
Abstract
Key message
We have constructed a densely populated, saturated genetic linkage map of black raspberry and successfully placed a locus for aphid resistance.
Abstract
Black raspberry (Rubus occidentalis L.) is a high-value crop in the Pacific Northwest of North America with an international marketplace. Few genetic resources are readily available and little improvement has been achieved through breeding efforts to address production challenges involved in growing this crop. Contributing to its lack of improvement is low genetic diversity in elite cultivars and an untapped reservoir of genetic diversity from wild germplasm. In the Pacific Northwest, where most production is centered, the current standard commercial cultivar is highly susceptible to the aphid Amphorophora agathonica Hottes, which is a vector for the Raspberry mosaic virus complex. Infection with the virus complex leads to a rapid decline in plant health resulting in field replacement after only 3–4 growing seasons. Sources of aphid resistance have been identified in wild germplasm and are used to develop mapping populations to study the inheritance of these valuable traits. We have constructed a genetic linkage map using single-nucleotide polymorphism and transferable (primarily simple sequence repeat) markers for F1 population ORUS 4305 consisting of 115 progeny that segregate for aphid resistance. Our linkage map of seven linkage groups representing the seven haploid chromosomes of black raspberry consists of 274 markers on the maternal map and 292 markers on the paternal map including a morphological locus for aphid resistance. This is the first linkage map of black raspberry and will aid in developing markers for marker-assisted breeding, comparative mapping with other Rubus species, and enhancing the black raspberry genome assembly.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genetic linkage map construction of rosaceous crops has been used to understand genetics and as a precursor to enabling molecular breeding for about 20 years. The earliest maps made during the 1990s were constructed mainly by using isozymes, random amplification of polymorphic DNA (RAPD), restriction fragment length polymorphism (RFLP), and morphological markers (Chaparro et al. 1994; Foolad et al. 1995; Hemmat et al. 1994; Rajapakse et al. 1995; Stockinger et al. 1996; Viruel et al. 1995). Advancements in DNA technology in the 2000s led to the rapid development of simple sequence repeat (SSR) markers for de novo map construction (Castro et al. 2013; Celton et al. 2009; Dirlewanger et al. 2004; Fernández-Fernández et al. 2008; Gisbert et al. 2009; Graham et al. 2004; Hibrand-Saint Oyant et al. 2008; Olmstead et al. 2008) as well as their incorporation into existing maps (Aranzana et al. 2003; Dirlewanger et al. 2006; Etienne et al. 2002; Liebhard et al. 2003; Paterson et al. 2013; Pierantoni et al. 2004; Silfverberg-Dilworth et al. 2006; Stafne et al. 2005; Vilanova et al. 2008; Woodhead et al. 2008, 2010). Additional technological advances in high-throughput detection of single-nucleotide polymorphic (SNP) loci using arrays, or genotyping by sequencing (GBS), and the associated improvement of data analysis have made SNP markers increasingly useful for genetic map construction. Recently, linkage maps for several members of the Rosaceae have been constructed using SNP array technology (Antanaviciute et al. 2012; Clark et al. 2014; Frett et al. 2014; Klagges et al. 2013; Montanari et al. 2013; Pirona et al. 2013; Yang et al. 2013).
The genus Rubus L. (Rosaceae, Rosoideae) has an estimated 750 species distributed world-wide (Alice and Campbell 1999; Thompson 1995). Of these, three are of particular commercial importance, red raspberry (R. idaeus L., subgenus Idaeobatus Focke), blackberry (Rubus sp., subgenus Rubus L.), and black raspberry (subgenus Idaeobatus). Genetic linkage maps have been constructed for tetraploid blackberry (Castro et al. 2013), diploid red raspberry (Sargent et al. 2007; Ward et al. 2013; Woodhead et al. 2010), and an interspecific cross between diploid red raspberry and diploid black raspberry (Bushakra et al. 2012). While blackberry and red raspberry are highly heterozygous, black raspberry, particularly R. occidentalis, is highly homozygous (Dossett et al. 2012b). Genetic improvement of blackberry and red raspberry through breeding has been a continual process for decades. For example, from 1994 to 2014, the American Pomological Society’s Fruit and Nut Variety Registry Lists 38–47 (Clark and Finn 1999, 2002, 2006; Clark et al. 2008, 2012; Daubeny 1997a, b, 1999, 2000, 2002, 2004, 2006, 2008; Finn and Clark 2000, 2004, 2014; Finn et al. 2010; Moore and Kempler 2010, 2012, 2014) records the release of 75 blackberry and hybrid berry and 189 red raspberry cultivars and only three black raspberry cultivars (‘Pequot’, ‘Niwot’, and ‘Explorer’). In addition, ‘Earlysweet,’ a selection derived from a purported cross between R. occidentalis and the western black raspberry, R. leucodermis Dougl. ex Torr. & Gray, was released in 1998 (Galletta et al. 1998). Black raspberry figures prominently in the pedigrees of many of the red raspberry cultivars released between 1994 and 2014. Difficulties in improving black raspberry through breeding were first reported by Slate (1933) while attempting to improve purple raspberries. Crossing with other species was proposed as a way to increase genetic diversity in cultivated black raspberry (Drain 1956; Hellman et al. 1982; Slate and Klein 1952), but has met with limited success. Low genetic diversity was proposed by Ourecky (1975) as the main reason for lack of development of improved cultivars.
More recent interest in improving black raspberry has been driven by research and commercial interest into its bioactive compounds and their influence on human health, specifically modulation of cancer cell proliferation, inflammation, cellular death, oxidation, etc. (Stoner et al. 2007). Since the 1940s, Oregon has been the primary commercial production region of black raspberry in North America. In 2014, 1650 acres were harvested that earned growers a utilized production value of over US$16.8 million (Anonymous 2015). One hindrance to expanding production is susceptibility of the predominant cultivar Munger to the Raspberry mosaic virus complex vectored by the North American large raspberry aphid, Amphorophora agathonica Hottes (Dossett and Finn 2010). Infection causes rapid decline of plantings, often with field replacement necessary after only three or four growing seasons (Halgren et al. 2007). In contrast, under perennial production in open fields for processed fruit, plantings of current cultivars of red raspberry are typically kept in the field for 7–8 growing seasons, and plantings of blackberry cultivars can last many decades (C.E. Finn, personal communication). Selection for cultivars with resistance to A. agathonica could significantly increase the longevity of the plants, reduce insecticide use, and therefore improve profitability for the grower and quality of the environment.
A low level of genetic diversity in cultivated black raspberry has been found using molecular tools. Weber (2003), using RAPD markers in 16 black raspberry cultivars, determined a level of similarity of 81 %. Two wild accessions and five elite genotypes accounted for more than 50 % of the similarity, while the remaining 11 cultivars shared 92 % similarity compared to 70 % similarity among red raspberry cultivars found by Graham et al. (1994). In 2005, Lewers and Weber used SSR markers from red raspberry and strawberry to evaluate an F2 population of a red raspberry × black raspberry cross and found that the homozygosity of the black raspberry clone used was 80 % and only 40 % in the red raspberry clone used. However, wild populations of black raspberry show greater genetic diversity. For example, Nybom and Schaal (1990) sampled black raspberry plants along a roadside in Missouri that were then analyzed by RFLP. They found 17 informative fragments that identified 15 genotypes in the 22 samples collected. Dossett et al. (2012b) used SSR markers to examine the genetic diversity among cultivars and wild germplasm. They found that the diversity at 21 loci was much higher among wild germplasm than in the elite cultivars, and that six elite cultivars were identical at these 21 loci.
