
Abstract
To dissect the genetic factors controlling naturally occurring variation of heading date in Asian rice cultivars, we performed QTL analyses using F2 populations derived from crosses between a japonica cultivar, Koshihikari, and each of 12 cultivars originating from various regions in Asia. These 12 diverse cultivars varied in heading date under natural field conditions in Tsukuba, Japan. Transgressive segregation was observed in 10 F2 combinations. QTL analyses using multiple crosses revealed a comprehensive series of loci involved in natural variation in flowering time. One to four QTLs were detected in each cross combination, and some QTLs were shared among combinations. The chromosomal locations of these QTLs corresponded well with those detected in other studies. The allelic effects of the QTLs varied among the cross combinations. Sequence analysis of several previously cloned genes controlling heading date, including Hd1, Hd3a, Hd6, RFT1, and Ghd7, identified several functional polymorphisms, indicating that allelic variation at these loci probably contributes to variation in heading date. Taken together, the QTL and sequencing results indicate that a large portion of the phenotypic variation in heading date in Asian rice cultivars could be generated by combinations of different alleles (possibly both loss- and gain-of-function) of the QTLs detected in this study.
Similar content being viewed by others

Introduction
Heading date is one of crucial factors determining regional and seasonal adaptation in rice and has been a major target of selection in breeding programs. Heading date is a complex trait that is governed by multiple genes and environmental factors, such as daylength, temperature, and soil conditions. A wide range of variation in heading date has been observed among rice cultivars. During the last decade, genetic studies using DNA markers have facilitated the genetic dissection of heading date, and many quantitative trait loci (QTLs) for heading date have been identified using several mapping populations (reviewed by Yano et al. 2001; Hittalmani et al. 2003; Lin et al. 2002, 2003; Gu and Foley 2007; Uga et al. 2007; Nonoue et al. 2008; Matsubara et al. 2008a; Maas et al. 2010).
The detection of QTLs for heading date has allowed further genetic analyses, such as the development of nearly isogenic lines (NILs), analysis of epistatic interactions among QTLs, and map-based cloning. Hd1, a major QTL for photoperiod sensitivity (daylength response), encodes a protein with zinc finger and CCT motifs and is an ortholog of CONSTANS, which is involved in long-day (LD) flowering promotion in Arabidopsis (Yano et al. 2000). Hd6 and Hd3a, genes involved in photoperiod sensitivity, encode a casein kinase 2 alpha and an Arabidopsis FT-like protein, respectively (Takahashi et al. 2001; Kojima et al. 2002). Hd1 protein activates transcription of the rice florigen genes Hd3a and RFT1 under short-day (SD) conditions while repressing transcription of these genes under LD conditions (Kojima et al. 2002; Tamaki et al. 2007; Komiya et al. 2008, 2009). Early heading date 1 (Ehd1), a promoter of heading, was detected on chromosome 10 by using a BC1F1 population derived from a cross between cultivar T65 and an accession of another cultivated species, Oryza glaberrima; further analysis revealed that Ehd1 encodes a B-type response regulator and activates Hd3a and RFT1 under SD conditions independently of Hd1 (Doi et al. 2004). Recently, Xue et al. (2008) demonstrated that a major flowering repressor, Ghd7, encodes a protein with a CCT motif that represses transcription of Ehd1, Hd3a, and RFT1. These genetic and molecular studies have contributed substantially to our understanding of heading date in rice (Izawa 2007; Tsuji et al. 2008).
Although knowledge about the genetic control of rice heading has accumulated rapidly, the genetic mechanisms underlying the wide range of heading date variation still remain to be clarified. During the last decade, genetic analyses have been performed on a limited number of cross combinations to identify QTLs involved in heading date (Yonemaru et al. 2010). Furthermore, it has been very difficult to precisely compare QTLs detected in different studies due to the different levels of mapping resolution in each study (Yonemaru et al. 2010). In general, since QTLs are detected based on the allelic differences between parental lines, it is difficult to determine whether a particular QTL is shared among different cross combinations. Therefore, to understand genetic factors controlling heading date in diverse germplasm, a common parental line should be used for the development of mapping populations.
To comprehensively dissect naturally occurring variation in rice heading date, we performed a QTL analysis in 12 populations derived from crosses of the japonica cultivar Koshihikari, as a common parental line, with diverse cultivars that originate from various regions in Asia. Some of the QTLs detected in these populations were shared among several cross combinations, and their chromosomal locations corresponded to those of QTLs reported in other studies. In addition, we performed sequence analysis of several heading date genes that had been previously cloned by a map-based strategy, to examine the basis of the variation detected as QTLs in these populations. Based on the data from the QTL analysis and the sequence variation in the cloned heading date genes, we concluded that a large portion of the wide range of phenotypic variation for heading date in Asian cultivars could be generated by combinations of alleles with loss or gain of function in those QTLs.
Materials and methods
Plant materials
Twelve Asian rice cultivars were selected based on their geographical origins and cultivar groups (Kojima et al. 2005) (Table 1). We made crosses between those cultivars and a japonica cultivar, Koshihikari, as a common parental line to produce F1s. Those F1s were self-pollinated to produce F2 progeny. For each population used in QTL mapping, 94 F2 plants and the two parental lines (24 plants of each) were raised in a paddy field at the National Institute of Agrobiological Sciences (NIAS) in Tsukuba, Japan, from April to November. The mean daylength during the cultivation period was 13.1 h in mid-April, 14.1 h in May, 14.6 h in June, 14.4 h in July, 13.5 h in August, and 12.4 h in September. Average temperature during the cultivation period was 17°C in May, 21°C in June, 24°C in July, 26°C in August, and 22°C in September. Cultivation management followed the standard procedures used at NIAS.
