
Abstract
Notch and Hedgehog signaling are involved in cancer biology and pathology, including the maintenance of tumor cell proliferation, cancer stem-like cells, and the tumor microenvironment. Given the complexity of Notch signaling in tumors, its role as both a tumor promoter and suppressor, and the crosstalk between pathways, the goal of developing clinically safe, effective, tumor-specific Notch-targeted drugs has remained intractable. Drugs developed against the Hedgehog signaling pathway have affirmed definitive therapeutic effects in basal cell carcinoma; however, in some contexts, the challenges of tumor resistance and recurrence leap to the forefront. The efficacy is very limited for other tumor types. In recent years, we have witnessed an exponential increase in the investigation and recognition of the critical roles of the Notch and Hedgehog signaling pathways in cancers, and the crosstalk between these pathways has vast space and value to explore. A series of clinical trials targeting signaling have been launched continually. In this review, we introduce current advances in the understanding of Notch and Hedgehog signaling and the crosstalk between pathways in specific tumor cell populations and microenvironments. Moreover, we also discuss the potential of targeting Notch and Hedgehog for cancer therapy, intending to promote the leap from bench to bedside.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Notch and Hedgehog (Hh) were first described in Drosophila by Thomas Hunt Morgan in 1917 and by Nusslein-Volhard and Wieschaus in 1980 [1, 2]. Both signaling pathways exist widely in vertebrates and invertebrates, are highly primordial and conserved in evolution, and are intimately involved in the fate determination of cells [3, 4], differentiation of tissues [5], development of organs [2, 6,7,8,9], formation of embryos [10, 11] and homeostasis in organisms [12]. Abnormalities in the modulation of both pathways during embryonic development may contribute to malformations [13,14,15] and disorders [16,17,18,19]. Notch signaling has multiple context-specific features and plays an essential role in the maintenance and balance of adult tissues [20]. In contrast, the Hh pathway is predominantly insufficiently active or even quiescent in mature organisms. As necessary, under certain conditions, it would be activated, such as wound healing and tissue recovery [21, 22] in bone [23, 24], intervertebral disc [25], heart, nerves [26,27,28,29], skin [30], muscle [31], and gastrointestinal mucosa [32, 33]. However, componential activation of the Hh signaling pathway is responsible for the formation and progression of diverse cancer types, i.e., basal cell carcinoma (BCC) [34, 35]. In addition, an interesting characteristic of Notch signaling is its high sensitivity to dose: both over- and under-activated Notch signaling generate phenotypes, which can partially account for mutations in the Notch pathway that can serve as both carcinogens and suppressors of tumors, relying on the cellular setting [36].
The Notch pathway is an architecturally linear signaling mechanism. Concisely, the intact Notch receptor is a heterodimer made up of an extracellular and a transmembrane subunit. Subsequent interaction with transmembrane ligands presented on the neighboring leads to subunit receptor segregation and causes the transmembrane subunit to be proteolytically hydrolyzed, releasing a Notch intracellular domain (NICD), which is subsequently transported to the nucleus to function as a transcriptional regulator [20, 37]. Pathways in addition to the canonical signaling pathway are also capable of initiating signaling and are categorized as noncanonical NOTCH signaling pathways [38, 39]. Analogously, activation of Hh signaling pathways also proceeds through both canonical and noncanonical routes [40, 41]. The former is activated by the binding of Hh ligand to Ptch1 protein to constitute a complex that relieves the inhibitory effect of smoothened (Smo), followed by a downstream cytoplasmic protein complex consisting of kinesin protein (Kif7), suppressor of fused (SUFU), and glioma-associated oncogene homolog (Gli), which transduces signals to the nucleus and regulates the expression of target genes [42]. Resembling the noncanonical initiation of the Notch pathway, the later activation of Hh signaling can be categorized into three kinds [40]. Nevertheless, contrary to Notch and other embryonic signaling pathways, the Hh signaling pathway is extremely dependent on the primary cilium [43], a single organelle, with the composition of vital signaling components varying in distribution across the cilium depending on whether Hh signaling is switched on or off [44,45,46].
Within this review, we summarize current advances in the understanding of Notch and Hh biology and crosstalk between pathways in cancer, which will assist in the design of new reasonable therapeutic options [36]. Recent drug development results and efforts to target Notch and Hh signaling are also outlined, and the major categories of investigational drugs thus far described that directly or indirectly alter Notch and Hh signaling are characterized.
Architecture of the Notch and Hh pathways
The canonical and noncanonical Notch signaling pathways
Notch signaling is an intercellular communication mechanism initiated by binding between a transmembrane receptor and a membrane-spanning ligand expressed on adjacent cells. The major components and distinct steps in the Notch pathway have been investigated widely and are summarized in brief (Fig. 1) [47, 48].

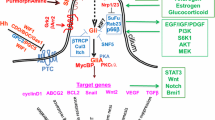
Overview of the Notch and Hedgehog signaling pathways. The blue arrows on the left side of the figure demonstrate the synthesis, modification, transfer and activation of Notch receptor precursors. The purple arrow on the right shows Hh ligand precursor synthesis, modification and translocation, and activation effect of Hh receptors. Crosstalk in the Hh and Notch signaling pathways is shown by red and yellow arrows
The canonical Notch signaling pathway is devoid of intermediate links, as the receptor is transduced straight into the nucleus after three cleavage events [12]. The precursor of the Notch receptor is a single-chain protein that is initially transported to the endoplasmic reticulum (ER) after production and extensively glycosylates future extracellular subunits, which modifies the affinity of the Notch receptor for various ligands (orange pentagons represent glycosylation) [49]. The glucosylated precursors are transmitted to the Golgi compartment and undergo the first proteolytic procedures at S1 sites in the Golgi network, yielding noncovalently bound Notch extracellular subunit (NEC)-transmembrane and intracellular domain (NTMIC) heterodimers to be shipped to the cell membrane as type I transmembrane proteins (NOTCH1-4 for mammals) [50]. The receptor interacts with canonical ligands (DLL1, 4, or Jagged 1, 2) located on juxtaposed cell membranes, exposing a cleavage site that is masked by the LNR domain in the silent phase and arousing dissociation of the receptor subunit by NEC subunit trans-endocytosis into the ligand-expressing cell [51,52,53]. This unveils the S2 site cleaved by an ADAM 10 or 17 (a disintegrin and metalloproteinase) on the extracellular residue of NTMIC [54]. Subsequent to S2 cleavage, another ligand processing step, the intermediate product of the transmembrane receptor called Notch extracellular truncation (NEXT) [54], is cleaved by the γ-secretase complex at the plasma membrane or in the endosome post-NEXT internalization (S3 cleavage) [55]. This phase releases NICD, which translocates to the nucleus, where it couples with CSL (CBF1/Suppressor of Hairless/LAG1; also named RBPJ), a DNA-binding protein, loosening chromatin [56] and leading to separation of the coinhibitory complex from CSL and facilitating coactivation complex recruitment [47, 57]. The interaction of NICD-CSL is stabilized by Mastermind-like (Maml), a triad of the NICD/CSL/Maml complex that amplifies downstream gene expression [56]. The pool of target genes varies broadly between cell types.
