
Abstract
Stroke can lead to cardiac complications such as arrhythmia, myocardial injury, and cardiac dysfunction, collectively termed stroke–heart syndrome (SHS). These cardiac alterations typically peak within 72 h of stroke onset and can have long-term effects on cardiac function. Post-stroke cardiac complications seriously affect prognosis and are the second most frequent cause of death in patients with stroke. Although traditional vascular risk factors contribute to SHS, other potential mechanisms indirectly induced by stroke have also been recognized. Accumulating clinical and experimental evidence has emphasized the role of central autonomic network disorders and inflammation as key pathophysiological mechanisms of SHS. Therefore, an assessment of post-stroke cardiac dysautonomia is necessary. Currently, the development of treatment strategies for SHS is a vital but challenging task. Identifying potential key mediators and signaling pathways of SHS is essential for developing therapeutic targets. Therapies targeting pathophysiological mechanisms may be promising. Remote ischemic conditioning exerts protective effects through humoral, nerve, and immune-inflammatory regulatory mechanisms, potentially preventing the development of SHS. In the future, well-designed trials are required to verify its clinical efficacy. This comprehensive review provides valuable insights for future research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A systematic global analysis reveals a rising incidence of stroke, and patients with stroke face a substantial risk of developing cardiac complications after onset [1,2,3]. Acute ischemic stroke (AIS) is the most common type of stroke that deserves special attention. In 1947, Byer et al. first reported that cerebrovascular disease could lead to arrhythmias and myocardial damage [4]. Subsequently, the interaction between brain injury and cardiac function has gained attention, with increasing clinical and experimental evidence supporting a causal relationship between stroke and cardiac dysfunction [5]. Cardiac complications after AIS can result not only from traditional vascular risk factors but also from mechanisms that remain poorly understood. The concept of stroke–heart syndrome (SHS) was proposed in 2018 by Scheitz et al., encompassing stroke-associated cardiac alterations [6].
SHS is categorized into (1) ischemic and non-ischemic acute myocardial injuries; (2) acute myocardial infarction (AMI) after stroke; (3) left ventricular dysfunction (LVD), heart failure (HF), and post-stroke Takotsubo syndrome (TTS); (4) sudden cerebral-cardiac death after stroke; and (5) electrocardiographic (ECG) changes and arrhythmias, including atrial fibrillation (AF) post-stroke [5]. Despite overwhelming evidence suggesting that SHS confers a poor prognosis, its potential pathophysiological mechanisms and therapeutic targets remain unknown.
Therefore, this review aims to present the latest summary of the epidemiology, clinical manifestations, and potential pathophysiological mechanisms of SHS following AIS.
Epidemiology of stroke–heart syndrome
Patients with AIS have an increased risk of cardiovascular complications. A trial demonstrated that up to one-fifth of patients with AIS developed major cardiovascular adverse events (MACEs) within 12 weeks of stroke onset [7]. A recent retrospective cohort study on more than 360,000 patients with AIS showed that over 27% of them experienced cardiac complications within 4 weeks after stroke [8]. Cardiac complications after stroke are common even in patients without any known history of heart disease. An analysis of 17 studies with 4869 patients with AIS without cardiac history revealed that the mean average incidence rate of asymptomatic coronary artery disease was 52% [9]. And 9.1% of patients with first-time AIS without known preexisting heart disease experienced MACE within 1 year. Compared with individuals without stroke, this population had a 25-fold increased risk of MACE, especially in the first month [10].
The hospitalization-related mortality rate of patients with AIS complicated by ischemic heart disease is approaching 30% [11]. Complex cardiovascular disease accounts for approximately 20% of post-stroke deaths, making it the second most frequent cause of post-stroke death after neurological death [7, 12]. In addition, it is reported that cardiac mortality is the highest in the first month after stroke, especially in the second week [7]. Approximately 4% of patients with AIS die due to cardiac causes, and up to 19% experience at least one MACE [7].
Manifestations of stroke–heart syndrome
The clinical manifestations of SHS, including asymptomatic ECG abnormalities, malignant arrhythmias, elevated cardiac biomarkers, LVD, and AMI, can develop continuously or occur simultaneously, coexist, or predispose patients to each other [13] (Table 1).
Electrocardiographic changes and cardiac arrhythmias
AIS frequently accompanies ECG changes. In patients without preexisting heart disease or clearly altered ECG, approximately one-third exhibit significantly abnormal ECG changes [14]. The most frequently observed post-AIS ECG abnormalities include QTc prolongation (20–65%), abnormal T-wave morphology (16–40%), and ST-segment changes (15–25%) [14,15,16,17,18,19,20,21]. Patients with severe neurological impairment and insular infarction are particularly prone to prolonged QTc [25]. Significant cardiac arrhythmias (CAs) can occur in up to 22% of patients with AIS, with right hemispheric infarcts (26.8%) associated with more frequent CA compared with left hemispheric infarcts (14.3%) [16]. Togha et al. reported that 16.5% of patients with AIS without prior cardiovascular diseases had CA [17]. ECG changes reach a peak early post-stroke, and CA has the highest probability of occurrence within the first 24 h [26]. Older age and higher National Institutes of Health Stroke Scale (NIHSS) score were reportedly independent predictors of CA within the first 3 days after stroke [26]. However, most ECG abnormalities are transient and disappear within 14 days [16, 59, 60]. Tachycardia-associated arrhythmias, especially AF, are more frequent than bradycardias [15, 26]. The incidence of AF following AIS is approximately 5–24% [21,22,23,24]. Consistent with the above-mentioned results, the incidence of AF detected after stroke (AFDAS) was higher in strokes involving the right insula (39%) than in strokes involving the left insula (4%) [61]. Notably, cardiac structure and function changes during AF in patients with stroke [62].
ECG abnormalities after AIS correlate strongly with neurological outcomes. Compared with that in patients with normal QTc, the mortality rate triples in patients with prolonged QTc during the early AIS [18]. AF, atrioventricular block, ST-segment changes, and inverted T-waves increase the mortality risk of patients within the subsequent 3 months [27]. AFDAS exhibits unique mechanisms and risk factors. Enhanced left atrial-pulmonary vein border fibrosis may serve as a structural substrate for AFDAS [63]. Patients with initial AFDAS diagnosis, as opposed to those with prior AF, may have a lower incidence of cardiac comorbidities and recurrent AIS [64, 65]. Nevertheless, Yang et al. reported similar AIS recurrence and mortality rates between these patient groups [66].
Cardiac troponin elevation
Increased cardiac troponin (cTn) levels are closely associated with acute myocardial injury, which is typically defined as a cTn level increase exceeding the reference superior limit, with a change of > 20% [67, 68]. Elevated cTn levels often occur without typical symptoms, emphasizing the importance of timely measurement upon admission for AIS [69]. The overall incidence of post-AIS myocardial injury is 25–60% with highly sensitive cardiac troponin (hs-cTn) [6, 28]. In addition, 25.3% of patients were diagnosed with acute myocardial injury after stroke [28]. Typically, elevated cTn levels appear transient, returning to normal range within hours to days. cTn assessment is pivotal in AMI diagnosis. The initial cTn level within 4.5-h post-stroke symptom onset demonstrates enhanced diagnostic efficiency for AMI, with 90.9% sensitivity and 74.8% specificity [70]. Previous studies reported that 2.3% of AIS patients were diagnosed with AMI during hospitalization, 1.6% of AIS patients were diagnosed with non-ST-elevation AMI, and 0.3% were diagnosed with ST-elevation AMI [29,30,31]. AMI occurs in 1–3% of patients with AIS within 1 year, with the highest risk in the first month and gradually decreasing thereafter [9, 32]. The cumulative 5-year incidence of post-stroke AMI is 2% in Korea [33]. Increased cTn levels may be associated with older age, a history of cardiac comorbidities, severe stroke, lower low-density lipoprotein (LDL) levels, renal insufficiency, and ECG abnormalities [25, 34, 35]. Stroke affecting the right insula is highly correlated with increased cTn levels [36, 37]. Extremely high cTn levels are also associated with heart dysfunction, decreased left ventricular ejection fraction (LVEF), and segmental ventricular hypokinesia [61]. Blaszczyk et al. observed that over 30% of patients with AIS developed focal myocardial fibrosis, primarily with an acute or subacute ischemic pattern, using cardiovascular magnetic resonance (CMR) imaging [71].
