
Abstract
Perilipin 5 (PLIN5/OXPAT) is a lipid droplet (LD) coat protein mainly present in tissues with a high fat-oxidative capacity, suggesting a role for PLIN5 in facilitating fatty acid oxidation. Here, we investigated the role of PLIN5 in fat oxidation in skeletal muscle. In human skeletal muscle, we observed that PLIN5 (but not PLIN2) protein content correlated tightly with OXPHOS content and in rat muscle PLIN5 content correlated with mitochondrial respiration rates on a lipid-derived substrate. This prompted us to examine PLIN5 protein expression in skeletal muscle mitochondria by means of immunogold electron microscopy and Western blots in isolated mitochondria. These data show that PLIN5, in contrast to PLIN2, not only localizes to LD but also to mitochondria, possibly facilitating fatty acid oxidation. Unilateral overexpression of PLIN5 in rat anterior tibialis muscle augmented myocellular fat storage without increasing mitochondrial density as indicated by the lack of change in protein content of five components of the OXPHOS system. Mitochondria isolated from PLIN5 overexpressing muscles did not possess increased fatty acid respiration. Interestingly though, 14C-palmitate oxidation assays in muscle homogenates from PLIN5 overexpressing muscles revealed a 44.8% (P = 0.05) increase in complete fatty acid oxidation. Thus, in mitochondrial isolations devoid of LD, PLIN5 does not augment fat oxidation, while in homogenates containing PLIN5-coated LD, fat oxidation is higher upon PLIN5 overexpression. The presence of PLIN5 in mitochondria helps to understand why PLIN5, in contrast to PLIN2, is of specific importance in fat oxidative tissues. Our data suggests involvement of PLIN5 in directing fatty acids from the LD to mitochondrial fatty acid oxidation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Perilipin 5 (aliases include OXPAT, lipid storage droplet protein 5, myocardial lipid droplet protein and PAT-1) is a lipid droplet (LD) coat protein that belongs to the perilipin family of proteins. Although the tissue distribution of this protein family differs considerably between members, all perilipin proteins are involved in accumulation and metabolism of lipids in LDs (Bickel et al. 2009; Brasaemle 2007). Perilipin 5 (PLIN5) expression is highest in tissues with a high fat-oxidative capacity, such as heart, oxidative (type I) skeletal muscles and brown adipose tissue (Yamaguchi et al. 2006; Wolins et al. 2006; Dalen et al. 2007). Recently, we showed that in rat and human skeletal muscle, PLIN5 protein content paralleled the LD content, with most abundant expression in muscle fibres that have highest fat content (type 1 fibres in humans, type 2a in rodents) (Minnaard et al. 2009).
PLIN5 has been found covering the lipid droplet and to be present in the cytosol (Wolins et al. 2006; Yamaguchi et al. 2006). PLIN5 overexpression in vitro in OP-9 and COS-7 cells increased cellular triglyceride storage in parallel with increases in fatty acid (FA) oxidation and induction of gene expression of mitochondrial enzymes involved in oxidative metabolism (Wolins et al. 2006). These results suggest involvement of PLIN5 in triglyceride storage as well as oxidative degradation of FAs released from the droplet, and suggest involvement of PLIN5 in lipid turnover. Recently, it has been shown that PLIN5 indeed has the potential to modulate the interaction of the major triacylglycerol lipase ATGL with the LD as well as with its co-factor CGI-58 (Lass et al. 2006) in a well orchestrated manner to modulate LD lipolysis (Granneman et al. 2009, 2011; Wang et al. 2011a).
To maintain cellular concentrations of cytotoxic FAs low, effective shuttling of FAs towards mitochondrial oxidation should be facilitated. In oxidative tissues this is in part facilitated by the close vicinity, and sometimes even physical interaction of LD with mitochondria. Functional interactions between LDs and mitochondria have long been reported (Blanchette-Mackie et al. 1995; Cohen et al. 2004; Hoppeler 1999; Hoppeler et al. 1973), and evidence is emerging that mitochondria are not only actively controlling FA levels in their vicinity by directing FAs towards β-oxidation, but the presence of the enzymatic machinery to re-esterify FA in mitochondria indicates that mitochondria may also have the option to redirect the fatty acids towards triacylglycerol synthesis in LD. Likewise, isoforms of acyl CoA:diacylglycerol acyltransferase (DGAT) (Stone et al. 2009), glycerol 3-phosphate acyl transferases (GPAT) (Hammond et al. 2002; Lewin et al. 2004) and long-chain acyl-CoA synthetases (Ellis et al. 2010) have been identified in mitochondria. Orchestrated interaction of processes at the LD and the mitochondria is hence of pivotal importance in tissues like cardiac and skeletal muscle and brown adipose tissue where large fluctuations in fatty acid oxidation are prominent. These tissues also typically express PLIN5 as a LD coat protein.
Imaging the LDs and PLIN5 by immunofluorescence revealed that the majority of PLIN5 is localised to the lipid droplet but also that—mainly in fat oxidative type I muscle fibres—PLIN5 could be readily detected in a staining pattern similar to recognised mitochondrial proteins (Minnaard et al. 2009). This staining pattern, along with the high expression of PLIN5 in tissues with high FA oxidation capacity (Dalen et al. 2007; Wolins et al. 2006; Yamaguchi et al. 2006) and the observation that overexpressing PLIN5 increased both palmitate oxidation and the expression of genes encoding mitochondrial proteins (Wolins et al. 2006) hints towards involvement of PLIN5 in mitochondrial fat oxidation. Likewise, it has recently been suggested that PLIN5 at the LD surface may recruit LD to the mitochondria (Wang and Sztalryd 2011).
We here show that PLIN5 is present within the mitochondria in cardiac and skeletal muscle. PLIN5 overexpression in skeletal muscle indicates that lipid droplets and mitochondria need interaction before PLIN5 possesses its facilitating role to augment mitochondrial fat oxidation.
