
Abstract
Objective
Ischemic stroke is a leading cause of death and disability in individuals worldwide. Cerebral ischemia–reperfusion injury (CIRI) typically results in severe secondary injury and complications following reperfusion therapy. Microglia play critical roles in the inflammatory reaction of CIRI. However, less attention has been given to microglial death in this process. Our study aims to explore microglial death in CIRI and the effects and mechanism of minocycline treatment on microglia.
Methods
A middle cerebral artery occlusion (MCAO) model was applied to induce CIRI in rats. At 0 h, 24 h and 48 h post-operation, rats were intraperitoneally injected with 45 mg/kg minocycline. Neurological deficit scoring, 2,3,5-triphenyltetrazolium chloride (TTC) staining, assessment of activated microglia and examination of mitochondrial structure were conducted and checked at 72 h after reperfusion. Additionally, an in vitro model of oxygen–glucose deprivation/reperfusion (OGD/R) model was established. BV-2 cells were treated with various pharmacological inhibitors of cell death or minocycline. Cell viability, lipid peroxidation, mitochondrial structure and function, and labile Fe2+ and ferroptosis-associated gene/protein levels were measured. Hemin was used for further validation after transcriptome analysis.
Results
In the MCAO and OGD/R models, ferroptosis was identified as a major form of microglial death. Minocycline inhibited microglia ferroptosis by reducing HO-1 expression. In addition, minocycline improved mitochondrial membrane potential, mitochondrial structures and microglial survival in vivo. Minocycline also decreased labile Fe2+ levels, lipid peroxidation, and expression of ferritin heavy chain (FTH) and it improved mitochondrial structure and function in vitro. Upregulation of HO-1 counteracted the protective effect of minocycline.
Conclusion
Ferroptosis is a major form of microglial death in CIRI. The protective mechanism of minocycline in CIRI partially hinges on its ability to effectively ameliorate microglia ferroptosis by downregulating HO-1 expression. Consequently, targeting microglia ferroptosis is a promising treatment for CIRI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke is one of the leading causes of death and disability globally [1]. Endovascular thrombectomy, with or without intravenous thrombolysis, represents an established therapy for patients undergoing treatment [2]. However, cerebral ischemia–reperfusion injury (CIRI) following reperfusion triggers numerous pathophysiological events, such as cerebral edema, neuroinflammation, blood–brain barrier (BBB) disruption, oxidative stress and various types of cell death [3,4,5,6]. Patients usually suffer from impairments in sensorimotor function, language problems, and cognitive faculties, including learning and memory disorders. Therefore, treatments directed at addressing these events remain the focal point of current research.
Microglia, the resident immune cells of the brain, constitute approximately 10% of brain cells [7]. Activated microglia can be classified into two phenotypes: M1 inflammatory microglia and M2 anti-inflammatory microglia. M2 microglia modulate inflammatory responses by releasing anti-inflammatory cytokines. CIRI triggers the activation, migration and proliferation of resident microglia to mitigate neuronal damage. Meanwhile, the cross-talk between microglia and neurons plays significant roles in CIRI [8]. Previous studies have established that neuronal death in the infarcted region manifests through various mechanisms, including autophagy, apoptosis, ferroptosis and other atypical forms of cell death, whether microglia in the infarcted area coincide with cell injury remains to be investigated.
Ferroptosis is a distinct form of regulated, iron-dependent cell death driven by lipid peroxidation, that has been widely implicated in neuronal damage during CIRI. Controlling ferroptosis has emerged as a potential therapeutic approach in CIRI [9, 10]. Microglia, belonging to the reticuloendothelial system, are cells that retain more iron compared to others [11]. In addition, CIRI results in oxidative stress that leads to lipid peroxidation in brain tissue [12]. Based on this premise, we hypothesize that ferroptosis may contribute to microglial death during CIRI.
Minocycline (Mino), a semisynthetic tetracycline derivative, has acute neuroprotective effects mainly through its anti-inflammatory, antioxidant, and neurogenesis effects, particularly its influence on microglial activation and polarization [13,14,15]. However, whether minocycline affects microglial death remains unclear. Hence in this study, utilizing the application of a middle cerebral artery occlusion (MCAO/R) rat model and oxygen–glucose deprivation/reperfusion (OGD/R) in vitro model, we aimed to investigate microglial death in CIRI, elucidate the impact and underlying mechanism of minocycline treatment on microglia.
Materials and methods
Reagents and experimental animals
2,3,5-triphenyl-tetrazolium chloride (TTC) was obtained from Sigma-Aldrich Co. (St. Louis, USA). Minocycline (Mino), Z-VAD-FMK (VAD), 3-methyladenine (3-MA), necrostatin-1(Nec-1), ferrostatin-1(Fer-1) and hemin were obtained from MedChemExpress (Shanghai, China). Calcein AM and propidium iodide (PI) were obtained from Beyotime Biotechnology (Shanghai, China). PBS and Hank’s Balanced Salt Solution (HBSS) were obtained from Solarbio Life Science (Beijing, China).
SPF male Sprague–Dawley rats (6–8 weeks old, 250–270 g) were obtained from Yangzhou University (Animal production licence number: SYXK (Su) 2017-0044). These rats were kept at 23 ± 2 °C with a 12-h of light/dark cycle and free access to rodent diet and water.
MCAO/R model establishment and treatment
The rats were anaesthetized by intraperitoneal injection of 3% sodium pentobarbital (50 mg/kg). An incision was made along the midline of the neck to expose the common carotid artery, followed by ligation of the external carotid artery and isolation of the internal carotid artery. A silicon-rubber tip-coated nylon monofilament was inserted through the external carotid artery into the internal carotid artery until slight resistance was felt [16]. After 1 h, the filament was gently removed for reperfusion. Rats in the sham group underwent only blood vessel separation and ligation without the insertion of a suture. Throughout and after the operation, a constant temperature heating pad at 37 °C was used to maintain the rats' body temperature at 37 ± 0.5 °C.
At 0 h, 24 h and 48 h post-operation, 45 mg/kg minocycline was intraperitoneally injected into both the minocycline and model group rats. To evaluate the effect of minocycline on microglia ferroptosis, we referred to previous research and upregulated HO-1 expression [17]. Hemin (50 mg/kg/d) was intraperitoneally injected into both the hemin and model group rats before the operation. The equal volume of saline was intraperitoneally injected into sham group rats.
