
Abstract
Key message
The gene underlying the melon fruit shape QTL fsqs8.1 is a member of the Ovate Family Proteins. Variation in fruit morphology is caused by changes in gene expression likely due to a cryptic structural variation in this locus.
Abstract
Melon cultivars have a wide range of fruit morphologies. Quantitative trait loci (QTL) have been identified underlying such diversity. This research focuses on the fruit shape QTL fsqs8.1, previously detected in a cross between the accession PI 124112 (CALC, producing elongated fruit) and the cultivar ‘Piel de Sapo’ (PS, producing oval fruit). The CALC fsqs8.1 allele induced round fruit shape, being responsible for the transgressive segregation for this trait observed in that population. In fact, the introgression line CALC8-1, carrying the fsqs8.1 locus from CALC into the PS genetic background, produced perfect round fruit. Following a map-based cloning approach, we found that the gene underlying fsqs8.1 is a member of the Ovate Family Proteins (OFP), CmOFP13, likely a homologue of AtOFP1 and SlOFP20 from Arabidopsis thaliana and tomato, respectively. The induction of the round shape was due to the higher expression of the CALC allele at the early ovary development stage. The fsqs8.1 locus showed an important structural variation, being CmOFP13 surrounded by two deletions in the CALC genome. The deletions are present at very low frequency in melon germplasm. Deletions and single nucleotide polymorphisms in the fsqs8.1 locus could not be not associated with variation in fruit shape among different melon accessions, what indicates that other genetic factors should be involved to induce the CALC fsqs8.1 allele effects. Therefore, fsqs8.1 is an example of a cryptic variation that alters gene expression, likely due to structural variation, resulting in phenotypic changes in melon fruit morphology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Melon (Cucumis melo L.) is a crop species that belongs to the Cucurbitaceae family, which also includes important cultivated species such as cucumber (Cucumis sativus L.), watermelon (Citrullus lanatus Thumb.), pumpkins and squashes (Cucurbita spp.), gourds [Momordica charantia L., Lagenaria siceraria Molina. Standley, Benincasa hispida (Thunb.) Cogn., Luffa aegyptiaca Mill. Luffa acutangula L. Roxb., Trichosanthes cucumerina L.] and other minor crops as Cucumis metuliferus Meyer ex Naudin and Cucumis anguria L. Together with watermelon and cucumber, melon is one of the cucurbit crops with the greatest economic impact. Worldwide production has increased since the beginning of the 21st Century from 20 million to more than 27 million tons (FAOSTAT 2021).
Melon was domesticated independently at least twice in Africa and Asia (Endl et al. 2018; Zhao et al. 2019), although the cultivars that are commonly grown worldwide derived from the Asian domestication through divergent diversification from the Indian subcontinent to Oriental and Occidental Eurasian regions (Gonzalo et al. 2019; Zhao et al. 2019). One of the consequences of this diversification was the development of a large number of landraces, traditional and modern cultivars that show an impressive diversity in fruit morphology, from the small (30–50 g) round fruit of wild melons to the large and extremely elongated fruit of flexuosus varieties (Monforte et al. 2014). Pitrat (2017) proposed an exhaustive horticultural classification of melon that reflects such phenotypic diversity, defining nineteen groups, one wild (agrestis), two semi‐domesticated or feral (kachri and chito) and sixteen domesticated and cultivated ones.
The genetic control of melon fruit morphology has been studied in several works, and quantitative trait loci (QTL) and consensus QTLs have been defined (reviewed by Monforte et al. 2014; Pan et al. 2020). The pleiotropic effects of two genes, a and p, on fruit shape are widely known (Perin et al. 2002): a gene controlling female flower sex expression, being strictly female or hermaphrodite in monoecious or andromonoecious varieties, respectively, whereas p controls the fruit carpel number (with three carpels being common for most varieties and five for a few other). Regarding the pleiotropic effects observed for a, fruit from strictly female flowers (in monoecious plants) are usually much more elongated than fruit from hermaphrodite flowers in andromonoecious plants (Monforte et al. 2005; Abdelmohsin and Pitrat 2008). Gene a encodes a 1-AMINOCYCLOPROPANE-1-CARBOXYLIC ACID SYNTHASE (CmACS-7), and a missense mutation in the gene produces a loss of CmACS-7 protein activity, leading to the development of stamens in the female flower (andromonoecy), which results in subsequent pleiotropic effects making a rounder fruit shape (Boualem et al. 2008). Whereas wild melons are monoecious, the andromonoecious mutation was selected during cultivar diversification. Traditional Indian landraces (the primary center of melon diversity) are monoecious, while andromonoecy is more common in Mediterranean and modern cultivars. Both andromonoecious and monoecious cultivars need pollinators to ensure proper fruit production, so the selection of the andromonoecious mutation does not seem to be related to an increase in fruit set. It is likely that the selection sought rounder fruit. On the contrary, the increase of in the fruit carpel number, controlled by p, has been associated with rounder fruits in several mapping populations (Perin et al. 2002; Monforte et al. 2004). Wild melons and most traditional and modern cultivars develop three carpels, while five carpels are found only in a small subset of Indian and Oriental cultivars. Strong evidences based on investigations performed in cucumber (Li et al. 2016), co-localization of candidate genes with the p locus in experimental mapping populations (Pan et al. 2020) and a genome-wide association study (GWAS) panel indicate that the gene p may be the melon homologue (CmCLV3) of CLAVATA3 which tomato homologue (SlCLV3) controls locule number in the tomato fruit (Xu et al. 2015). For the rest of the QTLs, candidate genes belonging to the CELL NUMBER REGULATOR (CNR/FW2.2), SlKLUH/CYP785A/FW3.2, OVATE, SUN, TON1 RECRUTING MOTIF (TRM), CELL SIZE REGULATOR (CSR/FW11.2), YABBY, and WOX families have been proposed based on the co-location of members from these families with melon fruit morphology QTLs (Monforte et al. 2014; Pan et al. 2020). Among them, OVATE, CSR and SUN showed a larger number of co-locations (Pan et al. 2020), although no functional verification has been carried out until now.
Diaz et al. (2014) mapped several fruit shape QTLs in a segregating population derived from the cross between the Indian landrace PI 124,112 (CALC, producing elongated fruit) and the Spanish “Piel de Sapo” (PS, producing oval fruit) cultivar. The QTL with the strongest effect was mapped to chromosome 8 and was named fsqs8.1. Surprisingly, round fruits were induced by the CALC allele, which explained the transgressive segregation observed for fruit shape in this population. The QTL was introgressed into PS by developing the introgression line (IL) CALC8-1, which carries a segment of chromosome 8 from CALC containing the fsqs8.1 locus, while the rest of the genome was PS. CALC8-1 produced round fruits, verifying the effect of fsqs8.1 (Diaz et al. 2014). Interestingly, fsqs8.1 maps to the same genome region as the consensus QTL FSQM8 (Monforte et al. 2014) and CmFSI8.3 (Pan et al. 2020). Additionally, Gur et al. (2017) showed a strong association of fruit shape in the same chromosome 8 genomic region in a diverse melon germplasm collection by GWAS. These works indicated that fsqs8.1 could be one of the major loci responsible for the enormous fruit shape variability observed in melon germplasm. Wu et al. (2018) fine-mapped fsqs8.1 between markers Sca76-197 and Sca76-45 on chromosome 8. Several genes were identified in that region, including a Serine/threonine kinase (MELOC025211), a 1-aminocyclopropane-1-carboxylate oxidase (MELO3C025210) and a member of the OFP family (MELO3C025206) that was previously named as CmOFP13 by Monforte et al. (2014). Among them, CmOFP13 was pointed out as the strongest candidate gene.
The objective of the present work is to identify the causal gene underlying fsqs8.1, to track the history of its variability during melon diversification, to shed light on its effects during ovary development, and to investigate its genetic effects in several genetic landscapes.
Material and methods
Plant material
Diaz et al. (2014) previously developed the introgression line CALC8-1 that carries an introgression on chromosome 8, covering the fsqs8.1 locus, from the traditional melon accession PI 124,112 (CALC) from Bihar, India, into the “Piel de Sapo” (PS) cultivar from Spain (Oliver et al. 2001). Eduardo et al. (2005) developed the introgression line SC8-3, which carries an introgression with fsqs8.1 locus from the Korean cultivar “Songwan Charmi” (SC) accession PI161375 in the same PS genetic background. The three genotypes are andromonoecious. Large F2 populations were developed from crosses between CALC8-1 and PS for fine mapping. SC8-3 and CALC8-1 were also crossed to study gene interaction between alleles.
