
Abstract
Heading date determines rice’s adaptation to its area and cropping season. We analyzed the molecular evolution of the Hd6 quantitative trait locus for photoperiod sensitivity in a total of 20 cultivated varieties and wild rice species and found 74 polymorphic sites within its coding region (1,002 bp), of which five were nonsynonymous substitutions. Thus, natural mutations and modifications of the coding region of Hd6 within the genus Oryza have been suppressed during its evolution; this is supported by low Ka (≤0.003) and Ka/Ks (≤0.576) values between species, indicating purifying selection for a protein-coding gene. A nonsynonymous substitution detected in the japonica variety “Nipponbare” (a premature stop codon and nonfunctional allele) was found within only some local Japanese japonica varieties, which suggests that this point mutation happened recently, probably after the introduction of Chinese rice to Japan, and is likely involved in rice adaptation to high latitudes. Phylogenetic analysis and genome divergence using the entire Hd6 genomic region confirmed the current taxonomic sections of Oryza and supported the hypothesis of independent domestication of indica and japonica rice.
Similar content being viewed by others

Introduction
Asian cultivated rice, Oryza sativa, is thought to have originated from the Asian wild rice Oryza rufipogon only about 10,000 years ago, although the place and time of its domestication are still debated [8, 18, 22, 28]. All wild Oryza species are distributed in the tropics and subtropics, whereas O. sativa has spread worldwide between 53°N and 40°S, and this wide range must have resulted from adaptive selection following its domestication [26]. Oryza comprises two cultivated and 22 wild species, with ten distinct genome types (AA, BB, BBCC, CC, CCDD, EE, FF, GG, HHJJ, and HHKK), which provide a good system for analyzing the mechanisms of evolution, speciation, and domestication by means of comparative genomics [1, 11]. Two major factors associated with climate change across the range of rice cultivation conditions (day length and temperature) are believed to have played a critical role in the spread of rice cultivation based on adaptation of the species to different areas and cropping seasons. Cultivated rice, known as a short-day plant in its original provenance in low-latitude areas, differentiates its panicle largely based on its photoperiod sensitivity. Heading of rice is promoted by short days and delayed by long days. In contrast with rice grown in the tropics and subtropics under warm conditions, rice grown at higher latitudes must respond to long-day conditions for its flowering during the summer because low autumn temperatures could greatly affect its pollen fertility. This indicates that early heading and weak or absent photoperiod sensitivity are essential for rice cultivars to be able to adapt to the climate changes that occur in northern areas of China and Japan. It is thereafter both agriculturally important and scientifically interesting to understand the control of genes involved in the flowering time of rice and its underlying evolutionary mechanisms.
O. sativa contains two traditional subspecies: indica and japonica. Varieties of indica rice are found throughout the tropical regions of Asia and are primarily grown under lowland conditions, whereas japonica varieties have differentiated into tropical japonica, distributed primarily in upland tropical regions, and temperate japonica, a recently derived group that is cultivated in temperate regions [27]. On the other hand, a current analysis of genetic distance and population structure based on DNA markers divided O. sativa into five variety groups: indica, aus, aromatic, temperate japonica, and tropical japonica [9]. Many genetic studies using different rice varieties have demonstrated the presence of significant variation in heading date within O. sativa populations and among variety groups. Recently, advanced quantitative trait loci (QTL) mapping and genomic sequencing technologies have revealed five genes involved in the flowering time of rice (Se5, Hd1, Hd3a, Hd6, and Ehd1), and these genes have been cloned at the molecular level [6, 15, 19, 33, 40].
To better understand the molecular and evolutionary mechanisms of functional rice genes related to speciation, domestication, and adaptation, we have established a system to conduct comparative genomics among the Oryza species and varieties. The present paper reports the results we have obtained using this system by sequencing and comparing the rice Hd6 gene from a total number of 20 cultivated varieties and wild rice species. Hd6 is a QTL related to heading date. It was detected on rice chromosome 3 in an analysis of advanced backcross progeny between the early-heading japonica variety “Nipponbare” and the late-heading indica variety “Kasalath” [39]. By means of map-based cloning and genetic complementation analysis, Hd6 was finally identified as a gene that encodes the α subunit of protein kinase CK2 (CK2α), in which the Kasalath allele functions to increase days-to-heading, whereas the Nipponbare allele is nonfunctional because it contains a premature stop codon caused by a nonsynonymous substitution in its coding region [33]. Our study demonstrated clearly that the coding region of Hd6 had an extremely high degree of sequence conservation within genus Oryza and that a nonsynonymous substitution of nucleotides in Nipponbare took place very recently, suggesting its possible role in the adaptation of rice cultivars to high latitudes.
Results
Sequencing the orthologous regions of Hd6 using BAC resources
We constructed eight new BAC libraries from the cultivated rice species O. sativa (indica and japonica subspecies) and Oryza glaberrima as well as from the wild rice species O. rufipogon, Oryza barthii, Oryza glumaepatula, Oryza meridionalis, and Oryza longistaminata, all of which had the AA genome (Table 1). Each of these BAC libraries contained more than 20,000 clones and covered their corresponding genomes by at least five times. Together with the Arizona Genomics Institute (AGI) BAC libraries, we were able to use a total of 18 cultivated varieties and wild rice species, 11 with an AA genome and seven with BB to GG genomes, for sequencing the Hd6 region.
