
Abstract
Myocardial infarction (MI) is the leading cause of death among ischemic heart diseases and is associated with several long-term cardiovascular complications, such as angina, re-infarction, arrhythmias, and heart failure. However, MI is frequently accompanied by non-cardiovascular multiple comorbidities, including brain disorders such as stroke, anxiety, depression, and cognitive impairment. Accumulating experimental and clinical evidence suggests a causal relationship between MI and stroke, but the precise underlying mechanisms have not yet been elucidated. Indeed, the risk of stroke remains a current challenge in patients with MI, in spite of the improvement of medical treatment among this patient population has reduced the risk of stroke. In this review, the effects of the signaling from the ischemic heart to the brain, such as neuroinflammation, neuronal apoptosis, and neurogenesis, and the possible actors mediating these effects, such as systemic inflammation, immunoresponse, extracellular vesicles, and microRNAs, are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent research in cardiological and neurological fields has shown that pathophysiological processes, once considered to lead solely cardiovascular or neurological manifestations respectively, may instead concomitantly affect both systems. Indeed, substantial available evidence has indicated a multitude bidirectional connection between the cardiovascular and central nervous systems [1,2,3]. In 1985, the term “neurocardiology” was coined for the first time to describe this new interdisciplinary area, which examines the interaction between the cardiovascular and autonomic nervous systems in pathological states [4].
In this context, accumulating clinical and experimental studies suggest a causal relationship between myocardial infarction (MI) and brain pathological alterations. MI and subsequent revascularization therapies could lead to transient reduction in cerebral blood flow, thereby damaging the brain [5]. It has also been argued that vascular inflammation, which is common features of MI patients, could be involved in the induction of depression symptoms [6]. Patients with MI have a high prevalence of behavioral disorders, such as anxiety and depression [7,8,9] and several associated symptoms, including cognitive deficits [10, 11]. In particular, a recent population-based cohort study showed that patients with MI exhibited a significantly higher risk of anxiety-like disorders (adjusted hazard ratio = 5.06) and depressive disorders (adjusted hazard ratio = 7.23) than those without MI, during the first 2 years of follow-up [9]. A large meta-analysis comparing patients with or without depression after MI showed that depression was associated with a 2.7-fold increased risk of cardiac-related death, a 2.3-fold increased risk of all-cause death, and a 1.6-fold increased risk of cardiovascular events in the 2 years after an acute MI [12]. Similarly, a recent meta-analysis, including 16 studies that enrolled patients with established acute MI, showed that MI patients with anxiety (prevalence ranged from 5.5 to 58.2%) had a significant long-term poorer prognosis (risk ratios = 1.27) and increased long-term major adverse cardiac events (MACEs) (risk ratios = 1.54) than those without anxiety [13]. On the other side, the casual relationship between MI and dementia is instead not fully demonstrated. In fact, only two studies have examined the risk of dementia after MI, but with equivocal findings [14, 15]. A case–control study failed to demonstrate a clear association between dementia and MI [14], whereas a cohort study showed an increased risk for patients with unrecognized MI, but not for patients with recognized MI [15].
Several studies have also demonstrated the association between MI and stroke [16]. These two diseases could evolve in parallel since they share the same risk factors, such as hypertension, diabetes, arrhythmias (including atrial fibrillation), high cholesterol levels, smoking, and chronic kidney disease [17]. At the same time, MI is considered one of the etiological causes of stroke [18]. It has been observed that patients with MI have a higher risk of stroke in the first 4 weeks immediately after acute MI than in the corresponding period of 4 weeks to 1 year after MI, and this risk remains high for the first 12 weeks [19, 20]. Of note, the risk of ischemic stroke was similarly elevated for up to 12 weeks for both ST-elevation myocardial infarction (STEMI) and non-STEMI [20], while the cumulative 3-month and 1-year ischemic stroke incidence were higher among patients with coronary artery bypass surgery (CABG) than among patients without CABG [21]. Over the last decade, the relative risk of ischemic stroke within 30 days [22] and 1 year [23] after acute MI has decreased by about 10% and 20% respectively, likely due to increased use of reperfusion therapies and more intense treatment with statins, acetylsalicylic acid, and P2Y12 inhibitors [21,22,23]. A recent study demonstrated that unrecognized MI, which makes up between one-third to one-half of all MI events [24], is also associated with an increased risk of stroke [25]. Relevantly, stroke following MI impairs the overall prognosis, since patients with ischemic stroke after acute MI have higher morbidity and mortality rates both in the short and long terms than patients without stroke [26]. In this respect, a recent large retrospective cohort study showed that the perspective of 1‐year mortality was about 15% higher for patients with acute MI plus stroke (51.5%) than for those with MI without stroke (37.1%) [20].
Despite this evidence, the precise underlying relationship between MI and stroke has not yet been elucidated. It is likely that the mechanisms could be different for early and late ischemic stroke after MI. Early ischemic stroke may be originated from embolization of blood clots in the left atrium after atrial fibrillation, or from mural thrombus formed in hypokinetic segments of the left ventricle [16], while late ischemic stroke may be caused by the presence of the mutual underlying risk factors. Instead, hemorrhagic stroke may be induced by antithrombotic medication to prevent the reoccurrence of MI. In addition, the incidence of both ischemic and hemorrhagic strokes was higher with percutaneous mechanical circulatory support device use in comparison to those without device in patients with STEMI and cardiogenic shock, suggesting that patients with devices may be hemodynamically sicker and require increased use of anticoagulation [27].
Overall, these findings suggest that the etiopathology of stroke after MI is a complex process. MI may result from a generalized and severe atherosclerotic disease associated with a systemic inflammation and alterations in the function of the neurocardiac axis [28] that, in turn, may increase the ischemic stroke risk. Although the improvement of medical treatment for hypercholesterolemia and pro-thrombotic status among patients with MI has reduced the risk of stroke, this one remains a current challenge. Growing evidence, especially from experimental studies and still to be consolidated, suggest that additional mechanisms could be involved in stroke development after MI. Thus, in this review, we will focus on these other possible signaling mechanisms relayed from the ischemic heart to the brain and the resulting alterations in brain functioning.
Mechanisms of heart–brain interaction after MI
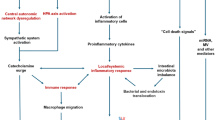
Figure 1 summarizes all the mechanisms discussed.

Mechanisms and cerebral effects of heart–brain interaction after MI. Brain alterations after MI may be caused by several mechanisms, including systemic inflammation, activation of the renin–angiotensin–aldosterone system (RAAS), circulating cardiac-derived DAMPs, EVs, and miRNAs, and reduced cardiac output
Immunoresponse and inflammation
It is widely documented that the immune system and inflammatory processes are activated following MI [29]. After MI, dying cardiomyocytes and the other cells populations in cardiac tissue release damage-associated molecular patterns (DAMPs) that can be recognized by pattern recognition receptors (PRRs) expressed by several immune cells, including neutrophils, monocytes/macrophages, and dendritic cells. In particular, cardiac resident CCR2+ macrophages are activated by DAMPs through binding with the toll-like receptor (TLR) 9, and, as a result, they increase the expression of the chemokine (C-X-C motif) ligand CXCL2 and CXCL5, stimulating the migration of neutrophils into cardiac ischemic tissue [30]. In turn, releasing their granule contents, neutrophils increase vessel permeability and promote, together with activated macrophages, the migration of monocytes to the site of inflammation [31]. The subsequent excessive production of the reactive oxygen species (ROS) and impairment of anti-oxidant system as well as enhanced production of matrix metalloproteinases (MMPs), pro-inflammatory cytokines, and chemokines, further aggravate inflammation [32]. Experimental and clinical studies showed that also the adaptive immune cells contribute to inflammation following MI. Indeed, lymphocytes, especially CD4+ T cells, are activated after acute coronary syndrome (ACS) and MI [33,34,35], and B cells can influence the monocyte migration after MI producing the chemokine CCL7 [36].
The activated immunoresponse and the inflammatory processes, necessary for cardiac remodeling and scar formation, are localized not only in the cardiac tissue, but also at the systemic level, as demonstrated by the high levels of circulating cytokines, such as TNF-α, IL-1, and IL-16, which persist for several weeks after the ischemic event and correlate with deteriorating cardiac function and increased mortality [37,38,39,40,41]. The circulating cytokines may evoke a cascade of events in the cerebral circulation, including thrombus formation [16]. In parallel, it has been suggested that systemic inflammation induce neuroinflammation, namely, increasing pro-inflammatory cytokines in the brain, within few minutes from MI [40, 42], and this inflammatory condition may persist even longer, approximately 6–8 weeks [40], after the initial peripheral inflammation has subsided [43]. The increased levels of cytokines in the brain may be due to the passage of peripheral cytokines through the blood–brain barrier (BBB). However, substantial increase in hypothalamic cytokines early after MI is not easily explained since cytokines are too large to readily cross the BBB. Thus, active transport of peripheral pro-inflammatory cytokines across the BBB or cytokines-mediated endothelial leakage and altered BBB permeability could be the possible routes [43, 44]. In this context, it was recently demonstrated that the pro-inflammatory DAMP, high mobility group box 1 (HMGB1), whose serum concentration increases early after the acute MI [45], dramatically enhances permeability in primary human brain microvascular endothelial cells and in human cerebromicrovascular endothelial cell line, a widely used model of human BBB in vitro [46]. This BBB alteration is matched by a significant downregulation of the zona occludin-1 (ZO-1) expression at intercellular at tight junctions [46]. Cytokines transport across the BBB could also be mediated by circulating EVs which are enriched with pro-inflammatory cytokines after MI [47]. Otherwise, the high level of pro-inflammatory cytokines may be originated from an increase in local production, but the underlying mechanism is not clearly elucidated. One hypothesis has suggested that pro-inflammatory cytokines upon crossing BBB could induce PGE2 production in endothelial cells of the cerebral blood vessels [48], leading to an increase in cerebral cytokine production. Furthermore, circulating cytokines could themselves stimulate microglia and astrocytes to produce cytokines. Another hypothesis suggested that elevated levels of angiotensin II and aldosterone following MI could initiate inflammatory response through induction of ROS [48]. The available evidence suggests that neuroinflammation after MI is a complex process in which several actors are involved in its occurrence and that integrates cellular and molecular responses, involving different cellular lineages (see “Neuroinflammation”).
EVs
EVs are nanometer-sized, lipid membrane-enclosed vesicles released by cells into the extracellular space to facilitate intercellular communication in diverse cellular processes [49]. EVs provide a unique mode of long-range delivery of lipids, metabolites, and proteins as well as ribonucleic acids (RNA) and deoxyribonucleic acids (DNA) from donor cells to distant recipient cells. A key role in regulating the EVs-mediated interactions is played by the membrane-bound signaling proteins of EVs, which interact with the extracellular environment determining the cell types they target. EVs have been traditionally subdivided into three major classes according to their diameter: exosomes (20–150 nm), microvesicles (also named microparticles; 100–1000 nm), and apoptotic bodies (> 500 nm) [50], but they could be often classified according to their surface proteins.