Neurological deficit scoring and TTC staining
Neurological function was evaluated at 72 h after MCAO/R using Longa’s method, as previously described [18]. Rats with no symptoms of nerve damage were assigned 0 points; rats whose contralateral forelimbs could not be fully extended when the tail was lifted were assigned 1 point; rats that turned to the temporal side while walking were assigned 2 points; rats that fell to the opposite side of the lesion while walking were assigned 3 points; and rats that could not walk and lost consciousness were assigned 4 points.
Brain slices, 2 mm in thickness, were obtained with a brain-cutting matrix. The slices were immersed in 1% TTC solution at 37 °C for 30 min, mounted on dry paper and photographed with a digital camera. The non-infarct area was represented by the red region, the infarcted area by the gray-white region, with the ischemic penumbra lying between the two. Infarct size and hemisphere size of each section were traced and quantified with ImageJ software (v1.52p). Infarct volume of each brain slice = infarct area × slice thickness, infarct volume percentage (%) = sum of infarct volumes/total brain tissue volume × 100%.
Measurement of brain water content
The brain tissues were collected without cardiac perfusion and cleaned with filter paper. The MCAO/R side was separated to measure wet weight with an analytical balance. Then, the brain tissues were dried at 105 °C for 24 h and weighed to obtain the dry weight. Brain water content (%) = (wet weight–dry weight)/wet weight × 100.
IHC and IF staining
For immunostaining, samples were stained using a primary antibody for Iba1 (17198, Cell Signaling Technology, 1:1500 dilution) and then analyzed using a streptavidin peroxidase detection system (Maixin) according to the manufacturer’s protocol. Diaminobenzidine (DAB) (Maixin) was used as the horseradish peroxidase (HRP)-specific substrate.
Formaldehyde-fixed brain sections were incubated with primary antibodies, including anti-Iba1(17198, Cell Signaling Technology, 1:100 dilution), anti-HO-1 (82206, Cell Signaling Technology, 1:100 dilution) and anti-HO-1 (66743-1-lg, Proteintech, 1:100 dilution), and then incubated with secondary antibodies. Cell nuclei were stained with DAPI (4′, 6-diamidino-2-phenylindole) (P0131, Beyotime, China). Immunostained samples were visualized under a fluorescence microscope (Eclipse TI-U, Nikon).
Enzyme-linked immunosorbent assay (ELISA)
Serum and cell culture supernatant were stored at − 80 °C after centrifugation. Tumor necrosis factor-α (TNF-α), Interleukin-1β (IL-1β), Interleukin-4 (IL-4) and Interleukin-10 (IL-10) were measured using ELISA kits (E-EL-R2856, E-EL-R0012, E-EL-R0014, E-EL-R0016, Elabscience, China). The inflammatory cytokine levels were determined using a microplate reader (Multiskan FC-51119080, Thermo Fisher Scientific) at 450 nm absorbance, following the manufacturer’s instructions.
Transmission electron microscopy (TEM)
The samples were fixed in 2.5% glutaraldehyde (pH 7.4) for 2 h, washed three times with 0.1 M phosphate buffer (pH 7.2) and fixed in 1% osmic acid at 4 °C for 2 h. The samples were gradient dehydrated in a graded series of ethanol. Subsequently, the samples were embedded in epon-araldite resin for penetration and placed in a mold for polymerization. After positioned, the ultrathin sections (50 nm) were made and collected for microstructure analysis. Following counterstaining with 3% uranyl acetate and 2.7% lead citrate, the sections were observed with a transmission electron microscope (HT7800, Hitachi).
Cell culture and OGD/R in vitro model
BV-2 cells (CL-0493, Procell Life Science & Technology Co., Ltd, Wuhan, China) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), with the medium was changed every 1–2 days. Cultures were maintained at 37 °C in a humidified atmosphere of 21% O2 and 5% CO2.
To establish the OGD/R model, BV-2 cells were initially cultured in glucose-free DMEM (Gibco, USA) without FBS in a hypoxia incubator (3131, Thermo Fisher Scientific) at 37 °C for 6 h (1% O2, 94% N2 and 5% CO2). Then, the glucose-free medium was replaced with DMEM containing 10% FBS. The cells were incubated under normal condition (as defined above) for 2 h. In the control group, BV-2 cells were incubated under normal conditions.
RNA-seq and genome-wide transcriptome analysis
After OGD/R and drug treatment, BV-2 cells were collected for RNA-seq (three biological replicates for each group). RNA-seq experiments were performed by Novogene (Beijing, China).
To preferentially select cDNA fragments 370–420 bp in length, the library fragments were purified with the AMPure XP system (Beckman Coulter, USA). Then PCR amplification was performed, and the PCR product was purified by AMPure XP beads to obtain the library. Reference genome and gene model annotation files were directly downloaded from the genome website. An index of the reference genome was built using HISAT2 (v2.0.5) and paired-end clean reads were aligned to the reference genome using HISAT 2 (v2.0.5).
Cell viability assay and LDH analysis
The Cell Counting Kit-8 (CCK-8) assay (Dojindo, Japan) was performed to assess cell viability. According to the manufacturer’s protocol, 10 µl of CCK-8 solution was added to 90 µl of the medium solution in each well of a 96-well plate and incubated for 2 h at 37 °C. The absorbance was measured at 450 nm using a microplate reader (Multiskan FC-51119080, Thermo Fisher Scientific).
LDH release was measured was detected by a LDH Cytotoxicity Assay Kit (C0017, Beyotime, China). Briefly, the supernatant from each well after different treatments was collected via centrifugation (400 × g, 5 min, 4 °C) and subjected to the assay according to the manufacturer's protocol for 15 min at 37 °C. The absorbance was measured at 490 nm using a microplate reader (Multiskan FC-51119080, Thermo Fisher Scientific).