Scoring of days-to-heading (DTH)
We recorded the DTH of each F2 plant as the number of days from seeding to the appearance of the first panicle. For the parental cultivars, DTH was scored in 10 plants per line and mean values were calculated for each line.
DNA marker analysis
Total DNA of individual F2 plants and parental lines was extracted from leaves by the CTAB method (Murray and Thompson 1980). Two types of DNA markers, simple sequence repeat (SSR) and single-nucleotide polymorphism (SNP) markers, were used for linkage map construction. For polymerase chain reaction (PCR) of the SSR markers, we used a 10-μl reaction volume containing 0.5 μl template DNA (20 ng μl−1), 5 μl 2× Go-Taq Green Master Mix (Promega, WI, USA) and 4.5 μl H2O. Amplification was performed for 35 cycles (1 min at 94°C, 1 min at 55°C, and 2 min at 72°C), followed by 7 min at 72°C. The amplified products were separated by electrophoresis in a 3% agarose gel to detect polymorphisms. SSR markers used in the primary F2 analysis were selected from those reported by McCouch et al. (2002). The 409 SNPs used for genotyping were selected at a spacing of around 1,000 kb from genome wide SNP data (Ebana et al. 2010) (Suppl Table S1). The SNPs were detected using the BeadStation 500G system (illumina, San Diego, USA). All experimental procedures for the SNP typing followed the manufacturer’s instructions. To obtain additional SSR markers showing polymorphism between Koshihikari and the other lines, we surveyed further genomic regions containing SSR motifs in the chromosomal regions of interest (Temnykh et al. 2000; McCouch et al. 2002; International Rice Genome Sequencing Project 2005). The resultant informative SSR markers were used for genotyping of F2 plants. Gene-specific markers for Hd1 and Hd6 were also used in the QTL analysis (Suppl Table S2).
Construction of linkage maps and QTL analyses
QTL analysis of the F2 populations was performed using genotype information based on the SSR and SNP markers. We used 86–164 SSR markers and 180–312 SNP markers distributed on the 12 rice chromosomes for the QTL analysis of each F2 population. Once a QTL was detected in a particular region of the genome, additional SSR markers were added to determine a more precise position and to increase the reliability of the estimated parameters for that putative QTL.
Linkage maps were constructed from the genotype data by using MAPMAKER/EXP 3.0 (Lander et al. 1987). The Kosambi map function was used to calculate genetic distances (Kosambi 1944). QTL analyses were performed using composite interval mapping (CIM) and interval mapping (IM) as implemented by the Zmapqtl program (model 6) provided in version 2.5 of Windows QTL Cartographer (Basten et al. 2005). Genome-wide threshold values (α = 0.05) were used to detect putative QTLs based on the results of 1,000 permutations. The additive and dominant effects and phenotypic variance explained by each QTL were estimated at the peak LOD score.
Sequence analysis
All exon regions of the Hd1, Hd3a, RFT1, and Ghd7 genes were amplified by PCR from the genomic DNA of each rice variety with unique primers, using AmpliTaq polymerase (Applied Biosystems, Foster, CA, USA) or LA Taq polymerase (TaKaRa, Kyoto, Japan) and a PTC-100 PCR machine (MJ Research, Waltham, MA, USA). Amplification was performed by using 35 cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, unless a different annealing temperature was determined (Suppl Table S3). In the case of Hd6, sequencing was conducted only for the third exon, in which a functional nucleotide polymorphism (FNP) was reported to exist within O. sativa (Yamane et al. 2009). Amplified DNA fragments were purified and sequenced with the Sanger dideoxy terminator method on capillary sequencers (Applied Biosystems, Foster City, CA, USA). To ensure that the sequence data were of high quality (phred score > 30), re-sequencing was performed when necessary. Each sequence read was individually mapped onto the Nipponbare reference coding region sequences to ensure that all exons of each gene were covered.
Results
Variations in heading date in 12 F2 populations
DTH of the 12 diverse cultivars and the common Koshihikari (KSH) parent varied from 91 (extremely early) to 191 (extremely late) under natural field conditions (Table 1). Among the accessions tested in this study, Qiu Zhao Zong (QZZ) showed the earliest heading. Four accessions, Deng Pao Zhai (DPZ), Khau Mac Kho (KMK), Bei Khe (BKH), and Naba (NAB), were relatively late heading, and two accessions, Khao Nam Jen (KNJ) and Bleiyo (BLE), showed extremely late heading (Table 1).
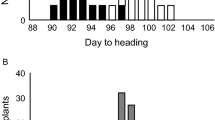
Frequency distributions of DTH for the 12 F2 populations are shown in Fig. 1. All 12 populations showed a wide range of DTH and a continuous distribution (Fig. 1). The segregation patterns were roughly classified into several categories. In the QZZ population, transgressive segregants for both early and late heading were observed (Fig. 1). In four F2 populations (TUP, MUH, DAV, and TOB), a wide range of variation was observed even though the parents of each population had very similar DTH under field conditions. In particular, the TOB F2 population showed an extreme amount of transgressive segregation toward late heading. In the BAS, DPZ, and KMK F2 populations, bimodal and transgressive segregation patterns were observed. In the BKH and NAB populations, both non-KSH parental lines showed relatively late heading, and extreme transgressive segregation toward late heading was observed in the F2 population. The most extreme transgressive segregation toward late heading was observed in the F2 population derived from BKH. In contrast, DTH of the two populations derived from parents with the longest DTH under field conditions (KNJ and BLE) were between those of the parental lines, and no transgressive segregation was observed.