An array of melon cultivars and accessions from different world regions and belonging to several melon horticultural groups was obtained from the Instituto de Conservación y Mejora de la Agrodiversidad Valenciana (COMAV, Polytechnic University of Valencia, Spain) germplasm bank (Supplementary Table S1) to study associations between Single Nucleotide Polymorphism (SNP) markers tightly linked to fsqs8.1 and fruit shape. A set of five of them were selected and crossed with CALC8-1 and PS to investigate interaction between fsqs8.1 and the genetic background. The cultivar ‘Mucha Nesvi’ (MN), a monoecious ameri cultivar, was crossed with CALC8-1 to verify the epistatic interaction for fruit shape between the sex expression gene a and fsqs8.1.
Positional cloning
Three thousand five hundred F2 seedlings from the CALC8-1 × PS cross were screened for the markers flanking the fsqs8.1 position Sca76-197 and Sca76-42 and the internal marker Sca76-140b (Supplementary Table S2). DNA was extracted according to Doyle and Doyle (1990) with minor modifications. Genotyping was carried out with the KASPAR technology (Smith and Maughan 2015) by the Genotyping Service at the John Innes Center (Norwich, UK). Recombinants were transferred to the greenhouse to obtain self-seed. The position of the recombinant events was established by genotyping the recombinant plants with an array of SNP markers (Supplementary Table S2) distributed along the fsqs8.1 locus by KASPAR and High Resolution Melting (HRM, Wittwer et al. 2003) with MELTDOCTOR™ (ThermoFisher Scientific, Waltham, MA) following the supplier recommendations. Seedlings from selected F3 families were genotyped with the appropriate markers to identify homozygous seedlings for CALC and PS for the corresponding segregating recombinant introgression. The selected seedlings were transferred to greenhouse. Plants from each family were planted in the same row in the greenhouse, randomly distributed within each family. The fruit shape index (FSI) was defined as the ratio between maximum fruit length and maximum fruit diameter. The mean FSI for the groups of plants with a contrasting allele composition at the segregating introgression within each F3 family were compared with a t-test to determine if fsqs8.1 was present or not in the respective introgressions.
Sequencing and genomic analysis of fsqs8.1 locus
Amino acid sequences of tomato, Arabidopsis thaliana and melon OVATE family protein (OFP) members, were retrieved from previous works (Monforte et al. 2014) and aligned with CLUSTAL. Evolutionary distances were computed using the Poisson correction method (Zuckerkandl and Pauling 1965) in units of the number of amino acid substitutions per site, removing ambiguous positions for each sequence pair. The tree was calculated with the neighbor-joining method (Saitou and Nei 1987), and the statistical support of the clusters was assessed by a bootstrap test with 2000 replicates (Felsenstein et al. 1985). All the analysis were conducted with MEGA X (Kumar et al. 2018).
MELO3C025206 was amplified from PS, CALC8-1 and SC8-3 using the Cmofp13 primers (Supplementary Table S2) with Taq I DNA polymerase (Biotools, Madrid, Spain). The PCR products were cleaned with ExoSap-IT™ (ThremoFisher Scientific) and sequenced by the Sequencing Service at the Instituto de Biología Molecular y Celular de Plantas (IBMCP; Valencia, Spain) with an ABI3130 Genetic analyzer (Applied Biosystems, Foster City, CA). The sequences were aligned with CLUSTAL implemented in MEGA X (Kumar et al. 2018).
Illumina raw sequences from melon accessions (Zhao et al. 2019) were retrieved from the Sequence Read Archive (SRA) under the NCBI BioProject with the accession ID PRJNA565104. Adaptors and low-quality regions were removed from the reads using cutadapt (Martin 2011). Clean, high-quality reads were then mapped to the melon reference genome v4.0 (Castanera et al. 2020) using BWA-MEM (Li 2013), and the mapping results were visualized with the Integrative Genomics Viewer IGV (Robinson et al. 2011).
The 10 × Chromium sequencing was performed at the GeT-PlaGe core facility, INRA Toulouse (France). The Chromium libraries were prepared according to 10X Genomics’ protocols using the Genome Reagent Kits v2. The samples quantity and quality controls were validated on Qubit, Nanodrop and Femto. The optimal performance was characterized on input gDNA with a mean length greater than 50 kb. The libraries were prepared from 3 µg of HMW gDNA (cut off at 50 Kb using BluePippin system) or 0.625–1.8 ng of HMW DNA. Briefly, in the microfluidic Genome Chip, a library of Genome Gel Beads was combined with HMW template gDNA in Master Mix and partitioning oil to create Gel Bead-In-EMulsions (GEMs) in the Chromium. Each gel bead was functionalized with millions of copies of a 10x™ Barcoded primer. Upon dissolution of the genome gel bead in the GEM, primers containing (i) an Illumina R1 sequence (Read 1 sequencing primer), (ii) a 16 bp 10 × Barcode, and (iii) a 6 bp random primer sequence were released. A read 1 sequence and the 10x™ Barcode were added to the molecules during GEM incubation. P5 and P7 primers, Read 2, and Sample Index were added during library construction. 10 cycles of PCR were applied to amplify libraries. Library quality was assessed using a fragment analyzer, and libraries were quantified by q-PCR using the Kapa Library Quantification Kit. The libraries were performed on an Illumina HiSeq3000 using a paired-end read length of 2 × 150 pb with the Illumina HiSeq3000 sequencing kits. The 10 × Genomics chromium libraries were assembled using the 10 × Genomics Supernova software (Weisenfeld et al. 2017), using default parameters.
Ovary development analysis and RT-PCR
For the development study, ovaries from the three genotypes CALC8-1, SC8-3 and PS were collected fresh at different stages: E1 (carpel primordial initiated), E2 (primordial stigmas appear), E3 (stigma and style defined and anthers with pollen) and E4 (ovule primordial initiated) that correspond to stages 6, 8-1, 8-2, 8-3, respectively, from Bai et al. (2004). The ovaries were fixed overnight in 4% (w/v) p-formaldehyde in 0.1 M sodium phosphate pH 7.2 with 0.05% (v/v) of Tween 20 at 4 °C and dehydrated in a graded ethanol series as described in Gomez et al. (2004, 2011). After dehydration, the samples were embedded in Paraffin wax (Paraplast Plus). To obtain the ovary sections, samples in paraffin were sectioned in a Leica RM2025 microtome. The sections were stained with 0.02 toluidine blue, and the images of ovaries were captured using a Leica DM5000 microscope equipped with the LAS X application at the microscopy services at the IBMCP.
Additionally, flowers from six days prior anthesis to full anthesis stages were collected freshly and sectioned longitudinally. The ovary hair was eliminated to facilitate the definition of ovary contour. Ovaries were observed with a Leica DMS 1000 stereomicroscopy equipped with the LAS X application for image capture at the microscopy service of the IBMCP.
Gene expression analysis during flower development was performed in the previously defined four development stages in PS, SC8-3 and CALC8-1. Five independent replicates were analyzed. RNA extractions were performed according to the protocol from the Aurum Total RNA Mini kit (Biorad, Hercules, CA)) and treated with Ambion DNAse I (RNAse-free) to eliminate DNA. cDNA was then synthesized from 1 µg of total RNA using PrimeScript 1st strand cDNA synthesis kit. The expression of genes was evaluated through quantitative RT-PCR (q-PCR) by using the TAKARA SBRY Premix ExTaq (Tli RNAseH Plus). Supplementary Table S2 shows the corresponding primers used for q-PCR. The q-PCR primers were designed from the coding regions of each gene by using the Primer3 software (Untergasser et al. 2012). To avoid possible cross-amplification, and before any q-PCR experiment, the homology of each primer was checked using the BLAST tool from MELONOMICS against the CM4.0 genome (www.melonomics.net) and the expected size tested in agarose gels. The q-PCR reactions consisted of two step of hold at 95 °C 30 s and 40 cycles of 3 s at 95 °C, and 30 s at 60 °C. The relative expression of each gene was determined by the comparative Ct (Cycle Threshold) method using C. melo ACTIN gene (MELO3C023264) as the internal standard. The efficiency of amplification for each amplicon was tested using increasing amounts of template cDNA, and were all roughly equivalent. The absolute value of the ΔCt slope (Ct of the target gene-Ct of the reference gene) versus serial dilutions of cDNA for a given sample must be less than 0.1. The relative expression of each gene was then calculated relative to a calibrator sample using the formula 2−ΔΔCt, where ΔΔCt is the difference between the ΔCt of each sample and the ΔCt of the calibrator sample. Means of relative expression between genotypes within each ovary development stage were compared with the nonparametric Kruskal–Wallis test.