Genetic diversity in wild black raspberry germplasm as detected by molecular tools (Dossett et al. 2012b; Nybom and Schaal 1990) and through breeding experiments (Dossett et al. 2008) is currently untapped. To address this, Dossett and Finn (2010) canvassed the native range of R. occidentalis collecting seed, which was subsequently germinated and evaluated for multiple traits including aphid resistance. From this study, three of 132 wild populations were determined to segregate for resistance to A. agathonica. Two populations, ORUS 3817 collected from Maine, and ORUS 3778 collected from Ontario, Canada, were subsequently used to develop populations for genetic mapping and phenotypic analysis. F1 progeny of susceptible cultivars Munger and Jewel crossed with individuals from ORUS 3778 and ORUS 3817 were all resistant to aphids under greenhouse conditions suggesting that the alleles for resistance are dominant and that ORUS 3778 (Ag 4 ) and ORUS 3817 (Ag 5 ) are homozygous for their respective alleles. Dossett and Finn (2010) originally identified one of the susceptible cultivars used in the crosses as ‘Black Hawk’, however, subsequent fingerprinting work found it to be ‘Jewel’ (Dossett et al. 2012a).
In this paper, we report the analysis of population ORUS 4305, an F1 black raspberry population, raised as one of several populations to investigate genetic sources of resistance to the aphid A. agathonica with the intent of mapping the aphid resistance allele Ag 4 . To quickly and efficiently generate markers for mapping we have employed GBS following the protocol established by Elshire et al. (2011) with modifications for Rubus (Ward et al. 2013), and anchored the map with SSR markers from a variety of sources. We have placed the phenotypic character of aphid resistance on this linkage map which covers the seven Rubus linkage groups (RLG) as defined by Bushakra et al. (2012).
Methods
Plant material
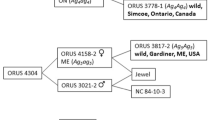
A full-sib (F1) family of 115 individuals was developed from the controlled cross of ORUS 3021-2 (female, susceptible to aphids, postulated genotype ag 4 ag 4 ) × ORUS 4153-1 (male, resistant to aphids, postulated genotype Ag 4 ag 4 ; Fig. 1). The source of this resistance is from ORUS 3778-1, an accession from wild seed collected in Ontario, Canada (Dossett and Finn 2010). Progeny from this cross were screened for aphid resistance as small seedlings in the greenhouse as described by Dossett and Finn (2010) and followed the expected 1:1 segregation ratio (56 resistant, 59 susceptible, χ 2 = 0.08, P = 0.78).

Pedigree of mapping population ORUS 4305. Population ORUS 4305 is derived from a wild-collected accession from Ontario, Canada (ORUS 3778-1), that exhibited resistance to the North American large raspberry aphid that was crossed with aphid-susceptible ‘Jewel.’ One of the progeny from that cross, ORUS 4153-1 with proposed genotype Ag 4 ag 4 representing the proposed gene conferring resistance, was used as the male parent and crossed with aphid-susceptible ORUS 3021-2
DNA extraction
Leaf samples were collected, bagged, kept cool, and transported to the laboratory. Leaf tissue aliquots of 30–50 mg were placed in a cluster tube (Corning Life Sciences, Tewksbury, MA, USA) containing a 4-mm stainless steel bead (McGuire Bearing Company, Salem, OR, USA). The samples were frozen in liquid nitrogen and stored at −80 °C until extraction. Frozen tissue was homogenized using the Retsch® MM301 Mixer Mill, (Retsch Inc., Hann, Germany) at a frequency of 30 cycles/s using three 30 s bursts. The E-Z 96® Plant DNA extraction kit (Omega Bio-Tek, Norcross, GA, USA) was used as previously described (Gilmore et al. 2011).
DNA quantification and quality
Genomic DNA was quantified using Quant-iT™ Picogreen® dsDNA Assay kit (Invitrogen, Eugene, OR, USA) following manufacturer’s instructions modified to 100 μl and compared against a λ standard DNA dilution series with a Victor3V 1420 Multilabel Counter (PerkinElmer, Downers Grove, IL, USA), followed by visualization on 1 % agarose gel in 1× TBE (Tris/Borate/EDTA) stained with ethidium bromide. Samples were stored at −20 °C prior to use.
Marker sources
SSR primer pairs were selected from multiple sources (Table 1). Markers derived from GBS were coded as S with a number indicating the scaffold followed by an underscore and a number indicating the physical SNP position on the scaffold (i.e., S75_381030) (Bryant et al. 2014). Markers developed from the sequencing of paired-end short reads were coded with Ro (R. occidentalis) or Ri (R. idaeus) followed immediately by a number (i.e., Ro11481, Ri13528) (Dossett et al. in press). All other markers are from published sources as indicated in Table 1. Ag4_AphidR is a phenotypic marker for aphid resistance.
An additional 26 SSR and two high-resolution melting (HRM) markers that mapped in multiple populations were identified from the literature (Bushakra et al. 2012; Castillo et al. 2010; Castro et al. 2013; Fernández-Fernández et al. 2013; Graham et al. 2004; Sargent et al. 2007) with the intention of anchoring and orienting the linkage groups to published maps (Table 2).
DNA amplification of SSR markers
DNA amplification was performed on a PTC-225 gradient thermal cycler (Bio-Rad, Hercules, CA, USA), a Dyad Peltier thermal cycler (Bio-Rad, Hercules, CA, USA), an Eppendorf Mastercycler (Eppendorf, Hamburg, Germany), or a Nexus (Eppendorf, Hamburg, Germany). A fluorescent labeling polymerase chain reaction (PCR) protocol (Schuelke 2000) was used for most SSR primer pairs. The forward (F) primer of each pair was extended on the 5′-end with an M13-TGTAAAACGACGGCCAGTAGC sequence tag to which a universal M13-tagged fluorescent dye label (WellRed D2, D3, D4; Integrated DNA Technologies, Inc., Coralville, IA, USA) annealed. The touch-down PCR protocol began with an initial denaturation for 3 min at 94 °C followed by 10 cycles of 94 °C for 40 s, 65 °C (decreasing 1 °C every cycle) for 45 s, 72 °C for 45 s; 20 cycles of 94 °C for 40 s, 52 °C for 45 s, 72 °C for 45 s; 10 cycles of 94 °C for 40 s, 53 °C for 45 s, 72 °C for 45 s; followed by a final extension of 72 °C for 30 min. Reactions were performed in a final volume of 15 μl consisting of 6 ng template DNA, 1× PCR buffer, 2 mM MgCl2, 200 μM dNTP, 0.5 μM reverse primer, 0.12 μM M13-tagged forward primer, 0.5 μM WellRed labeled M13 primer (D2, D3 or D4), and 0.025 U GoTaq® Hot Start Polymerase (Promega Corporation, Madison, WI, USA). For a few SSR primer pairs, the 5′-end of the F primer was fluorescently labeled (WellRed D2, D3, or D4). The PCR protocol used for labeled F-primers began with an initial denaturation for 3 min at 94 °C followed by 35 cycles of 94 °C for 40 s, appropriate annealing temperature for 40 s, 72 °C for 30 s; followed by a final extension of 72 °C for 30 min. The reverse primer for Rub1C6 was PIG-tailed with 5′-GTTT-3′ (Brownstein et al. 1996) to minimize the occurrence of split peaks and difficulties encountered in automated fragment analysis following capillary electrophoresis.