Frequency distributions of days to heading (DTH) in F2 populations derived from crosses between Koshihikari and 12 diverse Asian cultivars. DTH was scored at the Experimental Field of National Institute of Agrobiological Sciences, Tsukuba, Ibaraki, Japan (36°N). Arrows show the mean DTH in the parental lines. Parental lines are denoted by the abbreviations given in Table 1
QTLs involved in natural variation for heading date
QTLs detected in the 12 F2 populations are summarized in Table 2, Fig. 2, Suppl Figs. S1 and S2. Based on the CIM analysis, in the QZZ population, QTLs were detected on chromosome 7 (one near RM3670, another near RM5720), and chromosome 8 (RM5556); at each QTL, the KSH allele increased DTH. In the TUP population, QTLs were detected on chromosomes 2 (RM3789_1), 3 (RM5801), 6 (Hd1), and 8 (RM5432). The KSH allele increased DTH at the QTL on chromosome 6 and decreased DTH at the other three QTLs. In the MUH population, DTH variation could be explained by four QTLs: two on chromosome 3 (P1220 and P1710_1), one on chromosome 6 (RM3431), and one on chromosome 7 (P0388). The KSH alleles increased DTH at the QTL on chromosome 6 and at one of the two QTLs on chromosome 3 (P1220); at the other two QTLs, the KSH alleles decreased DTH. In the DAV population, variation for DTH could be explained by QTLs on chromosomes 6 (AH06000195 and P0013) and 8 (RM5556). The KSH allele decreased DTH at the QTL on chromosome 6 near AH06000195 and increased DTH at the other two QTLs. In the TOB population, we detected QTLs on chromosomes 6 (RM3805) and 8 (RM3374). The KSH allele at the QTL on chromosome 6 decreased DTH, whereas the KSH allele for the QTL on chromosome 8 increased DTH. In the BAS population, QTLs were mapped to chromosome 3 (Hd6) and chromosome 6 (RM6836_1). The KSH allele at the QTL on chromosome 3 decreased DTH, whereas the KSH allele at the QTL on chromosome 6 increased DTH. In the DPZ population, a single QTL was detected on chromosome 8 (RM5556); at this QTL, the KSH allele increased DTH. In the KMK population, QTLs were detected on chromosomes 2 (RM450), 3 (RM3867), and 6 (RM3431A). The KSH allele at the QTLs on chromosomes 2 and 3 decreased DTH; the KSH allele at the QTL on chromosome 6 increased DTH. In the BKH population, QTLs were detected on chromosomes 6 (RM3805) and 8 (RM5556). The KSH allele at the QTL on chromosome 6 decreased DTH; the KSH allele at the QTL on chromosome 8 increased DTH. In the NAB population, QTLs were detected on chromosomes 3 (RM6970), 6 (Hd1), 7 (RM5436), and 8 (RM3791); at these loci, only the KSH allele of the QTL on chromosome 6 increased DTH. In the KNJ population, QTLs were detected on chromosome 3 (RM6970) and chromosome 6 (RM3431A); at both QTLs, the KSH allele decreased DTH. In the BLE population, QTLs were detected on chromosome 2 (RM3316), chromosome 3 (RM6970), chromosome 6 (RM3805) and chromosome7 (RM1362_2); at all four QTLs, the KSH allele decreased DTH.

Chromosomal locations of QTLs detected in 12 F2 populations. Most likely QTL positions detected in each population (Table 2) are shown by arrowheads; confidence intervals (2-LOD reduction on each side) are indicated by bars extending from the arrowheads. Cultivar abbreviations are as shown in Table 1. The direction of additive effect of the Koshihikari (KSH) allele at each QTL detected is shown by a white (earlier heading) or black (later heading) arrowhead. Physical positions of Hd6 (Takahashi et al. 2001), Hd1 (Yano et al. 2000), Hd3a (Kojima et al. 2002), RFT1 (Kojima et al. 2002), and Ghd7 (Xue et al. 2008) are shown by horizontal bars. The positions of Hd7 (Yamamoto et al. 2000), Hd9 (Lin et al. 2002), Hd16 (Matsubara et al. 2008a), Hd2 (Yamamoto et al. 1998, 2000) and Hd5 (Lin et al. 2003) are shown as boxes defined by the positions of flanking markers
Of the 34 QTLs detected across the 12 F2 populations, 19 each explained more than 15% of the phenotypic variance in the populations where they were detected, indicating that those QTLs harbor genes with major effects. All 34 QTLs mapped within eight chromosomal regions (Fig. 2) corresponding to previously identified heading-date QTLs such as Hd1 (Yano et al. 2001), Hd2 (Yamamoto et al. 1998), Hd3a/RFT1 (Kojima et al. 2002), Ghd7 (Xue et al. 2008), Hd5 (Lin et al. 2003), Hd6/Hd16 (Takahashi et al. 2001; Matsubara et al. 2008a), Hd7 (Yamamoto et al. 2000), and Hd9 (Lin et al 2002) (Fig. 2; Table 2). It should be noted that three genomic regions (those containing Hd1, Hd5, and Hd6/Hd16) were each associated with DTH variation in seven populations (Fig. 2).