Arabidopsis thaliana transformation
MELO3C025206 open reading frame from both CALC8-1 was inserted into pDONR222 (Invitrogen, Waltham, MA) by PCR with two specific oligonucleotides (GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAGGAGATAGAACCATGAGAAATCACAAGTTCCGT and GGGGACCACTTTGTACAAGAAAGCTGGGTTTCAAAGAGGTGGAGGAGAAGG) using a BP Gateway reaction (Invitrogen). Upon confirmation of the sequence, cDNA was transferred to pGWB14 (Nakagawa et al. 2007) to render MPA635 (35S:OVATE_CAL8-1), and it was introduced in Arabidopsis thaliana (L.) Heynh. (ecotype Col-0) by Agrobacterium-mediated floral-dip transformation (Clough and Bent 1998). T1s, T2s, and T3s lines were selected by the presence of hygromycin (Duchefa Biochemie, Haarlem, The Netherlands) in Murashige and Skoog (MS) medium plates (Murashige and Skoog 1962) (Duchefa). Several T2 lines were inspected, and four representative lines were fixed for the transgene in homozygosis. Seeds were sterilized in ethanol and germinated in MS for 4 days at 4 °C in the dark, followed by 7–8 days at 22 °C under a long day photoperiod (16 h/8 h). Seedlings were then transferred to soil and grown in a chamber at 22 °C/19 °C under long day photoperiod (16 h/8 h). Pictures from seedlings at day 9, flowers, pistils, and fruits were taken with a SMZ1270 stereomicroscope (Nikon, Leuven, Belgium), whereas those from the rosettes of adult plants were obtained with an Alpha ILCE-6000L digital camera (Sony, Tokyo, Japan).
Association of fsqs8.1 locus haplotypes with fruit shape in melon germplasm
Sixty-six melon accessions (Supplementary Table S1) were grown in COMAV greenhouse facilities in 2014 and 2018 seasons in a completely randomized design with three biological replicates and the FSI was obtained for each one. Accessions were genotyped with SNPs Sca76-snp192, Sca76-snp155, Sca76-snp140 and the sca76-95indel-ov makers (Supplementary Table S2) located in the fsqs8.1 locus. Associations between the markers and the FSI were studied by ANOVA. Haplotypes were defined via visual inspection. FSI means of the haplotypes with at least five accessions were compared with Tukey’s multiple comparison test (p < 0.05). Statistical analyses were carried out with JMP® v. 11 (SAS Institute Inc. Cary, NC).
Interactions of fsqs8.1 in different genetic situations
To study the gene action between the CALC and SC fsqs8.1 alleles five plants of each, CALC8-1, SC8-3 and the hybrid CALC8-1-x-SC8-3 were grown in a completely randomized design in the COMAV greenhouse facilities. Fruits were collected at full mature stages and the FSI index recorded. The gene action (d/[a]), with d being the dominance deviation and [a] the absolute additive value) was calculated as:
d/[a] = [ CALC8-1-x-SC8-3-(CALC8-1-SC8-3)/2]/[(CALC8-1 + SC8-3)/2].
with the contrast 2*CALC8-1-x-SC8-3-(CALC8-1-SC8-3) = 0 to determine the statistical significance of the gene action.
Regarding the interaction of the fsqs8.1 alleles with the a sex expression gene, two hundred seedlings belonging to the F2 population derived from the cross CALC8-1-x- ‘Mucha Nesvi’ (MN) were genotyped using fsqs8.1 SNPs markers and the perfect marker for the a gene ALU-CAPS (Boualem et al. 2008) to select plants homozygous for the four possible allelic combinations at both genes. Plants were grown in the COMAV greenhouse facilities using a completely randomized design. The interaction between genes was contrasted by a two-way ANOVA.
Finally, interaction of fsqs8.1 alleles with different genetic backgrounds (GB) was performed in F1 hybrids from the crosses between CALC8-1 and PS with an array of six different melon cultivars (Supplementary Table S1), having, therefore, the CALC or PS fsqs8.1 alleles in heterozygous conditions. The hybrids were grown in two consecutive years (2016 and 2017) at the COMAV greenhouse facilities in a completely randomized design with ten replicated. The effect of the genetic background, fsqs8.1 allele, year and their respective interactions were assessed by the ANOVA model:
where FSQS8.1i indicates the CALC or PS fsqs8.1 allele in the hybrid in heterozygous conditions,GBj the effect of the different backgrounds, Yeark the effect of the year, and the respective two-way interactions. Statistical analyses were carried out with JMP® v. 11 (SAS Institute Inc. Cary, NC).
Results
Fine mapping of fsqs8.1
The genomic position of fsqs8.1 was previously defined between markers Sca76-197 and Sca76-45 on chromosome 8 of the melon genome (Wu et al. 2018). According to the CM4.0 melon genome version (Castanera et al. 2020), the physical distance of those markers is 169 kb (Fig. 1) between positions 26,839,235 and 27,008,844, which carries five annotated genes: MELO3C025211 (serine/threonine kinase), MELO3C025210 (gibberellin 2-beta-dioxygenase 2-like), MELO3C028356 (hypothetical unknown protein with some homology to a retrotransposon gag protein), MELO3C025206 (ovate family protein), and MELO3C025205 (RING-type E3 ubiquitin transferase). Interestingly, the 93 kb region between MELO3C025210 and MELO3C025206 is composed by long terminal repeats (LTR) and repetitive elements, characteristics of retrotransposons, and only contains one annotated gene, MELO3C028356.

High-resolution mapping of fsqs8.1. A schematic representation of the reference genome V4.0 (Castanera et al. 2020) based on the double haploid line DHL92 is depicted in the upper part of the figure, indicating the position of markers, annotated genes and the repetitive sequence rich region. The length of the genes and the direction of the transcription are indicated with arrows. The deduced genome of CALC8-1 based on re-sequencing data is depicted in the lower part. Blue dashed lines indicates the large deletions observed in CALC8-1. The position of fsqs8.1 based on the fine mapping experiments is indicated below the CALC8-1 genome. The candidate MELO3C025206 is highlighted in green. On the right, typical fruits of PI 124,112 (CALC, donor parent), PS (recipient parent) and CALC8-1 (introgression line carrying the fsqs8.1 locus from CALC in PS background) are also shown) (color figure online)
A fine mapping strategy was followed in order to reduce the number of candidate genes that might underlie fsqs8.1. Sequence reads previously obtained by Sanseverino et al. (2015) from PS, CALC, and SC accessions were mapped to the melon reference genome CM4.0 to identify SNPs for the subsequent fine mapping efforts. The reference genome was obtained from the double haploid line DHL92 derived from the cross between PS and SC. The genomic region including the fsqs8.1 locus in DHL92 corresponds to the PS genome. Twenty-five variant calls were identified; however, no variant calls were found in the 49 kb region between the positions from 26,898,531 to 26,947,890, corresponding to the previously mentioned LTR-rich region (Fig. 1). A visual inspection of PS, SC and CALC read mappings with IGV (Robinson et al. 2011) showed that no CALC Illumina reads mapped to this genomic region, except for some low-quality read mappings corresponding to repetitive sequences, while mapping reads were observed for PS and SC (Supplementary Fig. S1). Furthermore, a second smaller region lacking CALC Illumina reads at 5' of MELO3C025206 was also observed. In order to verify that this pattern was also present in the IL CALC8-1 genome, the IL and PS were re-sequenced using 10X Chromium technology. Although the assembly in this genomic region was not of sufficient quality, the read mapping pattern for both genotypes was similar as the one observed in the previous re-sequencings, confirming the presence of two deletions surrounding MELO3C025206 (Supplementary Fig. S1) in the CALC8-1 genome. A PCR marker was developed to amplify a sequence within the putative large deletion; the deletion-marker failed to amplify either CALC8-1 or CALC but produced a clear amplification product in PS (Supplementary Fig. S1). These results suggested a large 40 kb deletion, approximately from positions 26,900,000 to 26,938,000, and an smaller 3 kb deletion from positions 26,949,154 to 26,952,139 (Fig. 1) in the CALC fsqs8.1 locus.