Lipid peroxidation analysis with BODIPY 581/591-C11
The cells were subjected to various treatments in 6-well plates and then incubated with 10 μM BODIPY 581/591 C11 (Invitrogen, USA) at 37 °C for 30 min before being collected by trypsinization. After centrifugation at 500 × g for 5 min, the cells were resuspended in 500 μl of HBSS and analyzed using a flow cytometer (DxFLEX, Beckman Coulter). Each sample was analyzed with a minimum of 10,000 cells, and data analysis was performed using FlowJo software (v10.8.1).
Intracellular labile Fe2+ measurement
Labile Fe2+ was measured by BioTracker Far-red Labile Fe2+ Live Cell Dye (SCT037, Merck, USA). The cells were rinsed twice with PBS and incubated for 1 h at 37 °C with the staining solution (10 μM) in the plate. A 0.25% trypsin EDTA solution was used to detach the cells from the plate, and the cells were diluted with PBS on ice. After centrifugation at 500 × g for 5 min, the cells were resuspended in PBS and analyzed by flow cytometry.
MitoTracker red CMXRos staining
Cells were stained with MitoTracker Red CMXRos dye (C1035, Beyotime, China) and DAPI. Briefly, following treatment with various procedures, the cell culture medium was removed. Then 100 nM MitoTracker Red CMXRos was added, and the cells were incubated for 30 min at 37 °C. The cells were washed 3 times with HBSS and then incubated for 5 min at room temperature in DAPI labelling solution. The samples were visualized under a confocal microscope (TCS SP8 STED 3X, Leica).
Western blot
The protein lysates from BV-2 cells and brain tissues were prepared following standard protocols, and the protein content was determined using a BCA protein assay kit (Beyotime, China). Subsequently, the protein samples were separated by bis–tris gel (Invitrogen) and transferred onto polyvinylidene difluoride membranes (Roche) using a wet transfer system. The membranes were blocked in 5% bovine serum albumin (BSA) in Tris buffered saline with Tween 20 (TBST) for 1 h at room temperature and incubated with primary antibodies overnight at 4 °C. The primary antibodies used were anti-Gpx4 (ab125066, Abcam, 1:5000 dilution), anti-Fth (ab75973, Abcam, 1:2000 dilution), anti-Tfr (46222, Cell Signaling Technology, 1:1000 dilution), anti-4-HNE (ab46545, Abcam, 1:2000 dilution), anti-HO-1(82206, Cell Signaling Technology, 1:1000 dilution) and anti-β-actin (AC038, ABclonal Technology, 1:10,000 dilution). Then, the membranes were washed and incubated with HRP-conjugated secondary antibodies (AS003/AS014, ABclonal Technology, 1:5000 dilution) for 2 h at room temperature. Finally, the signals were detected using an enhanced chemiluminescence advanced system (iBright CL 1500, Thermo Fisher Scientific).
Quantitative real-time PCR assay
Total RNA from cells was extracted using TRIzol (Invitrogen), and complementary DNA was synthesized using a PrimeScript RT reagent kit (Takara Bio). Reverse transcription quantitative PCR (RT-qPCR) was performed using a real-time PCR system (Bio-Rad). The primers for RT-PCR were synthesized by Beijing Tsingke Biology Co., Ltd, and their sequences are listed as follows: HMOX1: forward, 5′-AAGGAGGTACACATCCAAGCCGA GA-3′, reverse, 5′-CATAGACTGGGTTCTGCTTGTTGCG-3′. Fth: forward, 5′-GGAGCAT GCCGAGAAACTGA-3′, reverse, 5′-TCTCCCAGTCATCACGGTCT-3′. Gpx4: forward, 5′-GCCAAAGTCCTAGGAAAC GC-3′, reverse, 5′-CCGGGTTGAAAGGTTCAGGA-3′. Tfr: forward, 5′-GTCTCATTGGTAGCCTCAGATG-3′, reverse, 5′-AGGGACAGTTCCA GGAGTAA-3′.
Tissue iron assay
The iron content in brain tissue from IP was measured by a Tissue Iron Content Colorimetric Assay Kit (A039-2-1, Nanjing Jiancheng Bioengineering Institute, China). According to the manufacturer’s instructions, brain samples were homogenized, and the supernatant was collected after centrifugation at 2000 × g for 10 min. The absorbance of each group was determined at 520 nm.
Tissue iron staining
The iron content in brain tissue from IP was assessed using the Lillie kit (G3310, Solarbio Life Science, China), which based on the Prussian blue reaction. After dewaxing the paraffin sections, staining solution was applied according to the manufacturer's instructions. Following dehydration and transparency, the sections were sealed with gum and subsequently examined and documented under a microscope.
Statistical analysis
All experiments were conducted at least three times. GraphPad Prism 8 was utilized for statistical analysis. Data were expressed as the means ± standard deviations (SDs). An unpaired t-test or one-way analysis of variance (ANOVA) was utilized for statistical analysis. A p < 0.05 was considered statistically significant.
Results
Minocycline alleviates the brain injury induced by MCAO/R in rats
To investigate the neuroprotective effects of minocycline on CIRI, we established a MCAO/R model in rats, and 45 mg/kg minocycline was intraperitoneally injected every 24 h after cerebral reperfusion (Fig. 1A). TTC staining of brain tissues showed that rats in the MCAO/R group had significantly larger infarcted volumes than those in the sham group. However, minocycline administration significantly reduced the percentage of infarcted volumes and brain water content (Fig. 1B–D). Additionally, neurological deficit scores (NDSs) were significantly lower in the minocycline-treated MCAO/R group compared to the MCAO/R group (Fig. 1E). These results collectively indicate that minocycline led to a favorable outcome phenotype in the MCAO/R rats.