Capillary electrophoresis of SSR markers
Success of the PCR was confirmed by 2 % agarose gel electrophoresis. Up to six fragments were pooled based on dye and predicted fragment size and separated on a Beckman CEQ 8000 capillary genetic analyzer (Beckman Coulter, Fullerton, CA, USA). Separation was followed by analysis of allele size and marker visualization using the fragment analysis module of the CEQ 8000 software.
High-resolution melting
The HRM technique (Wittwer et al. 2003) was used to amplify markers from Bushakra et al. (2012). Reactions were performed on PTC-225 gradient thermal cycler (Bio-Rad, Hercules, CA, USA), followed by HRM on the LightScanner® System (BioFire Defense, Salt Lake City, UT, USA). Reactions were performed in a final volume of 10 μl consisting of 6 ng DNA, 1× LightScanner Master Mix, 1 μM each forward and reverse primer. Each well was topped with one drop of mineral oil. The PCR amplification protocol was 94 °C for 30 s, followed by 30 s at the appropriate annealing temperature (57 or 58 °C) and extension at 72 °C for 30 s for 40 cycles. Following a final melting step at 95 °C for 30 s, the samples were cooled to 4 °C until HRM analysis. Amplicon melting occurred on the LightScanner where samples were heated to 98 °C over a period of 8 min with default settings. Analysis was performed using the LightScanner® Instrument & Analysis Software small amplicon genotyping module.
GBS library construction and sequencing
GBS libraries were constructed following Ward et al. (2013) and Elshire et al. (2011). Briefly, 100 ng of genomic DNA per sample were digested with 4 U of ApeKI (New England Biolabs, Ipswich, MA, USA) and then ligated with T4 ligase to 1.8 ng of combined common and unique barcode adapters (Elshire et al. 2011). Annealed and quantitated unique barcode and common adapters were provided by the Buckler Lab for Maize Genetics and Diversity, Cornell University (Ithaca, NY, USA) and Clemson University (Clemson, SC, USA) (Supplementary Table 1).
The GBS libraries were submitted to the Oregon State University Center for Genome Research and Biocomputing core facilities (Corvallis, OR, USA) for quantitation using a Qubit® fluorometer (Invitrogen, Carlsbad, CA, USA). The size distribution of the library was confirmed by checking 1000 pg of DNA with the Bioanalyzer 2100 HS-DNA chip (Agilent Technologies, Santa Clara, CA, USA). Libraries were diluted to 10 nM based on Qubit® readings and quantitative PCR (qPCR) was used to quantify the diluted libraries. For each pooled library, 15.5 pM were loaded for single-end Illumina® sequencing of 101 cycles with the HiSeq™ 2000 (Illumina, Inc.) and analyzed using the Version 3 cluster generation and sequencing kits (Illumina, Inc.).
The libraries were sequenced in three lanes at three different times. The first sequencing lane included 95 samples (91 progeny, and two replicated samples per parent). The second sequencing run included 88 samples (26 black raspberry including parents, grandparents, standards and progeny, and 62 unrelated strawberry samples). The third sequencing run included 64 samples (ORUS 3021-2 repeated 4 times, ORUS 4153-1 repeated 5 times and 55 progeny). Over all three runs, the parents ORUS 3021-2 and ORUS 4153-1 were sequenced at least twice in each lane. Forty-four progeny were sequenced more than once due to low initial quality and numbers of reads per individual.
GBS SNP calling
Version 3.0 of the TASSEL GBS discovery software pipeline (Li et al. 2009) was used to call SNP loci using a repeat-masked version of the genome sequence. Three GBS runs representing 112 individuals as described above were analyzed simultaneously. Data were initially subjected to sequence and nucleotide read quality control using Trimmomatic (Bolger et al. 2014) (http://www.usadellab.org/cms/?page=trimmomatic) and were then analyzed with TASSEL.
Genetic linkage map construction
All loci were converted into segregation codes for JoinMap® v. 4.1 (Van Ooijen 2006). Loci were then organized into parental sets and subjected to the maximum likelihood (ML) mapping algorithm. Independence Likelihood of Odds (LOD) threshold of 5 was used for establishing the linkage groups (LG). All other settings were default. Five progeny (ORUS 4305-38, 39, 41, 59, and 65) were excluded based on incongruous SNP data occurring from 30 (ORUS 4305-39) to 90 (ORUS 4305-65) times. GBS data were not available for ORUS 4305-7, 19, 45, 54, 58, 75, 95, 97, 103, and 110 due to poor DNA quality. The consensus map of seven linkage groups was generated by combining the parental linkage maps of ORUS 3021-2 and ORUS 4153-1 using the regression algorithm of the mapping software JoinMap v. 4.1. Linkage map visualization was accomplished with MapChart 2.2 (Voorrips 2002).
The quality of genotype calls and of each map were evaluated with a graphical genotyping approach in Microsoft Excel (Redmond, WA, USA) as previously described (Bassil et al. 2015; Young and Tanksley 1989).
Results
Transferable markers
In total, 552 SSR markers from new and published sources were evaluated for the amplification of polymorphic PCR products in the parents and one progeny. Of these, 118 failed to amplify, 235 were homozygous in both parents or gave ambiguous results, 138 were heterozygous in both parents, 29 were heterozygous in ORUS 3021-2, and 32 were heterozygous in ORUS 4153-1 (Table 2).
A total of 30 primer pairs (SSR and HRM) for 28 anchor loci were assessed for the production of a polymorphic PCR product in the parents and six progeny of population ORUS 4305. Twelve of these loci were successfully mapped (Table 2).
Eighty HRM primer pairs (Bushakra et al. 2012) were evaluated for the amplification of polymorphic PCR products on the parents and 14 progeny. Of these 80 HRM primer pairs, 57 were monomorphic, 12 were unclear or had poor amplification, and 11 were evaluated in the full population. Three of these HRM markers were mapped successfully, two in ORUS 3021-2 and one in ORUS 4153-1 (Table 2). Out of 660 transferable markers evaluated, a total of 72 (11 %) were successfully mapped. BLAST analysis (Altschul et al. 1990) of the forward and reverse primer and nucleotide sequences (when available), allowed scaffold assignment of most mapped transferable markers (Supplementary Table 2).