Animal experiments have demonstrated a fourfold increase in cTn level in mice 24 h after middle cerebral artery occlusion (MCAO), correlating with elevated mortality [72]. High initial and peak cTn levels are associated with poorer neurological outcomes and increased mortality, with dynamic cTn changes doubling in-hospital death risk [35]. Patients with AIS who have high cTn levels have a mortality rate of 14.7%, which is six times higher than that in patients with normal levels [34]. Elevated hs-cTn levels increased the incidence of death and major disability within 90 days [38]. Moreover, elevated cTn levels impact long-term prognosis, potentially increasing mortality related to stroke, heart disease, and cancer [39]. A recent study suggests that elevated hs-cTn levels are a valuable biomarker for cardio-cerebral vascular events recurrence and mortality [40].
Cardiac dysfunction
Numerous studies indicate that AIS can cause cardiac dysfunction. Min et al. induced left insular cortex ischemia in mice, resulting in cardiac dysfunction [73]. Veltkamp et al. observed a transient decrease in left ventricular contractility in early AIS stages in mice, recovering after 2 months [74]. Nevertheless, chronic systolic dysfunction was detected 8 weeks after focal cerebral ischemia in mice [75]. Clinical research suggests that approximately 10–24% of patients experience impaired left ventricular function with a low ejection fraction (EF) [41,42,43,44]. Approximately 13–15% of patients have moderate/severe left ventricular systolic dysfunction (EF < 40%) [43, 76]. Clinical HF is diagnosed in approximately 5–18% of patients with AIS, mostly with preserved EF [41,42,43,44]. Left ventricular diastolic dysfunction reportedly affects 23–59% of patients with AIS [44, 45]. Older age, serious stroke, history of prior cardiac diseases, and increased cTn levels are predictive factors for LVD. [44, 46,47,48] The size and area of stroke strongly correlate with the severity of cardiac dysfunction [49]. LVD is closely linked to damage in the right hemispheric central autonomic network (CAN) [50]. Infarcts in the right insula and left parietal cortex are more likely to result in LVD in patients with AIS without preexisting cardiac dysfunction [51].
The hospitalization mortality rate is 2.5 times higher in stroke patients with HF than in non-HF patients [52]. Regardless of EF reduction, HF correlates with poor neurological outcomes and 90-day mortality after stroke [41]. Lower LVEF independently predicts 90-day disability and increases the incidence of MACE and 1-year all-cause mortality post-stroke onset [42, 53]. However, abnormal echocardiographic results were not related to short-term mortality but significantly correlated with mortality 3 years after stroke [54].
Brain natriuretic peptide (BNP) and N-terminal pro-B-type natriuretic peptide (NT-proBNP) serve as predictors of post-stroke cardiac dysfunction [77]. NT-proBNP levels significantly increased within 24 h of AIS onset, whereas BNP correlated with left ventricular hypertrophy, left atrial dilatation, and LVEF [78, 79]. NT-proBNP levels are independently associated with all-cause mortality at 90 days [55].
TTS is a reversible form of HF, previously termed “stress-induced cardiomyopathy”, typically resolving within 1–6 months [5]. Post-stroke TTS involves transient ventricular dysfunction, with or without increased cTn and ECG changes [56]. Acute hemodynamic changes in TTS include depressed cardiac contractility, abbreviated systolic period, inefficient energetics, and extended active relaxation but unaltered diastolic passive stiffness [80]. TTS following AIS occurs in 0.4–2.7% of patients, with older, Caucasian, and female patients at increased risk [56,57,58]. Reduced LVEF and elevated neutrophil-to-lymphocyte ratio increase in-hospital complications and reduce long-term survival in TTS patients [81, 82]. TTS secondary to AIS predicts poorer short-term neurological functional outcomes and high mortality, with twice the inpatient mortality compared to patients without TTS [57, 58].
Pathophysiology of stroke–heart syndrome
SHS is caused by various pathophysiological mechanisms inductively defined as “stroke-induced heart injury (SIHI)” [5]. Evidence provides compelling support for CAN dysregulation, hypothalamic–pituitary–adrenal (HPA) axis activation, and inflammation in SHS [5, 6].
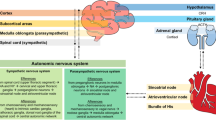
AIS can lead to damage to the CAN, resulting in overactivity of the sympathetic nervous system and HPA axis, further causing a surge in catecholamines [5, 6]. AIS leads to neuronal cell necrosis, activation of inflammatory cells, and the release of inflammatory response mediators, leading to local and systemic inflammation [83]. The peripheral immune system initiates after AIS, leading to macrophage migration [84]. There can be interactions between the autonomic nervous system, inflammatory response, and immune response [85]. Macrophages secrete cytokines and chemokines to promote the migration of inflammatory cells [84]. The surge in catecholamines enhances immune regulation and mediates an increase in inflammatory cytokines, aggravating the inflammatory response [85]. Simultaneously, inflammation also affects the release of catecholamines from the posterior hypothalamus and sympathetic nerve [85]. The increase in cortisol levels and inflammatory response caused by post-stroke stress response can increase intestinal barrier permeability, allowing the translocation of bacteria and endotoxins into the blood, further inducing an inflammatory response and the production of pro-inflammatory cytokines [84]. The interactions between these mechanisms jointly lead to cardiac damage (Fig. 1).

Mechanisms for stroke–heart syndrome. Stroke leads to central autonomic network dysregulation, HPA axis activation, inflammation, and immune response. These mechanisms interact with each other, ultimately resulting in cardiac damage. “Cell death signals”, miRNA, and MV are also involved in the development of SHS. HPA hypothalamic–pituitary–adrenal, MV microvesicle
Central autonomic network dysregulation and hypothalamic–pituitary–adrenal axis activation
The CAN involves various brain structures, including the insular cortex, anterior cingulate cortex, prelimbic and infralimbic areas, cingulate cortex, amygdala, and bed nucleus of the stria terminalis [86]. Damage to these sites disrupts the CAN structure and function and can result in a hyperactive stress response involving the sympathetic nervous system and HPA axis [5, 6]. Min et al. found that 64% of mice developed cardiac dysfunction after left MCAO, whereas those undergoing right MCAO did not [73]. Moreover, the severity of left insular cortex infarction in mice with impaired cardiac function was obviously greater [73]. Bieber et al. showed that mice undergoing right hemispheric MCAO developed mild cardiac dysfunction, whereas left MCAO-treated mice did not show such changes [75]. The mice with cardiac dysfunction had more severe insular cortex damage and increased sympathetic activity [75]. Moreover, larger insular infarct volumes correlate with more severe myocardial injury [87]. Meanwhile, this evidence supports the notion that the lateralization of the insula is closely connected with autonomic responses. Research suggests that the right insular cortex focuses on the sympathetic nervous system, whereas the left insular cortex is involved in parasympathetic function [88,89,90]. Heart rate variability (HRV) analysis studies reveal that patients with left hemisphere damage show enhanced parasympathetic regulation, whereas those with right hemisphere ischemia show the opposite effect [89, 90].