To examine validity of QTL detected in the CIM analysis, we also performed IM analysis to know a potential risk of a false positive detection of the QTLs. Based on IM analysis, several QTL, which were detected in the CIM analysis, were not detected (Table 2, Suppl Fig. S2). Of 34 QTLs detected in CIM, 11 QTLs on chromosome 2, 3, 6, 7 and 8 were not detected in IM analysis. Among them, only one QTL showing a relatively a large phenotypic contribution in the CIM analysis (more than 10% PVE) was not detected in the IM analysis (Table 2);
Sequence analysis of genes for heading date
Our QTL analyses strongly suggested that several previously cloned genes for heading date could explain a large portion of the phenotypic variation in each F2 population, indicating that there should be allelic differences for gene function at these loci. Therefore, to search for functional differences in these heading date genes, we sequenced the genomic regions of candidate genes Hd1, Hd6, Hd3a, RFT1, and Ghd7, which were located near many of the QTLs detected in this study. KSH is presumed to have a functional allele for each of these genes since it contains the same sequences as are found in the Nipponbare genome (Nonoue et al. 2008).
Within Hd1, a number of mutations were observed that would be expected to cause amino acid substitutions in the Hd1 protein. In addition, single-nucleotide polymorphisms (SNP) or indels (insertion/deletion mutations) that generated a premature stop codon in the exon region were observed in DAV, BAS, KMK, MUH, NAB, TUP, and QZZ (Fig. 3). These mutations would result in a full or partial deletion of the NLS domain at the C-terminus of the Hd1 protein. The most extreme case was found in QZZ, in which the amino acid sequences within the zinc finger motif were changed due to a frameshift caused by an indel. Based on these results, these seven accessions appear to contain alleles with a loss of function at Hd1.

Substitutions (boxed) and indels (-) of amino acids observed in heading date genes Hd1, RFT1, and Ghd7. The known domains within each gene are indicated as light-colored regions with labels above them. Numbers under the gene bars indicate sites of sequence changes; numbers on the right show the total length of each predicted amino acid sequence. The regions with amino acid changes due to frameshifts are labeled with an asterisk. Stop stop codon
In the predicted amino acid sequence of Hd3a, only one amino acid substitution (near the C-terminus) could be detected in TOB, NAB, DVA, DPZ, MUH, TUP, and BLE, as compared with the KSH allele (data not shown). In the RFT1 gene, on the other hand, a number of SNPs corresponding to amino acid substitutions were observed in KMK, DAV, TOB, BKH, NAB, DPZ, MUH, and TUP (Fig. 3). It should also be noted that DAV, TOB, BKH, BLE appear to have one allele; and NAB, DPZ, MUH, and TUP appear to have another (Fig. 3).
In Ghd7, several SNPs corresponding to amino acid substitutions were found in BLE, TOB, BKH, NAB, DAV, and DPZ, as compared with the KSH allele (Fig. 3); all of these except for DPZ had the same predicted amino acid sequence. In QZZ, no PCR fragments could be amplified from any Ghd7 exon region. Based on PCR examination of the regions flanking Ghd7 by using QZZ BAC clones covering the corresponding genomic region, it appeared that the genomic segment (~40 kb) normally harboring Ghd7 was completely deleted, as reported previously for the Chinese rice variety Zhenshan 97 (Xue et al. 2008).
For Hd6, it has been previously reported that the coding sequence is highly conserved and that natural mutations have been suppressed during its evolution in the genus Oryza (Yamane et al. 2009). A non-synonymous base substitution generating a premature stop codon was found in a limited number of Japanese japonica varieties, including KSH (Takahashi et al. 2001; Matsubara et al. 2008a; Yamane et al. 2009). Sequencing a 500-bp genomic region including the non-synonymous substitution site in the 12 other cultivars used in this study demonstrated that this functional nucleotide polymorphism (FNP) was not present in any of the lines (data not shown), suggesting that all of the cultivars except for KSH carry a functional allele at the Hd6 locus.
Discussion
Heading date in rice has been intensively analyzed over the last two decades. These efforts have contributed to our understanding of the genetic and molecular control of this trait (Izawa 2007; Tsuji et al. 2008). In particular, cloning of genes involving heading date has provided a unique opportunity to compare genetic control mechanisms between an LD plant, Arabidopsis, and an SD plant, rice (Izawa et al. 2003; Hayama and Coupland 2004); these analyses demonstrated the presence of unique genes and mechanisms for heading date regulation in rice (Izawa 2007). In spite of these achievements, the genetic factors underlying the wide range of naturally occurring variation in heading date among rice cultivars were not fully understood. This is partly due to the complex diversification of genetic control of heading date in rice accessions. Moreover, different accessions have been used to detect QTLs for heading date in different studies, making it difficult to systematically compare QTLs across studies.
In this study, we conducted QTL and sequence analyses using diverse Asian rice cultivars. We selected 12 diverse lines that originated from different geographical areas and compared them with the common japonica variety KSH. These 12 cultivars exhibited a wide range of DTH under natural field conditions at Tsukuba, Japan (Table 1). QTL analyses using F2 populations from 12 different crosses (KSH by each of the 12 diverse cultivars) revealed that one to four QTLs with major effects were involved in natural variation in heading date in each population. The chromosomal locations of these major genetic factors corresponded well with previously identified QTLs: Hd1, Hd2, Hd3a/RFT1, Ghd7, Hd5, Hd6/Hd16, Hd7, and Hd9. Although it could not be ruled out that other tightly linked genes controlling DTH exist in these chromosomal regions, we suggest that the majority of natural variation in flowering time is generated by the actions of multiple alleles at the QTLs detected in this study. Furthermore, we suggest that at least some of the QTLs detected in this study are identical to the previously identified QTLs.
More than 65% of the total phenotypic variance in 10 of the 12 populations could be explained by the QTLs detected in this study. However, less than 60% of the variance in F2 populations derived from DPZ and BKH could be explained by the QTLs detected in this study. In particular, although only one QTL was detected in the DPZ population, clear transgressive segregation for late heading was observed. This transgressive segregation could not be explained by the effect of a single QTL, indicating that additional QTLs must be involved in the variation in heading date in the DPZ population.