A growing number of experimental and clinical studies found that the level of microvesicles (MVs) in patients with coronary heart disease (CHD) increased significantly [47, 51, 52]. In particular, leukocyte-derived (CD45+lMVs), endothelium-derived (CD31+CD42−eMVs), platelet-derived (CD31+CD42+pMVs), erythrocyte-derived (CD235a+ ErMVs), and annexin-V+ MVs significantly increased in plasma of patients with subgroups of CHD, including stable angina (SA), unstable angina (UA), and MI (NSTEMI and STEMI) [53,54,55]. However, it is still controversial which EVs subpopulations are most useful for diagnostic or prognostic purposes. A recent meta-analysis, including 599 participants (137 healthy subjects, 148 patients with SA, 147 patients with UA, and 167 patients with MI), found that the level of MVs, especially CD31+CD42− and CD144+ eEVs, was higher in patients with CHD than in healthy subjects and had an increasing trend with the degree of CHD: SA < UA < MI [56]. Moreover, an increase in cardiomyocyte-derived EVs was found in plasma samples of STEMI patients, as well as in mice subjected to permanent left anterior descending (LAD) artery ligation [52]. EVs released from cardiomyocytes under pathophysiological conditions may convey “danger or inflammatory signals” to other cells. In vitro, hypoxia-induced released of EVs carrying TNF-α [57] and heat shock protein 60 (HSP60), a ligand of TLR4, which activates the innate immune response [58]. In mice with permanent LAD artery ligation, cardiac EVs were transiently accumulated in the infarcted heart, with a peak between 15 and 24 h post-MI, and originated mainly from cardiomyocyte (caveolin-3+; Troponin T+), cardiac fibroblast (CD90.2+), and endothelial cells (CD31+CD41−), while only a small population of leukocyte-derived CD45+ EVs was detected. These EVs were taken up by infiltrating monocytes/macrophages and regulated local inflammatory responses, leading an increased release of IL-6 and chemokines CCL2 and CCL7 [59]. After MI, circulating EVs could be also enriched with pro-inflammatory cytokines. Indeed, 24 h after MI in rats, plasma-derived EV were enriched with pro-inflammatory cytokines IL-1α, IL-1β, and Rantes. When added to the perfusates of isolated-perfused hearts, these EVs induced cardiomyocyte death and cardiac dysfunction through activation of the TLR4/NF-κB axis, whereas circulating EVs from healthy rats did not [47].
To date, it is widely accepted that EVs released by multiple cell types in response to MI participate both in the inflammatory injury and in tissue repair, but it is likely that they may influence other organs, such as the brain.
EVs were suggested as important signals mediating heart–brain interactions [1]. In fact, EVs released from the heart in normal or pathological conditions could influence both heart and brain, since some of molecular mechanisms and signaling pathways involving EVs were similar in MI and stroke. Although they are two distinct pathological conditions affecting different organs and type of cells, circulating EVs in patients after MI and stroke showed several similarities in proteins and microRNAs (miRNA) [60]. In details, these EVs have in common 14 proteins which were absent in healthy controls, such as apolipoprotein L1 (APOL1) and apolipoprotein C (APOC); involved in lipid metabolism; and complement C4 (C4) and C-reactive protein (CRP), factors, and activators of the complement system, which is activated after stroke and MI and contributes to tissue injury after ischemia. However, the hypothesis of circulating EVs as possible mediators of heart–brain axis was supported by two studies demonstrating that their passage through the BBB is very low in non-pathological conditions, but it was increased by orders of magnitude after chronic or acute systemic inflammation [61, 62]. Upon reaching the brain, likely via adsorptive-mediated transcytosis, EVs were able to transfer functional genetic material leading to concomitant changes in miRNA content of the receiving cells. Of note, erythrocyte-derived EVs in presence of systemic inflammation or obtained from Parkinson’s disease patients provoked an increase in microglial inflammatory responses [62]. In line, a more recent study demonstrated that peripheral circulating inflammatory exosomes induce neuroinflammation also in absence of systemic inflammation. In details, serum-derived exosomes purified from LPS-challenged mice or from mice fed high-fat diet induce microglial and astrocytic activation and increase the expression of inflammatory cytokines in the brain of recipient mice. The authors suggested that ependymal cells in the third and lateral ventricles may be the main entry sites via the blood cerebral spinal fluid brain barrier (BCSFB) for the exosomes to translocate into brain parenchyma, where are primarily taken up by microglial cells [63].
The mechanisms by which EVs cross the BBB are not fully elucidated. An endocytic, transcellular mechanism was reported to mediate the enhanced exosome transport through the BBB elicited by TNF-α in an in vitro BBB model [64]. Other studies suggested that clathrin-mediated transcytosis may play a role in EVs crossing the BBB [65, 66]. Finally, adsorptive mediated transcytosis and macropinocytosis were proposed as additional transcytotic mechanisms at the BBB that allow internalization of EVs without requiring them to be coated with specific transportation molecules [67, 68] (Fig. 2). Peripheral inflammation may also influence brain homeostasis through the activation of choroid plexus epithelium (CPE) and the subsequent release of choroid plexus-derived extracellular vesicles. In mice treated with LPS, EVs were secreted by CPE into the cerebrospinal fluid (CSF) and were taken up by astrocytes and microglia, which in turn respond with an inflammatory program. As a confirmation of the role of CPE, the effects of the peripheral inflammation-induced EV production by the CPE cells were reversed by the injection of an exosome inhibitor, and this was reflected by reduced upregulation of inflammatory genes [69]. However, we cannot rule out that systemic inflammation may stimulate the release of EVs by peripheral immune cells, which could directly cross the BBB through leaky tight junction, and target brain cells.

Potential pathways of cytokines, miRNAs, and EV passage across the BBB. Cytokines and miRNAs could cross BBB through EV-mediated transcellular routes, such as macropinocytosis, adsorptive mediated transcytosis, and clathrin-mediated transcytosis. In addition to transcellular routes, the breakdown of tight junction by DAMPs or inflammatory mediator may increase the permeability of cytokines and EVs at the paracellular route
Taken together, all these data suggest that circulating EVs released by peripheral organs under pathological conditions, including MI, could influence brain responses. However, to our knowledge, there are no studies that have assessed the cerebral effects of circulating EVs released after MI. Beyond reasonable hypotheses, future studies are needed to demonstrate whether circulating EVs is one of mechanisms through MI influences cerebral behavior.
miRNAs
MicroRNAs are bioactive small non-coding RNAs, which interact with the complementary sequences in the 3′ untranslated region (3′UTR) of protein-coding mRNAs, resulting in the inhibition of protein translation or mRNA degradation [70]. miRNAs are secreted into the extracellular space through three main mechanisms: (1) direct excretion from the cell upon binding to RNA-binding proteins, (2) budding off the cells through MVs formation, or (3) packaged into multivesicular bodies and released from the cells as exosomes [71].
miRNAs are involved in a myriad of biological processes, including proliferation, apoptosis, metabolism, differentiation, epithelial-to-mesenchymal transition, regulation of insulin secretion, division of stem cells, embryonic development and pattering, fetal growth, and immune system, including resistance to viral infection [72]. miRNAs may have cell-type-specific or tissue-specific expression patterns or may be expressed ubiquitously, but their expression could change in spatial as well as in temporal manner in pathological conditions, suggesting miRNA as potential biomarkers [73, 74].
Significant changes of miRNA expression in peripheral total blood samples of patients with MI were reported. Although 121 different miRNAs were observed to be dysregulated [75], robust evidence was found only for miR-1, miR-133a/b, miR-208a, and miR-499, whose serum levels increased in humans and animals following MI [76,77,78,79]. All these four miRNAs are regarded as heart-specific miRNAs [73] and found in plasma carried by exosomes (miR-1, miR-208, and miR-499) or as free-circulating compound (miR-133) [79].
In addition to modulating the signaling pathways after MI, several studies showed that these miRNAs are involved in cerebral physiological and pathological conditions, suggesting a possible mechanism through which MI affects the brain (Fig. 3). Circulating miRNAs are localized in MVs or bind to other plasma components such as high-density lipoprotein (HDL) particles and RNA-binding proteins [80]. Several evidences suggested that circulating EV-associated miRNAs are able to cross the BBB using exosomes [81,82,83]. However, more research must be done to elucidate how circulating cardiac-derived miRNA, in particular EV-free circulating miRNAs, cross BBB after MI.

The possible role of miRNAs released from the heart after MI in mediating cerebral effects. An overview of the molecular mechanisms and cerebral effects of the miRNAs whose expression are increased in the peripheral blood of patients with MI and for which it has been reported an involvement in cerebral physiological and pathological conditions
miR-1 is specifically expressed in adult cardiac and skeletal muscle tissues, and the increase of its serum level after MI suggests a necrotic death of cardiac myocytes as source [84]. Recently, it was proven that miR-1 might control the generation of synapses, brain growth, learning, and memory through regulation of brain-derived neurotropic factor (BDNF) and impact on target genes [85]. miRNA-1 also played a role in the damage induced by hypoxia in neurons affecting the expression of HSP-70 and consequently mediating hypoxia-induced apoptotic insults via an intrinsic Bax–mitochondria–caspase protease pathway [86]. In line, miRNA-1 knockdown by injections of anti-miR1 reduced the infarct volume in transient middle cerebral artery occlusion (MCAO) models induced by endothelin-1 in female rats [87] and by intraluminal filament in male rats [88], likely modulating the IGF signaling pathway. Two recent studies have clearly demonstrated the possibility that the heart may affect the brain through miR-1. Indeed, transgenic mice with cardiac-specific over-expression of miR-1 showed cognitive impairment that may be associated, at least in part, with the downregulation of BDNF expression in the hippocampus. The authors reported also an increased expression of miR-1 in the blood and hippocampus, although the expression of primary miR-1 was not changed. The latter data strongly suggested that circulating cardiac-derived miR-1 is very likely transport from the blood to brain, leading to regulation of cerebral target genes such as BDNF [81]. In mice with permanent LAD ligation, MI induced an increase of miR-1 levels in blood and hippocampus, likely originated from infarcted heart, leading neuronal microtubule damage and a decrease in the tubulin polymerization, inhibiting protein TPPP/p25 expression in the hippocampus. These changes were prevented by the selective knockdown of miR-1 in the hippocampus [82]. Relevantly, in transgenic mice with cardiac-specific over-expression of miR-1 and subjected to permanent LAD ligation, MI induced a reduction of learning, memory, and efficiency of synaptic transmission, likely through downregulation of BDNF, GluA1 subunit of the AMPA receptor, and dephosphorylation of GluA1. All these effects were prevented by the injection of a miR-1 antisense inhibitor in the CA1 area of the hippocampus [89]. Taken together, these results strongly indicate that the cardiac-originated miR-1 is a direct actor of brain dysfunction after MI.
Instead, miR‐133b has been suggested to regulate neurite outgrowth. Indeed, inhibition of miR‐133b expression by synthetic antisense oligonucleotides resulted in impaired locomotor recovery and reduced regeneration of axons after spinal cord injury (SCI) in adult zebrafish. The authors showed that miR-133b targets the Ras homolog gene family member A (RhoA), an inhibitor of axonal growth, as well as other neurite outgrowth‐related molecules [90]. Furthermore, in mice with SCI, the enhanced expression of miR-133b by lentiviral vector injection improved locomotor recovery by downregulation of the expression level of RhoA and chondroitin sulfate proteoglycans, and by decrease of infiltrating microglia/macrophage [91]. The same authors showed that transfection of miR-133b stimulated neurite outgrowth in cultured hippocampal neurons, likely decreasing the expression of RhoA, but also of xylosyltransferase 1 (Xylt1), an enzyme involved in the synthesis of chondroitin and dermatan sulfates, ephrin receptor A7 (Epha7), a key regulator of axon guidance, and purinergic receptor P2X ligand-gated ion channel 4 (P2RX4). In cell culture models of Parkinson’s disease, the overexpression of miR-133b ameliorated the MPP+-induced axon degeneration, blocking the MPP+-induced decrease in the Bcl-2/Bax ratio and increasing the activity of the pro-survival kinase Akt (p-Akt) [92], and was involved in the downregulating of α-synuclein [93]. Finally, the neuroprotective effect of miR-133b was also observed in a rat model of cardiac arrest [94]. In details, miR-133b incorporated in EVs, which were released from transplanted bone marrow mesenchymal stem cells (BMSCs), promoted survival of neuronal cells via regulation of JAK1 and AKT-GSK-3β-WNT pathway.
