 56–58]. Thus, these energy sensors provide a molecular explanation for how boosting energy turnover could be beneficial for the prevention and treatment of type 2 diabetes mellitus. They are also direct targets for pharmacological and nutritional approaches for treating/preventing diabetes. In fact, the most widely prescribed glucose-lowering drug, metformin, has been suggested to work via the induction of energy stress in the cell and thereby activation of AMPK [59], although the exact working mechanism is still the subject of debate [60, 61].

Schematic representation of cellular mechanisms linking energy-boosting interventions and metabolic health effects. Fatty acids and glucose are taken up into muscle via their respective transporters (CD36 [yellow transporter] and GLUT4 [red transporter]), which translocate from intracellular stores upon AMPK activation. Fatty acids and glucose can be used as substrates inside mitochondria for ATP generation, or be stored as glycogen and triacylglycerol. In the process of lipid storage, lipotoxic fatty acid intermediates can be formed that inhibit GLUT4 translocation. Mitochondrial uncoupling, via uncoupling proteins (UCP1), affects the efficiency by which ATP is formed. When ATP demand exceeds ATP formation, ADP levels increase and AMP may be formed, leading to activation of AMPK. Exercise, cold exposure and dietary resveratrol (found in red wine, among other things) may affect molecular pathways that activate AMPK and SIRT1, which in turn leads to the activation of PGC1, a transcription factor involved in the regulation of mitochondrial metabolism. As a result, mitochondrial uncoupling or enhanced fatty acid turnover may occur, thereby preventing the negative effects of cellular substrate overload
Human interventions that increase energy turnover
Exercise
Adding exercise to daily activities enhances whole-body energy expenditure and during exercise energy expenditure can be elevated several fold. It is also known that exercise training is one of the best strategies for the prevention or treatment of type 2 diabetes.
Acute exercise
Acute exercise increases glucose homeostasis through the activation of AMPK—the energy sensor of the cell—leading to the translocation of GLUT4 to the cell membrane, an alternative to insulin-induced GLUT4 translocation (Fig. 2) [62]. Furthermore, and in line with the previous outlined hypothesis, it was demonstrated by Schenk and Horowitz [63] that one bout of acute exercise could reverse lipid-induced insulin resistance in humans, and that this was accompanied by an increased partitioning of excess fatty acids towards triacylglycerol synthesis in muscle. Therefore, by increasing energy turnover and AMPK activation inside muscle, acute exercise can acutely and beneficially affect ‘insulin sensitivity’, at least in skeletal muscle.
Chronic exercise
Chronic exercise most likely leads to skeletal muscle adaptation via chronic activation of AMPK, although many other mechanisms have been suggested to explain why exercise-trained skeletal muscle has an improved insulin sensitivity. We will not review these mechanisms here, as they have been reviewed extensively elsewhere [64–67]. However, one of the best-described adaptations to chronic exercise training is an improvement in muscle oxidative capacity and mitochondrial function. Indeed, we [68, 69] and others [70–73] have shown that in type 2 diabetes patients an exercise-induced improvement in insulin sensitivity is associated with an improvement in mitochondrial function. In line with the hypothesis that elevated substrate turnover associates with insulin sensitivity, exercise training for weeks to months results in an improvement in insulin sensitivity not only in healthy individuals but also in individuals who are obese and have type 2 diabetes, even though levels of intramyocellular lipid are not lowered. In fact, intramyocellular lipid is often further increased [43]. Interestingly, endurance-trained athletes have higher levels of PLINs [43], suggesting improved regulation of substrate turnover with exercise training. Indeed, Bergman et al [74] showed that the fractional synthesis rate of intramyocellular triacyglycerol was significantly increased in endurance-trained male cyclists when compared with age- and BMI-matched sedentary men.