Minocycline alleviates the brain injury phenotype induced by MCAO/R in rats. A Timetable and experimental procedure of the MCAO/R rat model. B TTC staining photographs of coronal brain Sections (2 mm) in the four groups. C, D Analysis of infarct volumes and brain water content in the above four groups. E Analysis of NDSs in the above four groups. The results are presented as the means ± SDs (n = 3). ###p < 0.001, #p < 0.05 vs. the sham group; ***p < 0.001, **p < 0.01 vs. the MCAO/R group. MCAO/R middle cerebral artery occlusion/reperfusion; TTC 2,3,5-triphenyltetrazolium chloride; NDSs neurological deficit scores; Mino minocycline. (−) indicates that MCAO/R or Mino was not used, ( +) indicates that MCAO/R or Mino was used
Minocycline increases microglial number and reduces microglial damage in the ischemic penumbra
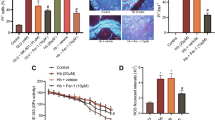
We conducted Iba-1 immunohistochemistry (IHC) and immunofluorescence (IF) staining to evaluate the number and morphology of microglia in the ischemic penumbra (IP). The results revealed that CIRI induced microglial activation and proliferation in IP of MCAO/R rats. Microglia exhibited enlargement and amoeboid morphology following MCAO/R induction. Interestingly, the number of microglia in the minocycline-treated MCAO/R group was significantly higher than that in the MCAO/R groups (Fig. 2A, S1). ELISA results showed that minocycline reduced the concentrations of TNF-α and IL-1β, and increased the concentrations of IL-4 and IL-10, indicating that minocycline selectively inhibited the proliferation of M1 microglia and promoted the proliferation of the M2 phenotype (Fig. 2B).

Minocycline increases the number of microglia and decreases microglial damage in the ischaemic penumbra. A Representative immunohistochemistry images of Iba-1-positive microglia. Top row scale bar: 100 μm, bottom row scale bar: 50 μm. B The levels of M1 marker TNF-α and IL-1β with M2 marker IL-4 and IL-10 in the serum were measured using ELISA. C Representative TEM images of microglia, the red arrow points to the mitochondria. Top row scale bar: 5 μm, bottom row scale bar: 1 μm. The results are presented as the means ± SDs (n = 3). ###p < 0.001, ##p < 0.01 vs. the sham group; ***p < 0.001, **p < 0.01 vs. the MCAO/R group. IP ischaemic penumbra; TEM transmission electron microscopy. (−) indicates that MCAO/R or Mino was not used, ( +) indicates that MCAO/R or Mino was used (color figure online)
To further investigate the ultrastructural changes in microglia within IP, we used TEM. In the sham group, mitochondrial structure in the cytoplasm of microglia appeared generally normal, characterized by prominent cristae and an intact membrane. However, CIRI caused remarkable microglial injury in the MCAO/R group, including outer mitochondrial membrane rupture, reduction or disappearance of mitochondrial cristae and vacuolation, without obvious alterations in the nucleus. Minocycline significantly ameliorated these morphological changes (Fig. 2C). Altogether, these data suggested that minocycline ameliorated microglial injury and promoted microglial numbers in IP.
Minocycline alleviates microglial death in vitro
To determine the reason for the increase in microglial numbers caused by minocycline treatment, we established an in vitro model of OGD/R using BV-2 cells to mimic microglial injury in CIRI. The results showed that minocycline could not induce microglial proliferation in the control group. Compared to the OGD/R group, cell viability significantly increased in the minocycline treatment group. To further clarify whether cell death was rescued by minocycline, we measured lactate dehydrogenase (LDH) release. The results demonstrated that LDH level in the minocycline treatment group was significantly lower than that in the OGD/R group (Fig. 3A). Moreover, costaining of PI and calcein-AM indicated that minocycline reduced the number of dead cells (Fig. 3B). The results of cytokine ELISA assays were similar to those in vitro; minocycline promoted the transformation of microglia from M1 to M2 phenotype (Fig. 3C). These results indicated that minocycline treatment ameliorated microglial death during CIRI in vitro.

Minocycline alleviates microglial death in vitro. A BV-2 cells were incubated with minocycline followed by OGD/R treatment for 8 h. The CCK-8 assay showed cell viability, and the LDH release assay showed cell damage. B Representative images of microglia after treatment with saline, minocycline (80 μM), OGD/R and OGD/R + minocycline (80 μM). Green fluorescent cells labelled with calcein-AM are alive; red fluorescent cells labelled with propidium iodide are dead. Scale bars: 100 μm. C The levels of M1 marker TNF-α and IL-1β with M2 marker IL-4 and IL-10 in the culture supernatant were measured using ELISA. The results are presented as the means ± SDs (n = 3). ###p < 0.001, ##p < 0.01, #p < 0.05 vs. the control group; ***p < 0.001, **p < 0.01, *p < 0.05 vs. the OGD/R group. OGD/R oxygen–glucose deprivation/reperfusion; CCK-8 cell counting kit-8; LDH lactate dehydrogenase. (−) indicates that OGD/R or Mino was not used, ( +) indicates that OGD/R or Mino was used (color figure online)
Ferroptosis is a major form of the microglial death induced by OGD/R in vitro
The major types of programmed cell death (PCD) include apoptosis, autophagy, necroptosis and ferroptosis. Various pharmacological inhibitors of cell death, including apoptosis (Z-VAD-FMK, VAD), autophagy (3-methyladenine, 3-MA), necroptosis (necrostatin-1, Nec-1) and ferroptosis (ferrostatin-1, Fer-1), were used to identify the PCD type occurring in OGD/R. The results indicated that Fer-1 treatment significantly improved cell viability and reduced LDH release compared to the OGD/R group (Fig. 4A). This result was consistent with costaining of PI and calcein-AM (Fig. 4B). These results indicated that ferroptosis might be the major form of microglial PCD in CIRI. To conform this, we measured intracellular labile Fe2+, which accumulates in ferroptotic cells. The results showed that the labile Fe2+ concentration was significantly higher in the OGD/R group than that in the control group, and this increase was rescued by Fer-1 treatment (Fig. 4C). Furthermore, lipid peroxidation was analyzed by detecting C11-BODIPY 581/591 and 4-HNE accumulation. ROS accumulation and lipid peroxidation increased in OGD/R, while Fer-1 significantly mitigated this increase (Fig. 4D–E). Glutathione peroxidase 4 (Gpx4), ferritin heavy chain (Fth) and transferrin receptor (Tfr) are commonly used as indicators of ferroptosis. Western blot results showed that, compared with the control group, Gpx4 level was lower in the OGD/R group, and Fer-1 increased its expression. Conversely, Fer-1 repressed Fth accumulation induced by OGD/R, but not the accumulation of Tfr (Fig. 4F).