We obtained seven highly informative recombinants in the fsqs8.1 locus after two rounds of recombination screening of 3500 F2 CALC8-1xPS seedlings, with recombination points among the annotated genes (Supplementary Table S3). The progeny analysis of those recombinants showed the fsqs8.1 locus expanded from positions 26,937,197 to 26,958,640. The only coding sequence in that region corresponded to MELO3C025206 (Fig. 1), strongly suggesting that this gene is underlying the fsqs8.1 QTL. BLAST analysis showed that MELO3C025206 encodes for a member of the ovate family proteins, previously named by Monforte et al. (2014) as CmOFP13. The direction of the translation was reverse to the reference genome, i. e., the sequence up to 26,937,197 was downstream to MELO3C025206, and sequence up to 26,958,640 upstream.
Ovary development and gene expression
The final melon fruit shape is usually determined pre-anthesis during ovary development (Perin et al. 2002; Eduardo et al. 2005). Ovary shape was studied in three genotypes: PS, CALC8-1 and SC8-3. SC8-3 is an IL that carries an introgression in the fsqs8.1 locus from SC (Eduardo et al. 2005). As fruits from SC8-3 are very elongated (Eduardo et al. 2007), it is likely that the three genotypes represent an allelic series for fsqs8.1 ranging from elongated (SC8-3), oval (PS) to round (CALC8-1). Differences in ovary shape at anthesis and pre-anthesis were observed among them (Fig. 2a, Supplementary Fig. S2), CALC8-1 showed a clear rounder shape four days before anthesis (DBA) and SC8-3 an elongated shape even at 6 DBA. To find the ovary developmental stage when the differences in ovary shape appeared, ovaries were collected in four early developmental stages: E1 (carpel primordial initiated), E2 (primordial stigmas appear), E3 (stigma and style defined and anthers with pollen) and E4 (ovule primordial initiated). Differences in ovary shape were already evident for SC8-3 at E2, with elongated ovaries compared to PS and CALC8-1, whereas CALC8-1 showed rounded ovaries compared to PS at E4 (Fig. 2b). Regarding ovary length and width, PS showed strong growth increase between E2 and E3 compared with CALC8-1, suggesting that there was already an inhibition of the growth in CALC8-1 at this stage that ended up with the variation in shape observed at E4 stage.

Developmental analysis and q-PCR of SC8-3, PS and CALC8-1 ovaries. a Ovaries from the three genotypes at anthesis. b Ovary length, width (both in µm) and shape index for the three genotypes through ovary development stages: E1 (carpel primordial initiated), E2 (primordial stigmas appear), E3 (stigma and style defined and anthers with pollen) and E4 (ovule primordial initiated). Asterisks (*) indicates means of CALC8-1 or SC8-3 ovaries statistically significant different (p < 0.05) from PS means c Accumulation of MELOC025206 transcripts estimated by qRT-PCR between genotypes and developmental stages. Different letters indicate statistically significant differences among genotypes within developmental stages (p < 0.05). d Paraffin sections of representative ovaries from the three genotypes at E4
Next, we wanted to correlate the expression of CmOFP13 with the variation of ovary shape through the ovary developmental stages among PS, SC8-3 and CALC8-1. Expression of CmOFP13 between the E2 and E4 stages showed different patterns among genotypes (Fig. 2c). CmOFP13 was residually expressed in SC8-3 at E2 and low levels at E3, whereas in PS the expression was low (although higher than SC8-3 in E2) in those stages, but, on the contrary, the expression was high in CALC8-1 in those stages. Finally, the expression was similar among genotypes in E4. Therefore, the correlation between CmOFP13 and ovary shape was observed: lack of expression in E2 for elongated shape, low expression in E2 and E3 with oval shape and high expression in E2 and E3 with round shape. Figure 2d depicts representative ovaries at E4, where the three different ovary shapes were evident among all three genotypes.
Characterization of CmOFP13 gene sequence and surrounding genomic region
CmOFP13 has a 999-nucleotide open reading frame, encoding for a protein compromised of 331 amino acids with no introns. CmOFP13 includes an ovate sequence motif, therefore likely being a member of the OVATE family proteins (OFP). This family has been extensively studied especially in A. thaliana (Wang et al. 2016), tomato (Rodriguez et al. 2011) and other plant species (Dangwal and Das 2018). Monforte et al. (2014) identified the melon gene members belonging to this family. A phylogenetic analysis was carried out with A. thaliana, tomato and melon OFPs (Fig. 3, Supplementary Fig. S3). CmOFP13 clustered with AtOFP1 and SlOFP20, and it is likely a homolog of these genes. More importantly, AtOFP1 and SlOFP20 induce organ shortening in their respective species (Hackbusch et al. 2005; Wu et al. 2018). Therefore, phylogenetic analysis reinforced the putative role of CmOFP13 in the regulation of organ shape in melon.

Phylogeny of Arabidopsis thaliana, tomato and melon ovate family protein genes based on Poisson distances (Zuckerkandl and Pauling 1965). Bootstrap values were based on 2000 replicates. Evolutionary analyses were conducted in MEGA X (Kumar et al. 2018). In order to simplify the figure, the nodes that do not include informative members for the current report are collapsed. The full phylogenetic tree is depicted in Fig. S2
CmOFP13 coding sequences from SC8-3, CALC8-1 and PS CmOFP13 were obtained with Sanger technology. Only one synonymous mutation was found between PS and CALC8-1. The same mutation was also present in SC8-3 plus two additional synonymous mutations (Supplementary Fig. S4). On the other hand, sequence variations were observed in the surrounding upstream and downstream non-coding regions (Supplementary Figure S5). In the downstream sequence, 24 SNPs were found in PI161375 and the deletion of approximately 40 kb at 3 kb from the stop codon of CmOFP13 coding sequence in PI124112 compared with the reference sequence (PS). In the case of upstream sequence, 123 and 114 SNPs were found in PI161375 and PI124112, respectively. The 3-kb deletion in PI124112 is located at −724 nucleotides from the transcription start point (according the annotation of accession XP_016903000 in www.ncbi.nlm.nih.gov). Therefore, these variations may affect the promoter sequences of CmOFP13 and most probably the expression profile of the gene but not preventing it completely. Thus, lack of non-synonymous mutations in the coding regions, the existing sequence variability in the surrounding regions and the differences of gene expression between PS and CALC8-1 strongly suggested that the changes in organ shape induced by CmOFP13 in CALC8-1 might be due to changes in gene expression, not differences in protein function between alleles.
The sequence variation in CMOFP13 across melon germplasm was studied in 1175 re-sequenced accessions (Zhao et al. 2019). Three synonymous mutations were identified in the CMOFP13 coding region (Fig. 4a). Haplotypes could be constructed for 1024 of them, showing a certain geographical distribution of the haplotypes (Fig. 4b). Haplotype A was characteristic of the China and Far East regions (what overlaps with the distribution of ssp. agrestis cultivars). Haplotype B (PS haplotype) was the most common haplotype in the Europe, America, Middle and Near East in regions, overlapping with the distribution of ssp. melo cultivars. Haplotype C (CALC haplotype) was characteristic of India and CALC, although was also present in America. Haplotype D had a very low frequency. In general, the haplotypes seemed to be mainly distributed geographically, nicely reflecting the history of melon diversification, with a primary center of diversification in India, a divergent diversification to the Western and Eastern regions and certain admixture due to migrations and breeding (Gonzalo et al. 2019). European cultivars have a highly diverse fruit morphology, and in fact, they have been classified in several horticultural groups based on this diversity (Pitrat 2017). However, a very low haplotypes diversity has been found among them. Therefore, the nucleotide variation at CMOFP13 coding sequence locus could not be related, at least directly, to the fruit morphology variation in melon germplasm.

Haplotypes found for CmOFP13 among 1175 re-sequenced melon accessions (Zhao et al. 2019). a SNPs defining the four haplotypes and their position on the melon genome v4.0 (Castanera et al. 2020). CALC8-1 shares the same haplotype as PI 124,112 (c), while PS shows the B haplotype. b Distribution of the four haplotypes through accessions belonging to different world regions
To further investigate the possible association between fsqs8.1 locus variability with FSI, 63 accessions from the COMAV’s germplasm bank representing diverse worldwide melon accessions were evaluated in two different experiments. FSI ranged from 0.8 (round fruit) to 4.5 (highly elongated) in the 2014 and 2018 experiments, respectively (Supplementary Fig. S6). Accessions showing very different FSI among experiments were removed. Accessions were genotyped with SNPs located in the fsqs8.1 locus, but no SNP was significantly associated with FSI. Eight haplotypes were found. However, B, D, E and G haplotypes presented four or less members, so they were removed for haplotype-based association analysis, remaining A, C, F, PS haplotypes. FSI mean for haplotype C (FSI = 1.04, 1.14 in 2014 and 2018 experiments, respectively) was significantly lower (rounder fruit) than the A and PS haplotypes means (ranging between 1.37 and 1.69). Most accessions with the haplotype C belong to the horticultural class cantalupensis, which characteristically produce round fruit (Pitrat 2017). Therefore, the association was not conclusive, as it could be due to the genetic structure of melon cultivars.