Materials and methods
PLIN5 antibody
The PLIN5 antibody used in this study is directed against amino acids 451–463 of the C-terminus and was derived from Progen (#GP31; guinea pig polyclonal; Progen, Heidelberg, Germany). This antibody, as well as other available antibodies against PLIN5, was tested in pilot experiments (Western blots and immunogold labelling for electron microscopy) in a variety of cells and tissues known to vary in endogenous PLIN5 protein expression (HEK and C2C12 cells, white adipose tissue, cardiac muscle, glycolytic skeletal muscle (EDL), oxidative skeletal muscle (soleus), liver and testis). Thus, PLIN5 expression was highest in oxidative tissues (heart and oxidative skeletal muscle). The antibody used in this study showed the expected tissue distribution, a double band of the expected size, and a low background and specific PLIN5 labelling on the transmission electron microscopy pictures. Negative controls using non-immune serum did not show any bands in Westerns nor did it result in immunogold or immunofluorescence staining.
Cell culture and treatments
HEK293 cells (LGC standards, Teddington, Middlesex, United Kingdom) were maintained in DMEM (GlutaMAX low glucose DMEM, Invitrogen, Breda, The Netherlands) containing antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin; Gibco-Invitrogen, Rockville, USA) supplemented with 10% FCS. Cells were transfected with Plin5 plasmid construct (pcDNA3.1-CMV (2 μg per well of a 12-well plate) using 10 μL/ml Lipofectamine 2,000 as transfection reagent (Invitrogen, Breda, The Netherlands). Cells were harvested 24 h posttransfection.
Human study
Eleven healthy, lean, male volunteers without a family history of diabetes or any other endocrine disorder participated in this study. The mean age of the subjects was 23.6 ± 1.0 years; mean BMI was 22.6 ± 0.5 kg/m2. Subjects were fed in energy balance for 60 h (50–35–15% of energy as carbohydrates, fat and protein, respectively). After 60 h, a muscle biopsy of the vastus lateralis was taken using the Bergstrom technique (Bergstrom 1975). Muscle samples were immediately frozen in melting isopentane and stored at 80°C until assayed. The study protocol was reviewed and approved by the Medical Ethical Committee of Maastricht University Medical Centre and all subjects gave their written informed consent before participating in the study.
ZDF rat study design
Twenty-six male, ZDF rats (ZDF/Gmi, fa/fa) were purchased from Charles River (Chatillon-sur-Chalaronne, France). The rats arrived at the age of 5 or 11 weeks and were housed in pairs with ad libitum access to tap water and chow. At 6, 12, and 19 weeks of age (n = 8, n = 8, and n = 10, respectively), rats were killed by cervical dislocation, and hind leg muscles were rapidly dissected. The tibialis anterior muscle was processed for mitochondrial isolation (Lenaers et al. 2010) and soleus and EDL muscles were processed for electron microscopy or rapidly frozen in melting isopentane (Fluka, Zwijndrecht, The Netherlands), and stored at −80°C. All experiments were approved by the The Animal Care and Use Committee of Maastricht University (approval number 2005-058) and the study complied with the principles of laboratory animal care.
Electroporation study design
A total of eighteen 8-week-old male Wistar rats were purchased from Charles River (Wilmington, Massachusetts, USA). Rats were housed individually on a 12:12 h light–dark cycle (light from 7:00 am to 7:00 pm), at room temperature (21–22°C) with ad libitum access to tap water. Rats were fed a high fat diet (45% energy from fat, D01060502, Research Diets, New Brunswick, NJ, USA) for the duration of the 3-week intervention. The Animal Care and Use Committee of Maastricht University approved the experiments (approval number 2010-036) and the study complied with the principles of laboratory animal care. During the experiments, all efforts were made to minimise suffering of the animals.
Electroporation
Two weeks after the start of the diet, overexpression of Plin5 in either the right or left tibialis anterior (TA) muscle of the rat was accomplished by an in vivo DNA electrotransfer technique to obtain overexpression of mouse perilipin 5 in one leg; the contralateral TA served as a sham-electroporated internal control. Plin5 was electroporated randomly in the left or the right TA. DNA electroporation was performed under isoflurane anaesthesia. TA muscles were transcutaneously injected with either 150 μg (2 μg/μl) pcDNA3.1-CMV-Plin5 or pcDNA3.1-empty vector in 0.9% sterile NaCl. Within 15 s after the last injection 5 electric pulses were applied by two stainless steel plate electrodes placed at the ventral and dorsal side of the leg. One high voltage pulse of 800 V/cm and four low voltage pulses of 80 V/cm at 1 Hz were generated by an ECM 830 electroporator (BTX, San Diego, CA, USA) as described previously (Bruce et al. 2007, 2009). Rats were sacrificed 8 days postelectroporation. TA muscles were excised and processed for mitochondrial isolations and electron microscopy. The remainder of the TA was rapidly frozen in melting isopentane for histology and protein isolations.
Mitochondrial isolations
After excision, the right and left TA muscles were placed into ice-cold medium containing 100 mM sucrose, 100 Mm KCl, 50 mM Tris–HCl, 1 mM KH2PO4, 0.1 mM EGTA, and 0.2% (wt/vol) FA-free bovine serum albumin (pH 7.4). The TA was immediately processed for mitochondrial isolation, essentially according to Tonkonogi and Sahlin (1997). The protein concentration in the mitochondrial pellet was measured using fluorescamine (Fluram; Fluka, Zwijndrecht, the Netherlands) with bovine serum albumin as a standard (Udenfriend et al. 1972). Freshly isolated mitochondria were used immediately for mitochondrial respirometry. Remaining mitochondria were stored at −80°C for further analysis.
High-resolution respirometry in isolated mitochondria
Mitochondrial oxygen consumption was measured essentially according to Hoeks et al. (2008) at 37°C using a two-chamber oxygraph (OROBOROS Instruments, Innsbruck, Austria) and expressed as pmol O2/mg mitochondrial protein per second. Mitochondria were characterised using a carbohydrate-derived substrate (5 mM pyruvate) and a FA-derived substrate (50 μM palmitoyl-CoA plus 2 mM carnitine). For this purpose, mitochondria (0.1 mg for pyruvate and 0.2 mg for palmitoyl-CoA plus carnitine) were incubated in a medium consisting of 100 mM sucrose, 20 mM K+-Tes (pH 7.2), 50 mM KCl, 2 mM MgCl2, 1 mM EDTA, 4 mM KH2PO4, 3 mM malate and 0.1% of FA-free BSA. Maximal coupled (state 3) respiration was initiated by addition of 450 μM of adenosine diphosphate (ADP). State 4o respiration, reflecting proton leak, was measured as the residual respiration following addition of 1 μg/ml oligomycin (an F1-F0 ATPase inhibitor). Maximal oxygen flux rates (state uncoupled, state U) were obtained by titration of the chemical uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP). Addition of excess cytochrome C to the chamber in the presence of pyruvate and ADP did not increase respiration (data not shown); indicating that during isolation outer mitochondrial membrane integrity was retained.
