
Abstract
Aims/hypothesis
We previously showed that type 2 diabetic patients are characterised by compromised intrinsic mitochondrial function. Here, we examined if exercise training could increase intrinsic mitochondrial function in diabetic patients compared with control individuals.
Methods
Fifteen male type 2 diabetic patients and 14 male control individuals matched for age, BMI and \( \dot{V}{{\hbox{O}}_{2{ \max }}} \) enrolled in a 12 week exercise intervention programme. Ex vivo mitochondrial function was assessed by high-resolution respirometry in permeabilised muscle fibres from vastus lateralis muscle. Before and after training, insulin-stimulated glucose disposal was examined during a hyperinsulinaemic–euglycaemic clamp.
Results
Diabetic patients had intrinsically lower ADP-stimulated state 3 respiration and lower carbonyl cyanide 4-(trifluoro-methoxy)phenylhydrazone (FCCP)-induced maximal oxidative respiration, both on glutamate and on glutamate and succinate, and in the presence of palmitoyl-carnitine (p < 0.05). After training, diabetic patients and control individuals showed increased state 3 respiration on the previously mentioned substrates (p < 0.05); however, an increase in FCCP-induced maximal oxidative respiration was observed only in diabetic patients (p < 0.05). The increase in mitochondrial respiration was accompanied by a 30% increase in mitochondrial content upon training (p < 0.01). After adjustment for mitochondrial density, state 3 and FCCP-induced maximal oxidative respiration were similar between groups after training. Improvements in mitochondrial respiration were paralleled by improvements in insulin-stimulated glucose disposal in diabetic patients, with a tendency for this in control individuals.
Conclusions/interpretation
We confirmed lower intrinsic mitochondrial function in diabetic patients compared with control individuals. Diabetic patients increased their mitochondrial content to the same extent as control individuals and had similar intrinsic mitochondrial function, which occurred parallel with improved insulin sensitivity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A low mitochondrial function has been implicated in the aetiology of insulin resistance and type 2 diabetes [1–4]. Several studies revealed compromised in vivo mitochondrial function in type 2 diabetic patients and first-degree relatives of type 2 diabetic patients [3–6], but it is still debated whether lower mitochondrial function is caused by a reduced mitochondrial content [7, 8] or an intrinsically lower mitochondrial function (i.e. mitochondrial respiration after adjustment for markers of mitochondrial density) [5, 9, 10]. Recently, we reported lower ex vivo mitochondrial function along with compromised in vivo mitochondrial function in type 2 diabetic patients compared with BMI- and age-matched overweight controls [5]. No difference in mitochondrial content between groups was observed. As a consequence, after normalisation for mitochondrial density, the ex vivo measurements on permeabilised muscle fibres revealed lower intrinsic mitochondrial function in type 2 diabetic patients. Using multiple substrates, we concluded that compromised mitochondrial function resides at the level of the phosphorylation system, as ADP-stimulated respiration of complex I and II and maximal carbonyl cyanide 4-(trifluoro-methoxy)phenylhydrazone (FCCP)-stimulated oxidative capacity was substantially lower in type 2 diabetic patients.
The beneficial effects of exercise training on metabolic risk factors, i.e. an increase in \( \dot{V}{{\hbox{O}}_{2{ \max }}} \) and insulin sensitivity, are well documented (reviewed by Zanuso et al. [11]). These exercise-induced metabolic improvements are also observed in individuals characterised by a sedentary lifestyle, as in type 2 diabetic patients, first-degree relatives of diabetic patients and obese control individuals [12–14]. It is well known that in healthy individuals exercise training stimulates mitochondrial biogenesis, indicated by an overall increase of markers for mitochondrial mass, like mitochondrial DNA (mtDNA) copy number and citrate synthase activity [14, 15]. It is, however, not known if diabetic patients, who are characterised by mitochondrial dysfunction, are also able to restore intrinsic mitochondrial function upon exercise training. Therefore, in this study we performed ex vivo mitochondrial measurements in muscle fibres of a subgroup of type 2 diabetic patients and control individuals who participated in a 12 week progressive supervised combined endurance and resistance training intervention programme, as published elsewhere [16]. We used high-resolution respirometry in permeabilised muscle fibres, using multiple substrate combinations, to examine intrinsic mitochondrial function before and after the training programme.