A recent study suggested that increased expression of miR-208 may augment susceptibility to schizophrenia by simultaneously conferring susceptibility to apoptosis and altering neural processing and connectivity through the suppression of BCL2 and calcium voltage-gated channel subunit alpha1 C (CACNA1C), respectively [95]. Furthermore, miR-208 reduced the expression of the RNA-binding protein quaking (QKI), whose suppression commonly contributes to demyelination of the neurons [96].
The serum level of miR-499 was markedly increased in the traumatic brain injury (TBI) patients compared with the healthy subjects and was associated with injury severity and clinical outcome, suggesting that miR-499 may serve as biomarker for the diagnosis and progression monitoring of TBI and that they may be involved in TBI pathogenesis [97]. However, the cerebral expression levels of miR-499-5p were gradually decreased after perinatal hypoxic-ischemic encephalopathy (HIE) in rats, while miR-499-5p injection significantly improved long-term neurological function recovery and decreased HIE-induced brain injury, reducing apoptotic neurons in the hippocampus, the infarct size, and level of CRP [98].
Cerebral alterations as a consequence of MI
Neuroinflammation
Neuroinflammation, defined as an inflammatory response within the brain, is mediated by the production of cytokines, chemokines, and ROS, which are produced by microglia, astrocytes, peripherally derived immune cells, and endothelial cells (Fig. 4). It is widely recognized that MI is able to induce neuroinflammation, which may persist even long after the initial peripheral inflammation has subsided [43], approximately 6–8 weeks after MI in the hypothalamus of rats [40].

Summary of mechanisms for neuroinflammation after MI. Circulating pro-inflammatory cytokines, DAMPs, and angiotensin II (Ang II) released from the ischemic heart reach the brain through the blood. Here, they can compromise integrity and enhance permeability of BBB, reducing the expression of junctional proteins such as ZO-1, occludin, and claudin-5. Ang-II and pro-inflammatory cytokines induce the activation of astrocytes and stimulate resting microglia to assume the pro-inflammatory M1 phenotype, which is induced also by the excessive release of ATP from activated astrocytes. In turn, M1 microglia and activated astrocytes produce large amounts of cytokines and ROS, which perpetuate neuroinflammation and lead to enhanced neuronal apoptosis and decreased neurogenesis. Furthermore, they produce an imbalance between excitatory and inhibitory neurotransmission, potentiating excitatory (glutamatergic and adrenergic) currents and inhibiting GABAergic currents
A recent study showed that MI, obtained in mice by permanent LAD coronary artery ligation, induced a cerebral increased expression of TNF precursor protein, a more than doubled TNFR1 expression, and almost 70% decline in TNFR2 expression, suggesting a shift toward a more pro-inflammatory state [41]. Increased brain cytokines can affect several processes involved in neuronal function, including apoptosis, oxidative stress, and metabolic processes [99]. In addition, neuroinflammation is thought to be a key for Alzheimer’s dementia progression [100], and it is able to influence behavioral aspects, including anxiety, cognitive deficit, and depression [101]. In this regard, sex and estrogen level could influence neuroinflammation and depression-like behavior. A recent study in male and female rats with permanent LAD artery ligation showed that cytokines (TNF-α, IL-1β, IL-2, and IL-6) increased significantly in the prefrontal cortex of MI male rats, while no changes were found in MI female rats [102].
Although circulating cytokines could cross BBB, as reported in “Immunoresponse and inflammation,” recent data indicate that the increase in pro-inflammatory cytokines involves the activation of cerebral cells [40, 103]. In response to injury or inflammation, context-specific signals can shape both astrocyte and microglial responses. In this context, for example, it has been shown that the blocking of TNF-α by its antagonist etanercept or by its genetic deletion causes a reduction in microglia activation, pointing the pivotal role of this cytokine in glial activation in mice with permanent LAD coronary ligation [104]. In light of these evidences, several studies have analyzed the effect of MI on microglia and astrocytes.
Infarcted rat, subjected to permanent LAD coronary ligation, showed microglial activation in the paraventricular nucleus (PVN) of the hypothalamus, in periaqueductal grey (PAG), in rostral ventrolateral medulla (RVLM), in nucleus tractus solitarius (NTS), and in area postrema (AP) [105, 106] during the late post-ischemia phases, but not in the earlier phase (within 1 week) [107]. In contrast, an early activation of microglia was observed in more recent studies [108,109,110]. In particular, cardiac ischemia/reperfusion (I/R) induced a shift of microglia phenotype from the beneficial M2 to the inflammatory harmful M1 at the end of 120 min of reperfusion period. Indeed, cardiac I/R induces an increase in Iba-1 positive cells, CD11b+/CD45+high microglia, and microglial dendritic volume, while filament length and dendrite complexity significantly decreased, suggesting that microglia tends toward an ameboid shape or M1 phenotype, indicating both activation and acquisition of phagocytic properties. These effects are, almost in part, mediated by the proprotein convertase subtilisin/kexin type 9 (PCSK9). Indeed, its cerebral expression is increased during cardiac I/R injury, and its inhibition reduces neuronal inflammation possibly because of a reduction of systemic inflammation [109].
However, the microglial activation was relatively selective within the PVN, since changes in the phenotype of microglia and a re-arrangement of its cytoskeleton occur in the PVN but not in the adjacent areas and in the cortex of MI rats [107, 111, 112]. Taken together, these results seem to suggest that, unlike other peripheral inflammatory conditions such as inflammatory bowel disease, MI not induces a generalized increase in the permeability of the BBB that could explain the activation of microglia, but only in selected brain regions that are centers for cardiovascular autonomic regulation [43, 103, 105]. This hypothesis has been challenged by recent studies showing a decreased expression of BBB tight junction proteins, claudin-5, and occludin, in whole brain [110], and an increased number of hypertrophic and dystrophic-like microglia in the prefrontal cortex, thalamus, and hippocampus of rats with permanent LAD coronary ligation [113].
Analyzing the results, another consideration to take into account is the responsiveness of microglia to MI that seems different between rats and mice. Contrary to mice, in rats, microglia remain active for a long time after MI, up to 16 weeks [107], whereas, after 18 days from permanent LAD artery ligation, in MI mice was reported alternative activated hyper-ramified microglia in PVN, that may represent a state of increased alertness rather than reflecting neuroinflammation [114]. Similarly, in a study using permanent ligation of the coronary artery in mice, no changes in reactive microgliosis were observed 3 months post-MI [115]. This discrepancy could be explained by the phasic pattern of microglia activation or the models used in mice. Indeed, a recent study, conducted in a mouse model of permanent cardiac ischemia, observed biphasic activation of microglia, with activity peaks after 1 and 8 weeks of ischemia, interspersed with a decline to 4 weeks [2]. The authors state that the systemic inflammatory response to acute MI may serve as the primer for the subsequent reoccurrence of neuroinflammation in the chronic phase of MI leading to heart failure.
It was speculated that the release of cytokines from activated microglia could stimulate neurons of PVN, contributing to the elevated sympathetic nerve activity seen in chronic heart failure [116]. In this context, two recent studies in rats with permanent LAD artery ligation showed that MI activates microglia which subsequently release cytokines in the PVN. In the first study, MI induced production of ATP which activates microglia through P2X7 receptor acutely promoting the synthesis of TNF-α and IL‐1β in the PVN [117]. The second study showed that MI induces, within the PVN, microglia stimulation through activation of macrophage-inducible C-type lectin (Mincle), a receptor primarily expressed in microglia which recognizes the DAMPs derived from dead cells [118], further causing sympathetic hyperactivity via NLRP3/IL‐1β dependent pathway [108]. The authors speculated that SAP130, a spliceosome-associated protein 130, and cytokines such as TNF‐α and IL‐6 derived from MI crossing through the BBB, could activate Mincle in microglia, resulting in pro‐inflammatory cytokines release and broader inflammatory response. However, how cytokines could affect sympathetic activity post‐MI is unknown. Previous studies have demonstrated that TNF-α and IL‐1β secretion promotes expression of activated nuclear transcription factor kappa B (NF‐kB) and ROS, which could enhance glutamatergic and adrenergic excitatory transmission and attenuate GABAergic inhibitory activity in the PVN [119, 120].
It is well documented that microglia activation occurs in animal models of stress-induced depression [121]. Thus, the activation of microglia was suggested to be involved in depression-like behavior developing in the chronic phase after MI. In the hippocampus of mice undergoing cardiac I/R, an increase in the number of microglial cells has been observed, and it is associated with a worsening of performance in learning tests involving hippocampus functionality [122]. Similarly, after permanent LAD coronary ligation, an increase in microglia and its activation, associated with a worsening of cognitive function, has been observed in the dentate gyrus of the hippocampus in mice [104, 114], and in dentate gyrus of the hippocampus and in the PVN of the hypothalamus in rats [106, 123]. It is likely that microglia activation, increasing local secretion of pro-inflammatory cytokines, could exacerbate neuronal activity [103, 105] and induce neuronal death [112] leading to behavioral signs of depression.
In additions to microglia, MI could activate also astrocytes. As observed in the amygdala of rats, MI induced morphological changes of astrocytes starting from 3 days after permanent LAD artery ligation, and 14 days post-MI, several GFAP+ astrocytes showed hypertrophied cytoplasm and highly ramified processes [123]. This evidence was confirmed by a more recent study showing this typical “activated” morphology of GFAP+ cells in the PVN of rats at 1 day after permanent LAD. Relevantly, astrocytes inhibition, by PVN injection of fluorocitrate, reduced the MI-induced expression of pro-inflammatory cytokines (TNF-α and IL-6), neuronal activation, and ventricular arrhythmia occurrence, and improved ventricular electrical instability [124]. Similarly, an early astrocytic activation was observed in rat model of cardiac I/R, as suggested by the increased numbers of GFAP+ cells, dendritic volume and complexity, and decreased filament length at the end of the 120 min of reperfusion period [109]. As for microglia, a recent study suggested that astrocytes regulate sympathetic activity via the release of ATP in the RVLM of rats with MI [125]. On the contrary, in MI mice with permanent LAD artery ligation were not observed an increased density of astrocytes in the hippocampus [114] and in the rostral ventrolateral medulla [126] after MI. This discrepancy in the effect of cardiac ischemia on astrocytes activation appears to be species-related, as seems also for microglia. Overall, these results suggest that astrocyte activation is involved in neuroinflammation and in the early phase of cardiac sympathetic hyper-activation following MI. This action seems to be mediated by the angiotensin II type 1 receptor (AT1R), which is weakly expressed in astrocytes under basal condition. The GFAP-specific AT1R deletion in mice with permanent LAD artery ligation inhibited the MI-induced upregulation of brain AT1R, despite the preservation of neuronal AT1R expression, and enhanced central sympathetic outflow, probably by inhibiting reactive oxygen species (ROS) [126].
Neuronal apoptosis, neuronal plasticity, and impaired neurogenesis
Inflammation is often associated with enhanced pro-apoptotic processes and altered neurogenesis, and these phenomena happen also in the brain after MI and could contribute to the development of the depressive-like behavior.