GBS SNP markers
The first sequencing run of 95 samples generated 596 K sequence clusters/mm2 (optimal density is 750–850 K clusters/mm2; MyIllumina Support Bulletin); the second and third sequencing runs were within the optimum range at 825 and 752 K clusters/mm2, respectively. These cluster densities provided raw reads ranging from approximately 165 million to 310 million. Over the three sequencing runs, 112 progeny and the two parents were sequenced to generate an average number of reads per individual of 3,105,333, with 20,317,182 (5.8 %) of reads unaligned. Default TASSEL filtering parameters using the parent information identified 57,238 SNP positions. Further filtering of the SNP data to remove those loci with more than 10 % missing data resulted in a data set of 7911 SNP loci, of which 3472 were monomorphic or ambiguous, 921 were heterozygous in both parents, 318 were heterozygous in ORUS 3021-2, and 326 were heterozygous in ORUS 4153-1 (Table 2).
Linkage mapping
Of the five progeny excluded based on incongruous SNP data, ORUS 4305-65 showed obvious phenotypic differences from the rest of the population and may be the result of a pollen contamination; however, the other four progeny were not phenotypically different from the rest of the population. A total of 100 progeny were used to construct the seven linkage groups for the parental linkage maps, the characteristics of which are summarized in Table 3. To construct the linkage map for ORUS 3021-2, five GBS-generated SNP markers were removed for skewed segregation ratios, four were removed for creating double recombination events within a distance of 10 cM or less, and one was removed due to unsuccessful linkage phase determination. For ORUS 3021-2 (Supplementary Fig. 1) the resulting 274 markers comprising the seven LGs spanned 779.4 centiMorgans (cM) with an average distance of 2.9 cM between markers. RLG7 had the greatest number of markers (56), and was also the longest (134.5 cM) with an average distance of 2.4 cM between markers. RLG2 was the shortest at 84.1 cM, with an average distance of 2.8 cM between the 30 markers, and two gaps of 11.4 and 11.9 cM. The largest gap for the map of ORUS 3021-2 was 22.2 cM on RLG6. Of the 222 GBS SNP markers used for map construction, 200 (90 %) segregated as expected, either 1:1 or 1:2:1; two loci (1 %) varied from expected at a significance level of 0.01, 11 loci (5 %) varied from expected at a significance level of 0.05, and nine loci (4 %) varied from expected at a significance level of 0.1.
To construct the linkage map for ORUS 4153-1, 18 GBS-generated SNP markers were removed for skewed segregation ratios, 14 were removed for creating double recombination events within a distance of 10 cM or less, and one SSR marker was removed due to unsuccessful linkage phase determination. For ORUS 4153-1 (Supplementary Fig. 2) the resulting 292 markers comprising the seven LGs spanned 892.1 cM with an average distance of 3.2 cM between markers. RLG7 had the greatest number of markers (64) and was also the longest (151.4 cM) with an average distance of 2.4 cM between markers, and three gaps greater than 10 cM, the largest of which was 12.2 cM; RLG1 was the shortest at 101.7 cM with 23 markers, an average distance of 4.4 cM between markers, and three gaps greater than 10 cM, the largest of which was 14.8 cM. The largest gap for the map of ORUS 4153-1 was 14.8 cM at the end of RLG1. Of the 249 GBS SNP markers used for map construction, 230 (92 %) segregated as expected, either 1:1 or 1:2:1; a single locus (0.4 %) varied from expected at a significance level of 0.01, nine loci (4 %) varied from expected at a significance level of 0.05, and nine loci (4 %) varied from expected at a significance level of 0.1.
Transferable markers for the parental maps ranged from a low of three markers on ORUS 3021-2 RLG7 and 4153-1 RLG6 to a high of 12 on ORUS 4153-1 RLG4. A total of 72 transferable markers were mapped in this population. BLAST analysis of the transferable markers against the draft genome assembly allowed scaffold assignment for 65 of 72 markers (90 %) so that 356 scaffolds were represented.
The phenotypic marker for aphid resistance, Ag4_AphidR, was located on RLG6 of the aphid-resistant parent ORUS 4153-1 and maps to the same location as S99_32802 (Fig. 2).

Rubus linkage group (RLG) 6 for black raspberry mapping population parent ORUS 4153-1. The morphological locus for Ag 4 aphid resistance against the North American large raspberry aphid is shown in blue bold font. The linkage map is constructed of single-nucleotide polymorphic (SNP) loci generated by genotyping by sequencing (GBS) (prefaced with S) and simple sequence repeat (SSR) loci from various Rubus sources (prefaced with Ro, Ri, Rh, Ru, Rub, and SQ). Transferable loci are indicated in bold font; anchor loci for comparisons with other Rubus linkage maps are indicated in bold italic font (color figure online)
The seven consensus RLGs (Table 3; Fig. 3) assembled from merging the parental maps consisted of 438 markers spanning 546.4 cM with an average distance between markers of 1.3 cM. Consensus RLG6 was the longest (90.2 cM) with an average distance between the 69 markers of 1.3 cM, and one gap of 10.4 cM. Consensus RLG7 had the most markers (77) that spanned 81.0 cM with an average distance of 1.1 cM between markers. RLG2 was the shortest at 70.8 cM with an average distance between the 59 markers of 1.2 cM. The 12 anchor markers identified from the literature (Table 1; Supplementary Figs. 1, 2, markers in italics) allowed the positive identification of consensus RLG 2-7, with the last, RLG1, identified by default.


Consensus linkage map for population ORUS 4305. Each of the linkage groups consists of single-nucleotide polymorphic (SNP) loci generated by genotyping by sequencing (GBS) (prefaced with S) and simple sequence repeat (SSR) loci from various Rubus sources (prefaced with Ro, Ri, Rh, Ru, Rub, and SQ). Transferable loci are indicated in bold font; anchor loci for comparisons with other Rubus linkage maps are indicated in bold italic font. The morphological locus for Ag 4 aphid resistance against the North American large raspberry aphid is shown in blue bold font (color figure online)
Thirteen of the 356 represented scaffolds (3.6 %) map to more than one linkage group (Table 4); 33 of the loci are SNPs and five are SSRs. Four scaffolds (S10, S26, S134, and S142) are represented by SNP loci on more than two linkage groups. Four scaffolds (S14, S71, S78, and S279) are represented by at least one SNP and a single SSR locus on more than one linkage group.
Discussion
We present the first linkage map constructed from a pure black raspberry cross. The first attempt at genetic linkage mapping using SSR markers on an F2 generation of a black raspberry × red raspberry cross identified high homozygosity as well as severe segregation distortion and did not result in a linkage map (Lewers and Weber 2005). The linkage map constructed using non-anonymous DNA sequences for black raspberry selection 96395S1 comprises 29 markers spaced on average at intervals of 10 cM over six LG spanning 306 cM (Bushakra et al. 2012). The first published red raspberry map of ‘Glen Moy’ × ‘Latham’ consisted of 273 markers derived from amplified fragment length polymorphic and genomic-SSR markers and spanned 789 cM over nine LG (Graham et al. 2004). Over the next 6 years as more markers were developed and added, the improved ‘Glen Moy’ × ‘Latham’ map reported by Woodhead et al. (2010) consisted of 228 markers over seven LG spanning 840.3 cM with transferable markers present on each LG. Paterson et al. (2013) subsequently added gene-based markers to the linkage map constructed by Woodhead et al. (2010) by mining Rubus transcriptome and EST databases for candidate genes in the fruit volatiles pathway. The efficiency of marker generation used here is a vast improvement over previous marker development techniques in Rubus. The saturated consensus linkage map presented here spans 546.2 cM and is composed of 374 GBS-generated SNP markers and 68 transferable markers with an average of 1.3 cM between markers. The transferable markers are distributed among the LG and can be used for alignment to other Rubus maps. The scaffold assignment allows for future fine mapping, QTL analysis, and improved black raspberry genome assembly.