Following overactivation of the sympathetic nervous system and HPA axis, catecholamines and cortisol are excessively released, impacting the heart [6]. The expression of norepinephrine in the serum and heart tissue significantly increases, with cardiac contraction band necrosis observed in mice experiencing cardiac dysfunction during the acute stage of stroke [73]. Cardiac dysfunction is not exclusive to the acute stage of AIS. Accumulating evidence indicates that cerebral ischemia can lead to chronic cardiac dysfunction [75, 91]. In an animal experiment, compared to 3 days after stroke, 1 month showed more significant and severe cardiac dysfunction and myocardial pathological remodeling [74]. Chronic systolic dysfunction was observed in both young and elderly mice at 8 weeks after MCAO. Elevated catecholamine and cortisol levels suggest the involvement of enhanced sympathetic activity and indicate that the influence of AIS on cardiac function is age-independent [75]. Hypertension may modify central control of the CAN, as seen in spontaneously hypertensive rats exhibiting significant decreases in arterial pressure and renal sympathetic nerve discharge 6 h after MCAO [92]. Excessive calcium influx into cardiomyocytes during catecholamine storms can induce cardiac electrical instability and CA [6]. Sun et al. noted higher L-type calcium current density in ventricular myocytes of MCAO rats with arrhythmias, attributed to upregulated mRNA and protein expression of α1C/Cav1.2, enhancing the L-type Ca2+ channel function [93]. Another study reported that significant CA primarily results from increased L-type Ca2+ current and reduced transient outward K+ current [94].
Immune response and inflammation
Inflammatory responses and immune regulation significantly impact stroke–cardiac interactions [72, 84, 85, 95]. Ischemic stroke (IS) induces systemic and local cardiac inflammation and causes acute HF with increased circulating cTn levels and bradycardia [72, 96, 97]. Interleukin (IL)-1 is pivotal in SIHI by enhancing the inflammatory response [5]. Cerebral ischemia can directly damage neurons, activate microglia, and release inflammatory factors such as IL-1, stimulating cardiac macrophage and myofibroblast [5, 98]. Splenectomy before cerebral infarction in mice alleviates early inflammatory responses and heart infiltration, suggesting the peripheral immune regulation of the spleen after stroke plays an important role in post-stroke cardiac dysfunction [84]. Persistent macrophage infiltration from acute to chronic stroke phase indicates its crucial role in brain–cardiac interactions [84]. Inflammatory mechanisms in chronic stroke may be attributed to the weakened protective benefit of the inflammatory reflex against the post-autonomic dysfunctional inflammatory response [63, 99].
Although inflammatory factors emerge rapidly after cerebral ischemia, their circulation is temporary, whereas myocardial remodeling can persist for weeks or months [5]. Systemic inflammation and sympathetic hyperactivity activate microvascular endothelial cells, causing coronary microvascular endothelial dysfunction (CMED) [100]. An experimental stroke model demonstrated the combined role of intercellular communication and pro-inflammatory signaling in brain–heart interactions [91]. Balint et al. detected increased endothelial nitric oxide synthase (eNOS)-expressing endothelial cells in the left atrium of mice 28 days after stroke, indicating CMED [63]. Insular cortical infarction causes myocardial inflammatory infiltration and fibrosis in the left atrial, with the severity of CMED and cardiac fibrosis being associated with the level of inflammation [63].
Potential molecular mechanism and signaling pathways of the stroke–heart syndrome
MicroRNA (miRNA)-126 regulates endothelial cell (EC) function, vascular remodeling, vascular integrity, and endovascular inflammation [101]. Deletion of miRNA-126 in mice resulted in impaired cardiac function, cardiomyocyte hypertrophy, fibrosis, inflammatory reactions, and oxidative stress after stroke [91]. Activation of the NLR pyrin domain-containing 3 (NLRP3) inflammasome also mediates brain–heart interactions post-stroke [102]. Elevated NLRP3 expression in diabetic stroke mice correlated with impaired cardiac function partially reversible by NLRP3 inhibitors [102]. Veltkamp et al. showed that left MCAO in mice exhibited rapid cardiomyocyte atrophy via upregulated E3-ligase atrogin-1 and proliferator-activated receptor gamma (Pparg)-dependent genes. Pparg regulates cardiomyocyte metabolic and structural remodeling, supporting the latent role of metabolic signaling pathways in SHS [74]. Notably, stroke diminishes cardiac function and increases cardiac vulnerability to ischemia [103]. Cerebral ischemia altered nitro-oxidative signaling in the heart, affecting the expression of eNOS and glutathione peroxidase 1 and modulating the survivor-activating factor enhancement cardioprotective signaling pathway [103].
Indirect “cell death signals” from the brain to the heart were confirmed in vitro and in vivo [104]. Supernatants from rat neuronal cells exposed to oxygen–glucose deprivation increased cell death markers in rat cardiac myocytes (RCMs), reducing their vitality and mitochondrial reductase activity. An in vivo study revealed RCM cell death in rats 3 months after MCAO and abnormal inflammatory responses and apoptosis in non-human primates 6 months after transient global ischemia [104, 105].
Future directions
Early prediction and identification of SHS
Approximately one-fifth of patients with AIS show persistent SHS symptoms [6, 106]. Prevention and early identification is crucial. Tumor necrosis factor-related apoptosis-inducing ligand expression correlates with premature ventricular extrasystoles after stroke [107]. Soluble ST2 predicts SHS severity, and a risk prediction scale incorporating various factors identifies high-risk patients with SHS [108].
Continuous cardiac monitoring is crucial for detecting serious CA and preventing sudden cardiac death. A retrospective study reported the effectiveness of continuous monitoring in detecting underlying clinical AF during the 6-month follow-up [109]. Dynamic analysis of the RR interval performed in the hyperacute phase of AIS can help identify high-risk paroxysmal AF episodes [110]. Several predictive models have been developed for AFDAS [23, 111]. Further, a study established a multimodal approach incorporating imaging, ECG, and original biomarkers [112].
In addition, CMR imaging may offer superior accuracy in detecting subtle cardiac changes related to cardiac ischemic processes [113]. The PRAISE (PRediction of acute coronary syndrome in Acute Ischemic Stroke) study is refining algorithms and developing guidelines based on CMR findings to improve the identification of AIS patients at risk of SHS [114].
Autonomic function assessment
Autonomic dysfunction, persisting for 6 months after stroke, plays a crucial role in SHS development [115]. Therefore, it is necessary to assess post-stroke cardiac dysautonomia. HRV analysis shows promise in predicting mortality and morbidity in stroke outcomes [90, 116,117,118,119]. Phase-rectified signal averaging is also an ECG-based method for evaluating cardiac autonomic function, which reflects sympathetic and vagal nerve activities by calculating the acceleration (AC) and deceleration (DC) capabilities of the heart rate [120]. Unfortunately, we are still far from realizing these approaches are very important unmet needs [121]. CORONA-IS (Cardiomyocyte injury following Acute Ischemic Stroke) study is addressing quantifying autonomic dysfunction [122].
Exploring underlying mechanisms and mediators
Brain-derived neurotrophic factor (BDNF) may impact SHS [123]. Circulating BDNF levels are decreased after stroke [124]. BDNF can bind to tropomyosin-related kinase receptor B and trigger a calmodulin-dependent protein kinase II-dependent signaling cascade, which enhances cardiomyocyte Ca2+ cycling [123]. MiRNA regulates various biological processes, including hypoxia responses, angiogenesis, and inflammation [13]. MiRNA-182 dysregulation is linked to multiple cardio-cerebrovascular diseases, with elevated levels in AIS patients and cardiovascular disease patients [125]. A relationship between BDNF and miRNA-182-5p has been reported in HF patients [126]. MiRNA-124-3p, a brain-enriched miRNA, is upregulated after cardiac arrest, correlating with neurological outcomes [127]. Circulating microvesicles (MVs) may also influence brain–heart interactions [13]. MVs are considered a potential biomarker for cardiovascular risk stratification [128]. In the initial stage of AIS, the endothelial MVs and leukocyte-derived MVs levels are elevated, and endothelial MVs were proved to be associated with stroke severity [129]. A prospective cohort found that elevated endothelial MV and leukocyte-derived MV levels were interrelated to the long-term risk of cardiovascular events in stroke patients [130]. Therefore, future studies are warranted on the effects of miRNAs and MVs in the development of SHS, which deserve further study.