More recently, attention has also shifted towards determining whether, and if so how, exercise can also be beneficial for other metabolic tissues. For example, it has been shown that exercise training can have beneficial effects on hepatic metabolism [75–77]. This may partly explain the beneficial effects of training on circulating triacylglycerol and cholesterol levels and on postprandial lipid metabolism, important factors in the development of obesity-related metabolic complications [78]. Interestingly, also in the liver a reduced energy metabolism has been linked to hepatic steatosis and hepatic insulin resistance, and it is tempting to speculate that exercise training beneficially affects these variables too [79]. Importantly, the field of exercise physiology has been boosted in recent years by the identification of the exercising muscle as an important endocrine organ that secretes so-called myokines, which are involved in inter-organ communication. Recent discoveries include the hormones irisin (although controversial), meteorin-like, Angptl4 and β-aminoisobutyric acid [80–82]. In accordance, these myokines have been suggested to affect whole-body energy metabolism, including activation of brown or beige adipocytes, reduce inflammation and improve hepatic fat oxidation. For example, Angptl4 was identified as a novel myokine that regulates excessive fat storage in non-exercising muscle and the heart [83], suggesting that increasing energy turnover in the active muscle may also affect substrate turnover in non-exercising muscles. Clearly, more studies are needed in humans to unravel how exercise-induced increases in energy turnover may affect metabolism in tissues and organs other than skeletal muscle. This may open an entirely new outlook on understanding how exercise may be able to improve health in general.
Exercise mimetics
The beneficial effects of exercise on metabolic health have stimulated the search for drugs or nutritional compounds that can mimic the effects of exercise. Although no pill could be expected to induce all health effects of exercise, opportunities lie in the molecular pathways that are central to much of the exercise-induced improvements in metabolic health. Resveratrol is one such nutritional compound, found in red wine, among other things, which has been extensively studied in pre-clinical experiments and has also been tested in humans. Resveratrol can activate the SIRT1–AMPK–PGC1 axis, and could thereby be described as an exercise-mimetic (Fig. 2). Although results are not wholly consistent, clinical trials in type 2 diabetes patients imply that resveratrol has a glucose-lowering effect (for review see [84]). Unfortunately, the number of human interventions with molecular details is so far limited. We have previously shown that resveratrol can indeed activate AMPK and increase SIRT1 and PGC1 levels in human skeletal muscle and results in elevated mitochondrial function and reduced hepatic fat content [40]. Interestingly and consistent with the outline above, resveratrol intake also resulted in increased intramyocellular lipid content and PLIN expression [85], suggesting that not only oxidative capacity but also the capacity for energy turnover is boosted and linked to improved metabolic health, as is the case with exercise training. However, resveratrol also reduces the RMR and the long-term consequences of this would need to be established. So far, the number of clinical trials using resveratrol or other AMPK/SIRT1 targets is still very small but this is expected to change rapidly in the coming years.
Cold exposure
Environmental temperature can have a major effect on whole-body and cellular energy expenditure (Fig. 1). When body heat loss is substantially increased by exposure to the cold, high rates of lipid and carbohydrate oxidation are essential to maintain an increased metabolic rate. Indeed, severe cold exposure in animals has been shown to increase lipolysis, lipid oxidation and NEFA turnover (among others: [86]), as well as glucose oxidation and turnover, and thereby improves glucose tolerance and peripheral glucose uptake (Fig. 2) [87, 88]. These findings indicate that an increase in cold-induced shivering thermogenesis can have pronounced effects on glucose homeostasis; however, these studies were conducted under severe cold exposure, a condition that cannot be sustained for long periods of time.
Role of NST
In contrast to extreme cold, mild cold exposure is an intervention that is feasible for application in humans for longer periods of time. When humans and animals are exposed to milder cold, NST increases to produce heat. Interestingly, this NST is blunted in obese individuals and is significantly reduced when compared with NST in lean counterparts [89]. This relatively low NST may be related to body composition, as overweight and obese individuals have much more (subcutaneous) body fat and have more tissue insulation. Therefore, in daily (indoor) situations obese people experience much less cold than their lean counterparts, thereby triggering NST to a lesser extent. Indeed, after weight loss, morbidly obese patients show an increased NST capacity [90]. In older people, NST is reduced and, together with reduced body temperatures in the cold, their net energy expenditure in the cold may even be lower than at thermoneutral conditions [17]. The extent to which reduced NST in the elderly is caused by habituation or biological factors related to ageing is not known. Older people in western society generally tend to spend more time indoors in relatively warm and stable environments and are less tolerant of lower ambient temperatures. Therefore they may have lost their NST capacity. Both by living in such a protective stable environment and by increased body fat, NST and related metabolic processes are diminished. On the other hand, biological ageing itself may also affect their metabolic cold responses. Intriguingly, NST is also blunted in patients with type 2 diabetes [91]. However, the lower NST in type 2 diabetes may be related to the fact that most type 2 diabetes patients are older and overweight. It is currently not known whether a low NST plays a role in the aetiology of type 2 diabetes.