The reduced-representation sequencing accomplished with GBS has generally been used in crop plants with high levels of heterozygosity. For example, Poland et al. (2012) were able to map 20,000 and 34,000 GBS-generated SNP loci in wheat and barley reference linkage maps, respectively; Lu et al. (2013) performed GBS in tetraploid switchgrass (Panicum virgatum) and were able to map 88,217 SNP loci; Truong et al. (2012) used GBS to generate SNP in Arabidopsis thaliana and lettuce (Lactuca sativa) and were able to map 1200 and 1113 SNP loci, respectively; Russell et al. (2014) mapped 790 SNP loci in blackcurrant (Ribes nigrum). This is the first use of GBS on black raspberry, a crop of relatively low genetic diversity. Even with an average number of reads per individual of 3,105,333 over the three sequencing runs, only 1545 SNP loci were found that met criteria for linkage mapping and of those 399 were mapped successfully (Table 2). While this is a sufficient number of markers to develop a well-populated map, it, along with the low mapping success rate of transferable markers, illustrates the low level of heterozygosity found in black raspberry. In contrast, the linkage maps constructed of GBS-derived SNP and SSR markers for red raspberry parents ‘Heritage’ and ‘Tulameen’ comprise 4521 markers spaced on average at intervals of 0.1 cM over seven LG spanning 462.7 cM and 2391 markers spaced on average at intervals of 0.1 cM spanning 376.6 cM, respectively (Ward et al. 2013). While digestion by a more frequent restriction enzyme cutter for GBS may be a way to increase the number of SNP loci identified, this does not guarantee mapping success as segregation within the population is essential for linkage mapping.
Up to 97 % of the mapped scaffolds were placed on a single linkage group indicating high quality assembly of the draft genome. The 13 scaffolds that map to multiple LGs will need to be investigated further to assess whether these inconsistencies represent errors in the genome assembly; however, initial observations could indicate regions of high chromosome homology or possible regions of genome duplication especially between RLG3 and RLG7.
The placement of the aphid-resistance morphological marker representing gene Ag 4 on RLG6 corresponds to the red raspberry genomic region found by Sargent et al. (2007) for A 1 . The only other aphid-resistance gene in Rubus that has been mapped is A 10 , which was found to be located on red raspberry RLG4 (Fernández-Fernández et al. 2013). A 1 originated from the old red raspberry ‘Baumforth’s A’ and confers race-specific resistance to three biotypes of the European large raspberry aphid, Amphorophora idaei Börner (biotypes 1, 3 and the A 10 -breaking; McMenemy et al. 2009), but is ineffective against the North American species A. agathonica. Ag4_AphidR maps to the same position as SNP S99_32802, providing us with a clearly defined region on which to focus our future fine-mapping efforts and comparative mapping to red raspberry. This linkage map region is associated with many quantitative trait loci (QTL) having to do with resistance to aphids (Sargent et al. 2007), and fungal (Graham et al. 2006) and fungal-like (Graham et al. 2011) pathogens in red raspberry and we hope to use our linkage map to better understand the underlying reasons for these associations.
Conclusions
We present here the first genetic linkage map of black raspberry comprised of GBS-generated SNP and transferable markers. The presence of SSR and HRM markers selected from the literature, along with the other transferable markers allowed us to positively identify all RLG as per Bushakra et al. (2012), and provide an opportunity to align all existing Rubus linkage maps. These maps will serve as a framework for anchoring scaffold sequences in the black raspberry draft genome sequence. Comparative mapping using the common markers and the draft genome sequence will be useful for aligning QTL among different species of Rubus. Future studies on the different sources of aphid resistance, including construction of densely populated linkage maps and cloning of loci associated with aphid resistance, will provide information on the loci and will result in the development of markers that can be used for marker-assisted breeding for aphid resistance in black raspberry.
Author contribution statement
JMB Project Coordinator performed marker screening, selected anchor markers, ran and scored all markers, constructed the genetic linkage map, and wrote the manuscript. DWB developed a custom pipeline for bioinformatic analyses, and performed GBS SNP calling. MD developed the mapping population, short-read Ro and Ri primers, performed the initial marker screening, and phenotyped aphid-resistance in the mapping population. KJV and RVB assisted in GBS SNP calling and other bioinformatic analyses, BLAST analyses, and linkage mapping. BSG assisted in developing and performed the initial marker screening. JL PI on NIFA SCRI grant (project main funding) and contributed to manuscript writing. TCM PI on NIFA SCRI grant (project main funding) and contributed computational resources and bioinformatics analysis. CEF PI on NIFA SCRI grant (project main funding), helped assemble and phenotype the germplasm, develop the mapping population, and contributed to manuscript writing. Primary advisor for the phenotyping portion of the NIFA SCRI grant. NVB PI on NIFA SCRI grant (project main funding), helped analyze short-read sequencing results, develop and test molecular markers, and contributed to manuscript writing. Primary advisor for the genomics portion of the NIFA SCRI grant.