In rats subjected to cardiac I/R, MI acutely decreased P13K activity and increased Bax/Bcl-2 ratio, caspase-3 activity, and numbers of TUNEL-positive cells in the amygdala, suggesting a possible link with the major depressive disorder observed following MI [127]. The same result on a caspase-3 activation in the amygdala of rats with cardiac I/R was observed in a recent study [128]. Interestingly, in the same animal model, an increased Bax/Bcl-2 ratio was observed in the hypothalamus and prefrontal cortex, but not in the amygdala and hippocampus, at subacute phase after MI. Together with the absence of enhanced caspase-3 activity, these results suggest a caspase-3 independent mechanism or different time of apoptosis activation in these cerebral structures after MI [129], as demonstrated by another study [130].
It was suggested that the MI-induced brain apoptosis may be ascribed to oxidative stress, mitochondrial dysfunction, and enhanced permeabilization of the mitochondrial outer membrane. The latter may lead to the release of pro-apoptotic proteins including Bax and cytochrome c, which activates caspase cascade [110]. Another study in a rat model of cardiac I/R also showed that MI decreased the expression of the receptor-interacting serine/threonine-protein kinase 1 (RIPK1), a protein implicated in the plasma membrane permeabilization and necrotic cell death, likely due to caspase-8-mediated cleavage, shifting the cell towards apoptosis [131]. An activation of apoptosis was also observed in rats with permanent LAD artery ligation. MI induced an increased mRNA expression of caspase-3, caspase-8, and caspase-9 and Bcl-2 in the hippocampus, which were associated with anxiety-like behavior [132]. A recent study demonstrated that enhanced plasma level of TNF-α contributes to apoptosis via activation of the extrinsic pathway in the limbic system after MI. The inhibition of TNF-α by PEG sTNFRI, a soluble p55 type 1 TNF receptor, reversed the MI-induced increase of caspase-3 and caspase-8 activity in medial amygdala, dentate gyrus, and hippocampus (CA1) [133].
The role of TNF-α signaling in the MI-mediated neurodegenerative processes is also supported by evidence showing that pharmacological blocking or genetic deletion of TNF-α ameliorated the reduction of cortical dendritic spines in mice with permanent LAD coronary ligation [104]. The dendritic spine density was also found to be reduced in the hippocampus of rats with transient LAD artery ligation [109, 110]. A decreased loss of dendritic spine density was observed after inhibition of PCSK9, suggesting its involvement in neuronal damage following cardiac I/R insults [109]. On the contrary, two studies showed no neurodegeneration and neuronal death in the dentate gyrus and hippocampus (CA1) of rats [134] and mice [135] with permanent LAD artery ligation. These contrasting results may be caused by the short time scale of apoptosis, the time of sampling, or the sensibility of techniques utilized.
In addition to apoptosis, MI could also affect neurogenesis. In mice with transient LAD artery ligation, neurogenesis in the granular zone of dentate gyrus was significantly decreased both acutely and chronically after MI, potentially contributing to the cognitive decline [122]. In contrast, in rats with permanent LAD artery ligation, MI enhanced cell proliferation and neuroblast differentiation in the subgranular zone of the dentate gyrus [134], while in mice with permanent LAD artery ligation, MI did not influence neurogenesis [41, 135]. In a more recent study using mice with permanent LAD artery ligation, neurogenesis slightly decreased in the hippocampus and in the piriform cortex [114]. These discrepancies may be ascribed to difference of the model, the species, and the age of animals.
Future prospective
As detailed in this review, the innate immune response and systemic inflammation could play a pivotal role in the development of neuroinflammation after MI. Thus, it would be rational to suppose that appropriate immunosuppressant and anti-inflammatory therapeutic strategies may have potential beneficial effects on the brain in post-MI conditions. Although there are no studies in this regard, some data obtained in different experimental or clinical contexts of systemic inflammation appear promising. In a model of chronic inflammatory disorder, as rheumatoid arthritis, the antagonism of TNF-α with infliximab reduced the infarct volume and the amount of microglia and activated macrophages in the ischemic hemisphere and improved the integrity of BBB and the neurological deficit in mice with ischemia/reperfusion (I/R) brain injury [136]. In patients with psoriasis, the antagonism of TNF-α with etanercept also reduced the circulating levels of inflammatory and cardiovascular proteins, such as TNF-α, IL-1β, IL-6, and IL-8 [137]. Moreover, the antagonism of IL-1β with canakinumab significantly reduced the evidence of residual inflammatory risk in patients with prior MI [138]. Relevantly, the systemic administration of interleukin-1 receptor antagonist (IL-1Ra) has shown to be neuroprotective and increased post-stroke neurogenesis in a murine model of atherosclerosis, obesity, and insulin resistance after cerebral ischemia, suggesting that this strategy as potential neuroprotective in patients with a raised inflammatory burden [139].
However, this anti-inflammatory strategy is far to be demonstrated safe in post-MI conditions, and instead could be contraindicated. Indeed, etanercept reduced systemic inflammation but increased platelet activation in MI patients [140], possibly leading a higher risk of cardiovascular events. In line, high doses of infliximab, a chimeric monoclonal antibody to TNF-α, increased the combined risk of death from any cause or hospitalization for heart failure (hazard ratio = 2.84) in patients with moderate-to-severe heart failure [141]. In rats, infliximab had a slight protective effect in the early hours after MI, but in the following days, it exacerbated the cardiac dysfunction, likely blocking the functions of compensatory mechanisms after MI such as cardiac remodeling, preventing tissue repair, and promoting further myocardial injury [142]. Altogether, these data suggest that a tolerable inflammatory process following MI could boost a healing procedure of heart tissue injuries and remodeling. Thus, anti-inflammatory strategies to reduce systemic inflammation after MI should be carefully balanced as they might interfere with cardiac tissue repair and healing.
In addition to cytokine antagonists, EVs could offer therapeutic chances for neuroprotection after MI, because of the low immunogenicity and toxicity, high blood circulation stability, and the unique ability of EVs to pass through the BBB. In particular, mesenchymal stem cell (MSC)derived EVs emerge as a potential candidate. This type of EVs could play a beneficial role in both the heart and brain in post-MI conditions. Indeed, the treatment with MSC-derived EVs was shown to reduce infarct size and enhance cardiac function and geometry, by decreasing oxidative stress and activating pro-survival signaling, in several animal models of MI [143,144,145]. Parallel to the effectiveness at cardiac level, MSC-derived EVs exerted also neuroprotective effects. In vitro studies have shown that MSC-derived EVs increase neuronal survival and stimulate neural cell regeneration, growth, and proliferation [146, 147]. Relevantly, MSC-derived EVs shifted microglia from activated pro-inflammatory states towards homeostatic and shriveling functions after cortical injury in aged monkeys [148]. In rats with focal brain injury, human bone marrow, MSC-derived EVs attenuated neuroinflammation, decreasing the level of pro-inflammatory cytokines and chemokines, and the number of activated immune cells, such as astrocytes, microglia, and infiltrating leucocytes, including T cytotoxic cells [149]. However, the neuroprotective effects of EVs are not exclusive of MSC-derived EVs. Indeed, treatment with platelet-derived EVs increased proliferation of neural progenitor cells, induced angiogenesis, and improved general motor and cognitive functions in rats after permanent ischemic stroke [150].
Several evidence reveal that the mechanisms of neuroprotective action of EVs might involve the transfer of specific miRNAs to resident cells [151, 152]. Various types of miRNAs could be involved in these processes, including miR-133b [153]. In vitro studies have proven that EVs from astrocytes, which were treated with MSC-derived EVs over-expressing miR-133b, significantly increase neurite growth in primary cortical neuronal cultures subjected to oxygen–glucose deprivation (OGD) as compared to EVs derived from untreated astrocytes [154]. In vivo studies confirmed that EVs modulate responses after ischemic stroke by transferring miR-133b. Indeed, MSC-derived EVs improved functional recovery and exhibited increased axonal plasticity and neurite remodeling in rats with transient cerebral ischemia. These neuroprotective effects were attenuated by the knocking-down of the miR-133b level in MSC-derived EVs, while were significantly enhanced by the miR-133b over-expression [83].
However, to translate MSC-derived EVs over-expressing miR-133b from the bench to the bedside, further studies evaluating the impact on the heart–brain axis, especially on the mutual interactions of these two organs in post-MI conditions, should be performed. Indeed, the same EV-miRNA may affect different signaling pathways in the heart or in the brain, or exert different effects depending on the stage of heart disease after MI, with opposite effects on disease outcomes. An example of this possible dual effect is provided by miR-1. In the early phases after MI, when the circulating level of miR-1 is increased, the antagonism of miR-1 with a specific antagomir exerted a significant protective effect on heart function, decreasing cardiomyocyte apoptosis and alleviating myocardial fibrosis and remodeling. The enhanced expression of miR-1 by a lentiviral vector exerted instead opposite effects [155]. In line, in an animal model of I/R injury using transgenic mice over-expressing miR-1, it was observed an increase of infarct size, apoptosis, and caspase-3 expression [156]. On the contrary, miR-1 expression was decreased in failing hearts [157]. The restoring of miR-1 expression was associated with normalized sodium–calcium exchanger (NCX)-1 expression and improved cardiac function in a chronic post-MI rat model of heart failure [157]. In mice with ligation of LAD, transplantation of MSCs over-expressing miR-1 was more effective for cardiac repair and for improved cardiac function, by enhancing cell survival and cardiomyocyte differentiation, compared to the MSCs without miR-1 over-expression [158]. In rat with MI, the upregulation of miR-1 expression partially contributed to the post-transcriptional repression of hyperpolarization-activated cyclic nucleotide-gated channel (HNC) protein expression, which may contribute to the effect of spironolactone to reduce the incidence of MI-associated ventricular arrhythmias [159].
Altogether, these data suggest a dual role of miR-1 in the phases of heart disease post-MI. In the early phase of MI, miR-1 may regulate cell death and oxidative stress, while in the later phase may contribute to post-MI remodeling or function as compensatory mechanisms.
However, the neuroprotective effects of the antagonism of miR-1 in post-MI conditions were provided by solid evidence (see “Mechanisms of heart–brain interaction after MI”) [81, 82, 87, 88]. To note, the heart-specific miR-1 over-expression was shown to directly mediate brain dysfunction [81]. Indeed, the transgenic mouse model of cardiac-specific over-expression of miR-1–2 showed increased miR-1 levels not only in the heart, but also in the blood and hippocampus, and cognitive impairment. It is reasonable to assume that, after crossing the BBB, the circulating heart-derived EVs release miR-1 to cerebral resident cells, which in turn inhibit the expression of BDNF, leading to the impairment of cognition [81]. Similarly, the increased miR-1 level observed in the hippocampus of MI mice [82, 89], in spite of unchanged endogenous biogenesis [82], suggests that this increase might arise directly from the infarcted heart through EV-mediated transfer. Indeed, the inhibition of EVs biogenesis prevented the MI-induced elevation of miR-1 levels in the blood and hippocampus, and the subsequent hippocampal microtubule damage [82]. In all these three studies, the knockdown of miR-1 reversed the cerebral dysfunctions, restoring the BDNF levels or the neuronal microtubules in the hippocampus.
In summary, due to the promising results obtained in experimental studies, an application of EVs in the management of brain complications in MI patients is of great interest from the clinical point of view. Although, no clinical trial of EVs transplantation has been performed to evaluate cerebral outcome in MI patients; these results will promote the development of protocols for the use of EVs in clinical trials.