Notch signaling is also initiated by atypical ligands, the deficiency of ligands, the absence of the cleaved Notch receptor, or interactions with other cytoplasmic or cytosolic effectors [38, 39]. It is known as noncanonical Notch signaling, which offers a fascinating new scope for exploration and potentially unmasks new interventional treatment targets. The ligand-independent activation pathway [58] denotes that due to metabolism, ligand-unbound NOTCH receptors can be endocytosed into endosomes containing ADAM and gamma enzymes, triggering S2 and S3 cleavage and thereby activating NOTCH signaling [59, 60]. This pathway is essential for T-cell development [61, 62]. In addition to combining with CSL, NICD crosstalks with signaling pathways such as NF-κB, mTORC, TGF-β, AKT, Wnt, or Hippo at the cytoplasmic or cytosolic level to modulate target gene transcription [63,64,65,66,67]. Classically, reciprocity between NICD and NF-κB affects the properties of multiple malignancies [68,69,70], suggesting new potential means of blocking noncanonical NOTCH signaling. Moreover, proper ligands and receptors direct cell proliferation, transition and apoptosis in a single/unbound form, and details deserve further elucidation.
Multiple factors act on different components of the pathway involved in regulating Notch signaling. Specific signals, such as AKT, CBFB, SIRT6, RUNX1, and DEC1, control the transcription of receptors that indirectly regulate the entire Notch signaling pathway [71,72,73,74]. Certainly, glycosylation of the NOTCH receptors on certain EGF-like repetitive sequences is essential for receptor maturation [75], ligand binding [76, 77], and S2 cleavage [78], and targeting glycosylation is also thought to be effective in inhibiting NOTCH signaling. Intracellular trafficking of receptors mediated by ubiquitination and specific distribution of receptors and ligands across the cell membrane also influence the regional intensity of NOTCH signaling [79, 80]. The balance between activation and degradation following endocytosis is critically associated with downstream signaling. Ligand ubiquitination in signal senders is a requisite for signal activation and mediates ligand endocytosis, facilitating exposure of the negative regulatory region (NRR) domain of the S2 cleavage receptor [81,82,83]. Notably, cis-inhibition has been observed in many tissues and organisms [84], significantly driving the horizontal suppression process and defining sharp boundaries [85]. Subsequent work is worth extending to explore the potential diversity of signaling states of cells by the combinatorial action of diverse receptors and ligands. Noncoding RNAs likewise occupy a place in the Notch signaling pathway by serving as regulators of respective target genes [86, 87]. Excluding ligands and receptors, the level of Notch signaling can be adjusted by posttranslational modifications of NICD, including hydroxylation, methylation, acetylation, phosphorylation, and sumoylation, which impact the intensity of Notch signaling [88].
The canonical and noncanonical Hh signaling pathways
The canonical Ptch1-Smo-Gli axis is achieved by autocrine or paracrine patterns. Many excellent publications have previously described in detail the processes that regulate Hh signaling (Fig. 1) [89,90,91]. Simply speaking, three mammalian counterparts of Hh were found: sonic hedgehog (SHh), Indian hedgehog (IHh) and desert hedgehog (DHh) [92]. Disparities in the timing of expression, spatial distribution and action characteristics among the different ligands were noticed [93]. The maturation and secretion of Hh ligands, glycoprotein precursors synthesized in the ER encoded by a set of vital Hh genes, undergo self-proteolysis, generating C-terminal fragments (Hh-Cs) that are transferred to the proteasome to experience rapid degradation [94], and the N-terminal peptides (Hh-Ns) continue to undergo dual lipidation in the ER by cholesterol and palmitic acid in the C-terminal and N-terminal domains, respectively [95, 96]. The modified Hh-N molecules are attached to the lipid bilayer in monomeric form until they are released by one of four pivotal mechanisms: 1. DISP (a transmembrane transporter-like protein)-mediated and SCUBE2 (a secreted glycoprotein)-coordinated release of Hh-Ns from the cell surface as a monomer [97,98,99,100]; 2. Self-aggregation of monomeric HH-Ns to form soluble multimers for liberation [101]; 3. Interaction of HH-N oligomers with heparan sulfate chains of glycoproteins, recruiting apolipoproteins to shape lipoprotein particles [102, 103]; 4. Formation of exovesicle [104]. Spontaneously, canonical Hh signaling is initiated by active Hh ligands. Mature Hh ligands bind to Ptch1/2 [105], a cholesterol transporter enriched in primary cilia (PC). HH-PTC conjugation, facilitated by growth arrest-specific 1 (GAS1), oncogenes (CDO), brother of CDO (BOC), and low-density lipoprotein receptor-related protein 2 (LRP2), induces Ptch to leave the cilium and inactivate, relieving inhibition of Ptch for a seven-pass transmembrane G protein-coupled receptor-like receptor known as Smo. The derepressed Smo subsequently moves to the proximal end of PC to enrich with Ellis‑van Creveld syndrome protein (EVC) and EVC2, suppressing critical protein kinases, including protein kinase A (PKA), casein kinase 1 (CK1), and synthase kinase 3β (GSK3β) [106, 107], depriving the ability to mediate phosphorylation of the SUFU-Gli complex. The role of SUFU as a key suppressor by sequestering Gli in the cytoplasm is dismantled, triggering the detachment of SUFU-Gli and releasing the full-length active Gli (Gli1/2/3-A). Subsequently, the translocation of Gli-A into the nucleus of cells is partially dependent on Kif7 [108], invoking Gli-mediated transcription [109, 110]. Hh target genes consist of Hh signaling components (Ptch, Gli1, GAS1, Hhip), cell cycle regulators (CCND2, CDK) [111, 112], and other transcription factors (VEGFA, PDGFA, BCL2, Wnt, TGF, SNAIL, ELK1, MSX2, BMI1, SOX2, NANOG, Oct4, FOXM1, NR2F1, c-Myc, etc.) [113].
In the absence of Hh ligands, Ptch impedes the transport and localization of Smo in PC [114], leading to phosphorylation and protein cleavage of Gli2 and Gli3 by PKA, CKI and GSK3β to produce repressed forms (Gli2-R and Gli3-R, respectively) [115,116,117,118,119]. Compared to Gli2 and Gli3, Gli1 is primarily regulated at the transcriptional level and is not cleaved into a repressed form. Gli1 can also be manipulated by the ubiquitin‒proteasome system (UPS) [120]. As a result, downstream Hh signaling is aborted. The current Gli pre-patterning model has suffered from challenges, with studies demonstrating that Gli inhibition is not the default state for the Hh pathway [121].