Intestinal microbiota imbalance is implicated in SHS development [13, 83]. AIS disrupts intestinal integrity, alters microbiota composition, disrupts immune homeostasis, and leads to the loss of intestinal neurons [131]. Increased intestinal permeability may trigger inflammatory reactions, exacerbating cardiac function [84]. Dysbiosis and gut microbiota translocation may promote inflammation post-myocardial ischemia/reperfusion, worsening heart damage [132]. Nevertheless, the detailed mechanism of gut microbiota in SHS remains unclear, necessitating future investigation.
Development of treatment strategies
AMI after AIS presents a crisis situation. Early-stage treatment of AIS and AMI primarily relies on intravenous thrombolysis therapy or endovascular therapy to achieve recanalization and salvage ischemic tissue, but the thrombolytic drug doses and treatment time window differ between these two cases [133]. For synchronous cardio-cerebral infarction, clear guidelines or robust evidence for formulating acute reperfusion strategies are lacking. According to the 2019 recommendations from the American Heart Association/American Stroke Association, administering intravenous thrombolysis with alteplase (AIS therapeutic dose) within 4.5 h, followed by percutaneous coronary intervention (if necessary), may be reasonable [134]. However, the recommendation lacks specific, and it is safer and more effective to provide individualized treatment schemes after evaluating the severity of cardio-cerebral events in patients with cardio-cerebral infarction.
At the same time, developing effective treatment strategies for SHS and its long-term consequences is vital [135]. Recently, researchers proposed a thorough and comprehensive care approach following the post-stroke ABC pathway to prevent, identify, and treat SHS [136]. The ABC pathway includes three prominent aspects: (A) appropriate antithrombotic therapy; (B) improvement in functional and psychological status; and (C) management of cardiovascular risk factors and comorbidities [137]. Stroke and cardiac diseases share various risk factors and pathophysiological mechanisms [138]. Although antithrombotic therapy is essential for both conditions, its benefits in preventing recurrent vascular events remain unclear [139]. A cohort study demonstrated similar long-term results of antithrombotic tactics in patients with AIS and HF (EF ≤ 40%) [140]. Adequate management of vascular risk factors is vital for preventing recurrent vascular events. Recent guidelines endorse targets for blood pressure, LDL cholesterol and glycated hemoglobin levels in patients with AIS, but optimal criteria for those with different cardiac complications remain unknown [141]. In addition, cardiac manifestations caused by non-atherosclerotic factors require a comprehensive examination to identify the etiology and subsequently treat the underlying causes [5] (Fig. 2).

Diagnosis and management of myocardial injury after acute ischemic stroke. Continuous cardiac monitoring, myocardial enzyme measurements, and autonomic function assessment are necessary after stroke onset. If no contraindications exist, patients with acute myocardial infarction after acute ischemic stroke (within the time window) can be treated with reperfusion therapy. Patients with other types of myocardial injury should undergo a comprehensive etiological examination followed by treatment of the underlying cause
SHS involves unique mechanisms beyond traditional risk factors, suggesting the benefits of therapies targeting sympathetic nervous system modulation, inflammation, vascular endothelial function improvement, and avoiding proarrhythmic drugs [100]. However, data on β-blockers, IL-1 receptor antagonists, and renin–angiotensin system inhibitors are insufficient for strong recommendations [5, 100]. Remote ischemic conditioning (RIC) has emerged as a safe and promising physical therapy for cardio-cerebrovascular diseases, with reported protective effects through humoral, nerve, and immune-inflammatory regulation mechanisms [142,143,144,145,146,147,148]. A recent study confirmed that RIC reduced the recurrence rate of vascular events and improved 90-day neurological outcomes in patients with AIS and AMI [149]. Therefore, early application of RIC post stroke onset may help prevent the development of SHS.
In conclusion, interdisciplinary cooperation is essential for the development and implementation of SHS treatment strategies. Future studies should focus on identifying key mediators and signaling pathways of brain–heart interaction to pinpoint therapeutic targets and improve patient outcomes. Recognizing SHS more deeply and conducting relevant clinical and animal studies are encouraging steps toward addressing this complex condition.
References
GBD (2019) Stroke Collaborators Global, regional, and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019 (2021). Lancet Neurol 20:795–820. https://doi.org/10.1016/S1474-4422(21)00252-0
Tsao CW et al (2023) Heart disease and stroke Statistics-2023 update: a report from the American Heart Association. Circulation 147:e93–e621. https://doi.org/10.1161/CIR.0000000000001123
Yang N, Lee H, Wu C (2023) Intravenous thrombolysis for acute ischemic stroke: from alteplase to Tenecteplase. Brain Circ 9:61–63. https://doi.org/10.4103/bc.bc_70_22
Byer E, Ashman R, Toth LA (1947) Electrocardiograms with large, upright T waves and long Q-T intervals. Am Heart J 33:796–806. https://doi.org/10.1016/0002-8703(47)90025-2
Sposato LA et al (2020) Post-stroke cardiovascular complications and neurogenic cardiac injury: JACC state-of-the-art review. J Am Coll Cardiol 76:2768–2785. https://doi.org/10.1016/j.jacc.2020.10.009
Scheitz JF, Nolte CH, Doehner W, Hachinski V, Endres M (2018) Stroke-heart syndrome: clinical presentation and underlying mechanisms. Lancet Neurol 17:1109–1120. https://doi.org/10.1016/S1474-4422(18)30336-3
Prosser J et al (2007) Predictors of early cardiac morbidity and mortality after ischemic stroke. Stroke 38:2295–2302. https://doi.org/10.1161/STROKEAHA.106.471813
Buckley BJR et al (2022) Stroke-heart syndrome: Incidence and clinical outcomes of cardiac complications following stroke. Stroke 53:1759–1763. https://doi.org/10.1161/STROKEAHA.121.037316
Gunnoo T et al (2016) Quantifying the risk of heart disease following acute ischaemic stroke: a meta-analysis of over 50,000 participants. BMJ Open 6:e009535. https://doi.org/10.1136/bmjopen-2015-009535
Sposato LA et al (2020) First-ever ischemic stroke and increased risk of incident heart disease in older adults. Neurology 94:e1559–e1570. https://doi.org/10.1212/WNL.0000000000009234
De Stefano F, Mayo T, Covarrubias C, Fiani B, Musch B (2021) Effect of comorbidities on ischemic stroke mortality: an analysis of the National Inpatient Sample (NIS) Database. Surg Neurol Int 12:268. https://doi.org/10.25259/SNI_415_2021
Scheitz JF, Nolte CH, Laufs U, Endres M (2015) Application and interpretation of high-sensitivity cardiac troponin assays in patients with acute ischemic stroke. Stroke 46:1132–1140. https://doi.org/10.1161/STROKEAHA.114.007858
Wang M, Peng Y (2022) Advances in brain-heart syndrome: attention to cardiac complications after ischemic stroke. Front Mol Neurosci 15:1053478. https://doi.org/10.3389/fnmol.2022.1053478