Conclusion
Recent evidence has led to consider myocardial infarction not only a mere disease of the heart, but a more complex disease mediating pathological response of many distant organs, including the brain. Myocardial infarction has a short- and long-term deleterious impact on brain homeostasis, which plays a causative role in occurrence in anxiety, depression cognitive deficits, and stroke in MI patients. In addition to the increased coagulation and thrombosis, other factors may favor brain damage after MI. In particular, enhanced systemic inflammation and changes in EVs and circulating miRNAs pattern released from heart and blood cells could also play a role in increase the risk of stroke in patients with MI. It is conceivable that these different pathways, likely interact closely with each other, could contribute to neuroinflammation and subsequent alteration of neuronal function, including apoptosis and neurogenesis, and oxidative stress. However, since the data are obtained from limited experiments studies, future research is required to precisely identify the further possible cardiac-specific mechanisms involved in facilitating the onset, or in affecting, the evolution of stroke in patient with MI. A better understanding of interactions within the heart–brain axis will improve the strategies, including novel neuroprotective approaches, to prevent or treat the brain dysfunctions of MI patients.
Availability of data and material
All data and materials are available and support the published claims and comply with field standards.
Code availability
Not applicable. No coding in the study.
References
Chen Z, Venkat P, Seyfried D, Chopp M, Yan T, Chen J (2017) Brain–heart interaction. Circ Res [Internet]. 121:451–68. Available from: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.117.311170
Thackeray JT, Hupe HC, Wang Y, Bankstahl JP, Berding G, Ross TL et al (2018) Myocardial inflammation predicts remodeling and neuroinflammation after myocardial infarction. J Am Coll Cardiol [Internet]. 71:263–75. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109717416408
Battaglini D, Robba C, Lopes da Silva A, dos Santos Samary C, Leme Silva P, Dal Pizzol F et al (2020) Brain–heart interaction after acute ischemic stroke. Crit Care [Internet]. 24:163. Available from: https://ccforum.biomedical.com/articles/10.1186/s13054-020-02885-8
Natelson BH (1985) Neurocardiology. Arch Neurol [Internet]. 42:178. Available from: https://archneur.jamanetwork.com/article.aspx?doi=10.1001/archneur.1985.04060020096022
Kaplan A, Yabluchanskiy A, Ghali R, Altara R, Booz GW, Zouein FA (2018) Cerebral blood flow alteration following acute myocardial infarction in mice. Biosci Rep [Internet]. 38. Available from: https://portlandpress.com/bioscirep/article/doi/10.1042/BSR20180382/88709/Cerebral-blood-flow-alteration-following-acute
Kalkman HO (2020) The association between vascular inflammation and depressive disorder. Causality, biomarkers and targeted treatment. Pharmaceuticals [Internet]. 13:92. Available from: https://www.mdpi.com/1424-8247/13/5/92
Guck TP, Kavan MG, Elsasser GN, Barone EJ (2001) Assessment and treatment of depression following myocardial infarction. Am Fam Physician [Internet]. 64:641–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11529263
Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG et al (2005) ACC/AHA 2005 Guideline update for the diagnosis and management of chronic heart failure in the adult. Circulation [Internet]. 112. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.105.167586
Feng H-P, Chien W-C, Cheng W-T, Chung C-H, Cheng S-M, Tzeng W-C (2016) Risk of anxiety and depressive disorders in patients with myocardial infarction. Medicine (Baltimore) [Internet]. 95:e4464. Available from: https://journals.lww.com/00005792-201608230-00028
Breteler MMB, Claus JJ, Grobbee DE, Hofman A (1994) Cardiovascular disease and distribution of cognitive function in elderly people: the Rotterdam study. BMJ [Internet]. 308:1604–8. Available from: https://www.bmj.com/;ookup/doi/10.1136/bmj.308.6944.1604
Wolters FJ, Segufa RA, Darweesh SKL, Bos D, Ikram MA, Sabayan B et al (2018) Coronary heart disease, heart failure, and the risk of dementia: a systematic review and meta-analysis. Alzheimer’s dement [Internet]. 14:1493–504. Available from: https://doi.wiley.com/10.1016/j.jalz.2018.01.007
van Melle JP, de Jonge P, Spijkerman TA, Tijssen JGP, Ormel J, van Veldhuisen DJ et al (2004) Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta-analysis. Psychosom Med [Internet]. 66:814–22. Available from: https://journals.lww.com/00006842-200411000-00004
Wen Y, Yang Y, Shen J, Luo S (2021) Anxiety and prognosis of patients with myocardial infarction: a meta-analysis. Clin Cardiol [Internet]. 44:761–70. Available from: https://onlinelibrary.wiley.com/doi/10.1002/clc.23605
Ikram MA, van Oijen M, de Jong FJ, Kors JA, Koudstaal PJ, Hofman A et al (2008) Unrecognized myocardial infarction in relation to risk of dementia and cerebral small vessel disease. Stroke [Internet]. 39:1421–6. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.107.501106
Bursi F, Rocca WA, Killian JM, Weston SA, Knopman DS, Jacobsen SJ et al (2006) Heart disease and dementia: a population-based study. Am J Epidemiol [Internet]. 163:135–41. Available from: https://academic.oup.com/aje/article/163/2/135/95837/Heart-Disease-and-Dementia-A-Populationbased-Study
Dutta M, Hanna E, Das P, Steinhubl SR (2006) Incidence and prevention of ischemic stroke following myocardial infarction: review of current literature. Cerebrovasc Dis [Internet]. 22:331–9. Available from: https://www.karger.com/Article/FullText/94847
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M et al (2016) Heart disease and stroke statistics—2016 update. Circulation [Internet]. 2016;133. Available https://www.ahajournals.org/doi./10.1161/CIR.0000000000000350
Adams HP, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL et al (1993) Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke [Internet]. 1993;24:35–41. Available from: https://www.ahajournals.org/doi/10.1161/01.STR.24.1.35
Witt BJ, Brown RD, Jacobsen SJ, Weston SA, Yawn BP, Roger VL (2005) A community-based study of stroke incidence after myocardial infarction. Ann Intern Med [Internet]. 143:785. Available from: http://annals.org/article.aspx?doi=https://annals.org/article.aspx?doi=10.7326/0003-4819-143-11-200512060-00006
Merkler AE, Diaz I, Wu X, Murthy SB, Gialdini G, Navi BB et al (2018) Duration of heightened ischemic stroke risk after acute myocardial infarction. J Am Heart Assoc [Internet]. 7. Available from: https://www.ahajournals.org/doi/https://www.ahajournals.org/doi/10.1161/JAHA.118.010782
Åström A, Söderström L, Mooe T (2020) Risk of ischemic stroke after acute myocardial infarction in patients undergoing coronary artery bypass graft surgery. Sci Rep [Internet]. 10:3831. Available from: http://www.nature.com/articles/s41598-020-60854-1
Kajermo U, Ulvenstam A, Modica A, Jernberg T, Mooe T (2014) Incidence, trends, and predictors of ischemic stroke 30 days after an acute myocardial infarction. Stroke [Internet]. 45:1324–30. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.113.001963
Ulvenstam A, Kajermo U, Modica A, Jernberg T, Söderström L, Mooe T (2014) Incidence, trends, and predictors of ischemic stroke 1 year after an acute myocardial infarction. Stroke [Internet]. 45:3263–8. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.114.005770
Pride YB, Piccirillo BJ, Gibson CM (2013) Prevalence, consequences, and implications for clinical trials of unrecognized myocardial infarction. Am J Cardiol [Internet]. 111:914–8. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0002914912024769
Merkler AE, Sigurdsson S, Eiriksdottir G, Safford MM, Phillips CL, Iadecola C et al (2019) Association between unrecognized myocardial infarction and cerebral infarction on magnetic resonance imaging. JAMA Neurol [Internet]. 76:956. Available from: https://jamanetwork.com/journals/jamaneurology/fullarticle/2733674
Brammås A, Jakobsson S, Ulvenstam A, Mooe T (2013) Mortality after ischemic stroke in patients with acute myocardial infarction. Stroke [Internet]. 44:3050–5. Available from: https://www.ahajournals.org/doi/10.1161/STROKEAHA.113.001434
Pahuja M, Chehab O, Ranka S, Mishra T, Ando T, Yassin AS et al (2021) Incidence and clinical outcomes of stroke in elevation myocardial infarction and cardiogenic shock. Catheter Cardiovasc Interv [Internet]. 97:217–25. Available from: https://onlinelibrary.wiley.com/doi/10.1002/ccd.28919
Fioranelli M, Bottaccioli AG, Bottaccioli F, Bianchi M, Rovesti M, Roccia MG (2018) Stress and inflammation in coronary artery disease: a review psychoneuroendocrineimmunology-based. Front Immunol [Internet]. 9. Available from: https://www.frontiersin.org/article/doi/10.3389/fimmu.2018.02031/full
Levine B, Kalman J, Mayer L, Fillit HM, Packer M (1990) Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med [Internet]. 323:236–41. Available from: http://www.nejm.org/doi/abs/https://www.nejm.org/doi/10.1056/NEJM199007263230405
Li W, Hsiao H-M, Higashikubo R, Saunders BT, Bharat A, Goldstein DR et al (2016) Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight [Internet]. 1. Available from: https://insight.jci.org/articles/view/87315
Soehnlein O, Lindbom L (2010) Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol [Internet]. 10:427–39. Available from: http://www.nature.com/articles/nri2779
Saparov A, Ogay V, Nurgozhin T, Chen WCW, Mansurov N, Issabekova A et al (2017) Role of the immune system in cardiac tissue damage and repair following myocardial infarction. Inflamm Res [Internet]. 66:739–51. Available from: https://link.springer.com/doi/10.1007/s00011-017-1060-4
Methe H, Brunner S, Wiegand D, Nabauer M, Koglin J, Edelman ER (2005) Enhanced T-helper-1 lymphocyte activation patterns in acute coronary syndromes. J Am Coll Cardiol [Internet]. 45:1939–45. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109705007552
Cheng X, Yu X, Ding Y, Fu Q, Xie J, Tang T et al (2008) The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol [Internet]. 127:89–97. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1521661608000120
Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G et al (2012) Activation of CD4 + T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation [Internet]. 125:1652–63. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.111.044164
Zouggari Y, Ait-Oufella H, Bonnin P, Simon T, Sage AP, Guérin C et al (2013) B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med [Internet]. 19:1273–80. Available from: http://www.nature.com/articles/nm.3284
Pudil R, Pidrman V, Krejsek J, Gregor J, Tichý M, Andrýs C et al (1999) Cytokines and adhesion molecules in the course of acute myocardial infarction. Clin Chim Acta [Internet]. 280:127–34. Available from: https://linkinghub.elsevier.com/retrieve/pii/S000989819800179X
Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C et al (2000) Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation [Internet]. 102:3060–7. Available from: https://www.ahajournals.org/doi/10.1161/01.CIR.102.25.3060
Mizia-Stec K, G??sior Z, Zahorska-Markiewicz B, Janowska J, Szulc A (2003) Jastrz??bska-Maj E, et al. Serum tumour necrosis factor-alpha, interleukin-2 and interleukin-10 activation in stable angina and acute coronary syndromes. Coron Artery Dis [Internet]. 14:431–8. Available from: http://journals.lww.com/00019501-200309000-00003