Other than canonical signaling, several pathways activated by Hh ligands have been recognized as "atypical" with no need for PC as a mandatory site [122, 123]. Hh signaling can be classified into three types involving Ptch-Hh-mediated (Type I), Smo-dependent (Type II), and Gli-activated (Type III) signaling according to their modulatory mechanisms [124]. The first type is further divided into two subcategories (Type IA and IB). Ptch1 in Type I can regulate apoptosis by recruiting proapoptotic elements such as caspase-9, dependence-associated receptor transmembrane (DRAL) and tumor-upregulated CARD (caspase-associated recruitment domain)-containing antagonist of caspase nine (TUCAN) to the C-terminal domain. Hitherto, the majority of the noncanonical Hh signaling axis in Type II has been achieved by coupling Smo as a G protein-coupled receptor (GPCR) to a heterotrimeric G protein of the G inhibitory (Gi) family [125, 126], altering several crucial protein kinase activities, including PKA, RhoA, Rac1, NF-1, Src, PI3K/PLCγ, and calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2) [127,128,129,130,131,132], and promoting actin rearrangement, immune synapse formation and altered glycolytic capacity [131, 133]. Furthermore, Smo-dependent noncanonical Hh signaling, such as the Gαi-LGN-NuMA-Dynein axis and LKB1/AMPK axis, can facilitate canonical signaling by fostering cilia formation [131]. As observed, various signaling cascades can be involved at the Gli level (Type III), such as RAS-RAF-MEK-ERK [134, 135], PI3K/AKT/mTOR [136,137,138], TGFβ [139, 140], DYRK family [141], BET proteins, and oncogenic drivers (EWS/FLI1, SOX9 FOXC1, c-MYC) [142,143,144,145], which are involved in optimizing Gli activity [146]. Apparently, the activity of Gli can be adversely affected by tumor suppressors (p53, NUMB, SNF5) [147,148,149], MAPKKK/MEKK [150, 151], and miRNAs (miR-324-5p, miR-361, miR-326) [152,153,154] in Smo-independent Hh signaling appear to be a fallback activation pathway in canonical Hh signaling and a way for cells to evade the classical Hh pathway to activate downstream gene expression in response to abnormal environmental stress.
Crosstalk between the Notch and Hedgehog signaling pathways in cancer
Studies have shown that Notch and Hh signaling function synergistically during cell development in several systems, such as the digestive system [155], the nervous system [156], and the immune system [157]. Some studies revealed crosstalk among the Hh, Notch and Wnt pathways that exert synergistic promotive effects in tumorigenesis [158, 159]. The developing nervous system has been more thoroughly studied (Fig. 1). Primarily, it is widely known that Notch signaling can modulate Hh [160]. Second, Hh signaling allows direct or indirect control of Notch through downstream activities [161]. Modulation of the Notch pathway by Hh includes the following: 1. Downstream effectors of Hh govern the transcription of ligands and regulators in the Notch pathway, such as Fringe proteins. 2. Gli proteins deliver immediate, Notch-independent transcriptional regulation of certain Notch target genes [162, 163]. Hh is controlled by Notch signaling in the following ways: 1. Downstream effectors of Notch control the transport of Hh components (e.g., Ptch and Smo) to PC [164]. 2. Downstream effectors of Notch manage Gli levels, irrespective of transcription. 3. NICD plays a direct role in controlling Gli gene transcription [165]. In addition, both Notch and Hh signaling are inhibited by NUMB [148], which contributes to the ubiquitination and degradation of Notch1 through its polypyrimidine tract-binding protein (PTB) structural domain to directly interact with the WW structural domain of Itchy E3 Ubiquitin Ligase (ITCH) [166]. Likewise, NUMB directly inhibits the Hedgehog pathway, while ITCH promotes ubiquitination of Glis1 [167].
Evidence indicates the significance of crosstalk between Hedgehog and Notch in cancer biology and the resistance of cancer stem cells (CSCs) to treatment [168]. However, little is known about the specific molecular mechanisms. A loss-of-function study on Ptch1 showed that it accelerates Notch1-induced T-ALL pathogenesis [169]. In contrast, a mutually exclusive interaction between the Hedgehog and Notch pathways has been found in skin cancer [170]. Notch1 expression deficiency in squamous cell carcinoma correlates with increased Gli2 expression, leading to tumorigenesis [171]. Similarly, the overall activation of Hedgehog and Notch has been described in the etiopathogenesis of pituitary adenoma and prostate cancer [172]. A better understanding and explorations of the regulatory mechanisms of crosstalk between Hh and Notch in the control of tumor onset and progression are needed.
Role in cancer and therapeutic strategies
Cancer inhibitor or promoter?
Undoubtedly, Notch signaling exerts bimodal effects in cancer [173]. The homologs of the specific Notch components involved are different in diverse cancers. Gain-of-function mutations involved in Notch genes have been reported in T-ALL [174], splenic marginal zone lymphoma [175], chronic lymphocytic leukemia [176], breast cancer [177], an [178]d adenoid cystic carcinoma [179]. In addition, aberrant activation of wild-type Notch signaling can be observed in lung adenocarcinoma [180], colorectal cancer [181], breast cancer [182], ovarian cancer [183], HCC [184], glioma [185] and other aggressive tumors. Carcinogenic roles of Notch signaling include, but are not limited to, prevention of apoptosis, preservation of a stem cell-like phenotype, induction of epithelial-mesenchymal transition (EMT) [186], generation of drug resistance, promotion of metastasis, facilitation of angiogenesis [187] and mediation of tumor-mesenchymal interactions. In some situations, Notch can manifest as a tumor suppressor, and its loss-of-function or conditional absence results in neuroendocrine tumors [188], squamous cell cancers [189], and pancreatic ductal carcinoma [190]. The mechanisms involved above are complex and context-dependent [173]. Intriguingly, in different subclonal populations within a single tumor, Notch can simultaneously act as both an oncogenic and tumor suppressor [191]. A subpopulation of non-neuroendocrine tumor cells with high Notch activity in mouse small cell lung cancer proliferates more slowly than neuroendocrine cells, in accordance with the tumor suppressive effects of Notch. However, this population of non-neuroendocrine cells is comparatively chemo-resistant and generates growth factors that proliferate neuroendocrine cells with low Notch activity, thus exerting a noncell-autonomous oncogenic effect. Does this suggest that the combination of chemotherapy and Notch inhibitors may be more effective than chemotherapy alone? Less clearly, Notch activity was upregulated in murine-derived HCCs with triple knockout of retinoblastoma protein (RB) and two related RB family members, p107 and p130 [192], but after a pan-Notch blockade was performed, accelerated development of HCC and elevated expression of Notch-related genes were associated with a good prognosis of HCC. This demonstrates that some of the Notch tumor suppressive effects in HCC may not be cell-autonomous, secondary to crosstalk with Wnt in liver-specific tumor-associated macrophages [193]. From the perspective of drug discovery, development, and combination therapy, pan-inhibition or activation of Notch signaling may not be the best strategy; tumor types, target cell populations, Notch paralogs, peripheral immunity, treatment timing, and combination dosing must be considered.