14C-palmitate oxidation assay
Ex vivo fat oxidation was measured as described (Frisard et al. 2010). Briefly, muscle was homogenised in a buffer (pH 7.4) containing 250 mM sucrose, 10 mM Tris–HCl, 2 mM ATP and 1 mM EDTA (Sigma, St. Louis, USA). Subsequently, muscle homogenates were incubated for 2 h with [1-14C]-labelled palmitate (1 μCi/ml, Perkin Elmer, Boston, USA) and 200 μM non-labelled (cold) palmitate in reaction media containing L-carnitine, ATP and coenzyme A (all from Sigma, St. Louis, USA) at 37°C. Palmitate was coupled to FA-free BSA in a molar ratio of 5:1. After incubation, reactions were terminated by adding 40 μl of 70% perchloric acid, and the CO2 produced during the incubation was trapped in 200 μl of 1 N sodium hydroxide that had been added to adjacent wells (Kim et al. 2002). The acidified medium was stored at 4°C overnight, and then acid soluble metabolites (ASMs) were assayed in supernatants of the acid precipitate. Radioactivity of CO2 and ASMs was determined by liquid scintillation counting by use of 5 ml of Hionic Fluor (Perkin Elmer, Boston, USA). Data were normalised to protein content.
Electron microscopy procedures
Ultrastructural morphology and PLIN5 localization were examined using transmission electron microscopy. For determinations of ultrastructural morphology, muscle tissue sections were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). Postfixation was performed in 1% osmium tetroxide in 0.1 M cacodylate buffer (pH 7.4) supplemented with 1.5% potassium ferrocyanide. The samples were then dehydrated and embedded in epon. Ultrathin sections were examined using a Philips CM100 electron microscope.
For determinations of subcellular localization of PLIN5, soleus, extensor digitorum longus (EDL) and heart muscle tissues 14-week old ZDF rats (ZDF/Gmi, fa/fa) and tibialis anterior muscle tissues from electroporated Wistar rats were cut into pieces with a maximum size of 1 mm3, fixed for 1 h at room temperature in a mixture of 2% formaldehyde and 0.2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), and stored in 1% formaldehyde in 0.1 M phosphate buffer (pH 7.4). Tissue pieces were immersed in a 2.3 M sucrose solution, frozen in liquid nitrogen and ultrathin cryo-sections were cut under liquid nitrogen, sections were transferred from the knife edge to a formvar and carbon-coated grid in 2.3 M sucrose/1.8% methylcellulose (1:1), as described by Tokuyasu (1973,1980,1986) Sections were immunogold labelled as described by Tokuyasu (1986) using a PLIN5 antibody (#GP31; Guinea pig polyclonal; Progen Biotechnik, Heidelberg, Germany) in a 1:100 dilution and protein A-gold (20 nm particles) (Slot and Geuze 1981). Guinea pig normal serum (Santa Cruz Biotechnology, Tebu-bio, Heerhugowaard, The Netherlands) was used as a negative control.
Oil-red-O stainings and immunofluorescence
Neutral lipids, mitochondria and PLIN5 in muscle sections were stained with a modified Oil-red-O staining for fluorescence microscopy as previously described (Koopman et al. 2001) using antibodies against PLIN5 (#GP31; Progen Biotechnik, Heidelberg, Germany), laminin (L-9,393; Sigma, St. Louis, USA), and OXPHOS (MS601; MitoSciences, Eugene, OR, USA) and the appropriate secondary Alexa-fluor conjugated antibodies (Invitrogen, Groningen, The Netherlands). Fluorescent images of Alexa-350 (laminin), Alexa-488 (PLIN5), Alexa-647 (OXPHOS), and Oil-red-O (visible in the red-594-channel) were acquired using an imaging system consisting of an IX81 inverted microscope (Olympus, Hamburg, Germany), an X-cite fluorescent light source (Lumen Dynamics group, Mississauga, Ontario, Canada), F-view II CCD camera (OSIS, Münster, Germany) and the Cell^F imaging software (OSIS). Fluorescent photomicrographs were made from Z-stacks consisting of 30-40 images with a distance of 0.28 μm using the maximum intensity projection method in the ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997–2009).
Western blots
Western blots were performed in protein lysates from whole muscle homogenates or mitochondrial fractions. Muscle samples and mitochondrial fractions were homogenised in ice-cold PBS containing 1% Nonidet-P40, 0.5% sodium dodecyl sulphate, 0.1 mM phenylmethylsulfonyl fluoride, complete inhibitor (Roche, Almere, The Netherlands) and processed for standard SDS-PAGE and Western blotting. Protein concentration was determined and equal amounts of protein were loaded per lane. The nitrocellulose membranes were incubated with antibodies against PLIN5 (GP31; Progen Biotechnik, Heidelberg, Germany), PLIN2 (GP40; Progen Biotechnik, Heidelberg, Germany), OXPHOS (MS601; MitoSciences, Eugene, OR, USA), PGC1α (516,557; Calbiochem; VWR International BV, Amsterdam, The Netherlands), VDAC (SC-8,828; Santa Cruz Biotechnology, Tebu-bio, Heerhugowaard, The Netherlands), and SR-actin (A-2,172; Sigma, St. Louis, USA).
Blots incubated with PLIN5, PLIN2, OXPHOS, PGC1α, VDAC and sr-actin were probed with appropriate IRDye800-conjugated or IRDye700-conjugated secondary antibodies (Rockland, Tebu-bio, Heerhugowaard, The Netherlands, and LICOR Biosciences, Westburg, Leusden, The Netherlands), and bands at a molecular weight corresponding to the control samples were quantified using the Odyssey infrared imaging system (LICOR Biosciences, Westburg, Leusden, The Netherlands).