Through Southern hybridization of BAC filters with the PCR product (156-bp) amplified from exon 3 of Hd6, candidate BAC clones were successfully screened from each BAC library. After confirming the chromosomal position by means of in silico mapping of end sequences to the public Nipponbare genome sequence, we selected one positive BAC clone that covered the Hd6 region from each variety or species for the construction of shotgun libraries and the selection of positive subclones through Southern hybridization. In the case of the tetraploid species Oryza minuta, we were able to choose two individual BAC clones (OM_Ba0079N19 and OM_Ba0144F13) for analysis because they revealed a length polymorphism of the intron between exons 9 and 10 that was amplified by PCR. By using 48 selected positive subclones for shotgun sequencing, assembly, and finishing, we successfully obtained complete genomic sequences from all varieties and species that ranged from 7,554 to 13,359 bp in length and covered each Hd6 region, including its two flanking sequences (Table 1). All sequences obtained from the different varieties and wild rice species in this study have been submitted to the DNA Data Bank of Japan. Together with the public sequences (7,288 bp) of Nipponbare and “93–11”, we were able to perform a comparative analysis of the orthologous sequences within the Hd6 region among a total number of 20 cultivated varieties and wild rice species.
Nucleotide variations in the entire region of Hd6
Nucleotide variations, including insertions and deletions (indels) and single-nucleotide polymorphisms (SNPs), occurred much more frequently in the two flanking regions and the introns but much less frequently in the exon regions (Fig. 1a). Entire regions (from the start to stop codons) of Hd6 annotated from the genomic sequences of all examined species revealed lengths ranging from 5,119 to 6,741 bp (Table 2). Because no indels of nucleotides were detected within the ten exons, the coding regions of Hd6 had an identical sequence length of 1,002 bp (including a 3-bp stop codon), which corresponds to 333 amino acids. Using the functional Kasalath Hd6 sequence as a reference, we found only seven independent SNPs within the exon regions of the AA genomes: one in Nipponbare, one in O. meridionalis, two in O. glumaepatula, and three in O. longistaminata (Fig. 1b, Table 2). Two of these seven SNPs that were detected in exon 3 of Nipponbare and O. glumaepatula were nonsynonymous substitutions. In particular, the nonsynonymous substitution in Nipponbare Hd6 resulted in a change of the predicted amino acid lysine (A271AG) to a premature stop codon (TAG) that made its mRNA encode a truncated protein with only 90 amino acids, as reported previously [33]. The nonsynonymous substitution in O. glumaepatula Hd6 resulted in a change of the predicted amino acid from leucine (C277TT) to isoleucine (ATT). The number of polymorphic nucleotide sites increased in the BB to GG genomes, with a maximum of 31 SNPs between the Kasalath and Oryza brachyantha sequences (Table 2). Surprisingly, only three nonsynonymous substitutions were observed within these SNPs: one in Oryza officinalis (proline TT949C→serine TCC), one in Oryza australiensis (alanine GGC944→glycine GGG), and one in O. brachyantha (alanine G751CT→serine TCT). Two sequences of Hd6 derived from the different genome type of tetraploid O. minuta revealed nucleotide variations at two sites, but both were synonymous substitutions. Consequently, except for the Nipponbare variety of O. sativa, O. glumaepatula, O. officinalis, O. australiensis, and O. brachyantha, all of the cultivated and wild rice species that we analyzed had a 100% identity in the amino acid sequence of Hd6, which reflects extremely high conservation of the gene composition within genus Oryza.

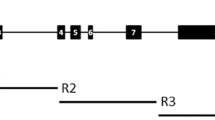
Analysis of sequence similarity within the Hd6 region of the studied species. a Conservation plot for nucleotides of the 20 cultivated varieties and wild rice species. The vertical axis represents the proportion (percent) of conserved nucleotides at each site (i.e., the proportion that are identical to the consensus sequence), and the values were calculated using a window size of 100 bp with a sliding 10-bp interval. In the diagram of Hd6 above the plot, the black boxes represent exons 1 to 10 from the 5′ (left) to the 3′ (right) end. b Distribution of SNPs between species (vertical white bars) detected within the coding region of Hd6 (using Kasalath as the reference sequence). Arrows indicate nonsynonymous substitution sites. Boxes above the graph represent conserved subdomains of CK2α numbered I to XI from the 5′ (left) to the 3′ (right) end of its nucleotide sequence. 1 O. sativa cv. Kasalath, 2 O. sativa cv. Shuusoushu, 3 O. sativa cv. 93–11, 4 O. sativa cv. Nipponbare, 5 O. sativa cv. Khau Mac Kho, 6 O. rufipogon W0106, 7 O. rufipogon W0574, 8 O. rufipogon W1943, 9 O. glaberrima, 10 O. barthii, 11 O. glumaepatula, 12 O. meridionalis, 13 O. longistaminata, 14 O. punctata, 15 O. minuta (OM_Ba0079N19), 16 O. minuta (OM_Ba0144F13), 17 O. officinalis, 18 O. alta, 19 O. australiensis, 20 O. brachyantha, 21 O. granulata.