OGD/R induces ferroptosis in microglia in vitro. A BV-2 cells were incubated with various pharmacological inhibitors of cell death, including Z-VAD-FMK (VAD, 40 mM), 3-methyladenine (3-MA,5 mM), necrostatin-1 (Nec, 50 mM), ferrostatin-1 (Fer-1, 400 μM), and then subjected to OGD/R treatment for 8 h. CCK-8 assays showed cell viability, and LDH release assays showed cell damage. B Representative images of microglia after treatment with saline, Fer-1 (400 μM), OGD/R and OGD/R + Fer-1 (400 μM). Green fluorescent cells labelled with calcein-AM are alive; red fluorescent cells labelled with propidium iodide are dead. Scale bars: 100 μm. C ROS generation was measured using BODIPY 581/591 C11 and flow cytometry. D Western blot analysis of 4-HNE in microglia. E Labile Fe2+ was measured using BioTracker Far-red and flow cytometry. F Western blot analysis of Gpx4, Fth and Tfr in microglia. G Quantitative analysis of MitoTracker Red CMXROS in microglia. Scale bars: 50 μm. H Representative TEM images of microglia. Top row scale bar: 2 μm, bottom row scale bar: 1 μm. The results are presented as the means ± SDs (n = 3). ###p < 0.001, ##p < 0.01, #p < 0.05 vs. the control group; ***p < 0.001, **p < 0.01 vs. the OGD/R group; NS not statistically significant. ROS reactive oxygen species. (−) indicates that OGD/R or Fer-1 was not used, ( +) indicates that OGD/R or Fer-1 was used (color figure online)
To further evaluate mitochondrial function, MitoTracker Red CMXROS staining and TEM were performed. Interestingly, the red fluorescence intensity of MitoTracker Red CMXROS in the OGD/R group declined greatly, and Fer-1 showed ability to reverse this decline (Fig. 4G). These data illustrated that Fer-1 significantly maintained the degree of mitochondrial membrane potential which was decreased by OGD/R treatment. Further morphological and microstructural details were obtained by using TEM, and Fer-1 alleviated the ruptures of the outer mitochondrial membrane and reduction or vanishing of mitochondrial cristae caused by OGD/R (Fig. 4H). In summary, these results indicate that ferroptosis is a major form of microglial death during OGD/R.
Minocycline inhibits ferroptosis of microglia with OGD/R in vitro
To analyze the effect of minocycline on ferroptosis of microglia, we added it into medium and mixed with a final concentration of 80 μM before routine culture or OGD/R treatment. The results showed that minocycline decreased the high labile Fe2+ level induced by OGD/R (Fig. 5A). Lipid peroxidation data indicated that minocycline decreased ROS accumulation and 4-HNE expression in microglia after OGD/R treatment (Fig. 5B–C). Similar to Fer-1, minocycline enhanced Gpx4 expression while suppressing Fth expression after OGD/R (Fig. 5D). Mito-Tracker Red CMXROS staining and TEM photos demonstrated that minocycline also contributed to the restoration of mitochondrial function, improving mitochondrial membrane potential and mitochondrial structures (Fig. 5E, F). Taken together, these data indicated that minocycline can effectively attenuate OGD/R-induced ferroptosis in microglia.


Minocycline alleviates the ferroptosis of microglia induced by OGD/R in vitro. A Labile Fe2+ was measured using BioTracker Far-red and flow cytometry. B ROS generation was measured using BODIPY 581/591 C11 and flow cytometry. C Western blot analysis of 4-HNE in microglia. D Western blot analysis of Gpx4, Fth and Tfr in microglia. E Quantitative analysis of MitoTracker Red CMXROS in microglia. Scale bars: 50 μm. F Representative TEM images of microglia. Top row scale bar: 2 μm, bottom row scale bar: 1 μm. The results are presented as the means ± SDs (n = 3). ###p < 0.001, ##p < 0.01, #p < 0.05 vs. the control group; ***p < 0.001, **p < 0.01, *p < 0.05 vs. the OGD/R group. NS not statistically significant. (−) indicates that OGD/R or Mino was not used, ( +) indicates that OGD/R or Mino was used (color figure online)
HO-1 is the potential target dictating to ferroptosis of microglia with OGD/R in vitro
To identify the mechanism by which minocycline ameliorates microglial ferroptosis, gene expression analyses were performed, and the results identified a total of 1509 differentially expressed genes between the control group and OGD/R group, with 645 genes downregulated, and 864 genes upregulated. In contrast, there were 456 downregulated and 144 upregulated genes between the OGD/R and minocycline treatment groups. The differential expression of the top 10 genes in our gene expression omnibus data was ranked. Intriguingly, the expression levels of heme oxygenase 1 (Hmox1) and Fth were significantly increased in the OGD/R group versus the control group, and the expression levels of both genes were significantly decreased in the minocycline treatment group (Fig. 6A). We then performed RT-qPCR for the Hmox1, Fth, Gpx4 and Tfr genes, and the results indicated that Hmox1 was repressed by minocycline (Fig. 6B).


Hemin alleviates the effect of minocycline on the microglia ferroptosis induced by OGD/R in vitro. A KEGG enrichment analysis of microglia after control treatment, OGD/R and OGD/R + minocycline. B RT-qPCR evaluation of Hmox1, Fth, Gpx4 and Tfr. C BV-2 cells were incubated with hemin and minocycline, and then subjected to OGD/R treatment for 8 h. The CCK-8 assay showed cell viability, and the LDH release assay showed cell damage. D Representative images of microglia after treatment with saline, hemin, OGD/R + hemin and OGD/R + henmin + minocycline. Green fluorescent cells labelled with Calcein-AM were alive; red fluorescent cells labelled with propidium iodide were dead. Scale bars: 100 μm. E Western blot analysis of HO-1 in microglia. F Labile Fe2+ was measured using BioTracker Far-red and flow cytometry. G ROS generation was measured using BODIPY 581/591 C11 and flow cytometry. H Western blot analysis of 4-HNE in microglia. I Western blot analysis of Gpx4, Fth and Tfr in microglia. J Quantitative analysis of MitoTracker Red CMXROS in microglia. Scale bars: 50 μm. K Representative TEM images of microglia. Top row scale bar: 2 μm, bottom row scale bar: 1 μm. The results are presented as the means ± SDs (n = 3). ###p < 0.001, ##p < 0.01, #p < 0.05 vs. the control group; ***p < 0.001, **p < 0.01, *p < 0.05 vs. the OGD/R group. NS not statistically significant. (−) indicates that OGD/R/Hemin/Mino was not used, ( +) indicates that OGD/R/Hemin/Mino was used (color figure online)
Hemin, a HO-1 (encoded by the Hmox1 gene) inducer, was then pretreated with BV-2 cells at 20 μM for 24 h. The protective effect of minocycline was significantly attenuated in microglia after OGD/R exposure. In the hemin treated group, cell viability was lower and LDH release was higher than those in the OGD/R group (Fig. 6C). This result was consistent with costaining of PI and calcein-AM (Fig. 6D). Western blot results revealed that HO-1 expression caused by OGD/R induction was increased following hemin treatment, and the minocycline response was not appreciable (Fig. 6E). The effect of minocycline on labile Fe2+ levels, ROS accumulation and 4-HNE expression was reversed by hemin (Fig. 6F–H). The Western blot results showed that minocycline increased Gpx4 and reduced Fth expression, with these effects being counteracted by hemin (Fig. 6I).