Statistical analysis
Results are presented as mean ± SE. Differences between groups were evaluated with paired t tests. Outcomes were considered statistically significant when P < 0.05 (two-tailed). Pearson’s correlation coefficients were used to describe the linear association between variables. All analyses were performed using the Statistical Package for the Social Sciences (SPSS 16.0, Nieuwegein, The Netherlands) software for Mac 16.0.
Results
Skeletal muscle PLIN5 protein content correlates with oxidative capacity
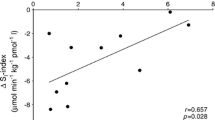
In order to explore the role of PLIN5 in skeletal muscle, we measured PLIN5 protein content in muscle biopsies of vastus lateralis muscle of young healthy male subjects. PLIN5 protein content correlated strongly with the protein content of structural components of the individual complexes of the electron transport chain; OXPHOS complexes I (r = 0.721, P = 0.012), III (r = 0.810, P = 0.003), IV (r = 0.704, P = 0.016) and V (r = 0.606, P = 0.048) but not with protein content of VDAC, an outer mitochondrial membrane protein without a recognised role in (fat) oxidation (r = 0.148, P = 0.664) (n = 11) (Table 1). These observations indirectly suggest a role of PLIN5 in mitochondrial fat oxidative metabolism. This led us to investigate the relationship between PLIN5 protein content in whole muscle homogenates with respiration rates of isolated muscle mitochondria on a fat-derived substrate (palmitoyl-CoA). Thus, we analysed correlations of skeletal muscle PLIN5 protein expression with mitochondrial function in a model of progressive myocellular fat deposition, the maturing Zucker diabetic fatty (ZDF) rats (fa/fa). In this model, PLIN5 protein content tracks with intramyocellular lipid content (Minnaard et al. 2009). We examined the correlation between PLIN5 content in whole muscle homogenates and intrinsic mitochondrial function in mitochondria isolated from these muscles by classical mitochondrial respirometry using palmitoyl-CoA as a FA-derived substrate. In these rats, PLIN5 protein content, but importantly not PLIN2 protein content, positively correlated with ADP-driven state 3 respiration (r = 0.713, P < 0.001) as well as respiration under maximal FCCP-mediated uncoupled conditions (state U, r = 0.663, P = 0.001) (Table 2). These observations may indicate that in muscles in which PLIN5 protein content scales with LD content, PLIN5 is involved in facilitating increased mitochondrial fat oxidation.
Perilipin 5 localizes to lipid droplets as well as to mitochondria in cardiac and skeletal muscle. The strong correlation of PLIN5 protein content with OXPHOS complexes and mitochondrial respiration on a FA-derived substrate led us to examine whether PLIN5, next to its recognised localisation to LD, is also present in mitochondria. Thus, we measured PLIN5 protein content in total muscle protein lysates as well as in mitochondrial fractions from rat skeletal muscle (Fig. 1a). A double band representing PLIN5 was observed in whole muscle lysates and more strikingly also in isolated mitochondria. To exclude the ability that the PLIN5 signal detected in the mitochondria originated from contamination of the mitochondrial fraction with ER or LD derived membrane fractions we examined protein expression of ADRP/PLIN2 in our mitochondrial fractions (Fig. 1b). The absence of an ADRP/PLIN2 band indicates that the mitochondrial fractions used were devoid of ER or LD membrane structures, substantiating our notion that PLIN5 indeed localizes to mitochondria.

Perilipin 5 protein expression in whole muscle homogenates, isolated mitochondria and muscle sections. a Western blotting of perilipin 5 in whole skeletal muscle lysates from heart muscle, extensor digitorum longus (EDL) and tibialis anterior muscles and mitochondria isolated from tibialis anterior muscle. b Western blots incubated with the PLIN2 antibody did not show any bands in isolated mitochondria, indicating that the mitochondrial fraction was devoid of lipid droplets and was not contaminated with fragments of the ER. HEK cells transfected with PLIN2 plasmid were used as positive control. c PLIN5 immunogold labelling in rat soleus muscle. PLIN5 coats the lipid droplets and is present at mitochondria but not at myofibrils. d PLIN5 immunogold labelling in rat left ventricle showing mitochondrial localization of PLIN5. LD lipid droplet, M mitochondrion
We further substantiated mitochondrial localisation of PLIN5 using immunogold electron microscopy, the gold standard methodology for subcellular localisation studies of single proteins. In all muscles examined (soleus, EDL and cardiac muscle) PLIN5 was present on the LDs, at the interface of LDs with mitochondria, as well as in the mitochondria (Fig. 1c, d), whereas other structures were almost completely devoid of gold particles. Negative controls by omission of the primary antibody or by using non-immune serum did not show any immunogold staining. Immunogold staining using the ADRP/PLIN2 antibody only revealed staining associated with LD (data not shown).
PLIN5 overexpression in rat tibialis anterior muscle
To examine the effect of overexpression of PLIN5 on its subcellular localisation, a CMV-driven full-length PLIN5 construct was used for unilateral overexpression of PLIN5 in rat tibialis anterior muscle by gene electroporation using the contralateral empty vector-electroporated leg as an internal control. Upon transfection in HEK cells the construct used resulted in the anticipated double band on the Western, representing two PLIN5 isoforms (Fig. 2a). In rat tibialis anterior muscle, PLIN5 overexpression doubled PLIN5 protein content in whole muscle homogenates (P = 0.0006, n = 14, paired t test) (Fig. 2b). Consistent with results obtained in vitro (Dalen et al. 2007; Wolins et al. 2006), PLIN5 overexpression profoundly augmented intramyocellular lipid storage in the transfected muscle (Fig. 2c). Immunohistochemistry revealed that PLIN5 was present on LDs and showed punctuate mitochondrial localisation. Double immunofluorescence using an antibody cocktail against structural components of all 5 OXPHOS complexes confirmed co-localisation of PLIN5 with mitochondria (Fig. 2d). Ultrastructural analysis by electron microscopy confirmed the light microscopy based observation of augmented lipid storage upon PLIN5 overexpression and clearly showed increased LD size (Fig. 3). In addition, almost all LD appear to interact tightly with mitochondria in the PLIN5-overexpressing muscle and sometimes even appear to envelop the lipid droplets (Fig. 3). Immunogold labelling of PLIN5 in electroporated muscle confirmed the localization of PLIN5 in mitochondria, on LDs and on the LD-mitochondrial interface (Fig. 4).