References
Alice LA, Campbell CS (1999) Phylogeny of Rubus (Rosaceae) based on nuclear ribosomal DNA internal transcribed spacer region sequences. Am J Bot 86:81–97
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Anonymous (2015) National Statistics for Raspberries. National Agricultural Statistics Service. www.nass.usda.gov. Accessed 27 May 2015
Antanaviciute L, Fernández-Fernández F, Jansen J, Banchi E, Evans K, Viola R, Velasco R, Dunwell J, Troggio M, Sargent D (2012) Development of a dense SNP-based linkage map of an apple rootstock progeny using the Malus Infinium whole genome genotyping array. BMC Genomics 13:203
Aranzana MJ, Pineda A, Cosson P, Dirlewanger E, Ascasibar J, Cipriani G, Ryder C, Testolin R, Abbott A, King G, Iezzoni A, Arús P (2003) A set of simple-sequence repeat (SSR) markers covering the Prunus genome. Theor Appl Genet 106:819–825
Bassil NV, Davis TM, Zhang H, Ficklin S, Mittmann M, Webster L, Mahoney L, Wood D, Alperin ES, Rosyara UR, Koehorst-van Putten H, Monfort A, Amaya I, Denoyes B, Sargent DJ, Bianco L, van Dijk T, Pirani A, Iezzoni A, Main D, Peace C, Yang Y, Whitaker V, Verma S, Bellon L, Brew F, Herrera R, van de Weg E (2015) Development and preliminary evaluation of a 90 K Axiom® SNP array for the allo-octoploid cultivated strawberry Fragaria × ananassa. BMC Genomics 16:30
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Brownstein M, Carpten J, Smith J (1996) Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20(1004–1006):1008–1010
Bryant D, Bushakra JM, Dossett M, Vining K, Filichkin S, Weiland J, Lee J, Finn CE, Bassil N, Mockler T (2014) Building the genomic infrastructure in black raspberry. HortScience 49:S233
Bushakra JM, Stephens MJ, Atmadjaja AN, Lewers KS, Symonds VV, Udall JA, Chagné D, Buck EJ, Gardiner SE (2012) Construction of black (Rubus occidentalis) and red (R. idaeus) raspberry linkage maps and their comparison to the genomes of strawberry, apple, and peach. Theor Appl Genet 125:311–327
Castillo NRF, Reed BM, Graham J, Fernández-Fernández F, Bassil NV (2010) Microsatellite markers for raspberry and blackberry. J Am Soc Hortic Sci 135:271–278
Castro P, Stafne ET, Clark JR, Lewers KS (2013) Genetic map of the primocane-fruiting and thornless traits of tetraploid blackberry. Theor Appl Genet 126:2521–2532
Celton J-M, Tustin DS, Chagné D, Gardiner SE (2009) Construction of a dense genetic linkage map for apple rootstocks using SSRs developed from Malus ESTs and Pyrus genomic sequences. Tree Genet Genomes 5:93–107
Chaparro JX, Werner DJ, O’Malley D, Sederoff RR (1994) Targeted mapping and linkage analysis of morphological, isozyme, and RAPD markers in peach. Theor Appl Genet 87:805–815
Clark JR, Finn CE (1999) Blackberry and hybrid berries. In: Okie WR (ed) Register of new fruit and nut varieties, list 39. HortScience 34:183–184
Clark JR, Finn CE (2002) Blackberry and hybrid berry. In: Okie WR (ed) Register of new fruit and nut varieties, list 41. HortScience 37:251–252
Clark JR, Finn CE (2006) Blackberry and hybrid berry. In: Clark JR, Finn CE (eds) Register of new fruit and nut cultivars, list 43. HortScience 41:1104–1106
Clark JR, McCall C, Finn CE (2008) Blackberry. In: Finn CE, Clark JR (eds) Register of new fruit and nut cultivars, list 44. HortScience 43:1323–1324
Clark JR, Sleezer SM, Finn CE (2012) Blackberry. In: Finn CE, Clark JR (eds) Register of new fruit and nut cultivars, list 46. HortScience 47:539
Clark MD, Schmitz CA, Rosyara UR, Luby JJ, Bradeen JM (2014) A consensus ‘Honeycrisp’ apple (Malus × domestica) genetic linkage map from three full-sib progeny populations. Tree Genet Genomes 10:627–639
Daubeny HA (1997a) Blackberry and hybrid berries. In: Okie WR (ed) Register of new fruit and nut varieties, list 38. HortScience 32:785–786
Daubeny HA (1997b) Raspberry. In: Okie WR (ed) Register of new fruit and nut varieties, list 38. HortScience 32:797–798
Daubeny HA (1999) Raspberry. In: Okie WR (ed) Register of new fruit and nut varieties, list 39. HortScience 34:196–197
Daubeny HA (2000) Raspberry. In: Okie WR (ed) Register of new fruit and nut varieties, list 40. HortScience 35:820–821
Daubeny HA (2002) Raspberry. In: Okie WR (ed) Register of new fruit and nut varieties, list 41. HortScience 37:264–265
Daubeny HA (2004) Raspberry. In: Okie WR (ed) Register of new fruit and nut varieties, list 42. HortScience 39:1516–1517
Daubeny HA (2006) Raspberry. In: Clark JR, Finn CE (eds) Register of new fruit and nut cultivars, list 43. HortScience 41:1122–1124
Daubeny HA (2008) Raspberry. In: Finn CE, Clark JR (eds) Register of new fruit and nut cultivars, list 44. HortScience 43:1336–1337
Dirlewanger E, Cosson P, Howad W, Capdeville G, Bosselut N, Claverie M, Voisin R, Poizat C, Lafargue B, Baron O, Laigret F, Kleinhentz M, Arús P, Esmenjaud D (2004) Microsatellite genetic linkage maps of myrobalan plum and an almond-peach hybrid—location of root-knot nematode resistance genes. Theor Appl Genet 109:827–838
Dirlewanger E, Cosson P, Boudehri K, Renaud C, Capdeville G, Tauzin Y, Laigret F, Moing A (2006) Development of a second-generation genetic linkage map for peach [Prunus persica (L.) Batsch] and characterization of morphological traits affecting flower and fruit. Tree Genet Genomes 3:1–13
Dossett M, Finn CE (2010) Identification of resistance to the large raspberry aphid in black raspberry. J Am Soc Hortic Sci 135:438–444
Dossett M, Lee J, Finn CE (2008) Inheritance of phenological, vegetative, and fruit chemistry traits in black raspberry. J Am Soc Hortic Sci 133:408–417
Dossett M, Bassil NV, Finn CE (2012a) Fingerprinting of black raspberry cultivars shows discrepancies in identification. Acta Hortic (ISHS) 946:49–53
Dossett M, Bassil NV, Lewers KS, Finn CE (2012b) Genetic diversity in wild and cultivated black raspberry (Rubus occidentalis L.) evaluated by simple sequence repeat markers. Genet Resour Crop Evol 59:1849–1865
Dossett M, Bushakra JM, Gilmore B, Koch C, Kempler C, Finn CE, Bassil N (in press) Development and transferability of black and red raspberry microsatellite markers using next generation sequencing. J Am Soc Hortic Sci
Drain BD (1956) Inheritance in black raspberry species. Proc Am Soc Hortic Sci 68:169–170
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
Etienne C, Rothan C, Moing A, Plomion C, Bodénès C, Svanella-Dumas L, Cosson P, Pronier V, Monet R, Dirlewanger E (2002) Candidate genes and QTLs for sugar and organic acid content in peach [Prunus persica (L.) Batsch]. Theor Appl Genet 105:145–159
Fernández-Fernández F, Evans K, Clarke J, Govan C, James C, Marič S, Tobutt K (2008) Development of an STS map of an interspecific progeny of Malus. Tree Genet Genomes 4:469–479
Fernández-Fernández F, Antanaviciute L, Knight VH, Dunwell JM, Battey NH, Sargent DJ (2013) Genetics of resistance to Amphorophora idaei in red raspberry. Acta Hort (ISHS) 976:501–508
Finn CE, Clark JR (2000) Blackberry and hybrid berries. In: Okie WR (ed) Register of new fruit and nut varieties, list 40. HortScience 35:814–815