Furthermore, the protective role of minocycline on mitochondrial potential and mitochondrial ultrastructure was inhibited by the administration of hemin (Fig. 6J, K). Collectively, the protective effect of minocycline on OGD/R-induced ferroptosis was inhibited by the upregulation of HO-1.
HO-1 upregulation reduces the protective effect of minocycline in MCAO/R rats
To evaluate the role of HO-1 upregulation in the protective effect of minocycline in vivo, rats were pretreated for 3 days with hemin 50 mg/kg/d (Fig. 7A). The increased expression of HO-1 protein was confirmed by Western blot (Fig. 7B). Strikingly, infarcted volumes and brain water content were significantly higher in the hemin treatment group compared to all other groups (Fig. 7C–E), and all NDSs of the groups reached 3 (Fig. 7F). Moreover, the concentration of tissue iron was significantly increased after MCAO/R, particularly in the hemin treatment group (Fig. 7G). This result was consistent with iron staining (Fig. 7H).

Hemin alleviates the protective effect of minocycline on the brain injury induced by MCAO/R rats. A Timetable and experimental procedure of rat treatment. B Western blot analysis of HO-1 in IP. C TTC staining photographs of coronal brain Sections (2 mm) in the above five groups. D, E Analysis of infarct volumes and brain water content in the above five groups. F Analysis of NDSs in the above five groups. G Analysis of tissue iron content in IP. H Iron staining in IP, blue points indicate iron deposition. Top row scale bar: 50 μm, bottom row scale bar: 5 μm. The results are presented as the means ± SDs (n = 3). ###p < 0.001, ##p < 0.01 vs. the sham group, **p < 0.01, *p < 0.05 vs. the MCAO/R group or hemin + MCAO/R group. NS not statistically significant. (−) indicates that MCAO/R, Hemin or Mino was not used, ( +) indicates that MCAO/R, Hemin or Mino was used (color figure online)
HO-1 upregulation counteracts the protective effect of minocycline on microglia ferroptosis in vivo
Using IHC and IF staining of IP, we observed a significant decrease in overall cellularity in the hemin treatment group during MCAO/R. Cell numbers, including microglia, showed little variation even when minocycline was injected promptly for 72 h (Figs. 8A, S2). Furthermore, upon pre-injection of hemin, minocycline was able to partially decrease the upregulation of HO-1 induced by MCAO/R (Fig. 8B). The mitochondrial damage in microglia after MCAO/R was more severe than that in the other groups, and the protective effect of minocycline on mitochondrial structure was not as strong as that in the previous steps. Ruptures of the outer mitochondrial membrane, reductions or vanishing of mitochondrial cristae and vacuolation were still present in many microglia (Fig. 8C). Consequently, the protective effect of minocycline on MCAO/R was also inhibited by the upregulation of HO-1.

Hemin alleviates the effect of minocycline on the microglia ferroptosis induced by MCAO/R rats. A Representative immunohistochemistry images of Iba-1-positive microglia. Top row scale bar: 100 μm, bottom row scale bar: 50 μm. B HO-1 immunofluorescence of microglia in IP. Scale bars: 100 μm. C Representative TEM images of microglia in IP, the red arrow points to the mitochondria. Top row scale bar: 5 μm, bottom row scale bar: 1 μm. The results are presented as the means ± SDs (n = 3). ###p < 0.001, ##p < 0.01 vs. the sham group. NS not statistically significant. (−) indicates that MCAO/R, Hemin or Mino was not used, ( +) indicates that MCAO/R, Hemin or Mino was used (color figure online)
Discussion
Microglia act as the main form of active immune defense in CIRI, but whether they suffer the reperfusion injury, like neurons, is unclear. In the present study, we demonstrated that microglia experienced ferroptosis during CIRI. Ischemia–reperfusion raises HO-1 levels in microglia with a subsequent increase in iron concentration, which results in the induction of microglia ferroptosis and weakens the neuroprotective effects. Furthermore, minocycline improved CIRI outcomes by ameliorating microglia ferroptosis through the downregulation of HO-1 expression (Fig. 9).

Putative mechanistic diagrams of minocycline alleviating the microglia ferroptosis induced by CIRI. CIRI raises HO-1 levels in microglia with a subsequent increase in iron concentration, which results in the induction of microglia ferroptosis. Minocycline improved CIRI outcomes by ameliorating microglia ferroptosis through decreasing HO-1 expression. CIRI cerebral ischemia–reperfusion injury
A direct result of CIRI is local blood supply disturbances in the brain that induce neuronal damage or cell death, thus resulting in nervous system dysfunction. If ischemia persists, IP will progress to irreversible damage, and reperfusion can aggravate neuronal injury and advance the process of neuronal death through a series of pathologies, such as excessive amounts of ROS, oxidative stress and inflammatory responses. Therefore, the primary goal of current treatments for CIRI is to salvage IP [19,20,21]. Microglia dominate the immune response, as the receptor-ligand pairs in microglia-neuron were more than other immune cells [22]. Consistent with previous research findings, minocycline promoted the transformation of microglia to the M2 phenotype, thereby playing a neuroprotective role [23, 24]. Our finding also indicated that CIRI leads to microglial death, and the reduced number of microglia is insufficient to perform their neuroprotective function, thereby influencing outcomes.