Due to the presence of indels between varieties or species, the total number of genomic sequences within intron regions of each Hd6 ranged from 4,117 to 5,739 bp (Fig. 1a, Table 2). Within the AA-genome species, Kasalath and two accessions of O. rufipogon (W0106, W0574) contained an identical 4,176-bp length for their intron regions, and only one SNP was detected between the sequences of Kasalath and W0106 (Fig. 2a). Four other Asian cultivated rice varieties (“Shuusoushu”, 93–11, Nipponbare, and “Khau Mac Kho”) and one accession of O. rufipogon (W1943) had the same 4,174-bp intron length and showed five to 16 SNPs compared with the Kasalath sequence. Although the total length of introns within the Hd6 region did not vary greatly, O. meridionalis and O. longistaminata had more indels and SNPs than the other AA species compared with the Kasalath sequence. Among the other species, Oryza alta had the longest intron region (5,739 bp) due to a specific insertion of an approximately 1-kbp segment found within the first intron region (Fig. 1a). Other species with intron regions outstandingly longer than that of Kasalath included O. australiensis, O. brachyantha, and Oryza granulata; of these, O. brachyantha and O. granulata also revealed the larger number of SNPs to the Kasalath sequence.

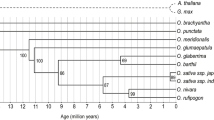
Comparison of the genomic sequences and phylogenetic relationships among the different species in the Hd6 region. a Sequence variations observed within the AA-genome species using Kasalath as a reference sequence. b A phylogenetic tree constructed using the aligned sequences from the entire gene region of all species. Only bootstrap percentage above 50% is shown. The scale bar indicates the genetic distance.
Nucleotide diversity of Hd6 within cultivated rice and its wild relatives
We found only five SNPs that led to a change in amino acid for all of the species (i.e., nonsynonymous polymorphisms), as mentioned above (Fig. 1b, Table 2). One of these SNPs, found in the exon 3 region of Nipponbare Hd6, resulted in a premature stop codon (from AAG to TAG) and has been shown to be involved in the genetic control of flowering time in rice [33]. We investigated the frequency of this point mutation within the two populations of O. sativa and O. rufipogon by using the world collections of rice varieties and wild rice accessions (Supplementary Table 1). Through direct PCR sequencing of amplified fragments (398-bp length) flanking the target site within these two collections, all (60 indica and japonica varieties from O. sativa and 19 accessions from O. rufipogon) contained the same type of nucleotide as in Kasalath. We further examined the point mutation within 51 local varieties collected in Japan and found that five local japonica varieties (“Koshihikari”, “Ginbouzu”, “Morita Wase”, “Ishijiro”, and “Himenomochi”) had the same mutation as that of Nipponbare.
Nucleotide diversity for the entire regions of Hd6 in five O. sativa varieties and three O. rufipogon accessions was detected by using the program DnaSP that showed π values of 0.0018 and 0.0017, respectively. In particular, a significantly low π value of 0.0004 was calculated from the exon regions of above samples. DnaSP analysis using the coding sequences revealed, moreover, a very low value (≤0.003) for the Ka (the rate of nonsynonymous substitutions) and a value ≤0.576 for the ratio Ka/Ks (the rate of synonymous substitutions) between the different varieties or species (Supplementary Table 2). In order to confirm the low nucleotide diversity of Hd6 gene within or between the two populations of O. sativa and O. rufipogon, additional analysis was performed using the above 398 bp (122-bp in exon 2, 80-bp in intron between the exon 2 and exon 3, 195-bp in exon 3) sequences from the world collections of rice varieties and wild rice accessions (Supplementary Table 1). As an expected result, similar and extremely low π values of 0.00016 and 0.00024 were achieved within the populations of O. sativa and its wild relatives of O. rufipogon (Supplementary Table 3).
Phylogenetic trees
Based on the nucleotide polymorphisms observed between each Hd6 region, we examined the phylogenetic relationships among the varieties and species. When only the coding regions were used for this analysis (a total of 74 polymorphic sites within the 1,002 bp of aligned sequences), all cultivated and wild rice containing the AA genome (except for O. longistaminata) were included in a single cluster (bootstrap percentage <50%) because of the extremely low nucleotide divergence, with only four variable sites (Table 2, Supplementary Fig. 1). Within the BB to GG genomes, Oryza punctata, O. minuta (two distinct sequences), and O. officinalis formed a single monophyletic clade close to that of the AA species. Below this were two monophyletic clades, one containing O. alta and O. australiensis and another containing O. brachyantha and O. granulata. We thus used the entire region of Hd6 (5′UTR, exons, introns, and 3′UTR), which had 1,285 polymorphic sites within a total of 4,507 bp of aligned sequences after removing indels, to construct the phylogenetic tree. In this tree, the cultivated indica and japonica varieties of O. sativa were separated into two clades (with a bootstrap percentage of 100%) that included different accessions or species of wild rice with an AA genome, although the genetic distance between the two clades was quite short (Fig. 2b). As an unexpected result, one indica variety (93–11) whose sequence was downloaded from the public database assembled from whole-genome shotgun sequences was included in the clade containing two japonica varieties and one wild rice O. rufipogon accession (W1943). Within the BB to GG genomes, the significance of the phylogenetic relationships (bootstrap percentage of 100%) between the species was recognized, except for O. alta (CCDD genome) and O. australiensis (EE genome), which formed a monophyletic clade, suggesting the O. alta sequence came from the DD genome. Obviously, two distinct sequences from the different BAC clones of O. minuta represented its different genomes: OM_Ba0079N19 from the BB genome and OM_Ba0144F13 from the CC genome.