In contrast to Notch signaling, inappropriate activation of Hh signaling leads to carcinogenesis. Deregulation of Hh signaling may be attributed to three key mechanisms: 1. Mutation driver: ligand-independent constitutive amplifications of signaling owing to deactivating mutations in patched receptor (Ptch1) or SUFU [194, 195], invoking mutations in Smo [196] (i.e., BCC [197, 198], subtyped medulloblastoma [199], rhabdomyosarcoma [200]). 2. Ligand dependency: (i) Auto-secretory ligand-dependent activation, in which tumor cells respond to increased expression of their HH ligands in a cell-autonomous manner (i.e., glioblastoma [201], neck squamous cell carcinoma [202, 203], and lung [204], breast [205, 206], ovarian [207], stomach [208, 209], esophageal [210], pancreatic [211], and prostate cancers); (ii) Paracrine-ligand-dependent model, in which Hh ligands produced and released by tumor cells switch on HH signaling in the peripheral stroma, in turn facilitating tumor expansion and metastasis, and creating a positive feedback loop (i.e., prostate, ovarian [212,213,214], pancreatic [215] and colorectal cancers [216, 217]); (iii) Reverse paracrine ligand-dependent signaling activation, indicates that Hh ligands are secreted from the stroma and received by tumor cells (i.e., multiple myeloma, lymphoma [218], and leukemia [219, 220]). 3. Noncanonical crosstalk: the specific signaling cascade described in the previous mechanism, like RAS-RAF-MEK-ERK and TGFβ [139, 140], upregulates Gli activity and promotes tumorigenesis (i.e., melanoma [221, 222], bladder cancer [223, 224], clear cell renal cell carcinoma [225], and HCC [226]). Although strategies to inactivate the Hh signaling pathway have been intensively studied, contradictory results have been obtained. The existence of interrelationships and cross-talk has pinned hopes for combination therapy. A cell type capable of both self-renewal by symmetric division and of generating more "differentiated" cells by asymmetric division is found in most types of liquid and solid cancers. The field of CSC has been reviewed extensively.
Cancer stem cells
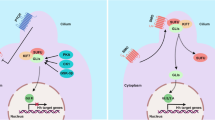
CSCs, a cell type capable of both self-renewal by symmetric division and of generating more "differentiated" cells by asymmetric division, are found in most types of liquid and solid cancers [227, 228]. and contribute to tumorigenesis, expansion, metastasis, drug resistance and recurrence. Wnt/β-catenin, TGF-β, Hedgehog and Notch are important signaling pathways for the sustainability of self-renewal in CSCs [206, 229](Fig. 2).

Overview of the Notch and Hedgehog signaling pathways in CSCs. Peripheral tumor cells, CSCs, and mesenchymal cells can initiate the transcription of stemness genes (NANOG, Oct4, Sox2, and Bmi1) in CSCs through Hedgehog signaling to maintain stem cell properties. The role of the Notch and Hedgehog signaling pathways is demonstrated in the zoomed-in CSC on the right side of the figure. S1P enhances CSC activity by binding S1PR secondary to Notch activation. The Hedgehog and Notch pathways also increase ABC-transporters, which increases drug efflux and leads to chemoresistance. The Hedgehog and Notch pathways also increase ROS further affecting the efficacy of subsequent drug and radiation therapy
Notch signaling plays an essential role in several cancer CSCs. In HER2 + breast cancers, membrane levels of Jagged 1 can be increased by HER2 inhibitors, thereby activating ligand-dependent Notch signaling in CSCs that are resistant to trastuzumab [227]. NOTCH1 activity was enhanced in triple-negative breast cancer (TNBC) CSC population after treatment with rapamycin complex 1/2 inhibitors, suggesting that combination therapy targeting the Notch pathway could be considered for TNBC with mutations leading to PI3K/mTOR pathway activation [230]. In estrogen receptor-positive (ER +) breast cancers, NOTCH1 activity is correlated with the risk of tumor recurrence [231]. More importantly, a number of ER + breast CSCs are NOTCH4 dependent, and elevated NOTCH4 expression relies on phosphorylation of Erα at Ser118 in cell lines containing ERα mutations [232,233,234]. Therefore, blocking ERα Ser118 phosphorylation or developing targeted Notch4 inhibitors may be valuable tools for the treatment of endocrine-resistant ER + breast cancers. In addition to receptor-associated Notch activation, sphingosine 1-phosphate induces highly oncogenic Notch activation via its own receptor in the ER + breast CSC population, which is also a potential therapeutic target [234]. Indeed, hypoxia, a feature of the CSC microenvironment, triggers Notch activation in glioblastoma CSCs by inducing the production of Vasorin-stabilized membrane-bound NOTCH1, which prevents degradation guided by the endocytic mediator NUMB [235].
Hh signaling is critical for the existence of subpopulations of tumor cells that exhibit stem cell characteristics [236, 237]. Hh signaling enables CSCs to maintain a stemness profile in multiple cancers by driving the expression of stemness-regulated genes such as NANOG, Oct4, Sox2, and Bmi1 [238,239,240]. CD200 + CD45 cancer cells have been determined to be a CSC population in BCC that expresses high levels of Gli1 and depends on Gli1 for survival [240]. Combination therapy with anti-CD200 + -neutralizing antibodies and Notch inhibitors is a new route to eradicate BCC. Noncanonical Ptch1-dependent Hh signaling is required for the survival of colon CSCs, particularly its regulation with crosstalk of the Wnt/β-catenin signaling pathway [241]. Suppression of Hh signaling was shown to inhibit pancreatic CSC growth in both in vitro and in vivo models, reversing the chemoresistance and stemness acquired by pancreatic cancer cells to the administration of gemcitabine [242]. The important role of Hh-Notch crosstalk in CSCs concerning cancer biology and chemoresistance has been confirmed, with conserved oncogenic properties associated with hypoxia and immunoevasion [243].
Migration and metastasis
Invasion and metastasis are unique properties of malignant tumors, and Notch and Hh are intertwined with other pathways that control tumor heterogeneity, stemness, EMT, angiogenic tumor cell dormancy, and other behaviors associated with tumor invasion and distant metastasis. Notch can interact directly or indirectly with ECM components [244]. In addition, cancer cell invasion into blood or lymphatic vessels relies on an actin-based structure called invadopodia that can be formed by Notch signaling mediated by activation upon hypoxia [245]. The coordinated response between NOTCH and EMT provides support for local angiogenesis and lymphangiogenesis [246, 247]. It is significant for the formation of circulating tumor cell (CTC) clusters, stabilization of CTCs in the bloodstream and extravasation of CTCs [248,249,250,251]. Several relatively isolated studies have also indicated a potential role of Notch in metastasis. Studies targeting Jagged ligands, their upstream regulators, or downstream effectors have proven that a high level of Jagged 1 is tightly associated with metastasis and recurrence of osteosarcoma [252]. Furthermore, TGFβ increases Jagged 1 expression in metastatic breast cancer cells, and Jagged 1 activates NOTCH1 in osteoblasts, causing IL-6 expression to support the survival of metastatic breast cancer cells and leading to the development of osteoclasts mediating bone breakdown. In chemotherapy-induced heterogeneous conditions within breast cancer, cancer cells evolve EMT by enhancing Notch signaling and have a greater ability to migrate and invade [253]. The relationship between Jagged-mediated Notch signaling and metastasis can also be observed in prostate cancer. Li et al. identified a novel FAS-ERK-JAG1-NOTCH1 axis that may be involved in the stemness and lung metastasis of oral squamous carcinoma [254]. In HCC, NET-4 promotes cell migration and tumor metastasis by increasing the enzymatic maturation of ADAM10, which in turn increases Notch signaling activated by cleavage of the Notch1 receptor catalyzed by the γ-secretase complex [255]. Activation of the Jagged-1/Notch/CXCR4 axis is also relevant to specific tumors [256]. NOTCH3 levels have also been shown to be associated with TNBC seeding and metastasis. Phosphoserine aminotransferase 1-specific upregulation intervenes in the metastasis of ER- breast cancer by prolonging the half-lives of NICD1 and β-catenin [257]. HOX transcript antisense intergenic RNA can positively correlate with the regulation of the Notch signaling pathway associated with tongue cancer invasion and metastasis [258].