Previous studies have confirmed that microglia undergo ferroptosis by different strategies. Graphene quantum dots, methamphetamine and RSL3 induce cell death in vitro [25,26,27], but whether microglia ferroptosis is involved in the development of CIRI has not been reported. We identified ferroptosis as the major form of microglial death in OGD/R. HO-1plays an important role by increasing the labile Fe2+ that triggers the upregulation of the iron storage protein FTH [28]. Combined with the induction of Tfr, these data illustrated that the main proportion of intracellular accumulated iron is endogenous Fe2+. Because of the high reactivity and instability, the excess irons produce free radicals through the Fenton reaction [29, 30]. Furthermore, ROS generated from the reaction between free radicals and polyunsaturated fatty acids (PUFAs) in the plasma membrane leads to mitochondrial damage. These changes may form the basis of microglia ferroptosis. CIRI is known to trigger the proliferation and migration of activated microglia [31, 32]. Our results showed that the microglia ferroptosis in IP aggravated CIRI, and that targeting microglia ferroptosis might be a new strategy for CIRI treatment.
Minocycline can easily cross the BBB and penetrate the central nervous system to play protective roles, such as stabilizing mitochondrial membranes and scavenging free radicals [14, 33]. Additionally, the phenolic ring structure of minocycline has radical scavenging properties [34]. In previous studies, minocycline significantly reduced infarct sizes, brain edema and improved neurological outcomes for up to 3 days of CIRI, such as the effects of anti-inflammatory and antioxidant [14, 35]. Combined with our data from the perspective of microglia, minocycline also inhibited the ferroptosis caused by CIRI though decreasing the microglial cellular HO-1 and Fe2+ levels, and improving mitochondrial injury.
Mechanically, hemin administration further confirmed that the protective effect of minocycline was diminished by upregulating the expression of HO-1 in vitro, but not completely in vivo. This discrepancy may be caused by minocycline playing a direct protective role in neurons [36], while its protective role in microglia is achieved through the amelioration of ferroptosis. Furthermore, the mechanism for the protective effect of surviving microglia that leads to good outcomes remains unknown. Following ischemia–reperfusion or OGD/R in microglia, the production of free radicals leading to lipid peroxidation may overload the antioxidant defense system, which plays an important role in reversing the destructive effects of ROS [37, 38]. However, whether the secretion of inflammatory factors from microglia or the cross-talk between microglia and neurons has an effect on the ferroptosis of microglia remains unclear. Exploring these aspects could be promising avenues for our future research.
In conclusion, our study reveals that microglia ferroptosis plays an important role in the process of CIRI, and that targeting microglia ferroptosis is a promising treatment for CIRI. Minocycline ameliorates microglia ferroptosis by reducing the HO-1 induced by CIRI. Our findings provide novel insights into the theoretical basis of minocycline treatment of CIRI.
Data availability
The datasets that support the conclusions of this article are included in the article. No datasets were generated or analysed during the current study.
References
Campbell BCV, De Silva DA, Macleod MR, Coutts SB, Schwamm LH, Davis SM, et al. Ischaemic stroke. Nat Rev Dis Primers. 2019;5:70.
Yang P, Song L, Zhang Y, Zhang X, Chen X, Li Y, et al. Intensive blood pressure control after endovascular thrombectomy for acute ischaemic stroke (ENCHANTED2/MT): a multicentre, open-label, blinded-endpoint, randomised controlled trial. Lancet. 2022;400:1585–96.
Shen L, Gan Q, Yang Y, Reis C, Zhang Z, Xu S, et al. Mitophagy in cerebral ischemia and ischemia/reperfusion injury. Front Aging Neurosci. 2021;13: 687246.
Mu Q, Yao K, Syeda MZ, Zhang M, Cheng Q, Zhang Y, et al. Ligustrazine nanoparticle hitchhiking on neutrophils for enhanced therapy of cerebral ischemia-reperfusion injury. Adv Sci (Weinh). 2023. https://doi.org/10.1002/advs.202301348.
Sun Y, Yang X, Xu L, Jia M, Zhang L, Li P, et al. The role of Nrf2 in relieving cerebral ischemia-reperfusion injury. Curr Neuropharmacol. 2023;21:1405–20.
Zhang Q, Jia M, Wang Y, Wang Q, Wu J. Cell death mechanisms in cerebral ischemia-reperfusion injury. Neurochem Res. 2022;47:3525–42.
Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50(253–71): e6.
Cramer T, Gill R, Thirouin ZS, Vaas M, Sampath S, Martineau F, et al. Cross-talk between GABAergic postsynapse and microglia regulate synapse loss after brain ischemia. Sci Adv. 2022;8:eabj0112.
Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–21.
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88.
Vakilian A, Khorramdelazad H, Heidari P, Sheikh Rezaei Z, Hassanshahi G. CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem Int. 2017;103:1–7.
Wu M, Gu X, Ma Z. Mitochondrial quality control in cerebral ischemia-reperfusion injury. Mol Neurobiol. 2021;58:5253–71.
Parvardeh S, Sheikholeslami MA, Ghafghazi S, Pouriran R, Mortazavi SE. Minocycline improves memory by enhancing hippocampal synaptic plasticity and restoring antioxidant enzyme activity in a rat model of cerebral ischemia-reperfusion. Basic Clin Neurosci. 2022;13:225–35.
Naderi Y, Panahi Y, Barreto GE, Sahebkar A. Neuroprotective effects of minocycline on focal cerebral ischemia injury: a systematic review. Neural Regen Res. 2020;15:773–82.
Lu Y, Zhou M, Li Y, Li Y, Hua Y, Fan Y. Minocycline promotes functional recovery in ischemic stroke by modulating microglia polarization through STAT1/STAT6 pathways. Biochem Pharmacol. 2021;186: 114464.
Nie QQ, Zheng ZQ, Liao J, et al. SPP1/AnxA1/TIMP1 as essential genes regulate the inflammatory response in the acute phase of cerebral ischemia-reperfusion in rats. J Inflamm Res. 2022;15:4873–90.