Discussion
Comparative analysis of orthologous gene sequences from multiple species can provide insights into the molecular evolution of their genes. It is particularly interesting and important to perform such analyses for functional genes related to the speciation, domestication, and adaptation of crops. In this study, we used enriched BAC library resources to sequence and compare the nucleotide variations within the genomic region of Hd6, a rice photoperiod sensitivity gene, in a total of 20 cultivated varieties and wild rice species. The results should provide important opportunities to understand the evolutionary and adaptive mechanisms of the rice genes involved in the genetic control of flowering time.
Enrichment of BAC resources for use in rice comparative genomics
A total of 12 deep-coverage BAC libraries representing ten genome types in genus Oryza have been constructed by AGI [2]. Within these libraries, however, only two species (O. rufipogon and O. glaberrima) have the AA genome. The wild rice species with an AA genome are most closely related to the cultivars in genus Oryza that constitute the primary gene pool that is considered useful for crop improvement. Based on the importance of both the molecular evolution and the functional genomics of rice, we set out to enrich the BAC resources by generating eight new libraries from the AA-genome species (Table 1). Positive BAC clones were successfully selected from each species, although the genome coverage of their BAC libraries is not as high as that of the AGI libraries. It is worth noting that three cultivated varieties (Shuusoushu, Kasalath, and Khau Mac Kho) correspond to the indica, aus, and tropical japonica groups previously revealed by the analysis of genetic distances and population structures in O. sativa using DNA markers [9]. Accession W1943 of the perennial O. rufipogon, on the other hand, is known to have a close phylogenetic relationship with the japonica varieties of O. sativa [5]. Consequently, these BAC libraries should be useful materials for the basic analysis of molecular and evolutionary mechanisms for the rice genes involved in the domestication and adaptation of the species.
Direct sequencing of PCR products using unique primer pairs is an effective way to obtain genomic sequences. This method, however, is not always useful, especially when the target region is long (more than 10 kbp) or contains sequences with reduced similarities, indels, multiple copies, and repetitive sequences. The system established in the present study through screening and physical mapping of candidate BAC clones, followed by sequencing a small number of shotgun subclones from a positive BAC clone, could thus provide a good platform for comparative analysis of orthologous genes because it does not require any special techniques or instruments and is cost effective and relatively fast.
Molecular evolution of Hd6
Hd6 encodes the α subunit of protein kinase CK2, which contains a number of conserved subdomains in its families and is known to be involved in a wide range of cellular functions, including the expression of some light-regulated genes in Arabidopsis [13, 21, 32]. The rice genome has a total of four copies of the putative gene encoding the α subunit of protein kinase CK2 (http://rapdb.dna.affrc.go.jp/). Although the deduced amino acid sequences of three copies (AK070271, Chr 7, 798,617 to 804,177 bp; AK120259, Chr 3, 5,644,400 to 5,648,028 bp; and AK120812, Chr 3, 32,416,746 to 32,421,167 bp) were 98%, 90%, and 61% identical to that of Hd6, only Hd6 has been reported thus far to be involved in determining the heading date of rice [33]. Among the 74 polymorphic sites observed in the Hd6 coding region (1,002 bp) of all species, we found only five nonsynonymous substitutions (in the cultivated rice Nipponbare and the wild rice O. glumaepatula, O. officinalis, O. australiensis, and O. brachyantha), of which two sites in Nipponbare and O. glumaepatula are located within the subdomain IV region (Fig. 1b). This indicates that conservation of the amino acid sequences of Hd6 within genus Oryza is nearly complete. This is supported by the DnaSP analysis using the coding sequences that revealed a very low value (≤0.003) for the Ka and a value ≤0.576 for the ratio Ka/Ks between the different varieties or species; this suggests purifying selection on this gene within genus Oryza (Supplementary Table 2). In plants, the alcohol dehydrogenase gene (Adh1) has been reported to be one of the most highly conserved genes [10, 11, 41]. For comparison, we also sequenced and calculated Ka and Ks from the coding sequences of Adh1 between all the species used in this study [38] and found a Ka value ≤0.013 and a Ka/Ks ratio ≤0.605. This demonstrates that the nonsynonymous substitution rate in Hd6 is even slower than that in Adh1. Although we do not know whether the change of one amino acid in the Hd6 region (except for Nipponbare) could affect the gene’s function, these results clearly reveal that natural mutations and modifications within the coding region of Hd6 were severely suppressed during the evolutionary history of Oryza genomes in tropical or subtropical regions under the natural conditions where all wild rice species are distributed.