Francis J, Chu Y, Johnson AK, Weiss RM, Felder RB (2004) Acute myocardial infarction induces hypothalamic cytokine synthesis. Am J Physiol Circ Physiol [Internet]. 286:H2264–71. Available from: https://www.physiology.org/doi/10.1152/ajpheart.01072.2003
Gouweleeuw L, Pol C, Simonides W, V de Kleijn D, Schoemaker R (2017) Evidence for neuroinflammation after myocardial infarction in a mouse model. Hear Mind [Internet]. 1:134. Available from: http://www.heartmindjournal.org/text.asp?2017/1/4/134/244372
Kang Y-M, Zhang Z-H, Johnson RF, Yu Y, Beltz T, Johnson AK et al (2006) Novel effect of mineralocorticoid receptor antagonism to reduce proinflammatory cytokines and hypothalamic activation in rats with ischemia-induced heart failure. Circ Res [Internet]. 99:758–66. Available from: https://www.ahajournals.org/doi/10.1161/01.RES.0000244092.95152.86
Liu H, Luiten PGM, Eisel ULM, Dejongste MJL, Schoemaker RG (2013) Depression after myocardial infarction: TNF-α-induced alterations of the blood–brain barrier and its putative therapeutic implications. Neurosci Biobehav Rev [Internet]. 37:561–72. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0149763413000390
Banks WA, Kastin AJ, Broadwell RD (1995) Passage of Cytokines across the Blood-Brain Barrier. Neuroimmunomodulation [Internet]. 2:241–8. Available from: https://www.karger.com/Article/FullText/97202
Raucci A, Di Maggio S, Scavello F, D’Ambrosio A, Bianchi ME, Capogrossi MC (2019) The Janus face of HMGB1 in heart disease: a necessary update. Cell Mol Life Sci [Internet]. 76:211–29. Available from: https://link.springer.com/doi/10.1007/s00018-018-2930-9
Festoff BW, Sajja RK, van Dreden P, Cucullo L (2016) HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J Neuroinflammation [Internet]. 13:194. Available from: https://jneuroinflamation.biomecentral.com/doi/10.1186/s12974-016-0670-z
Biemmi V, Milano G, Ciullo A, Cervio E, Burrello J, Dei Cas M et al (2020) Inflammatory extracellular vesicles prompt heart dysfunction via TRL4-dependent NF-κB activation. Theranostics [Internet]. 10:2773–90. Available from: http://www.thno.org/v10p2773.htm
Felder RB, Francis J, Zhang Z-H, Wei S-G, Weiss RM, Johnson AK (2003) Heart failure and the brain: new perspectives. Am J Physiol Integr Comp Physiol [Internet]. 284:R259–76. Available from: https://www.physiology.org/doi/10.1152/ajpregu.00317.2002
Zaborowski MP, Balaj L, Breakefield XO, Lai CP (2015) Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience [Internet]. 65:783–97. Available from: http://academic.oup.com/bioscience/article/65/8/783/240409/Extracellular-Vesicles-Composition-Biological
Ibrahim A, Marbán E (2016) Exosomes: Fundamental biology and roles in cardiovascular physiology. Annu Rev Physiol [Internet]. 78:67–83. Available from: https://www.annualreviers.org/doi/10.1146/annurev-physiol-021115-104929
Mallat Z, Benamer H, Hugel B, Benessiano J, Steg PG, Freyssinet J-M et al (2000) Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation [Internet]. 101:841–3. Available from: https://www.ahajournals.org/doi/10.1161/01.CIR.101.8.841
Rodriguez JA, Orbe J, Saenz-Pipaon G, Abizanda G, Gebara N, Radulescu F et al (2018) Selective increase of cardiomyocyte derived extracellular vesicles after experimental myocardial infarction and functional effects on the endothelium. Thromb Res [Internet]. 170:1–9. Available from: https://linkinghub.elsevier.com/retrieve/pii/S004938481830447X
Biasucci LM, Porto I, Di Vito L, De Maria GL, Leone AM, Tinelli G et al (2012) Differences in microparticle release in patients with acute coronary syndrome and stable angina. Circ J [Internet]. 76:2174–82. Available from: https://www.jstage.jst.go.jp/article/circj/76/9/76_CJ-12-0068/_article
Cui Y, Zheng L, Jiang M, Jia R, Zhang X, Quan Q et al (2013) Circulating microparticles in patients with coronary heart disease and its correlation with interleukin-6 and C-reactive protein. Mol Biol Rep [Internet]. 40:6437–42. Available from: https://link.springer.com/doi/10.1007/s11033-013-2758-1
Suades R, Padró T, Crespo J, Ramaiola I, Martin-Yuste V, Sabaté M et al (2016) Circulating microparticle signature in coronary and peripheral blood of ST elevation myocardial infarction patients in relation to pain-to-PCI elapsed time. Int J Cardiol [Internet]. 202:378–87. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0167527315304526
Wang B, Li T, Han X, Li Y, Cheng W, Wang L et al (2020) The level of circulating microparticles in patients with coronary heart disease: a systematic review and meta-analysis. J Cardiovasc Transl Res [Internet]. 13:702–12. Available from: https://link.springer.com/doi/10.1007/s12265-019-09945-7
Yu X, Deng L, Wang D, Li N, Chen X, Cheng X et al (2012) Mechanism of TNF-α autocrine effects in hypoxic cardiomyocytes: initiated by hypoxia inducible factor 1α, presented by exosomes. J Mol Cell Cardiol [Internet]. 53:848–57. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022282812003707
Gupta S, Knowlton AA (2007) HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Physiol Circ Physiol [Internet]. 292:H3052–6. Available from: https://www.physiology.org/doi/10.1152/ajpheart.01355.2006
Loyer X, Zlatanova I, Devue C, Yin M, Howangyin K-Y, Klaihmon P et al (2018) Intra-cardiac release of extracellular vesicles shapes inflammation following myocardial infarction. Circ Res [Internet]. 123:100–6. Available from: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.117.311326
Otero-Ortega L, Alonso-López E, Pérez-Mato M, Laso-García F, Gómez-de Frutos MC, Diekhorst L et al (2020) Similarities and differences in extracellular vesicle profiles between ischaemic stroke and myocardial infarction. Biomedicines [Internet]. 9:8. Available from: https://www.mdpi.com/2227-9059/9/1/8
Ridder K, Keller S, Dams M, Rupp A-K, Schlaudraff J, Del Turco D et al (2014) Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. Barres BA, editor. PLoS Biol [Internet]. 12:e1001874. Available from: https://dx.plos.org/doi/10.1371/journal.pbio.1001874
Matsumoto J, Stewart T, Sheng L, Li N, Bullock K, Song N et al (2017) Transmission of α-synuclein-containing erythrocyte-derived extracellular vesicles across the blood-brain barrier via adsorptive mediated transcytosis: another mechanism for initiation and progression of Parkinson’s disease? Acta Neuropathol Commun [Internet]. 5:71. Available from: https://actaneurocomms.biomedcentral.com/doi/10.1186/s40478-017-0470-4
Li JJ, Wang B, Kodali MC, Chen C, Kim E, Patters BJ et al (2018) In vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J Neuroinflammation [Internet]. 15:8. Available from: https://jneuroinflammation.biomedcentral.com/doi/10.1186/s12974-017-1038-8
Chen CC, Liu L, Ma F, Wong CW, Guo XE, Chacko J V et al (2016) Elucidation of exosome migration across the blood–brain barrier model in vitro. Cell Mol Bioeng [Internet]. 9:509–29. Available from: https://link.springer.com/doi/10.1007/s12195-016-0458-3
Skokos D, Botros HG, Demeure C, Morin J, Peronet R, Birkenmeier G et al (2003) Mast cell-derived exosomes induce phenotypic and functional maturation of dendritic cells and elicit specific immune responses in vivo. J Immunol [Internet]. 170:3037–45. Available from: https://www.jimmunol.org/doi/10.4049/jimmunol.170.6.3037
Zhang B, Sun X, Mei H, Wang Y, Liao Z, Chen J et al (2013) LDLR-mediated peptide-22-conjugated nanoparticles for dual-targeting therapy of brain glioma. Biomaterials [Internet]. 34:9171–82. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0142961213009873
Béduneau A, Saulnier P, Benoit J-P (2007) Active targeting of brain tumors using nanocarriers. Biomaterials [Internet]. 28:4947–67. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0142961207004656
Goulatis LI, Shusta EV (2017) Protein engineering approaches for regulating blood–brain barrier transcytosis. Curr Opin Struct Biol [Internet]. 45:109–15. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0959440X16301397
Balusu S, Van Wonterghem E, De Rycke R, Raemdonck K, Stremersch S, Gevaert K et al (2016) Identification of a novel mechanism of blood–brain communication during peripheral inflammation via choroid plexus‐derived extracellular vesicles. EMBO Mol Med [Internet]. 8:1162–83. Available from: https://onlinelibrary.wiley.com/doi/10.15252/emmm.201606271
Jonas S, Izaurralde E (2015) Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet [Internet]. 16:421–33. Available from: http://www.nature.com/articles/nrg3965
Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T (2010) Secretory Mechanisms and Intercellular Transfer of MicroRNAs in Living Cells. J Biol Chem [Internet]. 285:17442–52. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021925819354869
Vidigal JA, Ventura A (2015) The biological functions of miRNAs: lessons from in vivo studies. Trends Cell Biol [Internet]. 25:137–47. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0962892414001974
Wang G-K, Zhu J-Q, Zhang J-T, Li Q, Li Y, He J et al (2010) Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J [Internet]. 31:659–66. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehq013
Condrat CE, Thompson DC, Barbu MG, Bugnar OL, Boboc A, Cretoiu D et al (2020) miRNAs as biomarkers in disease: latest findings regarding their role in diagnosis and prognosis. Cells [Internet]. 9:276. Available from: https://www.mdpi.com/2073-4409/9/2/276
Meder B, Keller A, Vogel B, Haas J, Sedaghat-Hamedani F, Kayvanpour E et al (2011) MicroRNA signatures in total peripheral blood as novel biomarkers for acute myocardial infarction. Basic Res Cardiol [Internet]. 106:13–23. Available from: https://link.springer.com/doi/10.1007/s00395-010-0123-2
Boštjančič E, Zidar N, Štajer D, Glavač D (2010) MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology [Internet]. 115:163–9. Available from: https://www.karger.com/Article/FullText/268088
Devaux Y, Vausort M, Goretti E, Nazarov P V, Azuaje F, Gilson G et al (2012) Use of circulating microRNAs to diagnose acute myocardial infarction. Clin Chem [Internet]. 58:559–67. Available from: https://academic.oup.com/clinchem/article/58/3/559/5620606
Li C, Fang Z, Jiang T, Zhang Q, Liu C, Zhang C et al (2013) Serum microRNAs profile from genome-wide serves as a fingerprint for diagnosis of acute myocardial infarction and angina pectoris. BMC Med Genomics [Internet]. 6:16. Available https://bmcmedgenomics.biomedcentral.com/doi/10.1186/1755-8794-6-16
Cheng M, Yang J, Zhao X, Zhang E, Zeng Q, Yu Y et al (2019) Circulating myocardial microRNAs from infarcted hearts are carried in exosomes and mobilise bone marrow progenitor cells. Nat Commun [Internet]. 10:959. Available from: http://www.nature.com/articles/s41467-019-08895-7
van den Berg MMJ, Krauskopf J, Ramaekers JG, Kleinjans JCS, Prickaerts J, Briedé JJ (2020) Circulating microRNAs as potential biomarkers for psychiatric and neurodegenerative disorders. Prog Neurobiol [Internet]. 185:101732. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0301008219303508
Ma J-C, Duan M-J, Sun L-L, Yan M-L, Liu T, Wang Q et al (2015) Cardiac over-expression of microRNA-1 induces impairment of cognition in mice. Neuroscience [Internet]. 299:66–78. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0306452215004133