Khechinashvili G, Asplund K (2002) Electrocardiographic changes in patients with acute stroke: a systematic review. Cerebrovasc Dis 14:67–76. https://doi.org/10.1159/000064733
Zeng Z et al (2022) Assessing electrocardiogram changes after ischemic stroke with artificial intelligence. PLoS One 17:e0279706. https://doi.org/10.1371/journal.pone.0279706
Daniele O, Caravaglios G, Fierro B, Natalè E (2002) Stroke and cardiac arrhythmias. J Stroke Cerebrovasc Dis 11:28–33. https://doi.org/10.1053/jscd.2002.123972
Togha M, Sharifpour A, Ashraf H, Moghadam M, Sahraian MA (2013) Electrocardiographic abnormalities in acute cerebrovascular events in patients with/without cardiovascular disease. Ann Indian Acad Neurol 16:66–71. https://doi.org/10.4103/0972-2327.107710
Hjalmarsson C et al (2012) Can prolonged QTc and cTNT level predict the acute and long-term prognosis of stroke? Int J Cardiol 155:414–417. https://doi.org/10.1016/j.ijcard.2010.10.042
Hromádka M et al (2016) Prolonged corrected QT interval as a predictor of clinical outcome in acute ischemic stroke. J Stroke Cerebrovasc Dis 25:2911–2917. https://doi.org/10.1016/j.jstrokecerebrovasdis.2016.08.005
Amin OSM, Sheikhbzeni AS, Siddiq AN (2020) Relationship of QTc interval prolongation with acute ischemic stroke. Med Arch 74:195–198. https://doi.org/10.5455/medarh.2020.74.195-198
Fure B, Bruun Wyller T, Thommessen B (2006) Electrocardiographic and troponin T changes in acute ischaemic stroke. J Intern Med 259:592–597. https://doi.org/10.1111/j.1365-2796.2006.01639.x
Braillon A et al (2023) Incremental value of the combined brain-cardiac CT protocol on prediction of atrial fibrillation after stroke. Eur Stroke J 8:175–182. https://doi.org/10.1177/23969873221138197
Poh MQW et al (2023) Predicting atrial fibrillation after ischemic stroke: clinical, genetics and electrocardiogram modelling. Cerebrovasc Dis Extra 13:9–17. https://doi.org/10.1159/000528516
Baturova MA et al (2016) Electrocardiographic and echocardiographic predictors of paroxysmal atrial fibrillation detected after ischemic stroke. BMC Cardiovasc Disord 16:209. https://doi.org/10.1186/s12872-016-0384-2
Ahn SH et al (2016) Cardiac vulnerability to cerebrogenic stress as a possible cause of troponin elevation in stroke. J Am Heart Assoc. https://doi.org/10.1161/JAHA.116.004135
Kallmünzer B et al (2012) Serious cardiac arrhythmias after stroke: Incidence, time course, and predictors—a systematic, prospective analysis. Stroke 43:2892–2897. https://doi.org/10.1161/STROKEAHA.112.664318
Christensen H, Fogh Christensen A, Boysen G (2005) Abnormalities on ECG and telemetry predict stroke outcome at 3 months. J Neurol Sci 234:99–103. https://doi.org/10.1016/j.jns.2005.03.039
Stengl H et al (2022) Frequency, associated variables, and outcomes of acute myocardial injury according to the fourth Universal Definition of myocardial infarction in patients with acute ischemic stroke. Eur Stroke J 7:413–420. https://doi.org/10.1177/23969873221120159
Mehta S et al (2023) Prevalence and outcomes of patients with acute ischemic stroke and concomitant non-ST-elevation myocardial infarction (results from the national inpatient sample 2016 to 2019). Am J Cardiol 205:346–353. https://doi.org/10.1016/j.amjcard.2023.08.030
Liao J et al (2009) In-hospital myocardial infarction following acute ischaemic stroke: an observational study. Eur J Neurol 16:1035–1040. https://doi.org/10.1111/j.1468-1331.2009.02647.x
Mehta S, Kakouros N, Mir T, Loree S, Qureshi W (2024) Prevalence and outcomes of patients with acute ischemic stroke with concomitant ST-segment-elevation myocardial infarction (results from national inpatient sample 2016–2019). Stroke 55:1245–1253. https://doi.org/10.1161/STROKEAHA.123.044550
Boulanger M, Béjot Y, Rothwell PM, Touzé E (2018) Long-term risk of myocardial infarction compared to recurrent stroke after transient ischemic attack and ischemic stroke: systematic review and meta-analysis. J Am Heart Assoc. https://doi.org/10.1161/JAHA.117.007267
Lee KJ et al (2021) Five-year risk of acute myocardial infarction after acute ischemic stroke in Korea. J Am Heart Assoc 10:e018807. https://doi.org/10.1161/JAHA.120.018807
Alkhachroum AM, Miller B, Chami T, Tatsuoka C, Sila C (2019) A troponin study on patients with ischemic stroke, intracerebral hemorrhage and subarachnoid hemorrhage: type II myocardial infarction is significantly associated with stroke severity, discharge disposition and mortality. J Clin Neurosci 64:83–88. https://doi.org/10.1016/j.jocn.2019.04.005
Scheitz JF et al (2014) Prognostic relevance of cardiac troponin T levels and their dynamic changes measured with a high-sensitivity assay in acute ischaemic stroke: analyses from the TRELAS cohort. Int J Cardiol 177:886–893. https://doi.org/10.1016/j.ijcard.2014.10.036
Krause T et al (2017) Stroke in right dorsal anterior insular cortex is related to myocardial injury. Ann Neurol 81:502–511. https://doi.org/10.1002/ana.24906
Ay H et al (2006) Neuroanatomic correlates of stroke-related myocardial injury. Neurology 66:1325–1329. https://doi.org/10.1212/01.wnl.0000206077.13705.6d
He L, Wang J, Dong W (2018) The clinical prognostic significance of hs-cTnT elevation in patients with acute ischemic stroke. BMC Neurol 18:118. https://doi.org/10.1186/s12883-018-1121-5
Ahn SH et al (2017) Prognostic significance of troponin elevation for long-term mortality after ischemic stroke. J Stroke 19:312–322. https://doi.org/10.5853/jos.2016.01942
Kim BS et al (2023) Association of high-sensitivity troponin I with cardiac and cerebrovascular events in patient after ischemic Stroke. Cerebrovasc Dis 52:153–159. https://doi.org/10.1159/000525920
Ois A et al (2008) Heart failure in acute ischemic stroke. J Neurol 255:385–389. https://doi.org/10.1007/s00415-008-0677-1
Li Y et al (2019) Left ventricular ejection fraction and clinically defined heart failure to predict 90-day functional outcome after ischemic stroke. J Stroke Cerebrovasc Dis 28:371–380. https://doi.org/10.1016/j.jstrokecerebrovasdis.2018.10.002
Hays AG et al (2006) Left ventricular systolic dysfunction and the risk of ischemic stroke in a multiethnic population. Stroke 37:1715–1719. https://doi.org/10.1161/01.STR.0000227121.34717.40
Heuschmann PU et al (2021) Prevalence and determinants of systolic and diastolic cardiac dysfunction and heart failure in acute ischemic stroke patients: The SICFAIL study. ESC Heart Fail 8:1117–1129. https://doi.org/10.1002/ehf2.13145
Park HK et al (2016) Left ventricular diastolic dysfunction in ischemic stroke: functional and vascular outcomes. J Stroke 18:195–202. https://doi.org/10.5853/jos.2015.01697
Kim WJ, Nah HW, Kim DH, Cha JK (2016) Association between left ventricular dysfunction and functional outcomes at three months in acute ischemic stroke. J Stroke Cerebrovasc Dis 25:2247–2252. https://doi.org/10.1016/j.jstrokecerebrovasdis.2016.05.004
Oras J et al (2016) Elevated high-sensitive troponin T on admission is an indicator of poor long-term outcome in patients with subarachnoid haemorrhage: a prospective observational study. Crit Care 20:11. https://doi.org/10.1186/s13054-015-1181-5