To improve our understanding of the molecular evolution of genes related to heading date and the adaptation history of rice, we traced the origin of the nonsynonymous substitution site detected in exon 3 of the Nipponbare Hd6 gene that created a premature stop codon, leading to a nonfunctional allele in this japonica variety. Within the two world collections of O. sativa and O. rufipogon and the 51 local rice varieties collected in Japan (Supplementary Table 1), only five japonica varieties (Koshihikari, Ginbouzu, Morita Wase, Ishijiro, and Himenomochi) contained the same single mutation as that of Nipponbare. Koshihikari, registered during the 1950s, is considered the elite variety currently cultivated in Japan, largely because of its good quality and the preference for its taste by the Japanese people. According to their pedigrees (http://ineweb.narcc.affrc.go.jp/index.html, in Japanese), both Koshihikari and Nipponbare are considered to be closely related to the genetic background of two old local varieties of Ginbouzu (which was registered as a variety early in the twentieth century) and Morita Wase (registration information unknown). Moreover, we found that another local variety (“Aikoku”), which is known to be the progenitor of Ginbouzu, contained the wild-type gene at the same site. Unfortunately, we could not compare the genetic backgrounds of these varieties with those of Ishijiro and Himenomochi because there is no recorded pedigree for these two local varieties. Rice is thought to have been first introduced from China into Japan in the northern Kita-Kyushu area, at a latitude of about 34°N, approximately 3,000 years ago during the Jomon period [24]. All of the five japonica varieties containing the same point mutation as in Nipponbare (Koshihikari, Niigata Prefecture; Ginbouzu, Ishikawa Prefecture; Morita Wase, Yamagata Prefecture; Ishijiro, Toyama Prefecture; and Himenomochi, Akita Prefecture) are cultivated in the central and northern areas of Japan, at latitudes above 36°N. Our finding thus reveals strong evidence that the nonsynonymous substitution present in the Nipponbare Hd6 coding region might have originated from the Ginbouzu or Morita Wase local varieties and occurred very currently during the spread of rice cultivation from southern to northern areas of Japan.
Genome diversity within the Hd6 regions of different species
Recent researches from multiple gene loci reported that the average nucleotide diversity of O. sativa ranged 0.0023∼0.0024 [4, 43]. Obviously, the nucleotide diversity of Hd6 gene calculated using 398 bp PCR-amplified sequences from the both populations of O. sativa (π = 0.00016) and its wild relatives of O. rufipogon (π = 0.00024) in this study was much lower than that of the above researches (Supplementary Table 3). Negative values of Tajima’s D were observed in both populations suggesting an excess of low-frequency polymorphisms. These results consequently reveal the apparent presence of purifying selection for Hd6 gene (Supplementary Table 3). Nucleotide diversity (π) for the entire region of the Adh1 gene in O. rufipogon, on the other hand, is 0.0020 [41]. Although the number of varieties and accessions analyzed in the present study was limited, detection of the nucleotide diversity for the entire regions of Hd6 in five O. sativa varieties and three O. rufipogon accessions showed π values of 0.0018 and 0.0017, slightly lower than that of Adh1 gene (Table 3. Details on O. rufipogon are not shown). Further detection of the nucleotide diversity for Hd6 within all examined species revealed that the π values calculated from the entire genomic region and for the exon regions were 0.0524 and 0.0121, respectively, indicating that nucleotide diversity in the exon regions was low (less than one fourth that in the entire regions), again supporting the presence of strong suppression for natural mutations and modifications of the coding region of Hd6 within the genus Oryza.
We also estimated the DNA divergence (K) between each pair of species for the entire region of Hd6 (Table 3). The K values increased when comparing O. sativa with the other species that contained the AA, CC, BB, DD, EE, FF, and GG genomes, in that order. According to the divergence levels determined in this study, all rice species that we examined could be divided into three groups, with K values of 0.0011 to 0.0212 for the AA species, 0.0489 to 0.0675 for the BB, CC, DD, and EE species, and 0.1515 to 0.1662 for the FF and GG species. This result correlates well with the current taxonomic sections of Oryza classified based on the crossing ability between O. sativa and other rice species (http://www.shigen.nig.ac.jp/rice/oryzabase/wild/oryzaSpecies.jsp), thus confirming the categories of the O. sativa complex (AA genome), O. officinalis complex (BB, BBCC, CC, CCDD, and EE genomes), O. granulata complex (GG genome), and O. brachyantha (FF genome) as the primary, secondary, and tertiary gene pools for the cultivars [34, 35].
Phylogenetic relationships among the Oryza genomes analyzed using the entire region of Hd6 were consistent with that based on Adh1, again revealing the relatively close relationships between the CC and AA genomes and between the EE and DD genomes [11]. Within the five Asian rice cultivars included in our study, the indica varieties Kasalath and Shuusoushu were included in a cluster together with the two O. rufipogon accessions that originated in India and Malaysia (Fig. 2b). On the other hand, two japonica varieties (Nipponbare and Khau Mac Kho) formed another cluster with the indica variety (93–11) and one O. rufipogon accession (W1943). Based on a genome-wide investigation of retroposon and retrotransposon insertion patterns (J. Wu and T. Matsumoto, unpublished), indica variety 93–11 was considered to have a genomic background of the japonica variety around the Hd6 region, indicating the presence of gene flow between the two subspecies due to recent introgressions [36]. The above results support the hypothesis proposed as a result of studies using molecular markers such as isozymes, restriction fragment length polymorphisms (RFLPs), and short interspersed nuclear element insertions, as well as published genomic sequences, that Asian cultivated rice originated from two geographically distinct gene pools of the wild progenitor O. rufipogon [5, 12, 23, 36, 37].
Comparative analysis of orthologous gene sequences could help to reveal the mechanisms responsible for the molecular evolution and functions of rice genes. In the present study, we investigated sequence variations in Hd6 within several Oryza species and confirmed the presence of conservation and mutations of genomic sequences in this gene region that provided molecular evidence for the adaptation of rice to different environmental conditions in terms of day length. However, the mechanism that governs the response of heading date to photoperiod is very complicated, and flowering time is controlled by several numbers of QTL genes [6, 15, 19, 40]. To permit a comprehensive understanding of the molecular mechanisms and evolutionary development of the heading date genes involved in the domestication and adaptation process of rice, further comparative analysis of the genomic sequences and association of genotypes with phenotypes in other important genes, such as Hd1 and Hd3a, should also be conducted.