PLIN5 overexpression (gene electroporation) in rat muscle using an empty vector as internal control. a Specificity of the PLIN5 antibody (Progen). A full-length PLIN5 vector was transfected in cells without endogenous PLIN5 expression (HEK cells), this resulted in ectopic expression of PLIN5. b PLIN5 protein content in protein lysates from electroporated tibialis anterior muscle (n = 16). Error bars represent SEM, *P < 0.01. The samples were derived from the same experiment and were simultaneously processed in random order on two gels/blots. EV: empty vector. c Shows the effect of PLIN5 overexpression on lipid storage (upper right hand panel) and PLIN5 content (lower right hand panel) compared to the empty vector control (upper and lower left hand panels). LDs are stained in red), PLIN5 in green and cell membranes in blue. Bars represent 100 μm. d Overlay of PLIN5 and mitochondrial proteins upon PLIN5 overexpression. PLIN5 overexpression increases LD (in red) and PLIN5 (in green) content (upper right hand and middle left hand panel, respectively). Mitochondria are shown in magenta (middle left hand panel). The lower left hand panel shows LD decorated with PLIN5 while the lower right hand panel shows PLIN5 interaction with mitochondria. The bar represents 20 μm. Representative image are shown

PLIN5 overexpression results in large LDs. Left hand panels a and c show empty vector control data, right hand panels b and d show PLIN5 overexpression data. Note the apparent larger LD size upon PLIN5 overexpression. Bar represents 10 μm in the upper panel and 2 μm in the lower panels

PLIN5 labelling in mitochondria and on the lipid droplets. Immunogold staining of PLIN5 in tibialis anterior muscle of empty vector (control) (a) or PLIN5-electroporated muscle (b). PLIN5 overexpression b, c and d resulted in larger lipid droplets and intimate interaction of lipid droplets with mitochondria at PLIN5 positive sites (see arrowheads in c and d). Bars represent 1 μm. e Intramitochondrial PLIN5 labelling. Bar represents 0.5 μm
PLIN5 overexpression does not affect mitochondrial density
To examine the putative effect of PLIN5 overexpression on mitochondrial density, we measured in whole muscle homogenates the protein content of PGC1α (Fig. 5a), VDAC (Fig. 5b) and 5 distinct structural components of subunits of the individual OXPHOS complexes (complex I subunit NDUFB8, complex II subunit 30 kDa, complex III subunit Core 2, complex IV subunit I and complex V ATP synthase subunit alpha (Fig. 5c). Although we observed a non-significant (P = 0.14) increase in PGC1α, a key co-activator for mitochondrial biogenesis, protein content of VDAC and OXPHOS complexes remained unaltered, indicating that 8 days after overexpressing PLIN5 mitochondrial density was not affected.

Abundance of mitochondrial proteins upon PLIN5 overexpression. Western blots were performed for expression of PGC1α (a), VDAC (b) and five distinct structural components of the OXPHOS complexes (c) in lysates from whole muscle homogenates (n = 14). Error bars represent SEM, *P < 0.01. The samples were derived from the same experiment and were simultaneously processed in random order
PLIN5 overexpression does not improve respiration of isolated mitochondria
Mitochondrial respirometry assays in the mitochondria isolated from PLIN5 overexpressing muscles relative to muscles isolated from empty vector-electroporated muscles did not reveal significant differences in respiration, neither on pyruvate (empty vector vs. PLIN5 15.98 ± 1.17 vs. 15.48 ± 1.18 state 3/state 2, P = 0.83; 22.53 ± 0.88 vs. 22.29 ± 1.44 state U/state 2, P = 0.68), nor on palmitoyl-CoA (empty vector vs. PLIN5 6.76 ± 0.38 vs. 7.36 ± 0.30 state 3/state 2, P = 0.23; 8.56 ± 0.63 vs. 9.14 ± 0.49 state U/state 2, P = 0.37) (Fig. 6a, b). Thus, in a system in which PLIN5-coated LDs are dissociated from the mitochondria, PLIN5 overexpression does not augment mitochondrial (fat) oxidative capacity.

Effects of PLIN5 overexpression on mitochondrial respiration and FA oxidation. a and b State 3 (coupled) and state U (uncoupled) respiration on palmitoyl-CoA (a) or pyruvate (b), values are standardised to state 2 respiration (background respiration in the absence of ADP) (n = 9). c Total 14C-palmitate oxidation in muscle homogenates (CO2 plus ASMs). d The ratio of CO2 production (complete oxidation) to total FA oxidation (CO2 plus acid soluble metabolites) (n = 7). Error bars represent SEM, P values were derived from paired t tests
PLIN5 overexpression improved 14C-palmitate oxidation
Given the intimate association of LDs with mitochondria we performed complementary measurements of fat oxidation using 14C-palmitate in whole muscle homogenates when LDs and mitochondria were not dissociated. Total 14C-palmitate oxidation was not changed (empty vector 0.71 ± 0.06 nmol/2 h/mg protein, PLIN5 0.71 ± 0.03 nmol/2 h/mg protein, P = 0.99) (Fig. 6c). However, oxidation of 14C-palmitate to 14CO2 increased by 44.8% (empty vector 0.05 ± 0.008, PLIN5 0.07 ± 0.012 14CO2/total 14C-palmitate oxidation, P = 0.05) (Fig. 6d). The respiration data in isolated mitochondria and in muscle homogenates jointly indicate that overexpression of PLIN5 does not augment mitochondrial fat oxidative capacity per se but rather creates a situation in which shuttling of FAs towards mitochondria becomes more efficient, thereby protecting mitochondria against the deleterious effects of lipid overflow.