Ribeiro ASF, Zerolo BE, López-Espuela F, Sánchez R, Fernandes VS (2023) Cardiac system during the aging process. Aging Dis 14:1105–1122. https://doi.org/10.14336/AD.2023.0115
Hermanns N et al (2022) Molecular imaging of the brain-heart axis provides insights into cardiac dysfunction after cerebral ischemia. Basic Res Cardiol 117:52. https://doi.org/10.1007/s00395-022-00961-4
Winder K et al (2023) Acute right insular ischaemic lesions and poststroke left ventricular dysfunction. Stroke Vasc Neurol 8:301–306. https://doi.org/10.1136/svn-2022-001724
Chung D et al (2023) Topographical association between left ventricular strain and brain lesions in patients with acute ischemic stroke and normal cardiac function. J Am Heart Assoc 12:e029604. https://doi.org/10.1161/JAHA.123.029604
Divani AA, Vazquez G, Asadollahi M, Qureshi AI, Pullicino P (2009) Nationwide frequency and association of heart failure on stroke outcomes in the United States. J Card Fail 15:11–16. https://doi.org/10.1016/j.cardfail.2008.09.001
Lee JY et al (2018) Left ventricular ejection fraction predicts poststroke cardiovascular events and mortality in patients without atrial fibrillation and coronary heart disease. Korean Circ J 48:1148–1156. https://doi.org/10.4070/kcj.2018.0115
Wrigley P et al (2017) Prevalence of positive troponin and echocardiogram findings and association with mortality in acute ischemic stroke. Stroke 48:1226–1232. https://doi.org/10.1161/STROKEAHA.116.014561
Dieplinger B et al (2017) Prognostic value of inflammatory and cardiovascular biomarkers for prediction of 90-day all-cause mortality after acute ischemic stroke-results from the Linz stroke unit study. Clin Chem 63:1101–1109. https://doi.org/10.1373/clinchem.2016.269969
Ranieri M, Finsterer J, Bedini G, Parati EA, Bersano A (2018) Takotsubo syndrome: clinical features, pathogenesis, treatment, and relationship with cerebrovascular diseases. Curr Neurol Neurosci Rep 18:20. https://doi.org/10.1007/s11910-018-0833-7
Patel U et al (2022) Prevalence and impact of takotsubo syndrome in hospitalizations for acute ischemic stroke. Proc (Bayl Univ Med Cent) 35:156–161. https://doi.org/10.1080/08998280.2021.1995932
Jung JM et al (2016) Takotsubo-like myocardial dysfunction in ischemic stroke: a hospital-based registry and systematic literature review. Stroke 47:2729–2736. https://doi.org/10.1161/STROKEAHA.116.014304
Giammello F et al (2020) Isolated insular stroke: clinical presentation. Cerebrovasc Dis 49:10–18. https://doi.org/10.1159/000504777
Marafioti V, Turri G, Carbone V, Monaco S (2018) Association of prolonged QTc interval with takotsubo cardiomyopathy: a neurocardiac syndrome inside the mystery of the insula of Reil. Clin Cardiol 41:551–555. https://doi.org/10.1002/clc.22910
Min J et al (2022) Neurogenic cardiac outcome in patients after acute ischemic stroke: the brain and heart connection. J Stroke Cerebrovasc Dis 31:106859. https://doi.org/10.1016/j.jstrokecerebrovasdis.2022.106859
Kamyshnikova LA et al (2021) Structural and functional parameters of the cardiovascular system during atrial fibrillation in patients after stroke. Wiad Lek 74:465–470 (Warsaw, Poland 1960)
Balint B, Jaremek V, Thorburn V, Whitehead SN, Sposato LA (2019) Left atrial microvascular endothelial dysfunction, myocardial inflammation and fibrosis after selective insular cortex ischemic stroke. Int J Cardiol 292:148–155. https://doi.org/10.1016/j.ijcard.2019.06.004
Sposato LA et al (2018) Atrial fibrillation detected after stroke is related to a low risk of ischemic stroke recurrence. Neurology 90:e924–e931. https://doi.org/10.1212/WNL.0000000000005126
Roychoudhury R, Ma S, Qian C (2023) Stroke prevention and intracranial hemorrhage risk in atrial fibrillation management: a mini review. Brain Circ 9:148–153. https://doi.org/10.4103/bc.bc_22_23
Yang XM et al (2019) Atrial fibrillation known before or detected after stroke share similar risk of ischemic stroke recurrence and death. Stroke 50:1124–1129. https://doi.org/10.1161/STROKEAHA.118.024176
De Michieli L, Jaffe AS, Sandoval Y (2023) Use and prognostic implications of cardiac troponin in COVID-19. Heart Fail Clin 19:163–176. https://doi.org/10.1016/j.hfc.2022.08.005
Thygesen K et al (2018) Fourth universal definition of myocardial infarction (2018). Glob Heart 13:305–338. https://doi.org/10.1016/j.gheart.2018.08.004
Powers WJ et al (2018) 2018 Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 49:e46–e110. https://doi.org/10.1161/STR.0000000000000158
Sun Y, Miller MM, Yaghi S, Silver B, Henninger N (2020) Association of baseline cardiac troponin with acute myocardial infarction in stroke patients presenting within 4.5 hours. Stroke 51:108–114. https://doi.org/10.1161/STROKEAHA.119.027878
Blaszczyk E et al (2023) Myocardial injury in patients with acute ischemic stroke detected by cardiovascular magnetic resonance imaging. Eur J Radiol 165:110908. https://doi.org/10.1016/j.ejrad.2023.110908
Vornholz L et al (2021) Acute heart failure after reperfused ischemic stroke: association with systemic and cardiac inflammatory responses. Front Physiol 12:782760. https://doi.org/10.3389/fphys.2021.782760
Min J et al (2009) Cardiac dysfunction after left permanent cerebral focal ischemia: the brain and heart connection. Stroke 40:2560–2563. https://doi.org/10.1161/STROKEAHA.108.536086
Veltkamp R et al (2019) Experimental ischaemic stroke induces transient cardiac atrophy and dysfunction. J Cachexia Sarcopenia Muscle 10:54–62. https://doi.org/10.1002/jcsm.12335
Bieber M et al (2017) Stroke-induced chronic systolic dysfunction driven by sympathetic overactivity. Ann Neurol 82:729–743. https://doi.org/10.1002/ana.25073
Hassan MS et al (2023) Analysis of echocardiographic findings of patients with acute ischemic stroke admitted to a Tertiary Care Hospital in Mogadishu, Somalia. Int J Gen Med 16:2887–2895. https://doi.org/10.2147/IJGM.S414014
Xu C, Zheng A, He T, Cao Z (2020) Brain-heart axis and biomarkers of cardiac damage and dysfunction after stroke: a systematic review and meta-analysis. Int J Mol Sci. https://doi.org/10.3390/ijms21072347
Giannakoulas G et al (2005) N-terminal pro-brain natriuretic peptide levels are elevated in patients with acute ischemic stroke. Angiology 56:723–730. https://doi.org/10.1177/000331970505600610
von Rennenberg R et al (2023) Elevation of cardiac biomarkers in stroke is associated with pathological findings on cardiac MRI-results of the HEart and Brain interfaces in Acute Stroke study. Int J Stroke 18:180–186. https://doi.org/10.1177/17474930221095698
Stiermaier T et al (2023) Hemodynamic assessment in takotsubo syndrome. J Am Coll Cardiol 81:1979–1991. https://doi.org/10.1016/j.jacc.2023.03.398
Zweiker D et al (2022) Neutrophile-lymphocyte ratio and outcome in takotsubo syndrome. Biology. https://doi.org/10.3390/biology11081154
Santoro F et al (2020) Neutrophil/lymphocyte ratio predicts in-hospital complications in takotsubo syndrome. Results from a prospective multi-center registry. Clin Cardiol 43:1294–1300. https://doi.org/10.1002/clc.23442
Chen Z et al (2017) Brain-heart interaction: cardiac complications after stroke. Circ Res 121:451–468. https://doi.org/10.1161/CIRCRESAHA.117.311170