The presence of the deletions observed in the CALC and CALC8-1 genomes was also assessed in the 1,175 re-sequenced accessions. Raw sequence reads of 25 accessions (Supplementary Table S4) showing missing data for variants between chromosome 8 position 26,900,00 and 26,942,000 (Fig. 1) were retrieved and mapped de novo to the CM4.0 melon genome. All mappings showed the same pattern as previously observed with CALC and CALC8-1 (Supplementary Fig. S7). Interestingly, two independent replicates of CALC (MS-884 and MS-1155 in Zhao et al. 2019 coding) and MR-1 (ME-1018, a modern cultivar with CALC in its pedigree, Thomas 1986) showed the CALC8-1 read pattern, confirming the presence of the deletion in that genomic region. Most of the accessions showing the deletion read pattern belonged to India or had an Indian origin in the pedigree (MR-1). These results suggest that the origin of this structural variation occurred in an ancestral Indian accession, and it has not been dispersed with the diversification of melon cultivars, except its introduction into the MR-1 cultivar. Even though we did not have access to seeds of these accessions to evaluate their fruit, the Germplasm Resources Information Network (https://www.ars-grin.gov/) provides fruit evaluation data for nine of them. The FSI of those nine accessions ranged from round to elongated (Table 1), again suggesting the non-direct association of the genomic deletions with the variation in FSI. In fact, CALC, the accession donor of fsqs8.1 round allele, produces elongated fruit (Diaz et al. 2014).
Functional validation of CmOFP13 in Arabidopsis thaliana
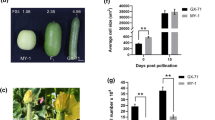
Thus, to verify whether CmOFP13 is indeed a functional homolog of AtOFP1 and SlOFP20, playing a role in the control of ovary and fruit shape, we over-expressed the allelic variant from CALC8-1 in A. thaliana, under the control of the 35S constitutive promoter from the plant pathogen Cauliflower Mosaic Virus (CaMV). Four transgenic lines were finally selected based on single locus segregation in hygromycin (Fig. 5A). All four lines showed a similar phenotype, although they had differences in penetration, with two mild (L1 and L8) and two strong (L6 and L11) phenotypes. All transgenic seedlings showed cotyledons with shorter petioles, and a protuberant proximal blade, which modified the normal oval shape of the Col0 accession to a kidney-shaped cotyledon. This phenotype continued during development, with round and curled leaves, resulting in compact plant rosettes. More importantly, the overexpression of CmOFP13 provoked a shortening of flower whorls, including pistils that resulted in shorter fruits. Pistil morphology was measured in the strong L6 line (Fig. 5b). Pistil length was reduced to 75%, whereas pistil width was increased by 43% of the Col-0 wild type. These changes resulted in a 50% reduction of the ratio length to width. Overall, these phenotypes strongly resembled those described upon overexpression of Class 1 OVATE genes from A. thaliana and tomato (Hackbusch et al. 2005; Wang et al. 2007, 2011; Wu et al. 2018). Therefore, a higher expression of CmOFP13 caused reduced ovary and fruit length in transgenic A. thaliana plants, which in agreement with the relationship found between the variation in expression of endogenous CmOFP13 and the variation in fruit shape in CALC8-1, PS and SC8-3. We interpreted that CmOFP13 was the functional homolog of AtOFP1 and SlOFP20, controlling ovary elongation in melon.

Alteration of organ shape due to overexpression of CmOFP13 in Arabidopsis thaliana. a Images of A. thaliana wild-type plants (Col0) and transgenic plants expressing CmOFP13 from CALC8-1 showing a mild (L1 and L8) or strong (L6 and L11) phenotype at the 9-day-old seedling stage, adult rosette, flowers, pistils, and mature siliques. Scale bars are depicted in each stage. b Comparison between ovary length, width and the length/width ratio between Col-0 and L6 transgenic line. (*) Indicates statistically significant differences (p < 0.001)
Genetic effects of fsqs8.1
Three different types of allelic interactions for fsqs8.1 were investigated: dominance between CALC and SC alleles, epistatic interaction with the a gene and interaction with different genetic background. For the dominance analysis, CALC and SC alleles were confronted in the hybrid CALC8-1-x-SC8-3. The gene action for FSI (d/[a]) was 0.43, and the contrast of FSI means between the hybrid and the expected mid-value from SC8-3 and CALC8-1 was statistically significant, with the hybrid being rounder than expected (p = 0.002, Fig. 6a). Therefore, the CALC allele had a partial dominant interaction over the SC allele.

Interactions of fsqs8.1 in different genetic situations. a Comparison of CALC8-1, SC8-3 and CALC8-1-x-SC8-3 hybrid fruit shape indexes. The blue bar indicates the expected mid-parent mean, and the asterisk the significance of the contrast (p = 0.0002). b Interaction between a and fsqs8.1 in an F2 population from the cross between CALC8-1 and ‘Mucha Nesvi’ (MN, a monoecious traditional cultivar). The means for the four homozygous allele combinations for the two loci (CALC/MN) for fsqs8.1, ANDRO/MONO for andromonoecious/monoecious (a), are shown on the left and the significance of the gene effects and interactions according two-way ANOVA on the right. c The fruit shape index of fruits from hybrids between accessions belonging to different melon horticultural groups with PS and CALC8-1 is depicted on the left. On the right, the significance of the effects (GB, genetic background, fsqs8.1 PS or CALC8-1 allele at the locus, and the two years) and their interactions from the three-way ANOVA, are shown
We next studied the epistatic interaction between the a gene, which controls sex expression (Boualem et al. 2008) and whether it had pleiotropic effects on FSI (Perin et al. 2002) with fsqs8.1 (Diaz et al. 2014). The interaction was evaluated in an F2 between CALC8-1 and ‘Mucha Nesvi’ (MN), a monoecious ameri cultivar that produces elongated fruit. Homozygous plants for the four genotypic combinations at these two loci were selected at seedling stage with AluCAPS (Boualamen et al. 2008) and fsqs8.1 SNPs. The effects of both genes on FSI were highly significant, with CALC fsqs8.1 allele inducing a round fruit and, and the MN a allele elongated ones (Fig. 6b). On the other hand, the interaction fsqs8.1-x-a was statistically significant, confirming the previous results by Diaz et al. (2014). All the plants homozygous for the CALC fsqs8.1 allele produced round fruit independently of their sex expression, whereas the effect of a on FSI was only evident in plants with the MN alleles at fsqs8.1.
Finally, the interaction of the fsqs8.1 alleles with different genetic background was studied by combining CALC8-1 with six melon accessions with contrasting fruit morphology belonging to different horticultural groups (cantalupensis, charentais, dudaim, flexuosus, inodorus and wild melon), and the hybrids were evaluated in two years (Fig. 6c). Hybrids with PS ranged from slightly round (FSI = 1.2) to very elongated (FSI = 4.2). A similar trend was observed in the CALC8-1 hybrids, although in all cases fruit were rounder compared with their respective PS hybrids. The effects of genetic background (GB) and fsqs8.1 were highly significant confirming the consistency of fsqs8.1 effects on diverse GBs. The interaction GB*fsqs8.1 was also significant, due to the variability in the magnitude of the effect across GBs, although fsqs8.1 induced rounder fruit in all cases. The larger effect was observed mostly in hybrids that included accessions that produced elongated fruit. It is likely that there is a limitation of the roundness of the melon fruit, so the effect of fsqs8.1 may be stronger in GBs that produce elongated melons.