Methods
In this study, 15 male type 2 diabetic patients and 14 normoglycaemic control individuals were included, all matched for age, BMI and \( \dot{V}{{\hbox{O}}_{2{ \max }}} \) (age: 59.5 ± 1.2 and 58.2 ± 1.1 years; BMI: 30.0 ± 0.9 and 30.2 ± 0.9 kg/m2; \( \dot{V}{{\hbox{O}}_{2{ \max }}} \): 28.2 ± 1.4 and 29.2 ± 1.2 ml kg−1 min−1; p > 0.05 for all variables). Patients had well-controlled type 2 diabetes (HbA1c 7.1 ± 0.2%) for at least 1 year before participation. All patients were on metformin only or used metformin in combination with sulfonylurea. Glucose-lowering medication was continued throughout the study except for the week preceding the actual tests and sampling of the biopsy. Participants were recruited via advertisements in local newspapers and gave their written informed consent. Before participation in the study, a medical history and physical examination was performed and a resting electrocardiogram was taken. Control individuals underwent a 2 h OGTT according to World Health Organization criteria and were excluded from participation if the test indicated a prediabetic state. Volunteers suffering from uncontrolled hypertension, cardiovascular disease or liver dysfunction, as well as control individuals using medication known to interfere with glucose metabolism, were excluded from participation.
Hydrostatic weighing to determine body composition and an incremental maximal aerobic cycling test to measure maximal oxygen consumption was performed before the exercise intervention. The incremental maximal cycling test was performed every 6 weeks, to measure participants’ physical fitness and to adjust training loads accordingly. This study was performed in a subgroup of a training intervention study that has been published elsewhere [16].
Hyperinsulinaemic–euglycaemic clamp
Before the exercise training programme, peripheral insulin sensitivity was measured using a 3 h hyperinsulinaemic–euglycaemic clamp (40 mU m−2 min−1), according to DeFronzo et al. [17], combined with primed continuous infusion of [6,6-2H2]glucose kinetics to measure rates of glucose disposal (R d) and appearance (R a), as previously described in detail elsewhere [5]. Participants arrived at 07:30 hours at the university after an overnight fast and baseline blood samples were drawn to measure glucose, insulin and NEFA followed by the start of the [6,6-2H2]glucose infusion. Insulin-stimulated blood samples were taken during the last 30 min of the clamp. Indirect calorimetry measurements were performed during baseline and insulin-stimulated conditions to measure carbohydrate and lipid oxidation and to calculate non-oxidative glucose disposal. Diabetic patients discontinued their glucose-lowering medication 1 week prior to the clamp, but restarted the use of medication after the test days. Muscle biopsies were taken before the clamp from the vastus lateralis muscle, under local anaesthesia (2% wt/vol. lidocaine) according the technique of Bergström et al. [18]. Following the exercise training programme, a second hyperinsulinaemic–euglycaemic clamp and muscle biopsy was performed, and again glucose-lowering medication was discontinued 1 week prior to this test day. The final exercise bout of the training programme was performed at least 2 days before the clamp.
Exercise training programme
Participants enrolled in a supervised combined endurance and resistance exercise training programme of 12 weeks. Endurance training consisted of ergometer cycling performed twice a week for 30 min at 55% of the predetermined maximal workload (W max) and resistance exercise consisted of one series of eight repetitions at 55% of their maximal voluntary contraction and two series of eight repetitions at 75% weekly, focusing on the large muscle groups. After 6 weeks, W max was re-measured, after which the training workload was adjusted. The same was done for the maximal voluntary contraction, which was re-measured every 4 weeks [16].
Plasma assays
Blood was collected in tubes containing EDTA and immediately centrifuged. Plasma was frozen in liquid nitrogen and stored at −80°C until assayed. Plasma NEFA, glucose and insulin concentrations, as well as isotopic enrichment of plasma glucose, were determined as previously described [5]. Plasma insulin concentrations were expressed as pmol/l [19].