Finn CE, Clark JR (2004) Blackberry and hybrid berries. In: Okie WR (ed) Register of new fruit and nut varieties, list 42. HortScience 39:1509
Finn CE, Clark JR (2014) Blackberry. In: Gasic K, Preece JE (eds) Register of new fruit and nut varieties, list 47. HortScience 49:399–400
Finn CE, Clark JR, Sleezer SM (2010) Blackberry. In: Clark JR, Finn CE (eds) Register of new fruit and nut cultivars, list 45. HortScience 45:720–721
Foolad MR, Arulsekar S, Becerra V, Bliss FA (1995) A genetic map of Prunus based on an interspecific cross between peach and almond. Theor Appl Genet 91:262–269
Frett TJ, Reighard GL, Okie WR, Gasic K (2014) Mapping quantitative trait loci associated with blush in peach [Prunus persica (L.) Batsch]. Tree Genet Genomes 10:367–381
Galletta GJ, Maas JL, Enns JM (1998) ‘Earlysweet’ black raspberry. Fruit Var J 52:123–124
Gilmore BS, Bassil NV, Hummer KE (2011) DNA extraction protocols from dormant buds of twelve woody plant genera. J Am Pomol Soc 65:201–207
Gisbert A, Martínez-Calvo J, Llácer G, Badenes M, Romero C (2009) Development of two loquat [Eriobotrya japonica (Thunb.) Lindl.] linkage maps based on AFLPs and SSR markers from different Rosaceae species. Mol Breed 23:523–538
Graham J, McNicol RJ, Greig K, Van de Ven WTG (1994) Identification of red raspberry cultivars and an assessment of their relatedness using fingerprints produced by random primers. J Hort Sci 69:123–130
Graham J, Smith K, MacKenzie K, Jorgenson L, Hackett C, Powell W (2004) The construction of a genetic linkage map of red raspberry (Rubus idaeus subsp. idaeus) based on AFLPs, genomic-SSR and EST-SSR markers. Theor Appl Genet 109:740–749
Graham J, Smith K, Tierney I, MacKenzie K, Hackett C (2006) Mapping gene H controlling cane pubescence in raspberry and its association with resistance to cane botrytis and spur blight, rust and cane spot. Theor Appl Genet 112:818–831
Graham J, Hackett C, Smith K, Woodhead M, MacKenzie K, Tierney I, Cooke D, Bayer M, Jennings N (2011) Towards an understanding of the nature of resistance to Phytophthora root rot in red raspberry. Theor Appl Genet 123:585–601
Halgren A, Tzanetakis IE, Martin RR (2007) Identification, characterization, and detection of Black raspberry necrosis virus. Phytopathology 97:44–50
Hellman EW, Skirvin RM, Otterbacher AG (1982) Unilateral incompatibility between red and black raspberries. J Am Soc Hortic Sci 107:781–784
Hemmat M, Weedon NF, Manganaris AG, Lawson DM (1994) Molecular marker linkage map for apple. J Hered 85:4–11
Hibrand-Saint Oyant L, Crespel L, Rajapakse S, Zhang L, Foucher F (2008) Genetic linkage maps of rose constructed with new microsatellite markers and locating QTL controlling flowering traits. Tree Genet Genomes 4:11–23
Klagges C, Campoy JA, Quero-García J, Guzmán A, Mansur L, Gratacós E, Silva H, Rosyara UR, Iezzoni A, Meisel LA, Dirlewanger E (2013) Construction and comparative analyses of highly dense linkage maps of two sweet cherry intra-specific progenies of commercial cultivars. PLoS ONE 8:e54743
Lewers KS, Weber CA (2005) The trouble with genetic mapping of raspberry. HortScience 40:1108
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Liebhard R, Koller B, Gianfranceschi L, Gessler C (2003) Creating a saturated reference map for the apple (Malus × domestica Borkh.) genome. Theor Appl Genet 106:1497–1508
Lu F, Lipka AE, Glaubitz J, Elshire R, Cherney JH, Casler MD, Buckler ES, Costich DE (2013) Switchgrass genomic diversity, ploidy, and evolution: novel insights from a network-based SNP discovery protocol. PLoS Genet 9:e1003215
McMenemy LS, Mitchell C, Johnson SN (2009) Biology of the European large raspberry aphid (Amphorophora idaei): its role in virus transmission and resistance breakdown in red raspberry. Agric For Entomol 11:61–71
Montanari S, Saeed M, Knäbel M, Kim Y, Troggio M, Malnoy M, Velasco R, Fontana P, Won K, Durel C-E, Perchepied L, Schaffer R, Wiedow C, Bus V, Brewer L, Gardiner SE, Crowhurst RN, Chagné D (2013) Identification of Pyrus single nucleotide polymorphisms (SNPs) and evaluation for genetic mapping in european pear and interspecific Pyrus hybrids. PLoS ONE 8:e77022
Moore PP, Kempler C (2010) Raspberry. In: Clark JR, Finn CE (eds) Register of new fruit and nut cultivars, list 45. HortScience 45:747–748
Moore PP, Kempler C (2012) Raspberry. In: Finn CE, Clark JR (eds) Register of new fruit and nut cultivars, list 46. HortScience 47:555–556
Moore PP, Kempler C (2014) Raspberry. In: Gasic K, Preece JE (eds) Register of new fruit and nut varieties, list 47. HortScience 49:415–416
Nybom H, Schaal BA (1990) DNA “fingerprints” reveal genotypic distributions in natural populations of blackberries and raspberries (Rubus, Rosaceae). Am J Bot 77:883–888
Olmstead J, Sebolt A, Cabrera A, Sooriyapathirana S, Hammar S, Iriarte G, Wang D, Chen C, van der Knaap E, Iezzoni A (2008) Construction of an intra-specific sweet cherry (Prunus avium L.) genetic linkage map and synteny analysis with the Prunus reference map. Tree Genet Genomes 4:897–910
Ourecky DK (1975) Brambles. In: Janick J, Moore JN (eds) Advances in fruit breeding. Purdue University Press, West Lafayette, pp 98–129
Paterson A, Kassim A, McCallum S, Woodhead M, Smith K, Zait D, Graham J (2013) Environmental and seasonal influences on red raspberry flavour volatiles and identification of quantitative trait loci (QTL) and candidate genes. Theor Appl Genet 126:33–48
Pierantoni L, Cho KH, Shin IS, Chiodini R, Tartarini S, Dondini L, Kang SJ, Sansavini S (2004) Characterisation and transferability of apple SSRs to two European pear F1 populations. Theor Appl Genet 109:1519–1524
Pirona R, Eduardo I, Pacheco I, Da Silva Linge C, Miculan M, Verde I, Tartarini S, Dondini L, Pea G, Bassi D, Rossini L (2013) Fine mapping and identification of a candidate gene for a major locus controlling maturity date in peach. BMC Plant Biol 13:166
Poland JA, Brown PJ, Sorrells ME, Jannink J-L (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 7:e32253
Rajapakse S, Belthoff LE, He G, Estager AE, Scorza R, Verde I, Ballard RE, Baird WV, Callahan A, Monet R, Abbott AG (1995) Genetic linkage mapping in peach using morphological, RFLP and RAPD markers. Theor Appl Genet 90:503–510
Russell J, Hackett C, Hedley P, Liu H, Milne L, Bayer M, Marshall D, Jorgensen L, Gordon S, Brennan R (2014) The use of genotyping by sequencing in blackcurrant (Ribes nigrum): developing high-resolution linkage maps in species without reference genome sequences. Mol Breed 33:835–849
Sargent D, Fernández-Fernández F, Rys A, Knight V, Simpson D, Tobutt K (2007) Mapping of A1 conferring resistance to the aphid Amphorophora idaei and dw (dwarfing habit) in red raspberry (Rubus idaeus L.) using AFLP and microsatellite markers. BMC Plant Biol 7:15