Discussion
In the current report, we combined high-resolution mapping together with expression and functional analysis to demonstrate that CmOFP13, a member of the melon OVATE gene family, is the causal gene underlying the melon fruit shape QTL fsqs8.1. The discovery of the OVATE gene family started with the study of the genetic control of the pear-shaped fruit in the tomato heirloom variety Yellow Pear (Ku et al. 1999). The pear shape of tomatoes was already studied by early tomato geneticists (Hendrick and Booth 1907; Price and Drinkard 1908) demonstrating that the pear shape was controlled by a single gene, which was named ovate (Lindstrom 1926, 1927). Liu et al. (2002) identified and cloned the first OVATE gene, which did not match with any known proteins at that time, thus the authors suggested the discovery of a new class of regulatory genes involved in plant growth. Further research demonstrated that OVATE was a member of a large gene family present in different plant species (Liu et al. 2014, Dangwai and Das 2018). Thus, Monforte et al. (2014) reported 21 OFP members of the OVATE gene family in the melon genome. The large number of members and the co-segregation of their position with fruit shape QTLs led to the hypothesis that OFP members other than OVATE could also be involved in the control of organ shape in different species (Rodriguez et al. 2011; Monforte et al. 2014).
Phylogenetic analysis showed that CmOFP13 is related to tomato SlOFP14, SlOF17 and SlOFP20 and the A. thaliana AtOFP1, but not to OVATE itself. Wu et al. (2018) demonstrated that SlOFP20 controlled the morphology of tomato fruit and potato tubers. More recently, the peach member PpOFP1, also likely homologue to SlOFP20, was found to control the flat fruit shape in this crop (Zhou et al. 2020). Evidences of the role of SlOFP20 homologues in fruit morphology have also been found in cucumber (Colle et al. 2017) and pepper (Tsaballa et al. 2011). Previously, Hackbusch et al. (2005) showed that the overexpression of AtOFP1 induced shortened siliques in transgenic A. thaliana plants. Even though the role of other OFP members on organ morphology cannot be ruled out (Snouffer et al. 2020), these results suggest strongly that the homologues of SlOFP20 are crucial for the establishment of organ shape in a large number of plant species.
In tomato and peach, the effect of SlOFP20 and PpOFP1 is a consequence of changes in gene expression, not changes in protein function (Wu et al. 2018; Zhou et al. 2020). Similarly, no amino acid changes were found between the CALC and PS CmOFP13 protein sequences. Therefore, the differences in fruit morphology between CALC8-1 and PS are likely caused by the early higher expression of the gene in CALC8-1, compared with PS, resulting in rounder fruit in CALC8-1. The role of gene expression variation in tomato fruit morphology was already pointed out for fw2.2, the first cloned fruit morphology QTL (Cong et al. 2002) and it has been observed in subsequent cloned morphology genes and QTLs: SUN (Xiao et al. 2008), lc (Munos et al. 2011), fas (Xu et al. 2015) and fw3.2 (Chakrabarti et al. 2013), among others. Therefore, fsqs8.1 is another example of this conserved mechanism that controls organ morphology.
The fsqs8.1 locus showed an important structural variation, with two deletions surrounding CmOFP13. The deletions were confirmed in the re-sequencings of CALC8-1, two independent CALCs (the donor of fsqs8.1 introgression in CALC8-1), and a small set of melon accession genomes. This structural variation may have affected regulatory sequences, which could explain the changes in the CmOFP13 expression patterns observed between genotypes in the current report. In tomato, hundreds of structural variations have been associated to changes in gene expression patterns (Alonge et al. 2020). Concomitantly, in the same species, Eshed and Lippman (2019) pointed out that mutations in 5’ and 3’ cis regulatory elements can alter gene expression, resulting in alleles with quantitative effects. In fact, both SlOFP20 and PpOFP1 genomic regions present structural variation (Wu et al. 2018, Zhou et al. 2020). In the case of fsqs8.1, the larger deletion region is rich in transposon-related sequences in the reference genome, thus it is likely that transposon activity has been relatively frequent in this region, and that their activity could have been responsible for the observed structural variation. The impact of transposons on the generation of phenotypic diversity underlies the maize domestication gene tb1 (Zhou et al. 2011). Insertions of retrotransposons in upstream regions of tb1 causes a differential expression that is responsible for the typical single branching of maize cultivars in contrast with the multiple branching of the wild ancestor teosinte. Recently, the role of transposon activity has garnered more attention. Domínguez et al. (2020) found associations of transposon element insertion polymorphisms with major agronomic traits and secondary metabolites in cultivated tomato. Similarly, evidence of the role of retrotransposons on the evolution of the genome and modification of gene expression has also been reported in melon (Demirci et al. 2021; Sanseverino et al. 2015; Yano et al. 2020). Thus, fsqs8.1 is perhaps another case of gene expression changes due to structural variation mediated by transposon activity.
Strikingly, the CmOFP13 coding sequence showed a very low variation across melon germplasm, with all mutations being silent, what suggested that the functionality of this protein is needed for proper melon development and only changes in expression patterns might be allowed. Attempts at finding an association between haplotypes in fsqs8.1 regions and fruit shape did not provide conclusive results. Previously, Gonzalo and van der Knaap (2008) reported that the effect of OVATE was dependent on the genetic background in tomato. Rodriguez et al. (2013) showed that SlOFP20 was a suppressor of OVATE, and that specific allele combinations between SlOFP20 and OVATE should coincide to produce the typical ovate elongated fruit. Moreover, Wu et al. (2018) demonstrated that slTRM5 (a member of TONNEAU1 Recruiting Motif family interacts with SlOFP20 and can modulate the effects on tomato fruit shape. In fact, Gur et al. (2017) found association between melon fruit shape and SNPs located in the fsqs8.1 region using a different GWAS panel. Therefore, the effects of OFP genes may depend on the genetic background, and the fact that the OFP family includes a large number of members may cause a buffering effect, thereby limiting the effects of a single OFP gene that would be apparent only in certain allelic combinations. Based on the deletion pattern on the fsqs8.1 locus, the CALC allele is present with low frequency in Indian germplasm, but it was not associated with fruit morphology in that germplasm. The CALC (and other accessions) genome may contain one or several suppressors of CmOFP13, or another gene/genes needed are not expressed at the proper time, resulting in elongated fruit. However, the introduction of the CALC CmOFP13 allele into the PS genetic background induced the development of round fruit, suggesting that the PS genome lacks the putative suppressors or the other needed gene/genes would be expressed at the proper time. Similarly, the effects of the CALC CmOFP13 allele on fruit shape were consistent when confronted with different genetic background. These experiments were also carried out in the hybrid PS-x-accessions genetic backgrounds (the genetic background of CALC8-1 is PS), so the putative suppressors might be suppressed or the other needed gene/genes would be expressed, and as consequence CALC CmOFP13 allele could induce its effects on fruit morphology.
An interesting practical breeding results from this work is the repression of fruit elongation induced by a by CmOFP13. The development of monoecious cultivars producing round fruits (characteristic of the common market class cantalupensis, galia and honeydew) is desirable, as it eliminates the need for emasculation for crossing make crosses to produce hybrid seeds. However, the effects of the a gene on fruit elongation makes difficult the combination of both properties. Therefore, CmOFP13 round alleles could be introduced in breeding programs to generate such cultivars.
In conclusion, we have demonstrated that the gene underlying the fruit shape QTL fsqs8.1 is CmOFP13, a member of OFP gene family, a likely homologue to the tomato SlOFP20 gene. The effects of CmOFP13 in ovary and fruit shape are most probably due to changes in its expression pattern, likely due to a structural variation mediated by transposon activity in the surrounding genomic region, and perhaps by promoting cell elongation by microtubule orientation (Zhang et al. 2020). The effects of CmOFP13 depend on the genetic background, being evident in the PS background, but not in the original Indian background. Even though no associations were found between SNPs or structural variation in the fsqs8.1 locus with fruit morphology in melon germplasm in this work, fsqs8.1 genomic region has been associated in fruit morphology in a wide range of melon germplasm (Monforte et al. 2014; Gur et al. 2017; Pan et al. 2020), actually the region is considered to include a consensus FSI QTL (Monforte et al. 2014; Pan et al. 2020). Other mutations should be involved in those alleles, likely regulatory mutations rather than coding mutations, or additional linked genes. Therefore, the CALC CmOFP13 allele is a cryptic variant that may have arisen by a structural variation, with a strong potential to become a new breeding tool for generating monoecious cultivars that produce round fruit.
Availability of data and material
Genome sequences from PS and CALC8-1 are available at NCBI repository BioProject ID PRJNA729905.