Role of brown adipose tissue
In rodents the main tissue responsible for NST is brown adipose tissue (BAT) [92]. Although NST in humans had been reported earlier, its relationship to functional BAT in adult humans was not revealed until 2009 [93–96]. In contrast to white adipose tissue, BAT burns triacylglycerol and glucose to generate heat through mitochondrial uncoupling [92]. Cold is the main stimulator of sympathetic nervous system-mediated BAT activation. Human brown fat is mainly studied by fluorodeoxyglucose–PET/CT imaging, which shows glucose uptake (rate) in those tissues that use glucose [97]. BAT is not activated in fasting and thermoneutral conditions [98], while it is activated by mild cold (i.e. without shivering) [94]. Using the appropriate individual cooling protocols [99], BAT glucose uptake was found to be significantly related to NST, indicating a role for BAT in whole-body thermogenesis, as shown in rodent studies. In parallel with NST, cold-induced BAT activation is reduced in obese and elderly individuals and those with type 2 diabetes [91, 100, 101]. Only recently, studies on the effect of cold acclimation on NST and BAT activity have been performed. Cold acclimation by intermittent exposure to a cool (14–17°C), or cold (10°C) environment resulted in significant increases in NST capacity [102]. A 10 day cold acclimation study with 6 h exposure to 14–15°C per day was enough to significantly increase NST by 65% on average [103]. A 6 week mild cold acclimation study (daily 2 h cold exposure at 17°C) also resulted in an increase in NST together with a concomitant decrease in body fat mass [104]. The latter two studies also revealed significant increases in BAT presence and activation [103, 104]. All in all, cold-induced BAT activity is significant in adults and parallels NST. The actual quantitative contributions of BAT and of other tissues (e.g. skeletal muscle) to whole-body NST are, however, not elucidated and await further studies. Furthermore, more information is needed on the duration, timing and temperatures to find out which treatments are most effective with respect to increasing NST.
Whether activation of BAT (potentially via elevating NST) affects glucose homeostasis and insulin sensitivity has not been studied extensively. Thus, glucose is oxidised in high amounts by BAT when activated, although the direct contribution of glucose oxidation to total thermogenesis in BAT is believed to be relatively small compared with that of fat oxidation, somewhere in the range of 10–16% [92, 105, 106]. It is likely that the glucose that is taken up is mainly used for the synthesis of glycerol-3-phosphate and triacylglycerols and also for the supply of extramitochondrial ATP through glycolysis to support fatty acid esterification to triacylglycerol and other cellular functions [107]. A study on noradrenaline (norepinephrine) stimulation of rat brown adipocytes revealed that glucose uptake and oxygen consumption were related. It has also been shown that increasing BAT by transplantation in mice has advantageous effects on body composition, insulin sensitivity and glucose metabolism [108].
In humans, retrospective patient studies show inverse relationships between BAT activity and diabetes and glycaemia [96, 109]. Prospective cold exposure studies show, as mentioned above, that glucose uptake in BAT is positively related to NST [103, 110]. These observations show that cold-induced thermogenesis goes hand in hand with increased glucose metabolism. The acute cold glucose uptake rate per unit of tissue mass as determined by dynamic PET/CT was higher in BAT than in skeletal muscle [110, 111]. Interestingly, insulin-stimulated glucose uptake in BAT in humans was positively related to the M value (a measure of whole-body insulin sensitivity derived from hyperinsulinaemic–euglycaemic clamps) [112].