Yan T et al (2020) Inflammatory responses mediate brain-heart interaction after ischemic stroke in adult mice. J Cereb Blood Flow Metab 40:1213–1229. https://doi.org/10.1177/0271678X18813317
Zou L, Han R (2022) Inflammatory response and immune regulation in brain-heart interaction after stroke. Cardiovasc Ther 2022:2406122. https://doi.org/10.1155/2022/2406122
Benarroch EE (1993) The central autonomic network: functional organization, dysfunction, and perspective. Mayo Clin Proc 68:988–1001. https://doi.org/10.1016/s0025-6196(12)62272-1
Bleilevens C et al (2015) Insular infarct size but not levosimendan influenced myocardial injury triggered by cerebral ischemia in rats. Exp Brain Res 233:149–156. https://doi.org/10.1007/s00221-014-4096-5
Xiong L et al (2013) Comprehensive assessment for autonomic dysfunction in different phases after ischemic stroke. Int J Stroke 8:645–651. https://doi.org/10.1111/j.1747-4949.2012.00829.x
Constantinescu V et al (2019) Heart rate variability analysis and cardiac dysautonomia in ischemic stroke patients. Clin Neurol Neurosurg 186:105528. https://doi.org/10.1016/j.clineuro.2019.105528
Constantinescu V, Matei D, Costache V, Cuciureanu D, Arsenescu-Georgescu C (2018) Linear and nonlinear parameters of heart rate variability in ischemic stroke patients. Neurol Neurochir Pol 52:194–206. https://doi.org/10.1016/j.pjnns.2017.10.002
Chen J et al (2017) MiR-126 affects brain-heart interaction after cerebral ischemic stroke. Transl Stroke Res 8:374–385. https://doi.org/10.1007/s12975-017-0520-z
Butcher KS, Hachinski VC, Wilson JX, Guiraudon C, Cechetto DF (1993) Cardiac and sympathetic effects of middle cerebral artery occlusion in the spontaneously hypertensive rat. Brain Res 621:79–86. https://doi.org/10.1016/0006-8993(93)90300-c
Sun L et al (2008) Aberration of L-type calcium channel in cardiac myocytes is one of the mechanisms of arrhythmia induced by cerebral ischemia. Cell Physiol Biochem 22:147–156. https://doi.org/10.1159/000149792
Wang L et al (2009) Ionic mechanisms underlying action potential prolongation by focal cerebral ischemia in rat ventricular myocytes. Cell Physiol Biochem 23:305–316. https://doi.org/10.1159/000218177
Li W et al (2020) Spleen associated immune-response mediates brain-heart interaction after intracerebral hemorrhage. Exp Neurol 327:113209. https://doi.org/10.1016/j.expneurol.2020.113209
Gao Y et al (2023) A narrative review of retinal vascular parameters and the applications (Part II): diagnosis in stroke. Brain Circ 9:129–134. https://doi.org/10.4103/bc.bc_9_23
Gao Y et al (2023) A narrative review of retinal vascular parameters and the applications (part I): measuring methods. Brain Circ 9:121–128. https://doi.org/10.4103/bc.bc_8_23
Li XH et al (2022) The signaling pathways and targets of natural compounds from traditional Chinese medicine in treating ischemic stroke. Molecules. https://doi.org/10.3390/molecules27103099
Olshansky B (2016) Vagus nerve modulation of inflammation: cardiovascular implications. Trends Cardiovasc Med 26:1–11. https://doi.org/10.1016/j.tcm.2015.03.016
Scheitz JF et al (2022) Stroke-heart syndrome: recent advances and challenges. J Am Heart Assoc 11:e026528. https://doi.org/10.1161/JAHA.122.026528
Wei XJ et al (2015) Biological significance of miR-126 expression in atrial fibrillation and heart failure. Braz J Med Biol Res 48:983–989. https://doi.org/10.1590/1414-431X20154590
Lin HB et al (2020) Macrophage-NLRP3 inflammasome activation exacerbates cardiac dysfunction after ischemic stroke in a mouse model of diabetes. Neurosci Bull 36:1035–1045. https://doi.org/10.1007/s12264-020-00544-0
Meloux A et al (2018) Ischemic stroke increases heart vulnerability to ischemia-reperfusion and alters myocardial cardioprotective pathways. Stroke 49:2752–2760. https://doi.org/10.1161/STROKEAHA.118.022207
Ishikawa H et al (2013) Ischemic stroke brain sends indirect cell death signals to the heart. Stroke 44:3175–3182. https://doi.org/10.1161/STROKEAHA.113.001714
Acosta SA, Mashkouri S, Nwokoye D, Lee JY, Borlongan CV (2017) Chronic inflammation and apoptosis propagate in ischemic cerebellum and heart of non-human primates. Oncotarget 8:102820–102834. https://doi.org/10.18632/oncotarget.18330
Xue F, Yun HJ, Peng L, Wu C (2023) Where are we heading in post-China angioplasty and stenting for symptomatic intracranial severe stenosis era? Brain Circ 9:3–5. https://doi.org/10.4103/bc.bc_68_22
Mihalovic M et al (2022) Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in patients after acute stroke: relation to stroke severity, myocardial injury, and impact on prognosis. J Clin Med. https://doi.org/10.3390/jcm11092552
Lian H et al (2020) Early prediction of cerebral-cardiac syndrome after ischemic stroke: the PANSCAN scale. BMC Neurol 20:272. https://doi.org/10.1186/s12883-020-01833-x
Khurshid S et al (2021) Usefulness of rhythm monitoring following acute ischemic stroke. Am J Cardiol 147:44–51. https://doi.org/10.1016/j.amjcard.2021.01.038
Adami A et al (2019) Electrocardiographic RR interval dynamic analysis to identify acute stroke patients at high risk for atrial fibrillation episodes during stroke unit admission. Transl Stroke Res 10:273–278. https://doi.org/10.1007/s12975-018-0645-8
Pang M, Li Z, Sun L, Zhao N, Hao L (2022) A nomogram for predicting atrial fibrillation detected after acute ischemic stroke. Front Neurol 13:1005885. https://doi.org/10.3389/fneur.2022.1005885
Garnier L et al (2022) Multimodal approach for the prediction of atrial fibrillation detected after stroke: SAFAS study. Front Cardiovasc Med 9:949213. https://doi.org/10.3389/fcvm.2022.949213
Rauseo E et al (2021) New imaging signatures of cardiac alterations in ischaemic heart disease and cerebrovascular disease using CMR Radiomics. Front Cardiovasc Med 8:716577. https://doi.org/10.3389/fcvm.2021.716577
Nolte CH et al (2020) PRediction of acute coronary syndrome in acute ischemic StrokE (PRAISE)—protocol of a prospective, multicenter trial with central reading and predefined endpoints. BMC Neurol 20:318. https://doi.org/10.1186/s12883-020-01903-0
Damkjær M et al (2023) Autonomic dysfunction after mild acute ischemic stroke and 6 months after: a prospective observational cohort study. BMC Neurol 23:26. https://doi.org/10.1186/s12883-023-03054-4
Constantinescu V, Matei D, Ignat B, Hodorog D, Cuciureanu DI (2020) Heart rate variability analysis: a useful tool to assess poststroke cardiac dysautonomia. Neurologist 25:49–54. https://doi.org/10.1097/NRL.0000000000000270
Guan L, Collet JP, Mazowita G, Claydon VE (2018) Autonomic nervous system and stress to predict secondary ischemic events after transient ischemic attack or minor stroke: possible implications of heart rate variability. Front Neurol 9:90. https://doi.org/10.3389/fneur.2018.00090
Constantinescu V et al (2016) Cortical modulation of cardiac autonomic activity in ischemic stroke patients. Acta Neurol Belg 116:473–480. https://doi.org/10.1007/s13760-016-0640-3
Buitrago-Ricaurte N, Cintra F, Silva GS (2020) Heart rate variability as an autonomic biomarker in ischemic stroke. Arq Neuro Psiquiatr 78:724–732. https://doi.org/10.1590/0004-282X20200087