References
Abdelmohsin ME, Pitrat M (2008) Pleiotropic effect of sex expression on fruit shape in melon. In: Pitrat M (ed) 9th EUCARPIA Meeting on Genetics and Breeding of Cucurbitaceae, INRA, Avignon, France, pp 551–555
Alonge M, Wang XG, Benoit M, Soyk S, Pereira L, Zhang L, Suresh H, Ramakrishnan S, Maumus F, Ciren D, Levy Y, Harel TH, Shalev-Schlosser G, Amsellem Z, Razifard H, Caicedo AL, Tieman DM, Klee H, Kirsche M, Aganezov S, Ranallo-Benavidez TR, Lemmon ZH, Kim J, Robitaille G, Kramer M, Goodwin S, McCombie WR, Hutton S, Van Eck J, Gillis J, Eshed Y, Sedlazeck FJ, van der Knaap E, Schatz MC, Lippman ZB (2020) Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182:145–161
Bai SL , Peng YB, Cui JX, Gu HT, Xu LY, Li YQ, Xu ZH, Bai SN (2004). Developmental analyses reveal early arrests of the spore-bearing parts of reproductive organs in unisexual flowers of cucumber (Cucumis sativus L.) Planta 220: 230–240
Boualem A, Fergany M, Fernandez R, Troadec C, Martin A, Morin H, Sari MA, Collin F, Flowers JM, Pitrat M, Purugganan MD, Dogimont C, Bendahmane A (2008) A conserved mutation in an ethylene biosynthesis enzyme leads to andromonoecy in melons. Science 321:836–838
Castanera R, Ruggieri V, Pujol M, Garcia-Mas J, Casacuberta JM (2020) An improved melon reference genome with single-molecule sequencing uncovers a recent burst of transposable elements with potential impact on genes. Front Plant Sci 10:1815
Chakrabarti M, Zhang N, Sauvage C, Munos S, Blanca J, Canizares J, Diez MJ, Schneider R, Mazourek M, McClead J, Causse M, van der Knaap E (2013) A cytochrome P450 regulates a domestication trait in cultivated tomato. Proc Natl Acad Sci USA 110:17125–17130
Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743
Colle M, Weng YQ, Kang YY, Ophir R, Sherman A, Grumet R (2017) Variation in cucumber (Cucumis sativus L.) fruit size and shape results from multiple components acting pre-anthesis and post-pollination. Planta 246:641–658
Cong B, Liu JP, Tanksley SD (2002) Natural alleles at a tomato fruit size quantitative trait locus differ by heterochronic regulatory mutations. Proc Natl Acad Sci USA 99:13606–13611
Dangwal M, Das S (2018) Identification and analysis of OVATE family members from genome of the early land plants provide insights into evolutionary history of OFP family and function. J Mol Evol 86:511–530
Demirci S, Fuentes RR, van Dooijeweert W, Aflitos S, Schijlen E, Hesselink T, de Ridder D, van Dijk A D J, Peters S (2021). Chasing breeding footprints through structural variations in Cucumis melo and wild relatives. G3 11: jkaa038
Diaz A, Zarouri B, Fergany M, Eduardo I, Alvarez JM, Pico B, Monforte AJ (2014) Mapping and introgression of QTL involved in fruit shape transgressive segregation into “Piel de Sapo” melon (Cucumis melo L.). PLoS ONE 9:e116098
Dominguez M, Dugas E, Benchouaia M, Leduque B, Jimenez-Gomez JM, Colot V, Quadrana L (2020) The impact of transposable elements on tomato diversity. Nat Commun 11:4058
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Eduardo I, Arus P, Monforte AJ (2005) Development of a genomic library of near isogenic lines (NILs) in melon (Cucumis melo L.) from the exotic accession PI161375. Theor Appl Genet 112:139–148
Eduardo I, Arus P, Monforte AJ, Obando J, Fernandez-Trujillo JP, Martinez JA, Alarcon AL, Alvarez JM, van der Knaap E (2007) Estimating the genetic architecture of fruit quality traits in melon using a genomic library of near isogenic lines. J Am Soc Hortic Sci 132:80–89
Endl J, Achigan-Dako EG, Pandey AK, Monforte AJ, Pico B, Schaefer H (2018) Repeated domestication of melon (Cucumis melo) in Africa and Asia and a new close relative from India. Am J Bot 105:1662–1671
Eshed Y, Lippman ZB (2019) Revolutions in agriculture chart a course for targeted breeding of old and new crops. Science 366:705–706
FAOSTAT (2021) https://www.fao.org/faostat/en/#home
Felsenstein J (1985) Confidence-limits on phylogenies–an approach using the bootstrap. Evolution 39:783–791
Gomez M, Beltran JP, Canas L (2004) The pea END1 promoter drives anther-specific gene expression in different plant species. Planta 219:967–981
Gomez MD, Urbez C, Perez-Amador MA, Carbonell J (2011) Characterization of constricted fruit (ctf) mutant uncovers a role for AtMYB117/LOF1 in ovule and fruit development in Arabidopsis thaliana. PLoS ONE 6:e18760
Gonzalo MJ, van der Knaap E (2008) A comparative analysis into the genetic bases of morphology in tomato varieties exhibiting elongated fruit shape. Theor Appl Genet 116:647–656
Gonzalo MJ, Diaz A, Dhillon NPS, Reddy UK, Pico B, Monforte AJ (2019) Re-evaluation of the role of Indian germplasm as center of melon diversification based on genotyping-by-sequencing analysis. BMC Genomics 20:448
Gur A, Tzuri G, Meir A, Sa’ar U, Portnoy V, Katzir N, Schaffer AA, Li L, Burger J, Tadmor Y (2017) Genome-wide linkage-disequilibrium mapping to the candidate gene level in melon (Cucumis melo). Sci Rep 7:9770
Hackbusch J, Richter K, Muller J, Salamini F, Uhrig JF (2005) A central role of Arabidopsis thaliana ovate family proteins in networking and subcellular localization of 3-aa loop extension homeodomain proteins. Proc Natl Acad Sci USA 102:4908–4912
Hedrick UP, Booth NO (1907) Mendelian characters in tomato. Proc Am Soci Hortic Sci 5:19–24
Ku HM, Doganlar S, Chen KY, Tanksley SD (1999) The genetic basis of pear-shaped tomato fruit. Theor Appl Genet 99:844–850
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Li S, Pan YP, Wen CL, Li YH, Liu XF, Zhang XL, Behera TK, Xing GM, Weng YQ (2016) Integrated analysis in bi-parental and natural populations reveals CsCLAVATA3 (CsCLV3) underlying carpel number variations in cucumber. Theor Appl Genet 129:1007–1022
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. http://arxiv.org/abs/arXiv: 1303.3997
Lindstrom EW (1926) Linked inheritance in tomatoes. Iowa State Coll J Sci 1:3–13
Lindstrom EW (1927) The inheritance of ovate and related shapes of tomato fruit. J Agric Res 34:961–985
Liu JP, Van Eck J, Cong B, Tanksley SD (2002) A new class of regulatory genes underlying the cause of pear-shaped tomato fruit. Proc Natl Acad Sci USA 99:13302–13306
Liu D, Sun W, Yuan YW, Zhang N, Hayward A, Liu YL, Wang Y (2014) Phylogenetic analyses provide the first insights into the evolution of OVATE family proteins in land plants. Ann Bot 113:1219–1233
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17:10–12
Monforte AJ, Oliver M, Gonzalo MJ, Alvarez JM, Dolcet-Sanjuan R, Arus P (2004) Identification of quantitative trait loci involved in fruit quality traits in melon (Cucumis melo L.). Theor Appl Genet 108:750–758
Monforte AJ, Eduardo I, Abad S, Arus P (2005) Inheritance mode of fruit traits in melon: heterosis for fruit shape and its correlation with genetic distance. Euphytica 144:31–38
Monforte AJ, Diaz A, Cano-Delgado A, van der Knaap E (2014) The genetic basis of fruit morphology in horticultural crops: lessons from tomato and melon. J Exp Bot 65:4625–4637
Munos S, Ranc N, Botton E, Berard A, Rolland S, Duffe P, Carretero Y, Le Paslier MC, Delalande C, Bouzayen M, Brunel D, Causse M (2011) Increase in tomato locule number is controlled by two single-nucleotide polymorphisms located near WUSCHEL. Plant Physiol 156:2244–2254
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Nakagawa T, Kurose T, Hino T, Tanaka K, Kawamukai M, Niwa Y, Toyooka K, Matsuoka K, Jinbo T, Kimura T (2007) Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J Biosci Bioeng 104:34–41