Whether the increased uptake of glucose by BAT significantly contributes to whole-body glucose metabolism in type 2 diabetes has not yet been substantiated, although it was recently shown that individuals with active BAT, when compared with individuals without BAT, showed significantly increased resting energy expenditure, whole-body glucose disposal, plasma glucose oxidation and insulin sensitivity [113]. In another study, Lee et al [114] showed that staying overnight in cold chambers (19°C) for 1 month increased BAT activity together with improved postprandial insulin sensitivity. In a recent study in individuals with type 2 diabetes we studied the effect of cold acclimation on BAT activity and insulin sensitivity using hyperinsulinaemic–euglycaemic clamp. The cold acclimation protocol was identical to that used by van der Lans et al [103], where we found significant increases in BAT and NST. In type 2 diabetes patients the amount of BAT at baseline was significantly lower than that in healthy lean individuals [91]. Acclimation increased BAT activity significantly but levels were still very low [91]. Very interestingly, insulin sensitivity increased after cold acclimation by 43% on average [91]. It is very unlikely that the small increase in BAT activity could be responsible for this improved insulin sensitivity. In fact, the study showed that the improved insulin sensitivity could be explained by enhanced GLUT4 translocation in skeletal muscle in the basal state, an effect that had been previously observed in cold-acclimated rats [115] and has been confirmed in obese humans [116]. Although the mechanisms responsible for GLUT4 translocation upon cold stimulation remain to be elucidated, these findings clearly demonstrate that the significant improvement in insulin sensitivity can be attributed to skeletal muscle tissue, rather than to BAT, and may involve increased energy turnover. However, since BAT increased in all participants after cold acclimation, an indirect role for BAT (e.g. by secreting BATokines) cannot be fully excluded.
Conclusions
In conclusion, energy metabolism in humans can be affected by exercise and cold-exposure interventions. Such interventions are associated with metabolic health effects that cannot be explained by effects on body-weight regulation. Rather, increasing energy turnover activates cellular energy sensors, such as AMPK, which trigger beneficial adaptive responses. Although the exact underlying mechanisms are still not fully understood in humans, exercise and mild cold exposure provide strong intervention strategies for the prevention and treatment of type 2 diabetes mellitus.
Abbreviations
- ANT1:
-
Adenine nucleotide translocase 1
- BAT:
-
Brown adipose tissue
- BMR:
-
Basal metabolic rate
- DIT:
-
Diet-induced thermogenesis
- DNP:
-
2,4-Dinitrophenol
- NEN:
-
Niclosamide ethanolamine salt
- NST:
-
Non-shivering thermogenesis
- PLIN:
-
Perilipin
- PGC1:
-
Peroxisome proliferator-activated receptor γ coactivator 1
- RMR:
-
Resting metabolic rate
- SIRT1:
-
Sirtuin 1
References
Krssak M, Falk Petersen K, Dresner A et al (1999) Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 42:113–116
Perseghin G, Scifo P, De Cobelli F et al (1999) Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 48:1600–1606
Slentz CA, Duscha BD, Johnson JL et al (2004) Effects of the amount of exercise on body weight, body composition, and measures of central obesity: STRRIDE--a randomized controlled study. Arch Intern Med 164:31–39
Tuomilehto J, Lindstrom J, Eriksson JG et al (2001) Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 344:1343–1350
Manson JE, Greenland P, LaCroix AZ et al (2002) Walking compared with vigorous exercise for the prevention of cardiovascular events in women. N Engl J Med 347:716–725
Hahn V, Halle M, Schmidt-Trucksass A, Rathmann W, Meisinger C, Mielck A (2009) Physical activity and the metabolic syndrome in elderly German men and women: results from the population-based KORA survey. Diabetes Care 32:511–513
Friedenreich CM, Neilson HK, Lynch BM (2010) State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 46:2593–2604
Dunstan DW, Salmon J, Healy GN et al (2007) Association of television viewing with fasting and 2-h postchallenge plasma glucose levels in adults without diagnosed diabetes. Diabetes Care 30:516–522