Discussion
PLIN5 was recently identified as a member of the perilipin family of lipid droplet coating proteins and was considered to be an exchangeable LD coat protein; unlike perilipins 1 and 2, PLIN5 is not exclusively found in the LD coat but also cytosolically (Dalen et al. 2007; Wolins et al. 2006; Yamaguchi et al. 2006). Using a variety of validated techniques, however, we here show that PLIN5 in muscle is found at the LDs as well as in mitochondria, this in contrast to PLIN2, the other major LD coat protein found in muscle. PLIN5 positive mitochondria were detected in oxidative (soleus), glycolytic (EDL), and mixed (TA) muscles as well as in cardiac muscle. Both subsarcolemmal mitochondria and intramyofibrillar mitochondria contained PLIN5. Gene electroporation mediated overexpression of PLIN5 doubled PLIN5 protein content and resulted in more numerous and apparently larger LDs in intimate interaction with mitochondria. Functional assays revealed no direct involvement of PLIN5 in mitochondrial respiration, as respiration in isolated mitochondria was not affected by PLIN5 overexpression. Interestingly though, fatty acid oxidation rates were higher in whole muscle homogenates of PLIN5 overexpressing muscles, suggesting a facilitating role of PLIN5 in augmenting fat oxidation situations when LDs are physically interconnected to the mitochondrial reticulum, possibly by shuttling or chaperoning of LD-derived FAs towards mitochondria for oxidation.
Recent reports have shown that PLIN5 localised to LDs in tissues rich in mitochondria (Dalen et al. 2007; Wolins et al. 2006; Yamaguchi et al. 2006), that overexpressing PLIN5 in OP-9 and COS-7 cells promoted fat oxidation (Wolins et al. 2006) and that the proteome of LDs contained multiple mitochondrial proteins (Brasaemle et al. 2004). In addition, PLIN5 interaction with ATGL on LDs and on undefined intracellular structures lacking neutral lipids (Granneman et al. 2011) has been reported. This has led us to investigate the possibility that PLIN5 also localizes to mitochondria.
Thus, we observed that PLIN5 protein content in vastus lateralis muscle of healthy young male subjects correlated positively and tightly with structural components of all five complexes of the electron transport chain, but not with mitochondrial proteins without a recognised role in fat oxidation. The lack of associations of PLIN5 with VDAC, a mitochondrial protein not involved in (fat) oxidation, narrows the interpretation of previous observations of PLIN5 being abundant in mitochondria rich tissues (Dalen et al. 2007; Wolins et al. 2006; Yamaguchi et al. 2006) and may hint towards a more direct involvement of PLIN5 in mitochondrial fat oxidation. Hence, we studied ADP-driven respiration rates on palmitoyl-CoA in mitochondria isolated from tibialis anterior muscles of ZDF rats with a range in myocellular fat content. We previously observed that muscles of these rats have increased PLIN5 and PLIN2 protein content which tracked with intramyocellular fat content (Minnaard et al. 2009). Fascinatingly, coupled respiration and maximal respiration on palmitoyl-CoA correlated tightly with muscle PLIN5 content but interestingly not with PLIN2 content. Thus, only PLIN5 paralleled changes in mitochondrial fat oxidative capacity. Upon Western blotting isolated mitochondria with a specific PLIN5 antibody, we were able to unmask PLIN5 as a mitochondrial protein. The mitochondrial isolation protocol used results in a very clean mitochondrial homogenate without contamination of other membrane structures of remnants of LD membranes. This was substantiated by the observation that the mitochondrial isolation was negative for PLIN2, while showing clear PLIN5 bands in mitochondria isolated from a variety of skeletal and cardiac muscles. To further substantiate these findings we performed double immunofluorescence microscopy with PLIN5 along with an antibody against structural components of the electron transport chain in rat tibialis anterior muscles. Confocal analysis of tissue sections indeed localised PLIN5 to the LD, but also to the mitochondria. For a more detailed confirmation of PLIN5 being expressed in mitochondria we applied post-embedding immunogold electron microscopy, the gold standard for high-resolution subcellular localisation studies of proteins (Zuber et al. 2005). Thus, we identified PLIN5 to the LD membrane as well as in mitochondria in sections from rat glycolytic (EDL), oxidative (soleus) and cardiac muscle. PLIN5 was consistently present in subsarcolemmal as well as in intermyofibrillar mitochondria of all muscle types examined. Again, no such observations were made for PLIN2, the other major LD coat protein in muscle. Except for LDs, other organelles and myofibrillar structures were almost devoid of any PLIN5 signal.
Having identified PLIN5 in mitochondria we set out to investigate the effect of PLIN5 overexpression on PLIN5 subcellular distribution and myocellular ultrastructure. We deliberately choose unilateral gene electroporation as the in vivo approach to overexpress PLIN5 in the tibialis anterior muscle of a rat. Thus, we were able to avoid lifelong adaptive responses while creating the ability to use the contralateral leg as an internal control. This resulted in a two-fold overexpression of PLIN5. Immunofluorescence microscopy and immunogold electron microscopy in tibialis anterior samples of the control and the overexpressing muscle confirmed specific localisation of PLIN5 to mitochondria and lipid droplets. No apparent changes in the subcellular distribution of PLIN5 were observed, i.e., subsarcolemmal and intermyofibrillar mitochondria pools both remained PLIN5 positive and qualitative examination of electron microscopical images did not indicate selective upregulation of PLIN5 at the LD site or at mitochondria. Using a variety of mitochondrial proteins we did not obverse indications of increased mitochondrial density upon PLIN5 overexpression during the 8 day time span of PLIN5 overexpression.
Moreover, PLIN5 overexpression augmented intramyocellular lipid storage and LD size, possibly reflecting the involvement of PLIN5 in ATGL-mediated lipolysis (Granneman et al. 2011; Wang et al. 2011a). The localization of PLIN5 on the LDs and in mitochondria suggests a role for PLIN5 in directing or binding fatty acids—or fatty acid derivatives—for mitochondrial oxidation. Measurements of respiration in isolated mitochondria did not reveal any differences between mitochondria derived from the sham- and the PLIN5-electroporated muscle, which does not support a direct role for PLIN5 in mitochondrial respiration. Interestingly though, when examining 14C-palmitate oxidation in muscle homogenates—when mitochondria are not examined in isolation but in context of their native subcellular milieu—we observed increased complete oxidation of 14C-palmitate to CO2. This occurred in the absence of an increase in accumulation of acid soluble metabolites, indicating that the supply of fatty acids and/or the rate of β-oxidation were better matched with the capacity for downstream FA oxidative metabolism. It should be noted that, albeit unlikely, we could not exclude the possibility that, during the isolation of mitochondria, the stronger physical interaction between mitochondria and lipid droplets upon PLIN5 overexpression resulted in a loss of mitochondria intimately connected to the large lipid droplets. This could explain the discrepancy in the results of the experiments in muscle homogenates and isolated mitochondria. Nevertheless, the increase 14C-palmitate to CO2 points towards a beneficial effect of PLIN5 overexpression on mitochondrial fatty acid oxidation.