Transgressive segregation was observed in most of populations; the exceptions were the populations derived from crosses with the latest-heading cultivars, KNJ and BLE. The transgressive segregation could be explained by the combination of KSH and other parental alleles having both positive and negative effects on DTH. In fact, both directions of effect were detected in the 10 F2 populations that showed transgressive segregation. In the KNJ and BLE F2 populations, a decrease in DTH was associated with the KSH allele for all QTLs detected in those populations. On the other hand, although an increase in DTH was associated with the KSH alleles of all three QTLs detected in QZZ, transgressive segregation was observed toward both late and early heading. This segregation pattern could not be clearly explained by the QTLs detected in the QZZ F2 population.
In two F2 populations (TOB and BKH), the same patterns of allelic differences were detected at Hd3a/RFT1 and Hd5: the TOB and BKH parents both contributed late-heading alleles at Hd3a/RFT1 and early-heading alleles at Hd5. These two populations showed a very wide range of DTH, and extremely late transgressive segregants were observed. However, variation in DTH was different between the two populations: BKH showed late heading compared with that of TOB, and the distribution of DTH was shifted toward late heading in the BKH population compared with that of TOB. This observation may imply that additional unknown late-heading factors in BKH or early-heading alleles in TOB might be involved in the variation observed in each population.
In the DAV and TOB F2 populations, two of the same QTLs, Hd3a/RFT1 and Hd5, were detected. The additive effects of RFT1 were almost same between DAV and TOB, and both accessions harbored an early allele at Hd5, although different magnitudes of additive effects for Hd5 were observed. The main difference detected between the cultivars was that the DAV population contained an early-heading allele of Hd1 (contributed by DAV), whereas no QTL was detected near Hd1 in the TOB population.
In a comparison between KNJ and BLE, both F2 populations showed a similar pattern of non-transgressive segregation. However, the only QTL in common between the populations was that detected near Hd6/Hd16. These results indicated that the strong daylength response in each cultivar was controlled by a different combination of alleles at QTLs for heading date.
For QZZ, extremely early-heading plants were observed in the F2 population. This segregation pattern was very similar to that of an F2 population derived from a cross between Hayamasari and Kasalath (Nonoue et al. 2008). The same two QTLs involved in early heading were detected in both analyses. It has also been reported that allelic variation in the Hd2 region on the distal end of the long arm of chromosome 7 could be involved in extremely early heading in rice (Fujino and Sekiguchi 2005; Gu and Foley 2007). In our study, a QTL near Hd2 was detected in the QZZ population, with the QZZ allele contributing to early heading (Table 2; Fig. 2). Therefore, we conclude that Ghd7, Hd2, and Hd5 play important roles in the genetics of extremely early heading.
The Hd6 QTL is located in the distal end of the long arm of chromosome 3. KSH possesses a loss-of-function allele at Hd6 (Matsubara et al. 2008a). Recently, a new QTL, Hd16, was also detected near Hd6. In this region, it was difficult to distinguish between Hd6 and Hd16 due to their close linkage. However, two LOD peaks were detected in BAS and KNJ, suggesting that both loci might contribute to the variation for heading date. This assumption will need to be tested by performing high-resolution mapping of this region.
In QTL analysis, QTLs are detected based on the effects of different alleles on the target traits being measured. At each QTL detected in this study, we could compare the effects of alleles from different cultivars by using the allele of KSH, a common parent, as a standard. For example, KSH has a functional allele of Hd1 that is the same as that found in Nipponbare, with a 36-bp deletion in the exon (Yano et al. 2000; Nonoue et al 2008). Depending on whether the effect of the KSH allele was positive or negative in a given population, one could predict whether the Hd1 allele in the other parent would have reduced or elevated function, respectively. Based on this assumption, we speculated that TUP, MUH, DAV, BAS, KMK, and NAB would possess a reduced-function or loss-of-function allele at Hd1, and that KNJ would possess an Hd1 allele with an enhanced level of function. Sequence analysis of Hd1 strongly supports the above assumption. A premature stop codon was detected in the Hd1 alleles of TUP, MUH, DAV, BAS, KMK, and NAB, indicating that these are very likely to be loss-of-function alleles. On the other hand, it is difficult to explain how the amino acid changes seen in the KNJ allele would lead to a gain-of-function compared with the KSH allele.
Xue et al. (2008) identified a repressor of heading, Ghd7: loss of Ghd7 function results in early heading. In QZZ, we detected a large deletion that appeared to include the entire Ghd7 sequence. The allelic effect of the KSH allele at the QTL near Ghd7 was positive in the QTL analysis of the QZZ F2 population (Table 2), consistent with loss of function of Ghd7 in QZZ.
Recently, Hagiwara et al. (2009) reported that an amino acid change (E105 K) in the coding region of RFT1 was involved in a significant loss of function in some Asian cultivated rice and its wild relatives, resulting in late heading. In the present study, QTLs with a large effect were detected in the region of Hd3a/RFT1 in four populations (DAV, TOB, BKH, and BLE). In each case, the KSH allele decreased DTH, suggesting that the function of RFT1 in the four diverse parents might have been decreased or lost. We could not detect any clear polymorphism affecting the function of Hd3a (data not shown), whereas functional polymorphism (loss-of-function mutation E105 K) was detected in the predicted RFT1 protein sequence in DAV, TOB, BKH, and BLE. These results clearly suggest that RFT1 may have a significant role in generating natural variation for heading date.