Materials and methods
Preparation of BAC libraries of cultivars and wild rice species
BAC libraries from four rice varieties and 14 wild rice accessions were used as primary materials for sequencing the whole genomic regions of the Hd6 gene (Table 1). BAC resources from nine accessions of the following wild Oryza species: O. rufipogon (two accessions of W0106 and W0574), O. punctata, O. minuta, O. officinalis, O. alta, O. australiensis, O. brachyantha, and O. granulata were supplied by the Arizona Genomics Institute (http://www.genome.arizona.edu/orders; [2] in the form of filters and individual clones). The BAC library of the indica variety Kasalath was constructed previously under the Rice Genome Research Program [3]. Eight new BAC libraries from the AA-genome species were constructed in the present study, including two O. sativa varieties (Shuusoushu, indica and Khau Mac Kho, japonica), one African cultivated rice (O. glaberrima), and five wild Oryza species (O. rufipogon, O. barthii, O. glumaepatula, O. longistaminata, and O. meridionalis). All of these BAC libraries were created using the conventional method, through a partial DNA digest of the MboI enzyme, size fractionation of high-molecular-weight DNA in a pulsed-field gel electrophoresis (CHEF, Bio-Rad Laboratories, Hercules, USA), as well as vector ligation (pIndigo BAC-5, EPICENTRE Biotechnologies, Madison, USA) and transformation of high-molecular-weight DNA into Escherichia coli (DH10B strain). High-density BAC filters from these libraries were also prepared using the Hybond-N+ nylon membrane (Amersham, Little Chalfont, UK) and a BioGrid robot BG600 (BioRobotics, Cambridge, UK). Each 8 × 12-cm filter contained 6,144 clones arrayed in 4 × 4 spotted pattern.
Selection of BAC candidates covering the genomic region of the Hd6 gene
A 156-bp PCR product amplified from the exon 3 region of Hd6 using Kasalath genomic DNA (see Supplementary Table 3 for the primer sequences) was prepared as the probe for Southern hybridization to screen candidate BAC clones from each library. Hybridization of the DNA probe with high-density BAC filters was conducted using the enhanced chemiluminescence system (ECL, Amersham). BAC-end sequences of candidate clones were downloaded, if available, from the Arizona Genomics Institute Web site (http://www.genome.arizona.edu/) or analyzed using ABI3700 capillary sequencers (Applied Biosystems, Foster, USA) in the Rice Genome Research Program, as described previously [16]. These BAC-end sequences were mapped in silico to the Nipponbare genome (Build 4.0 of pseudomolecules, http://rgp.dna.affrc.go.jp/E/IRGSP/Build4/build4.html) through a BLAST search (using BLASTN 1.5.6). A second primer pair (see Supplementary Table 3 for the primer sequences) that could amplify the intron sequence between exons 9 and 10 of Hd6 was also prepared to define positive BAC clones from among the different genome types of a tetraploid species by means of polymorphism detection of the PCR product length. All of the PCR amplification was performed using AmpliTaq polymerase (Applied Biosystems) in a PTC-100 PCR machine (MJ Research, Waltham, USA) under the following conditions: 35 cycles of 94°C for 30 s, 60°C for 60 s, and 72°C for 60 s.
Selective sequencing and assembly
DNA samples of each of the selected BAC clones that covered the Hd6 region were prepared according to the standard alkaline lysis procedure. To construct the shotgun sublibrary, BAC DNA was sonicated into fragments using the Sonifier 250 apparatus (Branson, Danbury, USA), with each fragment 5 to 10 kb in length and end-blunted using T4 DNA polymerase (TaKaRa, Kyoto, Japan), then randomly cloned into E. coli (DH10B strain) using pUC18 vectors (TaKaRa). Subclones containing the Hd6 gene sequence were enriched according to the following procedure. About 3,840 recombinants from each sublibrary were cultured in 384-well plates and dotted onto a membrane filter at a high density (8 × 12 cm, 6,144 clones arrayed in 4 × 4 pattern). After the filter was hybridized with the DNA probe, as described above, 48 positive subclones were selected and re-arrayed. After DNA isolation, two termini of each subclone were sequenced using the custom primers (see Supplementary Table 3 for the primer sequences) using ABI3700 capillary sequencers. Sequences were assembled and completed, if gaps remained, using the Phred/Phrap software and the Consed finishing tool, as described previously [31].