Thus, given the dual localization of PLIN5 to the LD and to mitochondria, it is tempting to speculate that next to a role for PLIN5 in releasing fatty acids from the LD by co-regulating lipolysis, PLIN5 is also involved in shuttling/chaperoning/binding fatty acids or fatty acid derivatives from LD origin to mitochondria for sake of oxidation. While this suggestion clearly needs further experimental substantiation, it is well in line with previous observations by others that overexpressing PLIN5 augmented fat oxidation (Wolins et al. 2006). In the process of finalising this manuscript, Wang et al. (Wang et al. 2011b) showed that PLIN5 is an important mediator of the release of fatty acids for mitochondrial β-oxidation in liver cells. Upon PKA stimulation, PLIN5 increases LD hydrolysis thus making fatty acids available for downstream mitochondrial metabolism (Wang et al. 2011b). Whether the same holds true for muscle deserves further investigation.
Although our data clearly support mitochondrial localization of PLIN5 it remains to be solved how PLIN5 gets into mitochondria. We were unable to find evidence for an N-terminal mitochondrial targeting pre-sequence following targeted database searches using PSORT (Horton et al. 2007) and SignalP (Dyrløv Bendtsen et al. 2004). Although such a pre-sequence is common for many mitochondrial proteins (van der Laan et al. 2010), it should be noted that a variety of other mitochondrial proteins do not contain such a pre-sequence either (Dukanovic and Rapaport 2011). Rather, the mitochondrial localization of these proteins is determined by structural characteristics of the protein. It has been postulated that protein localization to LDs is determined by basic and hydrophobic domains, with remarkable similarities to the signals that target proteins to mitochondria (Ingelmo-Torres et al. 2009). Interestingly, we did find structural homology of PLIN5 with the alpha and beta chains of the mitochondrial protein ATP synthase (PropSearch) (Hobohm and Sander 1995), providing additional support for structural characteristics in the PLIN5 sequence that could determine mitochondrial localization. Proteome analysis of the LD coat interestingly revealed that the mitochondrial protein ATP synthase was present in LD fractions of lipolytically stimulated adipocytes (Brasaemle et al. 2004). Thus, PLIN5 would not be the first protein localised to mitochondria and lipid droplets while lacking a mitochondrial targeting pre-sequence. Moreover, recent work revealed that the last 20 amino acids of the C-terminal end of the protein were identified to be essential for recruitment of mitochondria to the lipid droplet surface (Wang et al. 2011b).
In conclusion, we provide evidence that PLIN5 is abundantly expressed in subsarcolemmal and intermyofibrillar mitochondria of glycolytic, oxidative and cardiac muscle as well as on the interface of mitochondria and lipid droplets. PLIN5 overexpression augmented fat oxidation in muscle homogenates containing both LDs and mitochondria but not in isolated mitochondria. These findings unmask PLIN5 as a protein localised to LD and mitochondria with a putative role in channelling LD derived fatty acids towards mitochondrial oxidation. Interaction of PLIN5 coated LD with mitochondria appears more frequent and more intimate upon skeletal muscle PLIN5 overexpression.
References
Bergstrom J (1975) Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest 35:609–616
Bickel PE, Tansey JT, Welte MA (2009) PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta 1791:419–440
Blanchette-Mackie EJ, Dwyer NK, Barber T, Coxey RA, Takeda T, Rondinone CM, Theodorakis JL, Greenberg AS, Londos C (1995) Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes. J Lipid Res 36:1211–1226
Brasaemle DL (2007) Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res 48:2547–2559
Brasaemle DL, Dolios G, Shapiro L, Wang R (2004) Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3–L1 adipocytes. J Biol Chem 279:46835–46842
Bruce CR, Brolin C, Turner N, Cleasby ME, van der Leij FR, Cooney GJ, Kraegen EW (2007) Overexpression of carnitine palmitoyltransferase I in skeletal muscle in vivo increases fatty acid oxidation and reduces triacylglycerol esterification. Am J Physiol Endocrinol Metab 292:E1231–E1237
Bruce CR, Hoy AJ, Turner N, Watt MJ, Allen TL, Carpenter K, Cooney GJ, Febbraio MA, Kraegen EW (2009) Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high fat diet-induced insulin resistance. Diabetes 58:550–558
Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, Brasaemle DL, Scherer PE, Lisanti MP (2004) Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes 53:1261–1270
Dalen KT, Dahl T, Holter E, Arntsen B, Londos C, Sztalryd C, Nebb HI (2007) LSDP5 is a PAT protein specifically expressed in fatty acid oxidizing tissues. Biochim Biophys Acta 1771:210–227
Dukanovic J, Rapaport D (2011) Multiple pathways in the integration of proteins into the mitochondrial outer membrane. Biochim Biophys Acta 1808:971–980
Dyrløv Bendtsen J, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795
Ellis JM, Frahm JL, Li LO, Coleman RA (2010) Acyl-coenzyme A synthetases in metabolic control. Curr Opin Lipidol 21:212–217
Frisard MI, McMillan RP, Marchand J, Wahlberg KA, Wu Y, Voelker KA, Heilbronn L, Haynie K, Muoio B, Li L, Hulver MW (2010) Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am J Physiol Endocrinol Metab 298:E988–E998
Granneman JG, Moore HP, Mottillo EP, Zhu Z (2009) Functional interactions between MLDP (LSDP5) and ABHD5 in the control of intracellular lipid accumulation. J Biol Chem 284:3049–3057
Granneman JG, Moore H-PH, Mottillo EP, Zhu Z, Zhou L (2011) Interactions of perilipin-5 (PLIN5) with adipose trigylceride lipase (ATGL). J Biol Chem 286:5126–5135