Tronel C, Rochefort GY, Arlicot N, Bodard S, Chalon S, Antier D. Oxidative stress is related to the deleterious effects of heme oxygenase-1 in an in vivo neuroinflammatory rat model. Oxid Med Cell Longev. 2013;2013: 264935.
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91.
Patel SD, Liebeskind D. Collaterals and elusive ischemic penumbra. Transl Stroke Res. 2023;14:3–12.
Ermine CM, Bivard A, Parsons MW, Baron JC. The ischemic penumbra: from concept to reality. Int J Stroke. 2021;16:497–509.
Xiao L, Dai Z, Tang W, Liu C, Tang B. Astragaloside IV alleviates cerebral ischemia-reperfusion injury through NLRP3 inflammasome-mediated pyroptosis inhibition via activating Nrf2. Oxid Med Cell Longev. 2021;2021:9925561.
Zeng F, Cao J, Hong Z, Liu Y, Hao J, Qin Z, et al. Single-cell analyses reveal the dynamic functions of Itgb2+ microglia subclusters at different stages of cerebral ischemia-reperfusion injury in transient middle cerebral occlusion mice model. Front Immunol. 2023;14:1114663.
Lu Y, Zhou M, Li Y, et al. Minocycline promotes functional recovery in ischemic stroke by modulating microglia polarization through STAT1/STAT6 pathways. Biochem Pharmacol. 2021;186: 114464.
Yang Y, Salayandia VM, Thompson JF, et al. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J Neuroinflamm. 2015;12:26.
Wu T, Liang X, Liu X, Li Y, Wang Y, Kong L, et al. Induction of ferroptosis in response to graphene quantum dots through mitochondrial oxidative stress in microglia. Part Fibre Toxicol. 2020;17:30.
Lin S, Cheng H, Yang G, Wang C, Leung CK, Zhang S, et al. NRF2 antagonizes HIV-1 tat and methamphetamine-induced BV2 cell ferroptosis by regulating SLC7A11. Neurotox Res. 2023;41:398–407.
Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16:278–90.
Wang P, Cui Y, Ren Q, Yan B, Zhao Y, Yu P, et al. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021;12:447.
Henning Y, Blind US, Larafa S, Matschke J, Fandrey J. Hypoxia aggravates ferroptosis in RPE cells by promoting the Fenton reaction. Cell Death Dis. 2022;13:662.
He YJ, Liu XY, Xing L, Wan X, Chang X, Jiang HL. Fenton reaction-independent ferroptosis therapy via glutathione and iron redox couple sequentially triggered lipid peroxide generator. Biomaterials. 2020;241: 119911.
Surinkaew P, Sawaddiruk P, Apaijai N, Chattipakorn N, Chattipakorn SC. Role of microglia under cardiac and cerebral ischemia/reperfusion (I/R) injury. Metab Brain Dis. 2018;33:1019–30.
Gaire BP. Microglia as the critical regulators of neuroprotection and functional recovery in cerebral ischemia. Cell Mol Neurobiol. 2022;42:2505–25.
Du B, Li H, Zheng H, Fan C, Liang M, Lian Y, et al. Minocycline ameliorates depressive-like behavior and demyelination induced by transient global cerebral ischemia by inhibiting microglia activation. Front Pharmacol. 2019;10:1247.
Katayama S, Ota M. Minocycline-induced hyperpigmentation. N Engl J Med. 2021;385:2463.
Wang S, Wang C, Wang L, Cai Z. Minocycline inhibits mTOR signaling activation and alleviates behavioral deficits in the wistar rats with acute ischemia stroke. CNS Neurol Disord Drug Targets. 2020;19:791–9.
Zhao K, Wang P, Tang X, Chang N, Shi H, Guo L, et al. The mechanisms of minocycline in alleviating ischemic stroke damage and cerebral ischemia-reperfusion injury. Eur J Pharmacol. 2023;955: 175903.
Eshaghi Ghalibaf MH, Rajabian A, Parviz M, Akbarian M, Amirahmadi S, Vafaee F, et al. Minocycline alleviated scopolamine-induced amnesia by regulating antioxidant and cholinergic function. Heliyon. 2023;9: e13452.
Kryl’skii ED, Popova TN, Safonova OA, Stolyarova AO, Razuvaev GA, de Carvalho MAP. Transcriptional regulation of antioxidant enzymes activity and modulation of oxidative stress by melatonin in rats under cerebral ischemia/reperfusion conditions. Neuroscience. 2019;406:653–66.
Acknowledgements
We would like to thank The First Affiliated Hospital of Yangzhou University for its help in the detection of experimental samples.
Funding
This research was supported by the National Natural Science Foundation of China (82072130 and 82200749), Key Medical Research Projects in Jiangsu Province (ZD2022021), Key R&D Program Projects in Jiangsu Province (BE2023709), Natural Science Foundation of Basic Research Program in Science and Technology Department of Jiangsu Province (BK20221282), 2022 Municipal Planned Social Development of Yangzhou Science and Technology Bureau (YZ2022102), Health Talent Plan Project in Suzhou (GSWS2022007), Suzhou Key Laboratory of Anesthesiology (SZS2023013), Suzhou Clinical Medical Center for Anesthesiology (Szlcyxzxj202102), Medical Innovation and Transformation of Yangzhou University (AHYZUZHXM202101) and National Clinical Key Specialty for Anesthesiology.
Author information
Authors and Affiliations
Contributions
Conceptualization: FHJ, JYZ and LW; Methodology: MYW and YFC; Formal analysis and investigation: XJ and ZHG; Writing—original draft preparation: LW and YW; Writing—review and editing: LW, YW, JYZ and FHJ; Funding acquisition: YW, JYZ and FHJ; Resources: XWM; Supervision: JYZ and FHJ.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
All animal experiments were performed in accordance with standard guidelines for the care and use of laboratory animals and approved by the Experimental Animal Ethics Committee of Yangzhou University.
Additional information
Responsible Editor: Jianxiong Jiang.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Wang, Y., Wu, M. et al. Minocycline alleviates microglia ferroptosis by inhibiting HO-1 during cerebral ischemia–reperfusion injury. Inflamm. Res. (2024). https://doi.org/10.1007/s00011-024-01927-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00011-024-01927-z