Healy GN, Dunstan DW, Salmon J et al (2008) Breaks in sedentary time: beneficial associations with metabolic risk. Diabetes Care 31:661–666
Thorp AA, Healy GN, Owen N et al (2010) Deleterious associations of sitting time and television viewing time with cardiometabolic risk biomarkers: Australian Diabetes, Obesity and Lifestyle (AusDiab) study 2004-2005. Diabetes Care 33:327–334
Duvivier BM, Schaper NC, Bremers MA et al (2013) Minimal intensity physical activity (standing and walking) of longer duration improves insulin action and plasma lipids more than shorter periods of moderate to vigorous exercise (cycling) in sedentary subjects when energy expenditure is comparable. PLoS One 8, e55542
Ford ES, Li C, Zhao G, Pearson WS, Tsai J, Churilla JR (2010) Sedentary behavior, physical activity, and concentrations of insulin among US adults. Metabolism 59:1268–1275
Rolfe DFS, Brown GC (1997) Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 77:731–758
Ravussin E, Lillioja S, Anderson TE, Christin L, Bogardus C (1986) Determinants of 24-hour energy expenditure in man. Methods and results using a respiratory chamber. J Clin Investig 78:1568–1578
Gissane C, Corrigan DL, White JA (1991) Gross efficiency responses to exercise conditioning in adult males of various ages. J Sports Sci 9:383–391
Butte NF, Ekelund U, Westerterp KR (2012) Assessing physical activity using wearable monitors: measures of physical activity. Med Sci Sports Exerc 44:S5–S12
Kingma B, Frijns A, van Marken Lichtenbelt W (2012) The thermoneutral zone: implications for metabolic studies. Front Biosci (Elite Ed) 4:1975–1985
Mitchell P (1966) Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol Rev Camb Philos Soc 41:445–502
Benzinger TH (1969) Heat regulation: homeostasis of central temperature in man. Physiol Rev 49:671–759
Vosselman MJ, Vijgen GH, Kingma BR, Brans B, van Marken Lichtenbelt WD (2014) Frequent extreme cold exposure and brown fat and cold-induced thermogenesis: a study in a monozygotic twin. PLoS One 9, e101653
Dauncey MJ (1981) Influence of mild cold on 24 h energy expenditure, resting metabolism and diet-induced thermogenesis. Br J Nutr 45:257–267
Warwick PM, Busby R (1990) Influence of mild cold on 24 h energy expenditure in ʻnormallyʼ clothed adults. Br J Nutr 63:481–488
van Ooijen AMJ, van Marken Lichtenbelt WD, van Steenhoven AA, Westerterp K (2004) Seasonal changes in metabolic and temperature responses to cold air in humans. Physiol Behav 82:545–553
Wijers SL, Saris WH, van Marken Lichtenbelt WD (2009) Recent advances in adaptive thermogenesis: potential implications for the treatment of obesity. Obes Rev 10:218–226
Ravussin E, Lillioja S, Knowler WC et al (1988) Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med 318:467–472
Clapham JC, Arch JR, Chapman H et al (2000) Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature 406:415–418
Li B, Nolte LA, Ju JS et al (2000) Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat Med 6:1115–1120
Huppertz C, Fischer BM, Kim YB et al (2001) Uncoupling protein 3 (UCP3) stimulates glucose uptake in muscle cells through a phosphoinositide 3-kinase-dependent mechanism. J Biol Chem 276:12520–12529
Perry RJ, Kim T, Zhang XM et al (2013) Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab 18:740–748
Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI (2015) Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347:1253–1256
Befroy DE, Petersen KF, Dufour S, Mason GF, Rothman DL, Shulman GI (2008) Increased substrate oxidation and mitochondrial uncoupling in skeletal muscle of endurance-trained individuals. Proc Natl Acad Sci U S A 105:16701–16706
Andreyev A, Bondareva TO, Dedukhova VI et al (1989) The ATP/ADP-antiporter is involved in the uncoupling effect of fatty acids on mitochondria. Eur J Biochem 182:585–592
Skulachev VP (1991) Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett 294:158–162
Sparks LM, Gemmink A, Phielix E et al (2016) ANT1-mediated fatty acid-induced uncoupling as a target for improving myocellular insulin sensitivity. Diabetologia 59:1030–1039
Lebon V, Dufour S, Petersen KF et al (2001) Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest 108:733–737