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Silfverberg-Dilworth E, Matasci CL, van de Weg WE, Van Kaauwen MPW, Walser M, Kodde LP, Soglio V, Gianfranceschi L, Durel CE, Costa F, Yamamoto T, Koller B, Gessler C, Patocchi A (2006) Microsatellite markers spanning the apple (Malus × domestica Borkh.) genome. Tree Genet Genomes 2:202–224
Slate GL (1933) The best parents in purple raspberry breeding. Proc Am Soc Hortic Sci 30:108–112
Slate GL, Klein LG (1952) Black raspberry breeding. Proc Am Soc Hortic Sci 59:266–268
Stafne ET, Clark JR, Weber CA, Graham J, Lewers KS (2005) Simple sequence repeat (SSR) markers for genetic mapping of raspberry and blackberry. J Am Soc Hortic Sci 130:722–728
Stockinger EJ, Mulinix CA, Long CM, Brettin TS, Lezzoni AF (1996) A linkage map of sweet cherry based on RAPD analysis of a microspore-derived callus culture population. J Hered 87:214–218
Stoner GD, Wang L-S, Zikri N, Chen T, Hecht SS, Huang C, Sardo C, Lechner JF (2007) Cancer prevention with freeze-dried berries and berry components. Semin Cancer Biol 17:403–410
Thompson MM (1995) Chromosome numbers of Rubus species at the National Clonal Germplasm Repository. HortScience 30:1447–1452
Truong HT, Ramos AM, Yalcin F, de Ruiter M, van der Poel HJA, Huvenaars KHJ, Hogers RCJ, van Enckevort LJG, Janssen A, van Orsouw NJ, van Eijk MJT (2012) Sequence-based genotyping for marker discovery and co-dominant scoring in germplasm and populations. PLoS ONE 7:e37565
Van Ooijen JW (2006) JoinMap® 4, software for the calculation of genetic linkage maps in experimental populations. Kyazma B.V., Wageningen
Vilanova S, Sargent D, Arús P, Monfort A (2008) Synteny conservation between two distantly-related Rosaceae genomes: Prunus (the stone fruits) and Fragaria (the strawberry). BMC Plant Biol 8:67
Viruel MA, Messeguer R, De Vicente MC, Garcia-Mas J, Puigdomenech P, Vargas F, Arús P (1995) A linkage map with RFLP and isozyme markers for almond. Theor Appl Genet 91:964–971
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Ward J, Bhangoo J, Fernández-Fernández F, Moore P, Swanson J, Viola R, Velasco R, Bassil N, Weber C, Sargent D (2013) Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genomics 14:2
Weber CA (2003) Genetic diversity in black raspberry detected by RAPD markers. HortScience 38:269–272
Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ (2003) High-resolution genotyping by amplicon melting analysis using LCgreen. Clin Chem 49:853–860
Woodhead M, McCallum S, Smith K, Cardle L, Mazzitelli L, Graham J (2008) Identification, characterisation and mapping of simple sequence repeat (SSR) markers from raspberry root and bud ESTs. Mol Breed 22:555–563
Woodhead M, Weir A, Smith K, McCallum S, MacKenzie K, Graham J (2010) Functional markers for red raspberry. J Am Soc Hortic Sci 135:418–427
Yang N, Reighard G, Ritchie D, Okie W, Gasic K (2013) Mapping quantitative trait loci associated with resistance to bacterial spot (Xanthomonas arboricola pv. pruni) in peach. Tree Genet Genomes 9:573–586
Young ND, Tanksley SD (1989) Restriction fragment length polymorphism maps and the concept of graphical genotypes. Theor Appl Genet 77:95–101
Acknowledgments
The authors thank Dr. Jana Lee (USDA-ARS HCRU, Corvallis, OR, USA) for her help with rearing aphids, Dr. Eric van de Weg (University of Wageningen, The Netherlands) for his assistance with graphical genotyping and consultation, The Barley Lab at Oregon State University for access to JoinMap, Drs. Kim Lewers (USDA-ARS BARC, Beltsville, MD, USA) and Christopher Saski (Clemson University, Clemson, SC, USA) for access to unpublished SSR marker data, and Drs. Kim Lewers, James Olmstead (University of Florida, Gainesville, FL, USA) and two anonymous reviewers for their helpful suggestions. The authors thank the Washington Red Raspberry Commission, the Oregon Raspberry and Blackberry Commission, the North American Raspberry and Blackberry Growers Association, the USDA-ARS, and the Northwest Center for Small Fruit Research for providing matching funds or other support. This work was funded by the USDA-National Institute of Food and Agriculture (NIFA) Specialty Crop Research Initiative (SCRI) USDA-ARS CRIS 2072-21000-044-00D, 2072-21000-047-00D and 2072-21220-002-00D. Mention of trade names or commercial products in this publication is solely for the purpose of providing scientific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. A. Hackett.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
A genetic linkage map of female parent ORUS 3021-2 constructed using the maximum likelihood algorithm of the mapping software JoinMap v. 4.1 Each of the linkage groups consists of single nucleotide polymorphic (SNP) loci generated by genotyping by sequencing (GBS) (prefaced with S) and simple sequence repeat (SSR) loci from various Rubus sources (prefaced with Ro, Ri, Rh, Ru, Rub, and SQ). Transferable loci are indicated in bold font; anchor loci for comparisons with other Rubus linkage maps are indicated in bold italic font (DOCX 57 kb)
Supplementary Fig. 1
A genetic linkage map of male parent ORUS 4153-1 constructed using the maximum likelihood algorithm of the mapping software JoinMap v. 4.1. Each of the linkage groups consists of single nucleotide polymorphic (SNP) loci generated by genotyping by sequencing (GBS) (prefaced with S) and simple sequence repeat (SSR) loci from various Rubus sources (prefaced with Ro, Ri, Rh, Ru, Rub, and SQ). Transferable loci are indicated in bold font; anchor loci for comparisons with other Rubus linkage maps are indicated in bold italic font (DOCX 42 kb)
Supplementary Table 1
Genotyping by sequencing unique barcode sequences. Well indicates the location on the 96-well plate, each barcode sequence is complemented. aEach unique barcode sequence consists of two oligonucleotides 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTxxxx and 5′-CWGyyyyAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT, where ‘‘xxxx’’ and ‘‘yyyy’’ denote the barcode and barcode complement sequences, respectively. Common adapter sequences with an ApeKI-compatible sticky end are 5′-CWGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG and 5′-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCT (Elshire et al. 2011) (DOCX 14 kb)
Supplementary Table 2
Transferable locus basic linear alignment search tool (BLAST) results. Each primer sequence was compared to the black raspberry draft genome sequence using BLAST. Those loci for which a single hit was obtained were assigned a scaffold location. Those loci with both forward and reverse primers confirmed are indicated in bold font. Those loci with no confirmed scaffold location are in grey font (DOCX 47 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bushakra, J.M., Bryant, D.W., Dossett, M. et al. A genetic linkage map of black raspberry (Rubus occidentalis) and the mapping of Ag 4 conferring resistance to the aphid Amphorophora agathonica . Theor Appl Genet 128, 1631–1646 (2015). https://doi.org/10.1007/s00122-015-2541-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-015-2541-x