Compared to invasion and metastasis with Notch, Hh has been relatively poorly studied and poorly linked. A number of studies revealed that high Hedgehog-Gli characteristics are associated with metastasis in colon cancer [119, 259, 260]. Gli1 activity is relevant to tumor aggressiveness in papillary thyroid carcinoma. Hedgehog promotes EMT in a variety of solid tumors, including liver, pancreatic, colon, and breast cancers [261, 262]. Shh enhanced the locomotion and invasiveness of gastric and ovarian cancer cells, while no assistance was observed in cells treated with anti-Shh monoclonal [263, 264]. The growth, adhesion and migration of lung adenocarcinoma cells are associated with activation of the PI3K and SHh pathways [265]. Degalactotigonin suppresses human osteosarcoma metastasis by regulating GSK3β inactivation-mediated inhibition of the Hedgehog/Gli1 pathway [266]. Circ-Gli1 indirectly upregulated Cyr61 through the activation of the Hedgehog and Wnt pathways, thereby exacerbating melanoma metastasis and angiogenesis. In addition, circ-Gli1 induced GSK3β phosphorylation at Ser9 under certain conditions, preventing GSK3β from binding to Gli1 and β-catenin, thereby increasing Gli1 and β-catenin protein expression [267].
Treatment resistance and tumor recurrence
The prominent problem we face with current anticancer therapies, including chemotherapy, radiotherapy, targeted therapy and immunotherapy, is the emergence of drug resistance leading to the ineffective control of tumors. The mechanisms of drug resistance are often multifaceted and complicated, including altered drug transport, drug metabolism, altered drug targets, blockade of apoptosis, alteration of the cell cycle, formation of EMT, induction of the CSC phenotype, alteration of the tumor microenvironment (TME), etc [268, 269].
For chemotherapy, Notch and Hh signaling adjust the expression of membrane transporters to regulate drug efflux. Upregulation of Notch by doxorubicin in prostate and breast cancers increased the expression of adenosine triphosphate-binding cassette subfamily B member 1 (ABCB1) and drug resistance-associated protein 1 (MRP1), which increased drug efflux and led to chemoresistance [270, 271]. This chemo-resistant effect is reversed by Notch inhibitors. Notch controls apoptosis, which is reflected in ovarian cancer, pancreatic cancer, HCC, osteosarcoma and glioblastoma that are resistant to chemotherapy, and inhibition of Notch activity leads to apoptosis [272,273,274,275]. Notch controls the cell cycle by inducing the expression of several key cell cycle-related genes to develop drug resistance [276]. Notch affects chemoresistance by controlling reactive oxygen species (ROS) levels in CSCs [277]. Similarly, Hh directly modulates ABCB1 and ABCG2 expression, and inhibition of Gli1 improves the response to chemotherapeutic agents in ovarian cancer [278]. Inhibitors of Gli in combination with temozolomide for glioblastoma induce apoptosis in already resistant cells [275]. Hh delivers ROS-induced signaling to regulate chemoresistance in various cancers. ROS-associated activation of NRF2 upregulates SHh, leading to sorafenib resistance in HCC [279]. Drug resistance associated with EMT, CSCs and the TME is also closely linked to both pathways [280, 281]. Activation of SHh promotes migration and invasion and drives DDP resistance in bladder cancer [282].
In radiotherapy, Notch signaling is initiated upon irradiation and regulates DNA damage repair by directly reacting with ATM and disabling its kinase activity [283, 284]. Notch signaling can also influence radio-resistance by mediating the function of other molecules involved in cell survival, metabolism, ROS, and the cell cycle. In glioma CSCs, Notch inhibition does not change the DNA damage reaction in CSCs but inhibits Akt and Mcl1 activities after radiation, making CSCs more sensitive to radiation [285]. Notch signaling mediated by NRF2 associated with cellular oxidation has been an important determinant of radio-resistance in lung cancer cells. Notch signaling mediated by NRF2 associated with cellular oxidation has been an essential decision factor for radio-resistance in lung cancer cells. The role of Hh in tumor radio-resistance has also been mentioned. Gli1 is usually upregulated at the HNSCC tumor-stroma crossover after irradiation and assists in stroma-mediated radio-resistance, which can be reduced by pharmacological inhibition [286, 287].
In terms of targeted therapies, tumors resistant to tyrosine kinase inhibitors (TKIs) frequently exhibit Notch upregulation. In AML with FLT3 mutations treated with FLT3-TKI, Notch signaling is upregulated, leading to alternative ERK activation and resistance. This resistance can be eliminated by Notch inhibitors [288]. Similarly, resistance to TKIs in lung adenocarcinomas with EGFR mutations and to BRAFi in melanomas carrying activated BRAF mutations can be eliminated by Notch inhibitors [289, 290]. The interaction of Hh with the EGFR signaling pathway is evident in many embryonic developmental processes and is therefore relevant to the development of TKI resistance. NSCLC and HNSCC cells with EGFR-TKI resistance exhibit hyperactivation of Hh signaling [291]. Hh pathway inhibitors act synergistically with TKIs to increase tumor sensitivity to chemotherapy and prolong survival in tumor-bearing animals [292]. To date, several mechanisms of resistance to Smo antagonists targeting the Hh pathway have been successively uncovered, including the following. Hh pathway components include Smo mutations [293, 294], SUFU deletion [295, 296], and amplification of Gli or Hh target genes [296]. 2. activation of nonclassical Hh pathways [141, 297, 298]. 3. loss of primitive cilia [299].
In the context of immunotherapy, Notch signaling has a regulatory role in immune cells in the local or systemic TME, and activation of Notch facilitates immunotherapy [300]. A significant association between NOTCH1/2/3 mutations and better outcome with immune checkpoint inhibitors (ICIs) was found in wild-type genetic NSCLC patients [301]. Hh affects immune responses in a complicated and varied manner. Hh inhibitors in pancreatic ductal adenocarcinoma suppress cancer-associated fibroblast (CAF) proliferation and translation and diminish the barrier to immune cell infiltration into the tumor [302]. Tumor cell-secreted Shh induces tumor-associated macrophage (TAM) M2 polarization and restrains CD8 + T-cell recruitment to the TME, thereby mediating immunosuppression [303]. Hh activity is not only a predictor of resistance to ICIs as a biomarker, but in association with PD-L1 expression, it can better forecast clinical outcomes [304].
The generation of resistance to anticancer therapies is a major cause of failure of various treatments. Tumors evolve through these conserved developmental signaling pathways (Notch, Hh) under the selective pressure of therapy, resulting in drug resistance. Therefore, the exploration of these pathways is necessary to eradicate drug resistance.
Tumor microenvironment
Recent evidence suggests that there are intricate interactions between tumor cells and stroma that have a significant impact on both tumor progression and regression. In particular, the mutual interactions between tumor cells and immune cells and CAFs deserve deeper and more exploration.