Mitochondrial density
mtDNA copy number was determined in whole muscle as a measure of mitochondrial density, as previously described [5], and expressed as arbitrary units (AUs).
High-resolution respirometry
Ex vivo mitochondrial function was measured using high-resolution respirometry in a two-chamber oxygraph (Oroboros Instruments, Innsbruck, Austria) before and after the 12 week exercise training programme in permeabilised muscle fibres taken prior to each clamp. Detailed procedures for muscle fibre handling and respirometry measurements have been previously described [5]. In short, directly after the muscle biopsy was taken, ∼15 mg of the muscle tissue was directly placed in a conservation medium (BIOPS; Oroboros Instruments). In a Petri dish, muscle fibres were separated using small needles and the muscle membrane was permeabilised with saponin (stock solution at 5 mg/ml in BIOPS) according to Veksler et al. [20]. Subsequently to several washing steps with respiration medium (MiR05; Oroboros Instruments) to ensure removal of saponin, ∼4 mg wet weight of fibre was transferred into the oxygraph and respiratory measurements were performed at 37°C. The ex vivo mitochondrial respiration, or oxygen flux, was expressed as pmol mg−1 s−1, and after normalisation with respect to mtDNA content as (pmol mg−1 s−1)/(mtDNA copy number × 106). With the addition of several substrates, different states were reached to analyse mitochondrial function. State 3 respiration reflects ADP-stimulated respiration on glutamate (MG3) or on addition of glutamate and succinate (MGS3). Following MGS3, FCCP was titrated to determine maximal mitochondrial oxidative capacity, termed as state uncoupled (state u). In a separate trace, oligomycin was added after MGS3 to inhibit ATP synthase (state O4 respiration [O4]). Every protocol was applied with or without the addition of palmitoyl-carnitine (referred to as MPG3, MPGS3, O4-P and FCCP-P).
Statistics
Data are reported as means ± SE. Statistical analysis were performed using the statistical computer program SPSS 16.0 for Mac OS X. Differences between groups before and after the exercise training were statistically tested with an independent Student’s t test. Differences upon the exercise-training programme within groups were calculated with an ANOVA for repeated measurements. Differences were considered significant at p < 0.05.
Results
Participant characteristics
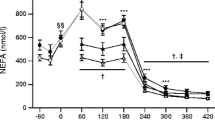
Groups were matched for age, BMI and maximal aerobic capacity. Changes in BMI, body weight and maximal aerobic capacity upon exercise training are shown in Table 1. Fasting plasma glucose concentrations were higher in diabetic patients (who were off medication at the time of sampling) compared with control individuals before training (8.9 ± 0.5 vs 5.9 ± 0.1 mmol/l; p < 0.05) and after training (8.8 ± 0.4 vs 5.4 ± 0.1 mmol/l; p < 0.05). Fasting plasma glucose levels did not change after exercise in diabetic patients, but were significantly lower in control individuals (p < 0.05). Fasting plasma insulin and NEFA concentrations did not differ between diabetic patients and control individuals before training (insulin: 93.0 ± 6.0 vs 120.6 ± 19.8 pmol/l; NEFA: 520 ± 29 vs 494 ± 30 μmol/l) and after training (insulin: 85.8 ± 5.4 vs 103.8 ± 7.4 pmol/l; NEFA: 502 ± 39 vs 429 ± 6 μmol/l). Training resulted in a significant decline in fasting plasma insulin levels in diabetic patients and control individuals (p < 0.05 and p < 0.01, respectively). Training did not affect fasting plasma NEFA levels in diabetic patients; however, NEFA tended to decrease in control individuals (p = 0.06).