Goodpaster BH, He J, Watkins S, Kelley DE (2001) Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 86:5755–5761
Shulman GI (2000) Cellular mechanisms of insulin resistance. J Clin Invest 106:171–176
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350:664–671
Amati F, Dube JJ, Alvarez-Carnero E et al (2011) Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: another paradox in endurance-trained athletes? Diabetes 60:2588–2597
Timmers S, de Vogel-van den Bosch J, Hesselink MK et al (2011) Paradoxical increase in TAG and DAG content parallel the insulin sensitizing effect of unilateral DGAT1 overexpression in rat skeletal muscle. PLoS One 6, e14503
Choi CS, Befroy DE, Codella R et al (2008) Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc Natl Acad Sci U S A 105:19926–19931
Summermatter S, Shui G, Maag D, Santos G, Wenk MR, Handschin C (2013) PGC-1alpha improves glucose homeostasis in skeletal muscle in an activity-dependent manner. Diabetes 62:85–95
Koves TR, Sparks LM, Kovalik JP et al (2013) PPARgamma coactivator-1alpha contributes to exercise-induced regulation of intramuscular lipid droplet programming in mice and humans. J Lipid Res 54:522–534
Bosma M, Hesselink MK, Sparks LM et al (2012) Perilipin 2 improves insulin sensitivity in skeletal muscle despite elevated intramuscular lipid levels. Diabetes 61:2679–2690
Bosma M, Minnaard R, Sparks LM et al (2012) The lipid droplet coat protein perilipin 5 also localizes to muscle mitochondria. Histochem Cell Biol 137:205–216
Bosma M, Sparks LM, Hooiveld GJ et al (2013) Overexpression of PLIN5 in skeletal muscle promotes oxidative gene expression and intramyocellular lipid content without compromising insulin sensitivity. Biochim Biophys Acta 1831:844–852
Listenberger LL, Han X, Lewis SE et al (2003) Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 100:3077–3082
Perreault L, Bergman BC, Hunerdosse DM, Playdon MC, Eckel RH (2010) Inflexibility in intramuscular triglyceride fractional synthesis distinguishes prediabetes from obesity in humans. Obesity (Silver Spring) 18:1524–1531
Perreault L, Bergman BC, Hunerdosse DM, Eckel RH (2010) Altered intramuscular lipid metabolism relates to diminished insulin action in men, but not women, in progression to diabetes. Obesity (Silver Spring) 18:2093–2100
Ravussin E, Smith SR (2002) Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann N Y Acad Sci 967:363–378
Adiels M, Taskinen MR, Boren J (2008) Fatty liver, insulin resistance, and dyslipidemia. Curr Diab Rep 8:60–64
Hardie DG (2014) AMPK--sensing energy while talking to other signaling pathways. Cell Metab 20:939–952
Hardie DG (2015) AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol 33:1–7
Hardie DG (2014) AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annu Rev Nutr 34:31–55
Michan S, Sinclair D (2007) Sirtuins in mammals: insights into their biological function. Biochem J 404:1–13
Chang HC, Guarente L (2014) SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 25:138–145
Ruderman NB, Carling D, Prentki M, Cacicedo JM (2013) AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest 123:2764–2772
Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13:225–238
Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B (2014) Metformin: from mechanisms of action to therapies. Cell Metab 20:953–966
Rena G, Pearson ER, Sakamoto K (2013) Molecular mechanism of action of metformin: old or new insights? Diabetologia 56:1898–1906
McCreight LJ, Bailey CJ, Pearson ER (2016) Metformin and the gastrointestinal tract. Diabetologia 59:426–435
Goodyear LJ (2000) AMP-activated protein kinase: a critical signaling intermediary for exercise-stimulated glucose transport? Exerc Sport Sci Rev 28:113–116
Schenk S, Horowitz JF (2007) Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest 117:1690–1698
Marcinko K, Steinberg GR (2014) The role of AMPK in controlling metabolism and mitochondrial biogenesis during exercise. Exp Physiol 99:1581–1585
Friedrichsen M, Mortensen B, Pehmoller C, Birk JB, Wojtaszewski JF (2013) Exercise-induced AMPK activity in skeletal muscle: role in glucose uptake and insulin sensitivity. Mol Cell Endocrinol 366:204–214