For Hd2 and Hd5, additive effects of the KSH alleles were seen in both directions (i.e., toward either increased or decreased DTH), depending on the F2 population. As mentioned above, this range of additive effects must reflect the functional status of gene(s) located within the QTLs. Unfortunately, neither Hd2 nor Hd5 have been cloned yet. The functional status of these genes will need to be tested by the cloning and sequencing of these two important QTLs for heading date.
Based on a previous sequence analysis, KSH has a non-functional allele at Hd6, which can be identified by a SNP (Matsubara et al. 2008a). In this study, it was revealed that none of the 12 other cultivars had this SNP at Hd6 (data not shown), suggesting that all 12 cultivars may have a functional allele at Hd6. Based on this sequence polymorphism, we expected to detect a QTL near Hd6 in all of the populations. However, no QTL was detected in the Hd6 region of QZZ, DAV, TOB, DPZ, or BKH (Fig. 2; Table 2). It is interesting to note that a KSH allele at Hd5 with a large positive effect on DTH was detected in all populations in which no QTL was detected in the Hd6 region. This indicates that genetic interaction between Hd6/Hd16 and Hd5 is likely to be involved in the expression of heading date. Alternatively, segregation of a large phenotypic effect of Hd5 may mask the action of Hd6.
Temperature and rainfall are major determinants of rice growing season, and DTH is important characteristic of rice cultivars. Natural or artificial selection of DTH has affected not only specific genes but also combinations of genes/QTLs. On the other hand, rice is an autogamous (self-fertilizing) crop in which natural crossing events among cultivars are extremely rare. Allele combinations at different QTLs that determine DTH and daylength response are expected to be characteristic of specific cultivar groups. The 12 cultivars used in this study have been classified into three cultivar groups, A, B, and C, which correspond to japonica, Aus, and indica, respectively (Table 1) (Kojima et al. 2005). The japonica cultivars studied here were KMK, KNJ, and KSH. The difference in DTH of KMK and KNJ is largely explained by the effect of the QTL near Hd1, with an early allele in KMK and a late allele in KNJ, and may reflect the needs of their respective growing environments. KMK is grown in mountainous areas of Vietnam, which may require photoperiod insensitive rice adapted to a relatively short growth period. On the other hand, KNJ seems to be grown in plains areas of tropical regions and a long growth period allows to raise high photoperiod sensitive rice. Three cultivars (TUP, MUH, and BAS) classified into group B by Kojima et al. (2005), exhibited relatively early heading among the cultivars in this study. This result is consistent with the typical Aus characteristics of photoperiod insensitivity and short growing period. The F2 populations derived from the three group B cultivars showed similar segregation patterns for DTH; the QTL and sequence analyses suggested that these patterns were mainly determined by allelic differences in Hd1 and Hd6/Hd16. On the other hand, although the seven cultivars classified into group C (QZZ, DAV, TOB, DPZ, BKH, NAB, KMK, KNJ, and BLE) showed a wide range of DTH under the natural field conditions (Table 1), less polymorphism was detected in Hd1 and Ghd7 in group C (indica) than within groups A or B. In addition, based on the QTL analysis, allelic differences in RFT1 and Hd5 were more likely to be involved in generation of natural variation in DTH among cultivars in group C than among the other two groups. Garris et al. (2005) suggested that cultivar groups Aus and Aman, other cultivar’s group in indica, had narrow and wide genetic diversity, respectively, based on SSR analysis. A similar result was observed in cultivar groups B and C (which correspond to Aus and Aman) based on their RFLPs (Kojima et al. 2005). Based on results in this study, it is likely that there is a relationship between allele combinations for the QTLs and cultivar groups or geographical distribution. However, the limited number of accessions in this study representing each cultivar group and geographical origin did not allow us to draw a clear conclusion. Future studies with more rice accessions will be needed to allow us to clearly establish the relationships between cultivar groups and genetic variation in QTLs for heading date.
Takahashi et al. (2009) demonstrated that variations in Hd1 proteins, Hd3a promoters, and Ehd1 expression levels largely contributed to the diversity of flowering time in cultivated rice. In this study, we confirmed that Hd1 could be one of the major factors determining heading date in Asian cultivars; moreover, RFT1, rather than Hd3a, contributed to heading date diversity in the rice cultivars included in this study. Furthermore, Ghd7, Hd5, and Hd6/Hd16 appeared to be important genetic factors for the determination of heading date in these rice cultivars. It should be noted that we did not detect any QTLs in the region of Ehd1 (chromosome 10) in this study. This finding implies that, despite its important role for the promotion of flowering (Doi et al. 2004; Xue et al. 2008, Matsubara et al. 2008b; Park et al. 2008), the Ehd1 gene does not contribute very much to heading date diversity in rice. Presumably, several of the QTLs detected in this study, such as Ghd7, Hd2 and Hd5, might modulate the expression of Ehd1, and consequently modify expression of florigen genes Hd3a and RFT1 (Xue et al. 2008).
Taking together the allelic differences found in the QTL analyses and the sequence variation at genes for heading date, our data strongly suggest that a large portion of the wide range of phenotypic variation in heading date and daylength response could be generated by combinations of different alleles, possibly both loss and gain of function, of QTLs Hd1, Hd2, RFT1, Ghd7, Hd5, and Hd6/Hd16. In general, it is difficult to detect all QTLs, particularly QTLs with minor effects, in primary mapping populations such as F2 populations (Ebitani et al. 2005; Uga et al. 2007; Takai et al. 2007; Ando et al. 2008). Although we successfully detected several major QTLs by using F2 populations, it is very likely that some QTLs with minor effects might be also involved in the phenotypic variation seen in these F2 populations. On the other hand, we also need to verify the effect of QTLs with relatively minor effects, which were not detected in the IM analysis. Therefore, it will be necessary to search again for heading date QTLs by using advanced progeny. In addition, DTH in the F2 plants was scored only under a single environment, natural field conditions at Tsukuba, Japan. It is possible that additional novel QTLs might be involved in differences in DTH under diverse environmental conditions, such as temperature and daylength, including artificial SD and LD conditions. Currently, we are developing populations of chromosome segment substitution lines (CSSLs) for 10 of the accessions (Fukuoka et al. 2010). It would be interesting to discover additional QTLs with minor or epistatic effects under diverse environmental conditions using these CSSLs, resulting in understanding more comprehensively the genetic factors that induce natural variation in heading date of Asian cultivated rice.