Insulin-mediated glucose disposal
Clamped plasma glucose concentrations were not different between type 2 diabetic patients and control individuals before the exercise training (5.7 ± 0.2 vs 5.7 ± 0.1 mmol/l) and after training (5.6 ± 0.2 vs 5.3 ± 0.1 mmol/l). Insulin sensitivity was expressed as the change in insulin-stimulated R d (ΔR d). Insulin-mediated glucose disposal was significantly lower in diabetic patients compared with control individuals before training (ΔR d: 8.1 ± 1.4 vs 16.0 ± 2.3 μmol kg−1 min−1; p < 0.01), and increased significantly after exercise training in diabetic patients to 12.0 ± 1.9 μmol kg−1 min−1 (p < 0.05) with a tendency for increase in control individuals (ΔR d 18.2 ± 2.5 μmol kg−1 min−1; p = 0.09). One control individual appeared to be a non-responder and showed an actual decrease in insulin sensitivity upon exercise training. Exclusion of this control individual resulted in a significant improvement in insulin sensitivity after training vs before training for the control group (ΔR d from 16.0 ± 2.6 to 18.7 ± 2.6 μmol kg−1 min−1; p < 0.05). The exclusion of the non-responder did not result in changes in non-normalised or normalised mitochondrial function between groups. Therefore, this individual was not excluded from the total analysis.
After training, insulin-mediated glucose disposal in type 2 diabetic patients remained significantly lower compared with control individuals (p < 0.05). The insulin-stimulated change in R a (ΔR a) was significant lower in type 2 diabetic patients compared with control individuals (ΔR a 9.1 ± 1.3 vs 18.6 ± 2.8 μmol kg−1 min−1; p < 0.05). The ΔR a upon exercise training increased to 12.3 ± 1.5 (p < 0.05) in diabetic patients and to 21.2 ± 3.0 μmol kg−1 min−1 (p < 0.05) in control individuals, with ΔR a remaining significantly lower in diabetic patients compared with control individuals.
Mitochondrial density
Mitochondrial density measured as mtDNA copy number was not different between diabetic patients and control individuals before training (2.0 ± 0.14 × 106 vs 1.8 ± 0.12 × 106 AU). After training, mtDNA copy number increased significantly to 2.7 ± 0.14 × 106 AU in diabetic patients (p < 0.01) and to 2.7 ± 0.13 × 106 AU (p < 0.01) in control individuals. After training, mtDNA copy number was not significantly different between control individuals and diabetic patients.
Ex vivo mitochondrial respirometry before training
Before training, respiratory values were different between diabetic patients and control individuals. State 3 respiration on MG3 and on MGS3 was significantly lower in diabetic patients compared with control individuals (MG3: 17.2 ± 1.4 vs 22.5 ± 2.0 pmol mg−1 s−1, p < 0.05 and MGS3: 28.1 ± 2.0 vs 33.8 ± 2.0 pmol mg−1 s−1, p = 0.05). No differences were found for O4 on oligomycin between groups (13.3 ± 0.8 vs 14.9 ± 1.2 pmol mg−1 s−1). Maximal respiratory capacity (FCCP-driven state u) in the presence of glutamate and succinate was significantly lower in diabetic patients compared with control individuals (41.2 ± 2.8 vs 53.3 ± 3.1 pmol mg−1 s−1; p < 0.05). When palmitoyl-carnitine was added as a substrate, state 3 respiration on a combination of palmitoyl-carnitine, glutamate and succinate (MPGS3) tended to be lower in diabetic patients compared with control individuals (31.2 ± 2.1 vs 39.1 ± 3.9 pmol mg−1 s−1; p = 0.08). O4 after the addition of oligomycin on MPGS3 was not different between diabetic patients and control individuals (20.4 ± 1.2 vs 22.2 ± 1.8 pmol mg−1 s−1). The maximal respiratory capacity (FCCP-driven state u) in the presence of palmitoyl-carnitine, glutamate and succinate was significantly lower in diabetic patients compared with control individuals (44.8 ± 3.1 vs 55.8 ± 3.1 pmol mg−1 s−1; p < 0.05).