Sun L-L, Duan M-J, Ma J-C, Xu L, Mao M, Biddyut D et al (2018) Myocardial infarction-induced hippocampal microtubule damage by cardiac originating microRNA-1 in mice. J Mol Cell Cardiol [Internet]. 120:12–27. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022282818301597
Xin H, Li Y, Liu Z, Wang X, Shang X, Cui Y et al (2013) MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells [Internet]. 31:2737–46. Available from: https://onlinelibrary.wiley.com/doi/10.1002/stem.1409
Chen J-F, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM et al (2006) The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet [Internet]. 38:228–33. Available from: http://www.nature.com/articles/ng1725
Varendi K, Kumar A, Härma M-A, Andressoo J-O (2014) miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cell Mol Life Sci [Internet]. 71:4443–56. Available from: https://link.springer.com/doi/10.1007/s00018-014-1628-x
Chang C-Y, Lui T-N, Lin J-W, Lin Y-L, Hsing C-H, Wang J-J et al (2016) Roles of microRNA-1 in hypoxia-induced apoptotic insults to neuronal cells. Arch Toxicol [Internet]. 90:191–202. Available from: https://link.springer.com/doi/10.1007/s00204-014-1364-x
Selvamani A, Sathyan P, Miranda RC, Sohrabji F (2012) An antagomir to MicroRNA Let7f promotes neuroprotection in an ischemic stroke model. Priller J, editor. PLoS One [Internet]. 7:e32662. Available from: https://dx.plos.org/doi/10.1371/journal.pone.0032662
Talebi A, Rahnema M, Bigdeli MR (2019) Effect of intravenous injection of antagomiR-1 on brain ischemia. Mol Biol Rep [Internet]. 46:1149–55. Available from: https://link.springer.com/doi/10.1007/s11033-018-04580-y
Ma J-C, Duan M-J, Li K-X, Biddyut D, Zhang S, Yan M-L et al (2018) Knockdown of microRNA-1 in the hippocampus ameliorates myocardial infarction induced impairment of long-term potentiation. Cell Physiol Biochem [Internet]. 50:1601–16. Available from: https://www.karger.com/Article/FullText/494657
Yu Y-M, Gibbs KM, Davila J, Campbell N, Sung S, Todorova TI et al (2011) MicroRNA miR-133b is essential for functional recovery after spinal cord injury in adult zebrafish. Eur J Neurosci [Internet]. 33:1587–97. Available from: https://doi.wiley.com/doi/10.1111/j.1460-9568.2011.07643.x
Theis T, Yoo M, Park CS, Chen J, Kügler S, Gibbs KM et al (2017) Lentiviral delivery of miR-133b improves functional recovery after spinal cord injury in mice. Mol Neurobiol [Internet]. 54:4659–71. Available from: https://link.springer.com/doi/10.1007/s12035-016-0007-z
Niu M, Xu R, Wang J, Hou B, Xie A (2016) MiR-133b ameliorates axon degeneration induced by MPP+ via targeting RhoA. Neuroscience [Internet]. 325:39–49. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0306452216300215
Zhang L-M, Wang M-H, Yang H-C, Tian T, Sun G-F, Ji Y-F et al (2019) Dopaminergic neuron injury in Parkinson’s disease is mitigated by interfering lncRNA SNHG14 expression to regulate the miR-133b/ α-synuclein pathway. Aging (Albany NY) [Internet]. 11:9264–79. Available from: https://www.aging-us.com/doi/10.18632/aging.102330
Li F, Zhang J, Chen A, Liao R, Duan Y, Xu Y et al (2021) Combined transplantation of neural stem cells and bone marrow mesenchymal stem cells promotes neuronal cell survival to alleviate brain damage after cardiac arrest via microRNA-133b incorporated in extracellular vesicles. Aging (Albany NY) [Internet]. 13:262–78. Available from: https://www.aging-us.com/doi/10.18632/aging.103920
Cao H, Baranova A, Yue W, Yu H, Zhu Z, Zhang F et al (2020) miRNA-coordinated schizophrenia risk network cross-talk with cardiovascular repair and opposed gliomagenesis. Front Genet [Internet]. 11. Available from: https://www.frontiersin.org/article/doi/10.3389/fgene.2020.00149/full
Panganiban CH, Barth JL, Darbelli L, Xing Y, Zhang J, Li H et al (2018) Noise-induced dysregulation of quaking RNA binding proteins contributes to auditory nerve demyelination and hearing loss. J Neurosci [Internet]. 38:2551–68. Available from: https://www.jneurosci.org/doi/10.1523/JNEUROSCI.2487-17.2018
Yang T, Song J, Bu X, Wang C, Wu J, Cai J et al (2016) Elevated serum miR-93, miR-191, and miR-499 are noninvasive biomarkers for the presence and progression of traumatic brain injury. J Neurochem [Internet]. 137:122–9. Available from: https://doi.wiley.com/doi/10.1111/jnc.13534
Jia H, Qu M, Fan G, Wu H, Wang L (2020) miR-499–5p suppresses C-reactive protein and provides neuroprotection in hypoxic-ischemic encephalopathy in neonatal rat. Neurosci Res [Internet]. 161:44–50. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0168010219303700
Anisman H, Merali Z, Poulter M, Hayley S (2005) Cytokines as a precipitant of depressive illness: animal and human studies. Curr Pharm Des [Internet]. 11:963–72. Available from: http://www.eurekaselect.com/openurl/content.php?genre=article&issn=1381-6128&volume=11&issue=8&spage=963
Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE et al (2001) In-vivo measurement of activated microglia in dementia. Lancet [Internet]. 358:461–7. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0140673601056252
Quan N (2014) In-depth conversation: Spectrum and kinetics of neuroimmune afferent pathways. Brain Behav Immun [Internet]. 40:1–8. Available from: https://linkinghub.elsevier.com/retrieve/pii/S088915911400052X
Najjar F, Ahmad M, Lagace D, Leenen FHH (2018) Sex differences in depression-like behavior and neuroinflammation in rats post-MI: role of estrogens. Am J Physiol Circ Physiol [Internet]. 315:H1159–73. Available from: https://www.physiology.org/doi/10.1152/ajpheart.00615.2017
Badoer E (2010) Microglia: Activation in acute and chronic inflammatory states and in response to cardiovascular dysfunction. Int J Biochem Cell Biol [Internet]. 42:1580–5. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1357272510002530
Meissner A, Visanji NP, Momen MA, Feng R, Francis BM, Bolz S et al (2015) Tumor necrosis factor-α underlies loss of cortical dendritic spine density in a mouse model of congestive heart failure. J Am Heart Assoc [Internet]. 4. Available from: https://www.ahajournals.org/doi/10.1161/JAHA.115.001920
Dworak M, Stebbing M, Kompa AR, Rana I, Krum H, Badoer E (2014) Attenuation of microglial and neuronal activation in the brain by ICV minocycline following myocardial infarction. Auton Neurosci [Internet]. 185:43–50. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1566070214000460
Wang H-W, Ahmad M, Jadayel R, Najjar F, Lagace D, Leenen FHH (2019) Inhibition of inflammation by minocycline improves heart failure and depression-like behaviour in rats after myocardial infarction. Bader M, editor. PLoS One [Internet]. 14:e0217437. Available from: https://dx.plos.org/doi/10.1371/journal.pone.0217437
Dworak M, Stebbing M, Kompa AR, Rana I, Krum H, Badoer E (2012) Sustained activation of microglia in the hypothalamic PVN following myocardial infarction. Auton Neurosci [Internet]. 169:70–6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1566070212000707
Wang Y, Yin J, Wang C, Hu H, Li X, Xue M et al (2019) Microglial Mincle receptor in the PVN contributes to sympathetic hyperactivity in acute myocardial infarction rat. J Cell Mol Med [Internet]. 23:112–25. Available from: https://doi.wiley.com/doi/10.1111/jcmm.13890
Apaijai N, Moisescu DM, Palee S, McSweeney CM, Saiyasit N, Maneechote C et al (2019) Pretreatment with PCSK9 inhibitor protects the brain against cardiac ischemia/reperfusion injury through a reduction of neuronal inflammation and amyloid beta aggregation. J Am Heart Assoc [Internet]. 8. Available from: https://www.ahajornals.org/doi/10.1161/JAHA.118.010838
Benjanuwattra J, Apaijai N, Chunchai T, Kerdphoo S, Jaiwongkam T, Arunsak B et al (2020) Metformin preferentially provides neuroprotection following cardiac ischemia/reperfusion in non-diabetic rats. Biochim Biophys Acta - Mol Basis Dis [Internet]. 1866:165893. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0925443920302416
Rana I, Stebbing M, Kompa A, Kelly DJ, Krum H, Badoer E (2010) Microglia activation in the hypothalamic PVN following myocardial infarction. Brain Res [Internet]. 1326:96–104. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0006899310003756
Pyner S (2014) The paraventricular nucleus and heart failure. Exp Physiol [Internet]. 99:332–9. Available from: https://doi.wiley.com/10.1113/expphysiol.2013.072678
Rinaldi B, Guida F, Furiano A, Donniacuo M, Luongo L, Gritti G et al (2015) Effect of prolonged moderate exercise on the changes of nonneuronal cells in early myocardial infarction. Neural Plast [Internet]. 2015:1–8. Available from: http://www.hindawi.com/journals/np/2015/265967/
Gouweleeuw L, Wajant H, Maier O, Eisel ULM, Blankesteijn WM, Schoemaker RG (2021) Effects of selective TNFR1 inhibition or TNFR2 stimulation, compared to non-selective TNF inhibition, on (neuro)inflammation and behavior after myocardial infarction in male mice. Brain Behav Immun [Internet]. 93:156–71. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0889159121000052
Hong X, Bu L, Wang Y, Xu J, Wu J, Huang Y et al (2013) Increases in the risk of cognitive impairment and alterations of cerebral β-amyloid metabolism in mouse model of heart failure. Yan R, editor. PLoS One [Internet]. 8:e63829. Available from: https://dx.plos.org/10.1371/journal.pone.0063829
Shi Z, Gan X-B, Fan Z-D, Zhang F, Zhou Y-B, Gao X-Y et al (2011) Inflammatory cytokines in paraventricular nucleus modulate sympathetic activity and cardiac sympathetic afferent reflex in rats. Acta Physiol [Internet]. 203:289–97. Available from: https://doi.wiley.com/10.1111/j.1748-1716.2011.02313.x
Du D, Jiang M, Liu M, Wang J, Xia C, Guan R et al (2015) Microglial P2X7 receptor in the hypothalamic paraventricular nuclei contributes to sympathoexcitatory responses in acute myocardial infarction rat. Neurosci Lett [Internet]. 587:22–8. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0304394014009719
Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T (2008) Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol [Internet]. 9:1179–88. Available from: http://www.nature.com/articles/ni.1651
Furukawa K, Mattson MP (2002) The transcription factor NF-κB mediates increases in calcium currents and decreases in NMDA- and AMPA/Kainate-induced currents induced by tumor necrosis factor-α in hippocampal neurons. J Neurochem [Internet]. 70:1876–86. Available from: https://doi.wiley.com/10.1046/j.1471-4159.1998.70051876.x
Díaz HS, Toledo C, Andrade DC, Marcus NJ, Del Rio R (2020) Neuroinflammation in heart failure: new insights for an old disease. J Physiol [Internet]. 598:33–59. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1113/JP278864
Jia X, Gao Z, Hu H (2020) Microglia in depression: current perspectives. Sci China Life Sci [Internet]. Available from: https://link.springer.com/10.1007/s11427-020-1815-6
Evonuk KS, Prabhu SD, Young ME, DeSilva TM (2017) Myocardial ischemia/reperfusion impairs neurogenesis and hippocampal-dependent learning and memory. Brain Behav Immun [Internet]. 61:266–73. Available from: https://linkinghub.elsevier.com/retrieve/pii/S088915911630410X
BAE E, HWANG IK, YOO K-Y, HAN TH, LEE CH, CHOI JH et al (2010) Gliosis in the amygdala following myocardial infarction in the rat. J Vet Med Sci [Internet]. 72:1041–5. Available from: http://www.jstage.jst.go.jp/article/jvms/72/8/72_09-0425/_article