Oliver M, Garcia-Mas J, Cardus M, Pueyo N, Lopez-Sese A, Arroyo M, Gomez-Paniagua H, Arus P, de Vicente MC (2001) Construction of a reference linkage map for melon. Genome 44:836–845
Pan YP, Wang YH, McGregor C, Liu S, Luan FS, Gao ML, Weng YQ (2020) Genetic architecture of fruit size and shape variation in cucurbits: a comparative perspective. Theor Appl Genet 133:1–21
Perin C, Hagen LS, Giovinazzo N, Besombes D, Dogimont C, Pitrat M (2002) Genetic control of fruit shape acts prior to anthesis in melon (Cucumis melo L.). Mol Genet Genom 266:933–941
Pitrat M (2017) Melon genetic resources: phenotypic diversity and horticultural taxonomy. In: Grumet R, Katzi N, Garcia-Mas J (eds) Genetics and genomics of the Cucurbitaceae. Springer, New York, pp 25–60
Price HC, Drinkard AW (1908) Inheritance in tomato hybrids. Va Agric Exp Station Bull 177:17–53
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29:24–26
Rodriguez GR, Munos S, Anderson C, Sim SC, Michel A, Causse M, Gardener BBM, Francis D, van der Knaap E (2011) Distribution of SUN, OVATE, LC, and FAS in the tomato germplasm and the relationship to fruit shape diversity. Plant Physiol 156:275–285
Rodriguez GR, Kim HJ, van der Knaap E (2013) Mapping of two suppressors of OVATE (sov) loci in tomato. Heredity 111:256–264
Saitou N, Nei M (1987) On the maximum-likelihood method for molecular phylogeny. Jpn J Genet 62:547–548
Sanseverino W, Henaff E, Vives C, Pinosio S, Burgos-Paz W, Morgante M, Ramos-Onsins SE, Garcia-Mas J, Maria Casacuberta J (2015) Transposon insertions, structural variations, and SNPs contribute to the evolution of the melon genome. Mol Biol Evol 32:2760–2774
SM Smith PJ Maughan 2015 SNP genotyping using KASPar assays Plant Genotyping: methods Protocols 1245 243 256
Snouffer A, Kraus C, van der Knaap E (2020) The shape of things to come: ovate family proteins regulate plant organ shape. Curr Opin Plant Biol 53:98–105
Thomas CE (1986) Downy and powdery mildew resistant muskmelon breeding line MR-1. HortScience 21:329–329
Tsaballa A, Pasentsis K, Darzentas N, Tsaftaris AS (2011) Multiple evidence for the role of an ovate-like gene in determining fruit shape in pepper. BMC Plant Biol 11:46
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3-new capabilities and interfaces. Nucleic Acids Res 40:e115
Wang S, Chang Y, Guo J, Chen JG (2007) Arabidopsis ovate family protein 1 is a transcriptional repressor that suppresses cell elongation. Plant J 50:858–872
Wang SC, Chang Y, Guo JJ, Zeng QN, Ellis BE, Chen JG (2011) Arabidopsis ovate family proteins, a novel transcriptional repressor family, control multiple aspects of plant growth and development. PLoS ONE 6:e23896
Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB (2017) Direct determination of diploid genome sequences. Genome Res 27:757–767
Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ (2003) High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem 49:853–860
Wu S, Zhang BY, Keyhaninejad N, Rodriguez GR, Kim HJ, Chakrabarti M, Illa-Berenguer E, Taitano NK, Gonzalo MJ, Diaz A, Pan YP, Leisner CP, Halterman D, Buell CR, Weng YQ, Jansky SH, van Eck H, Willemsen J, Monforte AJ, Meulia T, van der Knaap E (2018) A common genetic mechanism underlies morphological diversity in fruits and other plant organs. Nat Commun 9:4734
Xiao H, Jiang N, Schaffner E, Stockinger EJ, van der Knaap E (2008) A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 319:1527–1530
Xu C, Liberatore KL, MacAlister CA, Huang ZJ, Chu YH, Jiang K, Brooks C, Ogawa-Ohnishi M, Xiong GY, Pauly M, Van Eck J, Matsubayashi Y, van der Knaap E, Lippman ZB (2015) A cascade of arabinosyltransferases controls shoot meristem size in tomato. Nat Genet 47:784–792
Yano R, Ariizumi T, Nonaka S, Kawazu Y, Zhong SL, Mueller L, Giovannoni JJ, Rose JKC, Ezura H (2020) Comparative genomics of muskmelon reveals a potential role for retrotransposons in the modification of gene expression. Commun Biol 3:432
Zhang XW, Wu JL, Yu Q, Liu RY, Wang ZY, Sun Y (2020) AtOFPs regulate cell elongation by modulating microtubule orientation via direct interaction with TONNEAU2. Plant Sci 292:110405
Zhao GW, Lian Q, Zhang ZH, Fu QS, He YH, Ma S, Ruggieri V, Monforte AJ, Wang PY, Julca I, Wang HS, Liu JP, Xu Y, Wang RZ, Ji JB, Xu ZH, Kong WH, Zhong Y, Shang JL, Pereira L, Argyris J, Zhang J, Mayobre C, Pujol M, Oren E, Out D, Wang JM, Sun DX, Zhao SJ, Zhu YC, Li N, Katzir N, Gur A, Dogimont C, Schaefer H, Fan W, Bendahmane A, Fei Z, Pitrat M, Gabaldon T, Lin T, Garcia-Mas J, Xu YY, Huang SW (2019) A comprehensive genome variation map of melon identifies multiple domestication events and loci influencing agronomic traits. Nat Genet 51:1607–1615
Zhou LL, Zhang JY, Yan JB, Song RT (2011) Two transposable element insertions are causative mutations for the major domestication gene teosinte branched 1 in modern maize. Cell Res 21:1267–1270
Zhou H, Ma RJ, Gao L, Zhang JN, Zhang AD, Zhang XJ, Ren F, Zhang WH, Liao L, Yang QR, Xu SL, Ogutu CO, Zhao JB, Yu ML, Jiang Q, Korban SS, Han YP (2020) A 1.7-Mb chromosomal inversion downstream of aPpOFP1gene is responsible for flat fruit shape in peach. Plant Biotechnol J 19:192–205
Zuckerkandl E, Pauling L (1965) Molecules as documents of evolutionary history. J Theor Biol 8:357–366
Acknowledgements
The authors thank Ms. Soledad Casal and Dr. Gorka Perpiña for technical support, Dr. Javier Forment from the IBMCP Bioinformatics Facility for bioinformatics support. This work was supported by European Union and Spanish Ministry of Economy and Competitiveness /FEDER grants AGL2015-64625-C2-1-R to AJM/CR, by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe” grants AGL2017-85563-C2-1-R (BP), BIO2017-83138R (MAP-A) RTI2018-097665-B-C2 (AJM/CR), RTI2018-097665-B-C1 (JGM) and PROMETEO project 2017/078 and 2021/072 (to promote excellence groups) by the Conselleria d’Educació, Investigació, Cultura i Esports (Generalitat Valenciana) to BP. PS was supported by a scholarship from Bec.Ar of the National Ministry of Education, Argentina.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. European Union and Spanish Ministry of Economy and Competitiveness /FEDER grants AGL2015-64625-C2-1-R to AJM/CR, by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe” grants AGL2017-85563-C2-1-R (BP), BIO2017-83138R (MAP-A) RTI2018-097665-B-C2 (AJM/CR), RTI2018-097665-B-C1 (JGM) and PROMETEO project 2017/078 and 2021/072 (to promote excellence groups) by the Conselleria d’Educació, Investigació, Cultura i Esports (Generalitat Valenciana, ES) to BP. PS was supported by a scholarship from Bec.Ar of the National Ministry of Education, Argentina.
Author information
Authors and Affiliations
Contributions
CM-M, MJG, PS, MC, IM-F, CL, MD-G, CR conducted experiments related with the fine mapping, RT-PCR and development analysis. MZ, KGA, JG-M and AJM obtained and analyzed genomic data. PT and MAP-A performed the A thaliana experiments. CE and BP selected germplasm and performed greenhouse trials. AJM conceived and designed the study. AJM, CR and MAP-A wrote the manuscript. All authors reviewed and accepted the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
Authors declare no conflict of interest.
Additional information
Communicated by Herman J. van Eck.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martínez-Martínez, C., Gonzalo, M.J., Sipowicz, P. et al. A cryptic variation in a member of the Ovate Family Proteins is underlying the melon fruit shape QTL fsqs8.1. Theor Appl Genet 135, 785–801 (2022). https://doi.org/10.1007/s00122-021-03998-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-021-03998-6