Cancer-associated immune cells
Notch and Hh signaling play an important role in immune system development and homeostasis. The roles of Notch and Hh in immune cells have attracted much attention for immunotherapy reasons. Notch has been demonstrated to play a dual sword role in the regulation of immune responses in tumors, a role equal to that of regular hematopoiesis and the production of immune effector cells [305](Fig. 3). Upregulation of NOTCH1 and NOTCH3 (perhaps) in basal- like breast cancer cell(BLBCC) leads to secretion of cytokines, such as IL1β and CCL2, which recruit monocytes that mature into tumor-associated macrophages (TAMs) in stroma. TAM can secrete TGFβ, which combines with the receptor TGFβR1 in tumor cells to induce Jagged 1 via SMAD2/3. TAM can secrete TGFβ, which combines with the receptor TGFβR1 in tumor cells to induce Jagged 1 via SMAD2/3. Jagged 1 in turn induces NOTCH1 and perhaps NOTCH3, forming a Jagged-involved tumor-stromal paracrine signaling loop [306, 307]. Myeloid-derived suppressor cells (MDSCs) enhance the stemness of breast cancer cells by activating Notch and STAT3 [308]. Dysregulation of Notch signaling in immature T cells induces MDSCs in T-ALL-bearing mice [309]. The absence of NOTCH2 in CD8 + T cells compromises the antitumor response, which can be enhanced by Notch stimulation of DLL1-expressing dendritic cells [310,311,312]. An additional potential function of DLL1-mediated Notch activation is to coculture with activated CD4/8 + T cells to convert them into stem cell-like memory T cells, which upon restimulation leads to a positive antitumor response [313]. Jagged1-induced Notch stimulation increases the expression and secretion of multiple cytokines associated with increased macrophage infiltration and reduced T-cell activity [314]. High NOTCH3 expression is associated with low infiltration of CD8 + T cells and high infiltration of immunosuppressive cells in gastric cancer [315]. The Mechanism section mentions TCR-mediated ligand-independent Notch activation, the intensity of which is determined by the amount of NOTCH1 and NOTCH2 (possibly) interacting with the immune synapse [61]. Conditional expression of the intracellular domain of NOTCH1 under the granzyme B promoter in CD8 + T cells with antigen specificity enhanced the cytotoxic response and overcame the tolerogenic effect of MDSCs. This provides new insight into the enhanced anticancer activity of chimeric antigen receptor (CAR) T cells [316].

Notch-mediated paracrine cycle between BLBCC and TAMs. Activated NOTCH1 and NOTCH3 (possibly) upregulate the expression of IL-1β and CCL2 in tumor cells. IL-1β and CCL2 recruit monocytes into the tumor microenvironment to mature into TAMs. TAM-secreted IL-1β and CCL2 recruit more monocytes and TAMs additionally secrete TGFβ-pro. TGFβ-pro is activated by UPA After being TGFβ, it induces Jagged 1 expression in cancer cells through its receptor TGFβR1 in combination with SMAD2/3. Jagged 1 activates NOTCH1 and NOTCH3 (probably), thus upregulating IL-1β and CCL2 expression
Hh signaling facilitates the activation of lymphocyte functions such as migration, multiplication, and cytotoxicity [317]. However, Hh was previously proven to promote TAM polarization to restrain tumor-infiltrating CD8 T + cell recruitment (mentioned in the drug resistance section) [303]. Hh also promotes Th2 differentiation in naive human CD4 + T cells [318]. Gli1 evokes polarization of invading myeloid cells toward MDSCs by inducing Schlafen 4 [319]. Hh signaling is also implicated in the functions of dendritic cells under hypoxic conditions, including migration, chemotaxis, phagocytosis, and cytokine secretion [320]. The above results suggest that the up- or downregulation of immune cell function and immune response are dependent on Hh signaling.
Cancer-associated fibroblasts
CAFs are essential components of the TME, yielding and remodeling an extensive extracellular matrix, accounting for a substantial proportion of the tumor volume [321, 322]. Conventional fibroblasts are an important source of CAFs, and other cell types may play a partial role [323]. Notch is considered to function in CAF activation and in crosstalk between CAFs and mutant cancer cells. In ductal carcinoma in situ (DCIS) of the breast, direct Notch-mediated crosstalk between tumor cells and CAFs was found [324, 325]. Direct interaction between Jagged1 on breast DCIS and NOTCH2 receptors on peripheral fibroblasts is possible [326]. In lung cancer, Notch activation is induced in CAFs and is associated with poor prognosis [327, 328]. Myofibroblastic CAFs (myCAFs) may be activated by IL-6/IL-8-mediated Notch/HES1 and STAT3 pathways in the tumor microenvironment to increase CSCs [329]. Notch modulates two matrix-degrading enzymes, MMP2 and MMP4, via NF-κB signaling to remodel the extracellular matrix in the TME [330]. Similarly, urokinase PA can be directly regulated by Notch [331].
CAFs are not only a potential source of Hh ligands in the cancer stroma but may also respond to H signaling through nuclear Gli-1 activation [332]. However, it has been mentioned in reviews that CAFs that inhibit tumor progression through stroma-specific Hh stimulation have been detected in cancer-bearing mice, including tumor models of the colon, bladder, and pancreas [333,334,335]. Inhibition of Hh reduced the number of myCAFs and increased the number of inflammatory CAFs in pancreatic ductal adenocarcinoma, which correlated with a cytotoxic T-cell reduction and regulatory T-cell increment, consistent with increased immunosuppression [336]. Given the heterogeneity of CAFs, a number of studies have shown that CAF-mediated Hh signaling is biased toward pro-tumorigenicity. SHh is highly expressed in CAF lysate CAF-derived exosomes, improving the growth and migration ability of esophageal cancer cells in vitro and in vivo. In addition, CAFs secreted extracellular vesicle-encapsulated miR-10a-5p, which activated Hh signaling and promoted angiogenesis and tumorigenicity in cervical squamous cell carcinoma cells [337]. In prostate cancer, CAFs expressing high levels of CD90 play a key role in promoting tumor progression by activating Hh and altering other signaling pathways [211].
Tumor angiogenesis
Control of angiogenesis is considered a promising approach to limit tumor progression and metastasis (Fig. 4). During physiological angiogenesis, Activation of Dll4-Notch-VEGFR2 axis is essential for appropriate angiogenesis and couples germinating angiogenesis to arteriogenesis. Similarities are shown in tumorigenic angiogenesis, activation of VEGF signaling in the "tip cells" at the front of the new vascular buds induces the expression of DLL4, which subsequently triggers the activation of NOTCH1 in adjacent endothelial cells and suppresses the expression of VEGFR2 and VEGFR3 in adjacent endothelial cells, giving the cells a "stalk" phenotype and stopping the sprouting of new capillary vessel, and eventually forming new vessels [338]. Blockade of DLL4 causes vascular hypersprouting and disturbed angiogenesis. mAbs of DLL4 are effective in preclinical models of cancer [339]. Nevertheless, chronically inhibiting DLL4-NOTCH1 signaling induces hemangiomas and hemorrhages. Besides, Jagged 1 expressed by tumor cells mediates Notch activation and inhibits the expression of soluble VEGFR1, which traps VEGF outside the cell and antagonizes VEGF signaling activation [340]. It is suggested that selective intervention with Jagged 1 is an appealing potential strategy for regulating tumor angiogenesis.