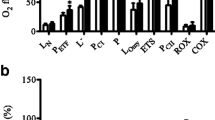
When respiratory values were normalised to mtDNA copy number, significant differences between groups for MG3 (p < 0.05), MGS3 (p < 0.05) and maximal respiratory capacity (FCCP-driven state u) (p < 0.05) remained, with a tendency towards lower O4 for diabetic patients compared with control individuals (p = 0.09) (Fig. 1a). Also, on palmitoyl-carnitine, respiratory values for MPG3 (p = 0.06), MPGS3 (p < 0.05) and maximal respiratory capacity (FCCP-driven state u) on MPGS3 remained significantly lower in diabetic patients compared with control individuals when normalised for mitochondrial content (p < 0.05) (Fig. 1b).

Intrinsic mitochondrial respiration ([pmol mg−1 s−1]/[mtDNA copy number × 106]) in 17 control individuals (white bars) vs 15 diabetic patients (black bars) before training. a In glutamate and succinate. Means ± SE. *p < 0.05; † p = 0.09. b In glutamate and succinate with the addition of palmitoyl-carnitine. Means ± SE. *p < 0.05; † p = 0.06. M, malate alone; MP, malate with palmitoyl-carnitine
Ex vivo mitochondrial respirometry: effect of training
After 12 weeks of exercise training, state 3 respiration on MG3 increased by 34% in diabetic patients (p < 0.01) and respiration on MGS3 increased by 33% (p < 0.01). Also, state 4 respiration and maximal respiratory capacity (FCCP-driven state u) levels increased by 28% (p < 0.01) and 33% (p < 0.01), respectively. Similarly, upon the addition of palmitoyl-carnitine, MPG3 showed an exercise-induced increase of 30% (p < 0.01) and MPGS3 of 28% (p < 0.01). O4 on MPGS3 significantly increased after training by 21% (p < 0.05) and FCCP-induced maximal respiration increased by 27% (p < 0.05). However, when respiration values were normalised to mtDNA copy number, significant differences reported for all states before and after training in diabetic patients no longer persisted (Fig. 2a,b), illustrating that the major part of the improvement in mitochondrial function was because of an increase in mitochondrial content.

Intrinsic mitochondrial respiration ([pmol mg−1 s−1]/[mtDNA copy number × 106]) for 14 control individuals and 15 diabetic patients before training and control individuals after training. a In glutamate and succinate. White and light-grey bars represent control individuals before and after training, respectively; black and dark-grey bars represent diabetic patients before and after training, respectively. Means ± SE. *p < 0.05. b In glutamate and succinate with the addition of palmitoyl-carnitine. Means ± SE. *p < 0.05. M, malate alone; MP, malate with palmitoyl-carnitine
Also, in control individuals, training improved state 3 respiration on MG3 by 26% (p < 0.05) and state 3 respiration on MGS3 increased by 28% (p < 0.01). O4 respiration on MGS3 increased by 32% (p < 0.01) after training; however, FCCP-driven maximal respiratory capacity in the presence of glutamate and succinate was similar before and after training. Similarly, upon the addition of palmitoyl-carnitine, MPG3 increased upon exercise training by 38% (p < 0.01), MPGS3 by 34% (p < 0.01) and O4 upon MPGS3 increased by 17% (p < 0.05). FCCP-driven maximal respiratory capacity measured in the presence of palmitoyl-carnitine, glutamate and succinate increased by 24% upon exercise training; however, this increase was not significant. Again, as in diabetic patients, the increase in mitochondrial respiration (with or without the addition of palmitoyl-carnitine) upon training was mainly accounted for by an increase in mitochondrial density, as no significant increase in intrinsic mitochondrial respiration was observed after adjustment for mtDNA copy number upon exercise training (Fig. 2a,b).
Before and after training, no correlations were found between ex vivo mitochondrial function and insulin-stimulated glucose disposal. Also, when ex vivo mitochondrial function was normalised for mitochondrial content, no correlations with markers of insulin sensitivity were found. Furthermore, body mass did not correlate with mitochondrial function. \( \dot{V}{{\hbox{O}}_{2{ \max }}} \) did correlate with non-normalised state 3 respiration on MPGS (p < 0.01) and tended to correlate with normalised state 3 on MPGS when data before and after training were included. \( \dot{V}{{\hbox{O}}_{2{ \max }}} \) did not, however, correlate with state 3 respiration during baseline measurements or after training.