Hammond LE, Gallagher PA, Wang S, Hiller S, Kluckman KD, Posey-Marcos EL, Maeda N, Coleman RA (2002) Mitochondrial glycerol-3-phosphate acyltransferase-deficient mice have reduced weight and liver triacylglycerol content and altered glycerolipid fatty acid composition. Mol Cell Biol 22:8204–8214
Hobohm U, Sander C (1995) A sequence property approach to searching protein databases. J Mol Biol 251:390–399
Hoeks J, Briedé JJ, de Vogel J, Schaart G, Nabben M, Moonen-Kornips E, Hesselink MKC, Schrauwen P (2008) Mitochondrial function, content and ROS production in rat skeletal muscle: Effect of high-fat feeding. FEBS Lett 582:510–516
Hoppeler H (1999) Skeletal muscle substrate metabolism. Int J Obes Relat Metab Disord 23(Suppl 3):S7–S10
Hoppeler H, Luthi P, Claassen H, Weibel ER, Howald H (1973) The ultrastructure of the normal human skeletal muscle. A morphometric analysis on untrained men, women and well-trained orienteers. Pflugers Arch 344:217–232
Horton P, Park K-J, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K (2007) WoLF PSORT: protein localization predictor. Nucleic Acids Res 35:W585–W587
Ingelmo-Torres M, Elena G-M, Adam K, Michael H-B, Francesc T, Albert H, Thomas G, John FH, Carlos E, Marta B, Steven PG, Robert GP, Albert P (2009) Hydrophobic and basic domains target proteins to lipid droplets. Traffic 10:1785–1801
Kim J-Y, Koves TR, Yu G-S, Gulick T, Cortright RN, Dohm GL, Muoio DM (2002) Evidence of a malonyl-CoA-insensitive carnitine palmitoyltransferase I activity in red skeletal muscle. Am J Physiol Endocrinol Metab 282:E1014–E1022
Koopman R, Schaart G, Hesselink M (2001) Optimisation of oil red O staining permits combination with immunofluorescence and automated quantification of lipids. Histochem Cell Biol 116:63–68
Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R (2006) Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab 3:309–319
Lenaers E, De Feyter HM, Hoeks J, Schrauwen P, Schaart G, Nabben M, Nicolay K, Prompers JJ, Hesselink MKC (2010) Adaptations in mitochondrial function parallel, but fail to rescue, the transition to severe hyperglycemia and hyperinsulinemia: a study in Zucker diabetic fatty rats. Obesity 18:1100–1107
Lewin TM, Schwerbrock NMJ, Lee DP, Coleman RA (2004) Identification of a new glycerol-3-phosphate acyltransferase isoenzyme, mtGPAT2, in mitochondria. J Biol Chem 279:13488–13495
Minnaard R, Schrauwen P, Schaart G, Jorgensen JA, Lenaers E, Mensink M, Hesselink MKC (2009) Adipocyte differentiation-related protein and OXPAT in rat and human skeletal muscle: involvement in lipid accumulation and type 2 diabetes mellitus. J Clin Endocrinol Metab 94:4077–4085
Slot JW, Geuze HJ (1981) Sizing of protein A-colloidal gold probes for immunoelectron microscopy. J Cell Biol 90:533–536
Stone SJ, Levin MC, Zhou P, Han J, Walther TC, Farese RV Jr (2009) The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J Biol Chem 284:5352–5361
Tokuyasu KT (1973) A technique for ultracryotomy of cell suspensions and tissues. J Cell Biol 57:551–565
Tokuyasu KT (1980) Immunochemistry on ultrathin frozen sections. Histochem J 12:381–403
Tokuyasu KT (1986) Application of cryoultramicrotomy to immunocytochemistry. J Microsc 143:139–149
Tonkonogi M, Sahlin K (1997) Rate of oxidative phosphorylation in isolated mitochondria from human skeletal muscle: effect of training status. Acta Physiol Scand 161:345–353
Udenfriend S, Stein S, Bohlen P, Dairman W, Leimgruber W, Weigele M (1972) Fluorescamine: a reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range. Science 178:871–872
van der Laan M, Hutu DP, Rehling P (2010) On the mechanism of preprotein import by the mitochondrial presequence translocase. Biochim Biophys Acta 1803:732–739
Wang H, Sztalryd C (2011) Oxidative tissue: perilipin 5 links storage with the furnace. Trends Endocrinol Metab 22:197–203
Wang H, Bell M, Sreenevasan U, Hu H, Liu J, Dalen K, Londos C, Yamaguchi T, Rizzo MA, Coleman R, Gong D, Brasaemle D, Sztalryd C (2011a) Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J Biol Chem 286:15707–15715
Wang H, Sreenevasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong D-W, Stanley WC, Sztalryd C (2011b) Perilipin 5, lipid droplet associated protein provides physical and metabolic linkage to mitochondria. J Lipid Res; In press. doi:10.1194/jlr.M017939
Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Croce MA, Gropler MC, Varma V, Yao-Borengasser A, Rasouli N, Kern PA, Finck BN, Bickel PE (2006) OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 55:3418–3428
Yamaguchi T, Matsushita S, Motojima K, Hirose F, Osumi T (2006) MLDP, a novel PAT family protein localized to lipid droplets and enriched in the heart, is regulated by peroxisome proliferator-activated receptor alpha. J Biol Chem 281:14232–14240
Zuber C, Fan J, Guhl B, Roth J (2005) Applications of immunogold labeling in ultrastructural pathology. Ultrastruct Pathol 29:319–330
Acknowledgments
Research presented in this article was financially supported by NUTRIM and the graduate school VLAG. A VICI (grant 918.96.618) and a VIDI (grant 917.66.359) for innovative research from the Netherlands Organization for Scientific Research (NWO) supports the work of P. Schrauwen and M. Hesselink, respectively. P. Bickel’s work was supported by NIH grant DK068046.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bosma, M., Minnaard, R., Sparks, L.M. et al. The lipid droplet coat protein perilipin 5 also localizes to muscle mitochondria. Histochem Cell Biol 137, 205–216 (2012). https://doi.org/10.1007/s00418-011-0888-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-011-0888-x