Notch- and Hh-mediated tumor neovascularization
The Hh pathway has been implicated in vascular development and neovascularization in human embryonic development, tissue injury and disease [341,342,343]. Indeed, the role of the Hh pathway in modulating tumor angiogenesis still deserves to be further explored [344]. There is a significant association between IHh and VEGF protein expression in HCC [345]. In pancreatic cancer, SHh indirectly promotes tumor angiogenesis by inducing Ptch1 and Gli1, thereby inhibiting two enriched stroma-derived antiangiogenic factors (THBS2 and TIMP2), and directly promotes tumor angiogenesis by activating small GTPases of the RHO family in the presence of VEGF [346]. Analogously, inhibition of nuclear translocation of Gli1 and transcriptional activity associated with Hh signaling can also inhibit angiogenesis in TNBC [347]. Moreover, extracellular vesicle-encapsulated microRNA-10a-5p detached from CAFs promotes cervical squamous cell carcinoma cell angiogenesis via the Hh signaling pathway [337].
Targeting Hedgehog and Notch signaling
Given the critical roles of Notch and Hh signaling in malignant transformation and oncogenic advancement, several agents targeting these pathways have been designed to potentiate antitumor effects and have been tested in animal models. An increasing number of targeted drugs have also been advanced to testing in clinical trials. In this section, we summarize current strategies targeting Notch and Hh signaling and discuss their anticancer effects, with the aim of promoting the leap from bench to bedside.
Therapeutic strategies based on Notch signaling
In light of the aberrant expression and wide participation of Notch signaling in tumor development, widespread efforts have been made to search for Notch-targeted therapeutic approaches, and numerous agents are being investigated in preclinical studies and undergoing clinical trials for cancer treatment [348, 349]. Current targeted strategies tested in the clinic principally converge on inhibiting or regulating γ-secretase and blocking the ligand‒receptor interaction via related monoclonal antibodies (Table 1) [350].
γ-Secretase inhibitors and modulators
Originally developed and used for the treatment of Alzheimer’s disease, γ-secretase inhibitors (GSIs) are currently extensively studied as anticancer drugs due to their potential to inhibit the activation of Notch signaling [351, 352]. In preclinical tumor models, GSIs have shown antitumor effects in a variety of tumor models, including lung [353], breast [354], glioma [355], pancreatic [356], colorectal [357], and ovarian cancers [358]. Although the application of pan-Notch GSI sometimes results in unexpected toxicity [359, 360], its antitumor activity has been shown in many cancer types through diverse mechanisms [361,362,363,364]. These promising findings motivated the initiation of a series of phased clinical trials. However, some of the GSIs (BMS-986115 [365], RO4929097 [366], MK0752 [367]) were discontinued in phase I clinical trials due to dose-limiting toxicity. Only RO4929097 and PF-03084014 have been included in phase II trials to date. Regrettably, the antitumor effects achieved were more limited, with no objective remission responses observed in the treatment of platinum-resistant ovarian cancer with RO4929097 [368]. Efficacy was also poor in metastatic melanoma [369], glioblastoma [370], and metastatic colorectal cancer [371]. Promisingly, PF-03084014 (nirogacestat) in desmoid tumors allowed 29% of patients to undergo a partial response lasting more than 2 years [372]. A phase III clinical trial of nirogacestat is expected. In contrast to the paninhibitory effect of GSIs on γ-secretase, γ-secretase modulators (GSMs) maintain a portion of Notch signaling function by altering the catalytic activity of γ-secretase [373]. Treatment of T-ALL cell lines with MRK-560 targeting the catalytic subclass of the γ-secretase complex has been demonstrated to decrease mutant NOTCH1 processing and lead to cell cycle arrest, and the toxicity can be tolerated [374].
Antibodies against ligands and receptors
Given that the location of extracellular ligands and receptors facilitates direct drug transport and targeted binding, enabling the development of antibodies targeting receptors and ligands was considered [375, 376]. Targeting antibodies against different receptors and ligands enables precise targeting and reduces serious adverse effects.
Antibodies against ligands
For the Notch ligands currently being explored and discovered, Jagged1, DLL3 and DLL4 are the most relevant and good quality studies. At the preclinical stage, 15D11 is one of the most prospective human monoclonal antibodies targeted against Jagged 1, with improved chemosensitivity, reduced bone metastases, and a high safety profile [377]. High expression of both Jagged 2 and DLL1 is positively associated with tumor progression and may also be promising targets, although agents aimed at these ligands have not been reported [328, 378]. DLL3 is an inhibitory ligand for NOTCH and is one of the ligands with positive results in clinical applications [379]. Rovalpituzumab tesirine (Rova-T) results revealed an objective remission rate of 18.3% and a 38% controlled incidence of severe drug-related AEs in patients with SCLC and large cell neuroendocrine carcinoma (LCNEC) [376]. Rova-T has a shorter OS and lower safety rate than standard second-line chemotherapy [380]. The mOS for SCLC patients receiving Rova-T after at least two lines of treatment was only 5.6 months, with an ORR of 12.4% [381]. Two clinical trials on SCLC, AM757 (bispecific antibody) and HPN328 (trispecific antibody), are still under recruitment. In addition, patient-derived glioma tumor spheroids with IDH mutations are susceptible to Rova-T in vitro, implicating the possibility of further exploration of indications and new drugs [382]. DLL4 is an important promoter of tumor angiogenesis and maintenance of CSCs [339]. It exhibits antitumor effects in ovarian cancer when blocked in combination with VEGF [383]. In phase I clinical studies, solid tumors exhibited a therapeutic response to both Enoticumab and Demcizumab, but the latter was associated with significant cardiotoxicity [384, 385]. Targeting DLL4 and VEGF dual variable domain immunoglobulins, ABT-165 demonstrated excellent efficacy and safety in preclinical models, and navicixizumab has shown modest antitumor efficacy and AEs in a phase I clinical trial in solid tumors [386, 387].
Antibodies aimed at receptors
NOTCH1 serves as a facilitator in various tumors, such as colorectal cancer and T-ALL, and is regarded as a possible antitumor target [388, 389]. In a phase I clinical trial for refractory solid tumors, the administration of brontictuzumab resulted in partial response in 2 patients and stable disease at ≥ 6 months in 4 patients [390]. Similarly, NOTCH2 and NOTCH3 act as promoters in breast cancer, pancreatic ductal adenocarcinoma [391], and lung cancer [392]. OMP-59R5, a NOTCH2 and NOTCH3 blocker, has been investigated in PDAC [393], SCLC, and other solid tumors in clinical trials [394]. However, OMP-59R5 in combination with chemotherapy has not achieved a good objective therapeutic response. PF-06650808 is a novel anti-NOTCH3 ADC that has achieved remission with a controlled safety profile in breast cancer patients with positive NOTCH3 expression [395]. The effect of NOTCH4 on tumors is context dependent, and there is a lack of well-studied agents.
ADAM inhibitors
ADAM10, or in some cases ADAM17, is the key protein for S2 cleavage of the ligand‒receptor binding domain in the Notch pathway. A phase I clinical trial of the bispecific ADAM10/17 inhibitor INCB7839 (aderbasib) is being conducted in recurrent, high-grade pediatric gliomas [396]. ZLDI-8, a novel ADAM17 inhibitor, was found to inhibit Notch and reverse rectal cancer resistance to 5-fluorouracil or irinotecan in vivo and in vitro [397]. Inhibitors of specific ADAM10 have not been reported.