Discussion
In the current study, type 2 diabetic patients and control individuals participated in a 12 week combined resistance and endurance exercise training programme to study the effects on mitochondrial function. In this group of individuals, who were carefully matched for age, BMI and aerobic capacity, we confirmed our previous finding [5] of lower intrinsic mitochondrial function in type 2 diabetic patients compared with control individuals on glutamate and succinate and also in the presence of palmitoyl-carnitine. Furthermore, we showed that the 12 week exercise training intervention increased mitochondrial content to a similar extent in diabetic patients as in control individuals, with a concomitant improvement of oxidative capacity as determined with ex vivo respiratory measurements in muscle biopsies taken from the vastus lateralis muscle. These improvements were paralleled by improved insulin-stimulated glucose disposal in type 2 diabetic patients and control individuals.
So far, exercise intervention studies in diabetic patients have reported an increase in mitochondrial content determined as increased mitochondrial enzyme activity (such as citrate synthase and NADH oxidase activity), mitochondrial DNA copy number or using electron microscopy [14, 21]. These studies, however, did not examine pretraining values of mitochondrial function relative to healthy controls. Thus it remained unclear if and to what extent improvements in mitochondrial function could be achieved by exercise training in patients with type 2 diabetes. The type 2 diabetic patients enrolled in this exercise intervention programme were recently reported to have lower in vivo mitochondrial function compared with control individuals, which was restored upon exercise training [16]. In vivo mitochondrial function is determined by mitochondrial content, intrinsic mitochondrial function and blood perfusion. Therefore, ex vivo determination of mitochondrial function before and after training is of particular interest, as it allows analysis of intrinsic mitochondrial capacity under a variety of substrates to reveal which mechanism at the mitochondrial level may underlie the improvement of in vivo mitochondrial function upon training.
ADP-stimulated state 3 respiration reflects the oxidative capacity of the mitochondria to reduce TCA cycle- and β-oxidation-derived equivalents as NADH and FADH2 by a convergent electron flow at complex I and II, resulting in oxidative phosphorylation of ADP into ATP at the level of ATP synthase. State 3 respiration is under control of components of the phosphorylation system, such as inorganic phosphate (Pi), adenine nucleotide transporter (ANT) and ATP synthase. On the other hand, maximal mitochondrial oxidative respiration is initiated by titration of the chemical uncoupler FCCP, thereby bypassing the control of the phosphorylation system. This therefore reflects the maximal oxidative capacity of the enzymes of the electron transport chain (ETC) and also of the upstream dehydrogenases (such as malate, pyruvate, glutamate and succinate dehydrogenases). In agreement with earlier results [5], we showed a ∼25% lower state 3 respiration and a ∼31% lower FCCP-induced maximal oxidative capacity in type 2 diabetic patients compared with control individuals, even when normalised to mitochondrial content. Upon exercise training, type 2 diabetic patients increased both mitochondrial state 3 respiration and FCCP-induced maximal oxidative capacity by ∼33%, and this improvement was accompanied by a ∼27% increase in mitochondrial content. Also, state 3 respiration in control individuals increased by ∼28% with a concomitant ∼33% rise in mitochondrial content, although FCCP-induced maximal oxidative capacity did not increase upon exercise training. Therefore, in both groups the increase in oxidative capacity was mainly accounted for by a training-induced increase in mitochondrial content. Importantly, after exercise training, ADP-stimulated state 3 and state u respiration were not significantly different between type 2 diabetic patients and control individuals, irrespectively of correction for mtDNA copy number. These improvements in mitochondrial function occurred in parallel with changes in insulin sensitivity, although the latter was not restored to control values in diabetic patients. This suggests that for complete restoration of insulin sensitivity in patients with type 2 diabetes an increase in muscle mitochondrial content alone is not sufficient.