Chen J, Yin D, He X, Gao M, Choi Y, Luo G et al (2020) Modulation of activated astrocytes in the hypothalamus paraventricular nucleus to prevent ventricular arrhythmia complicating acute myocardial infarction. Int J Cardiol [Internet]. 308:33–41. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0167527319358292
Marina N, Tang F, Figueiredo M, Mastitskaya S, Kasimov V, Mohamed-Ali V et al (2013) Purinergic signalling in the rostral ventro-lateral medulla controls sympathetic drive and contributes to the progression of heart failure following myocardial infarction in rats. Basic Res Cardiol [Internet].108:317. Available from: https://link.springer.com/10.1007/s00395-012-0317-x
Isegawa K, Hirooka Y, Katsuki M, Kishi T, Sunagawa K (2014) Angiotensin II type 1 receptor expression in astrocytes is upregulated leading to increased mortality in mice with myocardial infarction-induced heart failure. Am J Physiol Circ Physiol [Internet]. 307:H1448–55. Available from: https://www.physiology/doi/10.1152/ajpheart.00462.2014
Wann BP, Boucher M, Kaloustian S, Nim S, Godbout R, Rousseau G (2006) Apoptosis detected in the amygdala following myocardial infarction in the rat. Biol Psychiatry [Internet]. 59:430–3. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0006322305009066
Gilbert K, Godbout R, Rousseau G (2016) Caspase-3 activity in the rat amygdala measured by spectrofluorometry after myocardial infarction. J Vis Exp [Internet]. Available from: http://www.jove.com/video/53207/caspase-3-activity-rat-amygdala-measured-spectrofluorometry-after
Wann BP, Bah TM, Boucher M, Courtemanche J, Le Marec N, Rousseau G et al (2007) Vulnerability for apoptosis in the limbic system after myocardial infarction in rats: a possible model for human postinfarct major depression. J Psychiatry Neurosci [Internet]. 32:11–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17245469
Kaloustian S, Wann BP, Bah TM, Girard SA, Apostolakis A, Ishak S et al (2008) Apoptosis time course in the limbic system after myocardial infarction in the rat. Brain Res [Internet]. 1216:87–91. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0006899308009359
Lin Y, Devin A, Rodriguez Y, Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev [Internet]. 13:2514–26. Available from: http://www.genesdev.org/cgi/doi/https://www.genesdev.org/cgi/doi/10.1101/gad.13.19.2514
Wang J, Li P, Qin T, Sun D, Zhao X, Zhang B (2020) Protective effect of epigallocatechin-3-gallate against neuroinflammation and anxiety-like behavior in a rat model of myocardial infarction. Brain Behav [Internet]. 10. Available from: https://onlinelibrary.wiley.com/doi/10.1002/brb3.1633
Kaloustian S, Bah TM, Rondeau I, Mathieu S, Lada-Moldovan L, Ryvlin P et al (2009) Tumor necrosis factor-alpha participates in apoptosis in the limbic system after myocardial infarction. Apoptosis [Internet]. 14:1308–16. Available from: https://link.springer.com/10.1007/s10495-009-0395-x
Hwang IK, Yoo K-Y, Han TH, Lee CH, Choi JH, Yi SS et al (2009) Enhanced cell proliferation and neuroblast differentiation in the rat hippocampal dentate gyrus following myocardial infarction. Neurosci Lett [Internet]. 450:275–80. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0304394008016509
Frey A, Popp S, Post A, Langer S, Lehmann M, Hofmann U et al (2014) Experimental heart failure causes depression-like behavior together with differential regulation of inflammatory and structural genes in the brain. Front Behav Neurosci [Internet]. 8:376. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25400562
Bonetti NR, Diaz-Cañestro C, Liberale L, Crucet M, Akhmedov A, Merlini M et al (2019) Tumour necrosis factor-α inhibition improves stroke outcome in a mouse model of rheumatoid arthritis. Sci Rep [Internet]. 9:2173. Available from: http://www.nature.com/articles/s41598-019-38670-z
Kim J, Tomalin L, Lee J, Fitz LJ, Berstein G, Correa-da Rosa J et al (2018) Reduction of inflammatory and cardiovascular proteins in the blood of patients with psoriasis: differential responses between tofacitinib and etanercept after 4 weeks of treatment. J Invest Dermatol [Internet]. 138:273–81. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022202X17329482
Everett BM, MacFadyen JG, Thuren T, Libby P, Glynn RJ, Ridker PM (2020) Inhibition of interleukin-1β and reduction in atherothrombotic cardiovascular events in the CANTOS trial. J Am Coll Cardiol [Internet]. 76:1660–70. Available from: https://linkinghub.elsevier.com/retrieve/pii/S073510972036294X
Pradillo JM, Murray KN, Coutts GA, Moraga A, Oroz-Gonjar F, Boutin H et al (2017) Reparative effects of interleukin-1 receptor antagonist in young and aged/co-morbid rodents after cerebral ischemia. Brain Behav Immun [Internet]. 61:117–26. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0889159116305153
Padfield GJ, Din JN, Koushiappi E, Mills NL, Robinson SD, Cruden NLM et al (2017) Cardiovascular effects of tumour necrosis factor α antagonism in patients with acute myocardial infarction: a first in human study. Heart [Internet]. 99:1330–5. Available from: https://heart.bmj.com/look-up/doi/10.1136/heartjnl-2013-303648
Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT (2003) Anti-TNF therapy against congestive heart failure investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (AT. Circulation [Internet]. 107:3133–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12796126
Maleki Dizaji N, Garjani A, Mousavi S, Mohammadi M, Vaez H (2021) Time-dependent influence of infliximab on hemodynamic responses and cardiac injuries of isoproterenol-induced myocardial infarction in rats. Eur J Pharmacol [Internet]. 903:174122. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0014299921002752
Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor ENE et al (2013) Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res [Internet]. 10:301–12. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1873506113000032
Lai RC, Arslan F, Lee MM, Sze NSK, Choo A, Chen TS et al (2010) Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res [Internet]. 4:214–22. Available from: https://linkinghub.elsevier.com/retrieve/pii/S187350610900141X
Charles CJ, Li RR, Yeung T, Mazlan SMI, Lai RC, de Kleijn DP V et al (2020) Systemic mesenchymal stem cell-derived exosomes reduce myocardial infarct size: characterization with mri in a porcine model. Front Cardiovasc Med [Internet]. 7. Available from: https://www.frontiersin.org/articles/10.3389/fcvm.2020.601990/full
Hao P, Liang Z, Piao H, Ji X, Wang Y, Liu Y, et al (2014) Conditioned medium of human adipose-derived mesenchymal stem cells mediates protection in neurons following glutamate excitotoxicity by regulating energy metabolism and GAP-43 expression. Metab Brain Dis [Internet]. 29:193–205. Available from: https://link.springer.com/10.1007/s11011-014-9490-y
Lin S-S, Zhu B, Guo Z-K, Huang G-Z, Wang Z, Chen J et al (2014) Bone marrow mesenchymal stem cell-derived microvesicles protect rat pheochromocytoma PC12 cells from glutamate-induced injury via a PI3K/Akt dependent pathway. Neurochem Res [Internet]. 39:922–31. Available from: https://link.springer.com/10.1007/s11064-014-1288-0
Go V, Bowley BGE, Pessina MA, Zhang ZG, Chopp M, Finklestein SP et al (2020) Extracellular vesicles from mesenchymal stem cells reduce microglial-mediated neuroinflammation after cortical injury in aged Rhesus monkeys. GeroScience [Internet]. 42:1–17. Available from: https://link.springer.com/10.1007/s11357-019-00115-w
Dabrowska S, Andrzejewska A, Strzemecki D, Muraca M, Janowski M, Lukomska B (2019) Human bone marrow mesenchymal stem cell-derived extracellular vesicles attenuate neuroinflammation evoked by focal brain injury in rats. J Neuroinflammation [Internet]. 16:216. Available from: https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-019-1602-5
Hayon Y, Dashevsky O, Shai E, Brill A, Varon D, R. Leker R (2012) Platelet microparticles induce angiogenesis and neurogenesis after cerebral ischemia. Curr Neurovasc Res [Internet]. 9:185–92. Available from: http://www.eurekaselect.com/100235/article
Zhang Y, Chopp M, Liu XS, Katakowski M, Wang X, Tian X et al (2017) Exosomes derived from mesenchymal stromal cells promote axonal growth of cortical neurons. Mol Neurobiol [Internet]. 54:2659–73. Available from: https://link.springer.com/10.1007/s12035-016-9851-0
Qiu G, Zheng G, Ge M, Wang J, Huang R, Shu Q et al (2018) Mesenchymal stem cell-derived extracellular vesicles affect disease outcomes via transfer of microRNAs. Stem Cell Res Ther [Internet]. 9:320. Available from: https://stemvellres.biomedcentral.com/articles/10.1186/s13287-018-1069-9
Xin H, Li Y, Buller B, Katakowski M, Zhang Y, Wang X et al (2012) Exosome-mediated transfer of miR-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells [Internet]. 30:1556–64. Available from: https://onlinelibrary.wiley.com.doi/10.1002/stem.1129
Xin H, Wang F, Li Y, Lu Q-E, Cheung WL, Zhang Y et al (2017) Secondary Release of exosomes from astrocytes contributes to the increase in neural plasticity and improvement of functional recovery after stroke in rats treated with exosomes harvested from MicroRNA 133b-overexpressing multipotent mesenchymal stromal Ce. Cell Transplant [Internet]. 26:243–57. Available from: https://journals.sagepub.com/doi/10.3727/096368916X693031
Wei L, Zhang Y, Qi X, Sun X, Li Y, Xu Y (2019) Ubiquitin‑proteasomes are the dominant mediators of the regulatory effect of microRNA‑1 on cardiac remodeling after myocardial infarction. Int J Mol Med [Internet]. Available from: https://www.spandidos-publications.com/10.3892/ijmm.2019.4330
Pan Z, Sun X, Ren J, Li X, Gao X, Lu C et al (2012) miR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. Salloum FN, editor. PLoS One [Internet]. 7:e50515. Available from: hhttps://dx.plos.org/10.1371/journal.pone.0050515
Kumarswamy R, Lyon AR, Volkmann I, Mills AM, Bretthauer J, Pahuja A et al (2012) SERCA2a gene therapy restores microRNA-1 expression in heart failure via an Akt/FoxO3A-dependent pathway. Eur Heart J [Internet]. 33:1067–75. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehs043
Huang F, Li M-L, Fang Z-F, Hu X-Q, Liu Q-M, Liu Z-J et al (2013) Overexpression of microRNA-1 improves the efficacy of mesenchymal stem cell transplantation after myocardial infarction. Cardiology [Internet]. 125:18–30. Available from: https://www.karger.com/Article/FullText/347081
Yu H-D, Xia S, Zha C-Q, Deng S-B, Du J-L, She Q (2015) Spironolactone regulates HCN protein expression through micro-RNA-1 in rats with myocardial infarction. J Cardiovasc Pharmacol [Internet]. 65:587–92. Available from: http://journals.lww.com/00005344-201506000-00011
Author information
Authors and Affiliations
Contributions
P.G and L.S. had the idea for the article and directed the drafting and revising of the work, P.G performed the literature search, and L.C., J.R., M.M, and M.C. critically revised the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Human and animal rights and informed consent
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gelosa, P., Castiglioni, L., Rzemieniec, J. et al. Cerebral derailment after myocardial infarct: mechanisms and effects of the signaling from the ischemic heart to brain. J Mol Med 100, 23–41 (2022). https://doi.org/10.1007/s00109-021-02154-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-021-02154-3