References
Ando T, Yamamoto T, Shimizu T, Ma XF, Shomura A et al (2008) Genetic dissection and pyramiding of quantitative traits for panicle architecture by using chromosomal segment substitution lines in rice. Theor Appl Genet 116:881–890
Basten CJ, Weir BS, Zeng ZB (2005) QTL Cartographer, version 1.17. Department of Statistics, North Carolina State University, Raleigh, p 189
Doi K, Izawa T, Fuse T, Yamanouchi U, Kubo T et al (2004) Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Genes Dev 18:926–936
Ebana K, Yonemaru J, Fukuoka S, Iwata H, Kanamori H, Namiki N, Nagasaki H, Masahiro Y (2010) Genetic structure revealed by a whole-genome single-nucleotide polymorphism survey of diverse accessions of cultivated Asian rice (Oryza sativa L.). Breed Sci 60:390–397
Ebitani T, Takeuchi Y, Nonoue Y, Yamamoto T, Takeuchi Y et al (2005) Construction and evaluation of chromosome segment substitution lines carrying overlapping chromosome segments of indica rice cultivar ‘Kasalath’ in a genetic background of japonica elite cultivar ‘Koshihikari’. Breed Sci 55:65–73
Fujino K, Sekiguchi H (2005) Identification of QTLs conferring genetic variation for heading date among rice varieties at the northern-limit of rice cultivation. Breed Sci 55:141–146
Fukuoka S, Nonoue Y, Yano M (2010) Germplasm enhancement by developing advanced plant materials from diverse rice accessions. Breed Sci 60:509–517
Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch SR (2005) Genetic structure and diversity in Oryza sativa L. Genetics 169:1631–1638
Gu XY, Foley ME (2007) Epistatic interactions of three loci regulate flowering time under short and long daylengths in a backcross population of rice. Theor Appl Genet 114:745–754
Hagiwara WE, Uwatoko N, Sasaki A, Matsubara K, Nagano H et al (2009) Diversification in flowering time due to tandem FT-like gene duplication, generating novel Mendelian factors in wild and cultivated rice. Mol Ecol 18:1537–1549
Hayama R, Coupland G (2004) The molecular basis of diversity in the photoperiodic flowering responses of Arabidopsis and rice. Plant Physiol 135:677–684
Hittalmani S, Huang N, Courtois B, Venuprasad R, Shashidhar HE et al (2003) Identification of QTL for growth- and grain yield-related traits in rice across nine locations of Asia. Theor Appl Genet 107:679–690
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Izawa T (2007) Adaptation of flowering-time by natural and artificial selection in Arabidopsis and rice. J Exp Bot 58:3091–3097
Izawa T, Takahashi Y, Yano M (2003) Comparative biology comes into bloom: genomic and genetic comparison of flowering pathways in rice and Arabidopsis. Curr Opin Plant Biol 6:113–120
Kojima S, Takahashi Y, Kobayashi Y, Monna L, Sasaki T et al (2002) Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol 43:1096–1105
Kojima Y, Ebana K, Fukuoka S, Nagamine T, Kawase M (2005) Development of an RFLP-based rice diversity research set of germplasm. Breed Sci 55:431–440
Komiya R, Yokoi S, Shimamoto K (2008) Hd3a and RFT1 are essential for flowering in rice. Development 135:767–774
Komiya R, Yokoi S, Shimamoto K (2009) A gene network for long-day flowering activates RFT1 encoding a mobile flowering signal in rice. Development 136:3443–3450
Kosambi DD (1944) The estimation of map distance from recombination values. Ann Eugen 12:172–175
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ et al (1987) Mapmaker: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Lin HX, Ashikari M, Yamanouchi U, Sasaki T, Yano M (2002) Identification and characterization of a quantitative trait locus, Hd9, controlling heading date in rice. Breed Sci 52:35–41
Lin HX, Liang ZW, Sasaki T, Yano M (2003) Fine mapping and characterization of quantitative trait loci Hd4 and Hd5 controlling heading date in rice. Breed Sci 53:51–59
Maas LF, McClung A, McCouch SR (2010) Dissection of a QTL reveals an adaptive, interacting gene complex associated with transgressive variation for flowering time in rice. Theor Appl Genet 120:875–1071
Matsubara K, Kono I, Hori K, Nonoue Y, Ono N et al (2008a) Novel QTLs for photoperiodic flowering revealed by using reciprocal backcross inbred lines from crosses between japonica rice cultivars. Theor Appl Genet 117:935–945
Matsubara K, Yamanouchi U, Wang ZX, Minobe Y, Izawa T et al (2008b) Ehd2, a rice ortholog of the maize INDETERMINATE1 gene, promotes flowering by up-regulating Ehd1. Plant Physiol 148:1425–1435
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K et al (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucl Acids Res 8:4321–4325
Nonoue Y, Fujino K, Hirayama Y, Yamanouchi U, Lin SY et al (2008) Detection of quantitative trait loci controlling extremely early heading in rice. Theor Appl Genet 116:715–722
Park SJ, Kim SL, Lee S, Je BI, Piao HL et al (2008) Rice Indeterminate 1 (OsId1) is necessary for the expression of Ehd1 (Early heading date 1) regardless of photoperiod. Plant J 56:1018–1029