Data analysis
Genomic sequences of Hd6, including its flanking regions in Nipponbare and 93–11, were obtained from the International Rice Genome Sequencing Project (http://rgp.dna.affrc.go.jp/E/IRGSP/Build4/build4.html) and the Beijing Genomics Institute (http://rise.genomics.org.cn/rice/index2.jsp), respectively [14, 42]. Annotation and structural analysis of Hd6 in all cultivated varieties and wild rice accessions were performed using the est2genome (http://emboss.sourceforge.net/) and sim4 (http://globin.cse.psu.edu/) software using the completed sequence (GenBank accession AB036788) of Kasalath Hd6 as a reference [7, 29]. For comparison of the Hd6 genome among all cultivated varieties and wild rice accessions, sequences were aligned using the MAFFT multiple sequence alignment program (http://align.bmr.kyushu-u.ac.jp/mafft/software/) to investigate nucleotide differences such as substitutions, insertions, and deletions [17]. A conservation plot was generated based on the similarity of aligned sequences in this analysis. We also used the DnaSP program (Ver 4.20, http://www.ub.es/dnasp/) to analyze sequence variations for the number of polymorphic sites and nucleotide diversity (π, the average number of nucleotide differences per site) of the Hd6 region within O. sativa varieties and O. rufipogon accessions [30]. We also detected the nucleotide divergence (K, also called the genetic distance) between each pair of Oryza species and calculated the Ka/Ks ratio (where Ka represents the rate of nonsynonymous substitutions, and Ks represents the rate of synonymous substitutions) using DnaSP to provide an indication of positive selection for a protein-coding gene. Phylogenetic trees were also constructed based on nucleotide polymorphisms within the aligned sequences using the CLC Free workbench (http://www.clcbio.com/) using the unweighted pair-group with arithmetic mean method with 1,000 replicates.
Sequence analysis of Hd6 using populations of O. sativa and O. rufipogon
We prepared 60 rice varieties (WRC) of O. sativa and 19 accessions (W, AS, IRGC) of O. rufipogon from the world core collections of the National Institute of Agrobiological Sciences (NIAS), National Institute of Genetics, and International Rice Research Institute so that we could survey the sequence diversity including the frequency and distribution of a single mutation in the exon 3 region of Nipponbare Hd6 within the above two populations (Supplementary Table 1). The world collection of rice varieties was developed previously based on a genome-wide RFLP survey of 332 varieties representing a wide range of geographic provenances to analyze rice genomic diversity [20]. The world collection of O. rufipogon was selected based on differences in their geographic provenances and typical phenotypes (http://www.shigen.nig.ac.jp/rice/oryzabase/wild/coreCollection.jsp). A total of 51 Japanese local rice varieties (JRC) obtained from the DNA bank of NIAS were further used to trace the history of the single mutation (Supplementary Table 1). Total genomic DNA of each variety and accession was extracted from the leaves using the cetyltrimethyl ammonium bromide method [25]. Specific primer pairs (see Supplementary Table 3 for the primer sequences) were designed from the exon 2 and exon 3 regions of the mutation site for PCR amplification under the same conditions described above. The resulting PCR products were sequenced with the above primers using ABI3700 capillary sequencers.
References
Aggarwal RK, Brar DS, Khush GS. Two new genomes in the Oryza complex identified on the basis of molecular divergence analysis using total genomic DNA hybridization. Mol Gen Genet. 1997;254:1–12.
Ammiraju JSS, Luo M, Goicoechea JL, Wang W, Kudrna D, Mueller C, et al. The Oryza bacterial artificial chromosome library resource: Construction and analysis of 12 deep-coverage large-insert BAC libraries that represent the 10 genome types of the genus Oryza. Genome Res. 2006;16:140–7.
Baba T, Katagiri S, Tanoue H, Tanaka R, Chiden Y, Saji S, et al. Construction and characterization of rice genomic libraries: PAC library of japonica variety, Nipponbare and BAC library of indica variety, Kasalath. Bulletin of the NIAR. 2000;14:41–9.
Caicedo AL, Williamson SH, Hernandez RD, Boyko A, Fledel-Alon A, York TL, et al. Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genet. 2007;3(9):e163. doi:10.1371/journal.pgen.0030163.
Cheng C, Motohashi R, Tsuchimoto S, Fukuta Y, Ohtsubo H, Ohtsubo E. Polyphyletic origin of cultivated rice: based on the interspersion pattern of SINEs. Mol Biol Evol. 2003;20:67–75.
Doi K, Izawa T, Fuse T, Yamanouchi U, Kubo T, Shimatani Z, et al. Ehd1, a B-type response regulator in rice, confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Genes Devel. 2004;18:926–36.
Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–74.
Gao L, Innan H. Nonindependent domestication of the two rice subspecies, Oryza sativa ssp. indica and ssp. japonica, demonstrated by multilocus microsatellites. Genetics. 2008;179:965–76.
Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch S. Genetic structure and diversity in Oryza sativa L.. Genetics. 2005;169:1631–8.
Gaut BS, Morton BR, McCaig BC, Clegg MT. Substitution rate comparisons between grass and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc Natl Acad Sci USA. 1996;93:10274–97.
Ge S, Sang T, Lu B-R, Hong D-Y. Phylogeny of rice genomes with emphasis on origins of allotetraploid species. Proc Natl Acad Sci USA. 1999;96:14400–5.
Glaszmann JC. Isozymes and classification of Asian rice varieties. Theor Appl Genet. 1987;74:21–30.
Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52.
IRGSP, International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature. 2005;436:793–800.
Izawa T, Oikawa T, Tokutomi S, Okuno K, Shimamoto K. Phytochromes confer the photoperiodic control of flowering time in rice (a short-day plant). Plant J. 2000;22:391–9.
Katagiri S, Wu J, Ito Y, Karasawa W, Shibata M, Kanamori H, et al. End sequencing and chromosomal in silico mapping of BAC clones derived from an indica rice cultivar, Kasalath. Breeding Sci. 2004;54:273–9.
Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–8.
Khush GS. Origin, dispersal, cultivation and variation of rice. Plant Mol Biol. 1997;35:25–34.
Kojima S, Takahashi Y, Kobayashi Y, Monna L, Sasaki T, Araki T, et al. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 2002;43:1096–105.
Kojima Y, Ebana K, Fukuoka S, Nagamine T, Kawase M. Development of an RFLP-based rice diversity research set of germplasm. Breeding Sci. 2005;55:431–40.
Lee Y, Lloyd AM, Roux SJ. Antisense expression of the CK2-subunit gene in Arabidopsis. Effects on light-regulated gene expression and plant growth. Plant Physiol. 1999;119:989–1000.
Londo JP, Chiang Y-C, Hung K-H, Chiang T-Y, Schaal BA. Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proc Natl Acad Sci USA. 2006;103:9578–83.
Ma J, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci USA. 2004;101:12404–10.
Matsuo T. Origin and differentiation of cultivated rice. In: Matsuo T, Futsuhara Y, Kikuchi F, and Yamaguchi H, editors. Science of the rice plant, vol. 3, Genetics. Tokyo: Food and Agriculture Policy Research Center; 1997. p. 69–110.
Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980;8:4321–5.
Nanda JS. Antiquity and spread of rice cultivation. In: Nanda JS and Sharma SD, editors. Monograph on Genus Oryza. Science Publishers, Inc. Enfield, New Hampshire, USA; 2003. p. 331–346.
Oka H-I. Origin of cultivated rice. Tokyo: Japan Scientific Societies Press; 1988.
Oka H-I, Chang W-T. The impact of cultivation on populations of wild rice, O. sativa f. spontanea. Phyton. 1959;13:105–17.
Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends in Genetics. 2000;16:276–7.
Rozas J, Rozas R. DnaSP, DNA sequence polymorphism: an interactive program for estimating population genetics parameters from DNA sequence data. Comput Applic Biosci. 1995;11:621–5.
Sasaki T, Matsumoto T, Yamamoto K, Sakata K, Baba T, Katayose Y, et al. The genome sequence and structure of rice chromosome 1. Nature. 2002;420:312–6.
Sugano S, Andronis C, Green RM, Wang Z-Y, Tobin EM. Protein kinase CK2 interacts with and phosphorylates the Arabidopsis circadian clock-associated 1 protein. Proc Natl Acad Sci USA. 1998;95:11020–5.
Takahashi Y, Shomura A, Sasaki T, Yano M. Hd6, a rice quantitative trait locus involved in photoperiod sensitivity, encodes the a subunit of protein kinase CK2. Proc Natl Acad Sci USA. 2001;98:7922–7.
Tateoka T. Taxonomic studies of Oryza. III. Key to the species and their enumeration. Bot Mag Tokyo. 1963;76:165–73.
Vaughan D, Morishima H, Kadowaki K. Diversity in the Oryza genus. Curr Opin Plant Biol. 2003;6:139–46.
Vitte C, Ishii T, Lamy F, Brar D, Panaud O. Genomic paleontology provides evidence for two distinct origins of Asian rice (Oryza sativa L.). Mol Gen Genomics. 2004;272:504–11.
Wang ZY, Tanksley SD. Restriction fragment length polymorphism in Oryza sativa L. Genome. 1989;32:1113–8.
Wu J, Mizuno H, Sasaki T, Matsumoto T. Comparative analysis of rice genome sequence to understand the molecular basis of genome evolution. Rice. 2008;2:119–26.
Yamamoto T, Lin H, Sasaki T, Yano M. Identification of heading date quantitative trait locus Hd6 and characterization of its epistatic interactions with Hd2 in rice using advanced backcross progeny. Genetics. 2000;154:885–91.
Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell. 2000;12:2473–83.
Yoshida K, Miyashita NT, Ishii T. Nucleotide polymorphism in the Adh1 locus region of the wild rice Oryza rufipogon. Theor Appl Genet. 2004;109:1406–16.
Yu J, Hu S, Wang J, Wong GK-S, Li S, Liu B, et al. A draft sequence of the rice genome (Oryza sativa L. ssp indica). Science. 2002;296:79–92.
Zhu Q, Zheng X, Luo J, Gaut BS, Ge S. Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol Biol Evol. 2007;24:875–88.
Acknowledgments
We thank Dr. N. Kurata of the National Institute of Genetics; Drs. D. A. Vaughan, K. Ebana, and T. Izawa of the National Institute of Agrobiological Resource Sciences; and Dr. R. A. Wing of the Arizona Genomics Institute for providing the plant materials and BAC libraries used in this study. This work was supported by grants from the Ministry of Agriculture, Forestry and Fisheries of Japan (Green Technology Project GD-2007, Genomics for Agricultural Innovation QTL5003).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
(XLS 510 KB).
Supplementary Table 2
(XLS 345 KB).
Supplementary Table 3
(XLS 200 KB).
Supplementary Table 4
(XLS 340 KB).
Supplementary Figure 1
(PPT 597 KB).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yamane, H., Ito, T., Ishikubo, H. et al. Molecular and Evolutionary Analysis of the Hd6 Photoperiod Sensitivity Gene Within Genus Oryza . Rice 2, 56–66 (2009). https://doi.org/10.1007/s12284-008-9019-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12284-008-9019-2