Xu YH, Wang XD, Yang JJ, Zhou L, Pan YC (2016) Changes of deceleration and acceleration capacity of heart rate in patients with acute hemispheric ischemic stroke. Clin Interv Aging 11:293–298. https://doi.org/10.2147/CIA.S99542
Silvani A, Calandra-Buonaura G, Dampney RAL, Cortelli P (2016) Brain-heart interactions: physiology and clinical implications. Philos Trans A Math Phys Eng Sci. https://doi.org/10.1098/rsta.2015.0181
Stengl H et al (2021) Cardiomyocyte injury following acute ischemic stroke: protocol for a prospective observational cohort study. JMIR Res Protoc 10:e24186. https://doi.org/10.2196/24186
Feng N et al (2015) Constitutive BDNF/TrkB signaling is required for normal cardiac contraction and relaxation. Proc Natl Acad Sci USA 112:1880–1885. https://doi.org/10.1073/pnas.1417949112
Mojtabavi H, Shaka Z, Momtazmanesh S, Ajdari A, Rezaei N (2022) Circulating brain-derived neurotrophic factor as a potential biomarker in stroke: a systematic review and meta-analysis. J Transl Med 20:126. https://doi.org/10.1186/s12967-022-03312-y
Qi R, Liu H, Liu C, Xu Y, Liu C (2020) Expression and short-term prognostic value of miR-126 and miR-182 in patients with acute stroke. Exp Ther Med 19:527–534. https://doi.org/10.3892/etm.2019.8227
Fang F, Zhang X, Li B, Gan S (2022) miR-182-5p combined with brain-derived neurotrophic factor assists the diagnosis of chronic heart failure and predicts a poor prognosis. J Cardiothorac Surg 17:88. https://doi.org/10.1186/s13019-022-01802-0
Devaux Y et al (2016) Association of circulating MicroRNA-124-3p levels with outcomes after out-of-hospital cardiac arrest: a substudy of a randomized clinical trial. JAMA Cardiol 1:305–313. https://doi.org/10.1001/jamacardio.2016.0480
Ridger VC et al (2017) Microvesicles in vascular homeostasis and diseases. Position paper of the European Society of Cardiology (ESC) Working Group on Atherosclerosis and Vascular Biology. Thromb Haemost 117:1296–1316. https://doi.org/10.1160/TH16-12-0943
He Z, Tang Y, Qin C (2017) Increased circulating leukocyte-derived microparticles in ischemic cerebrovascular disease. Thromb Res 154:19–25. https://doi.org/10.1016/j.thromres.2017.03.025
Huo S et al (2021) Endothelial and leukocyte-derived microvesicles and cardiovascular risk after stroke: PROSCIS-B. Neurology 96:e937–e946. https://doi.org/10.1212/WNL.0000000000011223
Zhao L et al (2023) The interaction between intestinal microenvironment and stroke. CNS Neurosci Ther 29(Suppl 1):185–199. https://doi.org/10.1111/cns.14275
Zhao J et al (2023) Heart-gut microbiota communication determines the severity of cardiac injury after myocardial ischaemia/reperfusion. Cardiovasc Res 119:1390–1402. https://doi.org/10.1093/cvr/cvad023
de Castillo LLC et al (2021) Cardiocerebral infarction: a single institutional series. J Stroke Cerebrovasc Dis 30:105831. https://doi.org/10.1016/j.jstrokecerebrovasdis.2021.105831
Powers WJ et al (2019) Guidelines for the early management of patients with acute ischemic stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: a Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 50:e344–e418. https://doi.org/10.1161/STR.0000000000000211
Li F, Gao J, Kohls W, Geng X, Ding Y (2022) Perspectives on benefit of early and prereperfusion hypothermia by pharmacological approach in stroke. Brain Circ 8:69–75. https://doi.org/10.4103/bc.bc_27_22
Lip GYH et al (2023) Poststroke cardiovascular management: current concepts, integrated care, and future developments. Curr Probl Cardiol 48:101738. https://doi.org/10.1016/j.cpcardiol.2023.101738
Lip GYH et al (2022) Integrated care for optimizing the management of stroke and associated heart disease: a position paper of the European Society of Cardiology Council on Stroke. Eur Heart J 43:2442–2460. https://doi.org/10.1093/eurheartj/ehac245
Deng G et al (2023) Risk factors, pathophysiologic mechanisms, and potential treatment strategies of futile recanalization after endovascular therapy in acute ischemic stroke. Aging Dis 14:2096–2112. https://doi.org/10.14336/AD.2023.0321-1
Uzuner N, Uzuner GT (2023) Risk factors for multiple recurrent ischemic strokes. Brain Circ 9:21–24. https://doi.org/10.4103/bc.bc_73_22
Patel P et al (2022) Outcomes associated with antithrombotic strategies in heart failure with reduced ejection fraction and sinus rhythm following acute ischemic stroke. Front Neurol 13:1041806. https://doi.org/10.3389/fneur.2022.1041806
Kleindorfer DO et al (2021) 2021 Guideline for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline from the American Heart Association/American Stroke Association. Stroke 52:e364–e467. https://doi.org/10.1161/STR.0000000000000375
Cui Y, Chen YN, Nguyen TN, Chen HS (2023) Time from onset to remote ischemic conditioning and clinical outcome after acute moderate ischemic stroke. Ann Neurol 94:561–571. https://doi.org/10.1002/ana.26715
England TJ et al (2019) Remote ischemic conditioning after stroke Trial 2: a Phase IIb randomized controlled trial in hyperacute stroke. J Am Heart Assoc 8:e013572. https://doi.org/10.1161/JAHA.119.013572
Zhao W, Hausenloy DJ, Hess DC, Yellon DM, Ji X (2023) Remote ischemic conditioning: challenges and opportunities. Stroke 54:2204–2207. https://doi.org/10.1161/STROKEAHA.123.043279
Qin C et al (2020) Effects of remote ischemic conditioning on cerebral hemodynamics in ischemic stroke. Neuropsychiatr Dis Treat 16:283–299. https://doi.org/10.2147/NDT.S231944
Xu Y, Wang Y, Ji X (2023) Immune and inflammatory mechanism of remote ischemic conditioning: a narrative review. Brain Circ 9:77–87. https://doi.org/10.4103/bc.bc_57_22
Eren F, Yilmaz SE (2022) Neuroprotective approach in acute ischemic stroke: a systematic review of clinical and experimental studies. Brain Circ 8:172–179. https://doi.org/10.4103/bc.bc_52_22
Guan Y, Liu J, Gu Y, Ji X (2023) Effects of hypoxia on cerebral microvascular angiogenesis: benefits or damages? Aging Dis 14:370–385. https://doi.org/10.14336/AD.2022.0902
Li S et al (2024) Remote ischemic conditioning reduces adverse events in patients with acute ischemic stroke complicating acute myocardial infarction: a randomized controlled trial. Crit Care 28:5. https://doi.org/10.1186/s13054-023-04786-y
Acknowledgements
The authors thank Editage for English language editing and review services. This work was supported by the National Natural Science Foundation of China (Grant numbers 82371305 and 82274401), the National Key R&D Program of China (Grant numbers 2022YFC2408800 and 2022YFC3602401), Suzhou Municipal Health Commission (Grant number LCZX202028), and Suzhou Municipal Science and Technology Bureau (Grant number SS2019046).
Author information
Authors and Affiliations
Contributions
Formal analysis and investigation: Lanjing Wang; writing—original draft preparation: Lanjing Wang; writing—review and editing: Linqing Ma, Changhong Ren, Wenbo Zhao, Xunming Ji, Zhi Liu, and Sijie Li.
Corresponding authors
Ethics declarations
Conflicts of interest
The authors have no relevant financial or non-financial interests to disclose.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Ma, L., Ren, C. et al. Stroke–heart syndrome: current progress and future outlook. J Neurol (2024). https://doi.org/10.1007/s00415-024-12480-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00415-024-12480-4