Takahashi Y, Shomura A, Sasaki T, Yano M (2001) Hd6, a rice quantitative trait locus involved in photoperiod sensitivity, encodes the α subunit of protein kinase CK2. Proc Natl Acad Sci USA 98:7922–7927
Takahashi Y, Teshima KM, Yokoi S, Innan H, Shimamoto K (2009) Variations in Hd1 proteins, Hd3a promoters, and Ehd1 expression levels contribute to diversity of flowering time in cultivated rice. Proc Natl Acad Sci USA 106:4555–4560
Takai T, Nonoue Y, Yamamoto S, Yamanouchi U, Matsubara K et al (2007) Development of chromosome segment substitution lines derived from backcross between indica donor rice cultivar ‘Nona Bokra’ and japonica recipient cultivar ‘Koshihikari’. Breed Sci 57:257–261
Tamaki S, Matsuo S, Wong HL, Yokoi S, Shimamoto K (2007) Hd3a protein is a mobile flowering signal in rice. Science 316:1033–1036
Temnykh S, Park WD, Ayres N, Cartinhour S, Hauck N et al (2000) Mapping and genome organization of microsatellite sequences in rice (Oryza sativa L.). Theor Appl Genet 100:697–712
Tsuji H, Tamaki S, Komiya R, Shimamoto K (2008) Florigen and the photoperiodic control of flowering in rice. Rice 1:25–35
Uga Y, Nonoue Y, Liang ZW, Lin HX, Yamamoto S et al (2007) Accumulation of additive effects generates a strong photoperiod sensitivity in the extremely late-heading rice cultivar ‘Nona Bokra’. Theor Appl Genet 114:1457–1466
Xue W, Xing Y, Weng X, Zhao Y, Tang W et al (2008) Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet 40:761–767
Yamamoto T, Kuboki Y, Lin SY, Sasaki T, Yano M (1998) Fine mapping of quantitative trait loci, Hd-1, Hd-2 and Hd-3, controlling heading date of rice, as single Mendelian factors. Theor Appl Genet 97:37–44
Yamamoto T, Lin HX, Sasaki T, Yano M (2000) Identification of heading date quantitative trait locus Hd6 and characterization of its epistatic interactions with Hd2 in rice using advanced backcross progeny. Genetics 154:885–891
Yamamoto T, Yonemaru J, Yano M (2009) Towards the understanding of complex traits in rice: substantially or superficially? DNA Res 16:141–154
Yamane H, Ito T, Ishikubo H, Fujisawa M, Yamagata H et al (2009) Molecular and evolutionary analysis of the Hd6 photoperiod sensitivity gene within Genus Oryza. Rice 2:56–66
Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L et al (2000) Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12:2473–2483
Yano M, Kojima S, Takahashi Y, Lin HX, Sasaki T (2001) Genetic control of flowering time in rice, a short-day plant. Plant Physiol 127:1425–1429
Yonemaru J, Yamamoto T, Fukuoka S, Uga Y, Hori K et al (2010) Q-TARO: QTL annotation rice online database. Rice 3:194–203
Acknowledgments
We thank to the Technical Support Section of NIAS for the management of rice field. This work was supported by grants from the Ministry of Agriculture, Forestry and Fisheries of Japan (Integrated Research Project for Plant, Insect and Animal using Genome Technology QT-1005 and Genomics for Agricultural Innovation NVR-0001 to M. Yano, and Genomics for Agricultural Innovation QTL-5003 to J. Wu).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by E. Carbonell.
K. Ebana and T. Shibaya contributed equally to this work.
Nucleotide sequences from 12 accession are deposited in DNA Data Bank of Japan as Hd1 (AB564428–AB564439), Hd3a (AB564440–AB564451), RFT1 (AB564452–AB564463) and Ghd7 (AB564464–AB564474).
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2010_1524_MOESM4_ESM.ppt
Suppl Fig. S1. Log-likelihood (LOD) and additive-effect plots (upper figures) and linkage map (lower figures) from the QTL analyses of the 12 F2 populations. Each population is indicated by the abbreviation of its non-KSH parental line. The upper figure for each population is composed of the LOD plot (top) and an estimated additive-effect plot (bottom) across the 12 rice chromosomes. The LOD threshold used to declare putative QTLs in each population is indicated by the horizontal line in each upper figure. The name of the nearest marker is shown next to each LOD peak. Putative QTL locations are indicated by black blocks in the linkage maps (bottom part of each figure). (PPT 791 kb)
122_2010_1524_MOESM5_ESM.ppt
Suppl Fig. S2. Log-likelihood (LOD) (upper figure) and additive-effect plots (lower figures) in interval mapping (IM function) from the QTL analyses of the 12 F2 populations. Each population is indicated by the abbreviation of its non-KSH parental line. The LOD threshold used to declare putative QTLs in each population is indicated by the horizontal line in each upper figure. The name of the nearest marker is shown next to each LOD peak. Black arrow indicates position of putative QTL detected in CIM analysis, but not in IM analysis. (PPT 379 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ebana, K., Shibaya, T., Wu, J. et al. Uncovering of major genetic factors generating naturally occurring variation in heading date among Asian rice cultivars. Theor Appl Genet 122, 1199–1210 (2011). https://doi.org/10.1007/s00122-010-1524-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1524-1