The exercise training-induced improvements in ex vivo mitochondrial function were detected in nearly all respiratory states on glutamate and succinate and also in the presence of palmitoyl-carnitine. All substrates were added in excess to ensure that respiratory measurements were not limited by substrate availability influencing oxygen flux. A similar increase of the oxidation levels on glutamate and succinate, and as in the presence of fatty acid-derived substrates, were seen upon training in both groups, indicating that the exercise intervention did not lead to a preferential oxidation of fatty acids or glucose-derived substrates. From these results, it may be concluded that the increase of the respiratory states after exercise training in control individuals and type 2 diabetic patients is based at the level of the ETC or the upstream dehydrogenases.
However, it should be noted that substrate competition might differ between the ex vivo condition (in which an excess of substrates were available) and the in vivo situation. For instance, the fatty acid-derived substrate palmitoyl-carnitine used in the ex vivo measurements can enter the mitochondria independently of the carnitine shuttle system, in which carnitine palmitoyltransferase 1 (CPT1) is an important regulator. However, it could not be deduced from this study whether CPT1 or other substrate transporters in vivo are altered upon exercise training, influencing substrate competition.
Remarkably, in control individuals only, exercise training did not enhance FCCP-driven maximal respiratory capacity, as occurred in type 2 diabetic patients. As a result of the increase in mitochondrial content, FCCP-driven maximal respiratory capacity per mitochondrion was actually lower after training. The exact meaning of this finding cannot be deduced from the present study, and future studies are needed to confirm this finding. In both groups, exercise training stimulated mitochondrial biogenesis and enhanced mitochondrial function; however, a significant increase in insulin sensitivity was seen in type 2 diabetic patients, with a tendency in control individuals. From the present study, we cannot deduce if the training-induced increase in mitochondrial content and enhanced oxidative function is responsible for the improved insulin sensitivity as seen in type 2 diabetic patients and control individuals. However, we found no correlation between (changes in) insulin sensitivity and mitochondrial function, although a parallel increase of both variables was seen upon exercise training. These results might suggest that insulin sensitivity and mitochondrial function are two effects of exercise training that are not necessarily causally related. In that respect, insulin sensitivity remained lower upon training in type 2 diabetic patients compared with control individuals. This may suggest that improved mitochondrial function alone was not sufficient to restore insulin sensitivity, and that additional exercise-related factors could be responsible for the training-increased insulin sensitivity.
In this study, participants underwent a combined endurance and resistance exercise-training programme as a combined training strategy would result in greater adverse events of the metabolic profile as previously reviewed [11]. A combined aerobic and resistance training would improve oxidative capacity and in addition would enhance muscle quality (strength and mass) [22]. The absent correlation between improved insulin sensitivity and mitochondrial function could be partly caused by the effect of resistance training, in which increased insulin-sensitive muscle mass is a well known outcome.
In conclusion, in the present study we confirmed lower intrinsic mitochondrial function in male type 2 diabetic patients compared with control individuals matched for age, BMI and physical activity. Furthermore, we showed that type 2 diabetic patients increased their mitochondrial content to a similar extent as control individuals. Ex vivo mitochondrial function increased upon exercise at the level of ADP-stimulated oxidative phosphorylation and also FCCP-driven maximal oxidative capacity, both on glutamate and on glutamate and succinate and in the presence of palmitoyl-carnitine. Importantly, type 2 diabetic patients had similar intrinsic mitochondrial function compared with control individuals, paralleled by an improvement, but not restoration, of insulin sensitivity.