Fujii N, Jessen N, Goodyear LJ (2006) AMP-activated protein kinase and the regulation of glucose transport. Am J Physiol Endocrinol Metab 291:E867–E877
McGee SL, Hargreaves M (2006) Exercise and skeletal muscle glucose transporter 4 expression: molecular mechanisms. Clin Exp Pharmacol Physiol 33:395–399
Meex RC, Schrauwen-Hinderling VB, Moonen-Kornips E et al (2010) Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes 59:572–579
Phielix E, Meex R, Moonen-Kornips E, Hesselink MK, Schrauwen P (2010) Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia 53:1714–1721
Goodpaster BH, Katsiaras A, Kelley DE (2003) Enhanced fat oxidation through physical activity is associated with improvements in insulin sensitivity in obesity. Diabetes 52:2191–2197
Coen PM, Menshikova EV, Distefano G et al (2015) Exercise and weight loss improve muscle mitochondrial respiration, lipid partitioning, and insulin sensitivity after gastric bypass surgery. Diabetes 64:3737–3750
Konopka AR, Asante A, Lanza IR et al (2015) Defects in mitochondrial efficiency and H2O2 emissions in obese women are restored to a lean phenotype with aerobic exercise training. Diabetes 64:2104–2115
Hey-Mogensen M, Hojlund K, Vind BF et al (2010) Effect of physical training on mitochondrial respiration and reactive oxygen species release in skeletal muscle in patients with obesity and type 2 diabetes. Diabetologia 53:1976–1985
Bergman BC, Perreault L, Hunerdosse DM, Koehler MC, Samek AM, Eckel RH (2010) Increased intramuscular lipid synthesis and low saturation relate to insulin sensitivity in endurance-trained athletes. J Appl Physiol (1985) 108:1134–1141
Balducci S, Sacchetti M, Haxhi J et al (2015) The Italian Diabetes and Exercise Study 2 (IDES-2): a long-term behavioral intervention for adoption and maintenance of a physically active lifestyle. Trials 16:569
Hallsworth K, Thoma C, Hollingsworth KG et al (2015) Modified high-intensity interval training reduces liver fat and improves cardiac function in non-alcoholic fatty liver disease: a randomized controlled trial. Clin Sci (Lond) 129:1097–1105
Bacchi E, Negri C, Targher G et al (2013) Both resistance training and aerobic training reduce hepatic fat content in type 2 diabetic subjects with nonalcoholic fatty liver disease (the RAED2 Randomized Trial). Hepatology 58:1287–1295
Keating SE, Hackett DA, George J, Johnson NA (2012) Exercise and non-alcoholic fatty liver disease: a systematic review and meta-analysis. J Hepatol 57:157–166
Szendroedi J, Chmelik M, Schmid AI et al (2009) Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology 50:1079–1086
Roberts LD, Bostrom P, OʼSullivan JF et al (2014) β-Aminoisobutyric acid induces browning of white fat and hepatic β-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab 19:96–108
Rao RR, Long JZ, White JP et al (2014) Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 157:1279–1291
Bostrom P, Wu J, Jedrychowski MP et al (2012) A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481:463–468
Catoire M, Alex S, Paraskevopulos N et al (2014) Fatty acid-inducible ANGPTL4 governs lipid metabolic response to exercise. Proc Natl Acad Sci U S A 111:E1043–E1052
de Ligt M, Timmers S, Schrauwen P (2015) Resveratrol and obesity: can resveratrol relieve metabolic disturbances? Biochim Biophys Acta 1852:1137–1144
Timmers S, Konings E, Bilet L et al (2011) Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 14:612–622
Himms-Hagen J (1972) Lipid metabolism during cold-exposure and during cold-acclimation. Lipids 7:310–323
Martineau L, Jacobs I (1991) Effects of muscle glycogen and plasma FFA availability on human metabolic responses in cold water. J Appl Physiol (1985) 71:1331–1339
Vallerand AL, Zamecnik J, Jacobs I (1995) Plasma glucose turnover during cold stress in humans. J Appl Physiol (1985) 78:1296–1302
Wijers SL, Saris W, van Marken Lichtenbelt WD (2010) Cold induced adaptive thermogenesis in lean and obese. J Proteome Res 18:1092–1099
Vijgen GH, Bouvy ND, Teule GJ et al (2012) Increase in brown adipose tissue activity after weight loss in morbidly obese subjects. J Clin Endocrinol Metab 97:E1229–E1233
Hanssen MJ, Hoeks J, Brans B et al (2015) Short-term cold acclimation improves insulin sensitivity in patients with type 2 diabetes mellitus. Nat Med 21:863–865
Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359
Virtanen KA, Lidell ME, Orava J et al (2009) Functional brown adipose tissue in healthy adults. N Engl J Med 360:1518–1525
van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM et al (2009) Cold-activated brown adipose tissue in healthy men. N Engl J Med 360:1500–1508
Saito M, Okamatsu-Ogura Y, Matsushita M et al (2009) High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 58:1526–1531
Cypess AM, Lehman S, Williams G et al (2009) Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360:1509–1517
Nedergaard J, Bengtsson T, Cannon B (2007) Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab 293:E444–E452
Hanssen MJ, Wierts R, Hoeks J et al (2015) Glucose uptake in human brown adipose tissue is impaired upon fasting-induced insulin resistance. Diabetologia 58:586–595
van der Lans AA, Wierts R, Vosselman MJ, Schrauwen P, Brans B, van Marken Lichtenbelt WD (2014) Cold-activated brown adipose tissue in human adults: methodological issues. Am J Physiol Regul Integr Comp Physiol 307:R103–R113
Vijgen GH, Bouvy ND, Teule GJ, Brans B, Schrauwen P, van Marken Lichtenbelt WD (2011) Brown adipose tissue in morbidly obese subjects. PLoS ONE 6, e17247
Yoneshiro T, Aita S, Matsushita M et al (2011) Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. J Proteome Res 19:1755–1760
Blondin DP, Labbe SM, Tingelstad HC et al (2014) Increased brown adipose tissue oxidative capacity in cold-acclimated humans. J Clin Endocrinol Metab 99:E438–E446
van der Lans AA, Hoeks J, Brans B et al (2013) Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J Clin Invest 123:3395–3403
Yoneshiro T, Aita S, Matsushita M et al (2013) Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest 123:3404–3408
Ma SW, Foster DO (1986) Uptake of glucose and release of fatty acids and glycerol by rat brown adipose tissue in vivo. Can J Physiol Pharmacol 64:609–614
Isler D, Hill HP, Meier MK (1987) Glucose metabolism in isolated brown adipocytes under β-adrenergic stimulation. Quantitative contribution of glucose to total thermogenesis. Biochem J 245:789–793
Festuccia WT, Blanchard PG, Deshaies Y (2011) Control of brown adipose tissue glucose and lipid metabolism by PPARγ. Front Endocrinol 2:84
Stanford KI, Middelbeek RJ, Townsend KL et al (2013) Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest 123:215–223
Ouellet V, Routhier-Labadie A, Bellemare W et al (2011) Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J Clin Endocrinol Metab 96:192–199
Orava J, Nuutila P, Lidell ME et al (2011) Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab 14:272–279
Ouellet V, Labbe SM, Blondin DP et al (2012) Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest 122:545–552
Orava J, Nuutila P, Noponen T et al (2013) Blunted metabolic responses to cold and insulin stimulation in brown adipose tissue of obese humans. Obesity (Silver Spring) 21:2279–2287
Chondronikola M, Volpi E, Borsheim E et al (2014) Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes 63:4089–4099
Lee P, Smith S, Linderman J et al (2014) Temperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes 63:3686–3698
Gasparetti AL, de Souza CT, Pereira-da-Silva M et al (2003) Cold exposure induces tissue-specific modulation of the insulin-signalling pathway in Rattus norvegicus. J Physiol 552:149–162
Hanssen MJ, van der Lans AA, Brans B et al (2016) Short-term cold acclimation recruits brown adipose tissue in obese humans. Diabetes 65:1179–1189
Acknowledgements
We acknowledge the support from the Netherlands Cardiovascular Research Initiative: an initiative with support of the Dutch Heart Foundation (CVON2014-02 ENERGISE)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
Both authors were responsible for drafting the article and revising it critically for important intellectual content. Both authors approved the version to be published.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary material
(Downloadable slideset (ESM) (PDF 452 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schrauwen, P., van Marken Lichtenbelt, W.D. Combatting type 2 diabetes by turning up the heat. Diabetologia 59, 2269–2279 (2016). https://doi.org/10.1007/s00125-016-4068-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4068-3