Abbreviations
- AU:
-
Arbitrary unit
- ETC:
-
Electron transport chain
- FCCP:
-
Carbonyl cyanide 4-(trifluoro-methoxy)phenylhydrazone
- FCCP-P:
-
FCCP with the addition of palmitoyl-carnitine
- MG3:
-
Respiration on malate and glutamate
- MGS3:
-
Respiration on malate, glutamate and succinate
- MPG3:
-
Respiration on malate and glutamate with the addition of palmitoyl-carnitine
- MPGS3:
-
Respiration on malate, glutamate and succinate with the addition of palmitoyl-carnitine
- mtDNA:
-
Mitochondrial DNA
- O4:
-
State O4 respiration
- O4-P:
-
State O4 respiration with the addition of palmitoyl-carnitine
- R a :
-
Rate of glucose appearance
- R d :
-
Rate of glucose disposal
References
Lowell BB, Shulman GI (2005) Mitochondrial dysfunction and type 2 diabetes. Science 307:384–387
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350:664–671
Petersen KF, Dufour S, Shulman GI (2005) Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med 2:e233
Schrauwen-Hinderling VB, Roden M, Kooi ME, Hesselink MK, Schrauwen P (2007) Muscular mitochondrial dysfunction and type 2 diabetes mellitus. Curr Opin Clin Nutr Metab Care 10:698–703
Phielix E, Schrauwen-Hinderling VB, Mensink M et al (2008) Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 57:2943–2949
Szendroedi J, Schmid AI, Chmelik M et al (2007) Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med 4:e154
Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, Dela F (2007) Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia 50:790–796
Larsen S, Ara I, Rabol R et al (2009) Are substrate use during exercise and mitochondrial respiratory capacity decreased in arm and leg muscle in type 2 diabetes? Diabetologia 52:1400–1408
Mogensen M, Sahlin K, Fernstrom M et al (2007) Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 56:1592–1599
Kelley DE, He J, Menshikova EV, Ritov VB (2002) Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51:2944–2950
Zanuso S, Jimenez A, Pugliese G, Corigliano G, Balducci S (2010) Exercise for the management of type 2 diabetes: a review of the evidence. Acta Diabetol 47:15–22
Ostergard T, Andersen JL, Nyholm B et al (2006) Impact of exercise training on insulin sensitivity, physical fitness, and muscle oxidative capacity in first-degree relatives of type 2 diabetic patients. Am J Physiol Endocrinol Metab 290:E998–E1005
Toledo FG, Menshikova EV, Azuma K et al (2008) Mitochondrial capacity in skeletal muscle is not stimulated by weight loss despite increases in insulin action and decreases in intramyocellular lipid content. Diabetes 57:987–994
Toledo FG, Menshikova EV, Ritov VB et al (2007) Effects of physical activity and weight loss on skeletal muscle mitochondria and relationship with glucose control in type 2 diabetes. Diabetes 56:2142–2147
Lanza IR, Nair KS (2009) Muscle mitochondrial changes with aging and exercise. Am J Clin Nutr 89:467S–471S
Meex RC, Schrauwen-Hinderling VB, Moonen-Kornips E et al (2009) Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes 59:572–579
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237:E214–E223
Bergström J, Hermansen L, Hultman E, Saltin B (1967) Diet, muscle glycogen and physical performance. Acta Physiol Scand 71:140–150
Volund A (1993) Conversion of insulin units to SI units. Am J Clin Nutr 58:714–715
Veksler VI, Kuznetsov AV, Sharov VG, Kapelko VI, Saks VA (1987) Mitochondrial respiratory parameters in cardiac tissue: a novel method of assessment by using saponin-skinned fibers. Biochim Biophys Acta 892:191–196
Bordenave S, Metz L, Flavier S et al (2008) Training-induced improvement in lipid oxidation in type 2 diabetes mellitus is related to alterations in muscle mitochondrial activity. Effect of endurance training in type 2 diabetes. Diab Metab 34:162–168
Cuff DJ, Meneilly GS, Martin A, Ignaszewski A, Tildesley HD, Frohlich JJ (2003) Effective exercise modality to reduce insulin resistance in women with type 2 diabetes. Diab Care 26:2977–2982
Acknowledgements
E. Phielix was supported by a grant of the Dutch Diabetes Research Foundation (grant 2004.00.059). A VICI (grant 918.96.618) and a VIDI (grant 917.66.359) for innovative research from the Netherlands Organization for Scientific Research supported the work of P. Schrauwen and M. Hesselink, respectively.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Phielix, E., Meex, R., Moonen-Kornips, E. et al. Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia 53, 1714–1721 (2010). https://doi.org/10.1007/s00125-010-1764-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1764-2