Transcriptional complex inhibitors
Activation of transcription is the final step in signaling. NOTCH transcription is dependent on a complex composed of CSL, NICD, and Maml. The small molecule IMR-1 disrupts the recruitment of Maml1 and attenuates target gene transcription [398]. CB103 is described as an inhibitor of the Notch transcriptional complex via protein–protein interactions and is currently in clinical development with initial success in the inhibition of breast cancer and leukemia xenografts [399, 400].
Targeting Hh signaling for cancer therapy
It was elucidated that Hedgehog signaling could be blocked at various sites. Over the past decades, different methods aiming to inhibit the Hh pathway have been widely investigated in both solid and hematological cancers. Current targeted strategies mainly focus on inhibiting key components in the pathway cascade, such as SHh, Smo, and Gli [401,402,403,404].
Targeting SHh
The overexpression of SHh contributes to excessive Hh signaling and is positively correlated with tumor burden in nearly all cancer types [405]. As such, inhibiting SHh may represent an attractive therapeutic opportunity. Some SHh inhibitors have been developed to delay tumor progression. For example, 5E1 is a monoclonal anti-Shh antibody that can block the binding of SHh from interacting with Ptch1. The use of 5E1 was demonstrated to repress tumor proliferation and enhance the efficacy of radiation and cisplatin in cervical cancer [406,407,408]. RU-SKI 43, a dihydrothienopyridine derivative that was designed to target Hh acyltransferase [409], and previous studies confirmed its antitumor potential both in vivo and in vitro [410, 411]. Sulforaphane is the main ingredient in broccoli/broccoli sprouts and was recently found to inhibit leukemia stem-like cell growth and proliferation by modulating SHh signaling [412]. Furthermore, the N-terminal product of SHh (ShhN) has been shown to be overexpressed in cancer and thus can be recognized as a promising target that merits further consideration [413]. Despite these encouraging findings, none of these agents has been evaluated in clinical trials, and tremendous efforts are also needed to translate related basic research into clinical practice.
Targeting Smo
Smo represents a primary and promising target for blocking Hh signaling for a long time. Some Smo antagonists have been tested in the clinic. Mechanically, Smo antagonists specifically bind to the transmembrane domain and the pockets in the extracellular domain of Smo so as to inhibit Hh signal transduction [344, 414].
Vismodegib (GDC‑0449) was the first FDA-approved inhibitor in 2012 that can bind to SMO to disrupt its consistent activation, primarily for the treatment of patients with recurrent, locally advanced or metastatic basal cell carcinoma (BBC) [415, 416]. Several clinical trials have been conducted to test the anticancer effects of vismodegib as monotherapy or in combination with other agents in various conditions, including metastatic pancreatic cancer, prostate cancer, gastric cancer, recurrent glioblastoma, myelofibrosis, and acute myeloid leukemia [417,418,419,420,421]. Most of these trials are in stages I and II, as listed in Table 2. At present, when surgery and radiotherapy are inappropriate for anticancer therapy, vismodegib remains the major option for locally advanced BCC patients [422]. Although some complete or partial antitumor responses were obtained (NCT00607724, NCT02667574, NCT01815840, NCT01367665) [423,424,425,426,427], disappointing results remain (NCT01088815, NCT0160118, NCT01537107) [428,429,430], and unexpected adverse events associated with vismodegib frequently led to interruption of treatment, ultimately leading to cancer recurrence (NCT00957229) [431]. Moreover, in the course of vismodegib treatment, researchers found a novel Smo mutation that was typically accompanied by therapeutic resistance, which limited further clinical application of Smo-based therapy [432, 433]. Hence, how to reduce treatment-associated toxic side effects and Smo mutation is the next challenge we need to overcome.
Sonidegib, also known as LDE‑225, is a synthetically derived, new molecular entity that exerts its function by binding and inhibiting Smo receptors and is the second Smo inhibitor approved by the FDA in 2015 for the treatment of patients with locally advanced BCC not amenable to curative surgery or radiation therapy [434,435,436]. The use of sonidegib was typically well tolerated and yielded impressive clinical efficacy with reported objective responses, and there was a significant association between the anticancer efficiency and the Hh signaling activation monitored by gene expression (NCT01350115, NCT01327053, NCT00880308) [437,438,439,440,441,442]. Consistently, in a recent observational, retrospective, single-center study, researchers evaluated the clinical efficacy and safety of sonidegib in patients with BCC. The results showed that all enrolled subjects benefited from the treatment, either with a response or stabilization of the disease, supporting the further application of sonidegib in the clinic [443]. Nevertheless, in patients with BCC who were resistant to vismodegib, the same therapeutic resistance was also observed, thereby hampering sonidegib’s anticancer effect (NCT01529450) [444]. Future efforts in targeting Smo in cancer should focus on approaches addressing and overcoming these resistance mechanisms [42, 445]. In addition to BCC, previous and ongoing phased clinical trials have also been conducted to test the activity and response of sonidegib in other cancer types, including hepatocellular carcinoma, lung cancer, pancreatic adenocarcinoma, prostate cancer and esophageal cancer (Table 2).
PF-04449913 (Glasdegib) is another Smo inhibitor developed by Pfizer and has been approved by the FDA for treating both solid tumors and hematological malignancies [446,447,448,449]. Previous phased clinical trials identified its safety, tolerance, and potential antitumor efficacy in myeloid malignancies, glioblastoma and other advanced solid tumors [450,451,452]. Puhlished data support its further evaluation and motivate the subsequent initiation of phase III clinical trials as monotherapy or in combination with standard chemotherapy, which is summarized in Table 2.
Taladegib (LY-2940680) can bind to the Smo receptor, thereby blocking the propagation of Hh signaling. Currently, this synthetic small molecule inhibitor is being tested in several clinical trials to treat patients with advanced solid tumors [453]. TAK-441 is a highly potent and oral SMO inhibitor that has activity against both Hh ligand overexpression and mutation-driven Hh signaling pathway activation [454].
Furthermore, other Smo inhibitors, including LDE225, LEQ506, BMS-833923, IPI-926, LY2940680 and GDC0499, are currently under clinical investigation [455].
Targeting Gli
As previously mentioned, Gli represents a crucial transcription factor that can act as a downstream effector of canonical and noncanonical Hh signaling. Consequently, targeting Gli has the potential to overcome therapeutic resistance caused by Smo inhibitors [456, 457]. Simultaneously, extraordinary efforts have been made to discover and design reagents targeting Gli to achieve Hh pathway inhibition.
Initially, identified from cellular screening assays in 2007, GANT58 and GANT61 were demonstrated to be selective inhibitors of Hh-driven tumor growth because of their capacity to downregulate the transcriptional activity of Gli1 and Gli2 [458,459,460,461]. Since then, a substantial number of studies have explored their antitumor effects and have shown that GANT61 can inhibit malignant behavior, induce autophagy and apoptosis, and enhance the therapeutic sensitivity of tumor cells both in vitro and in vivo [462,463,464,465,466]. However, no clinical trials using GANT61 for treating human cancer are currently ongoing.
Arsenic trioxide (ATO) is another Gli inhibitor with the potential to decrease Gli transcriptional activity by binding to Gli1 and Gli2 and has been reported to delay tumor progression in preclinical studies [467, 468]. Clinically, ATO is evaluated for the treatment of different cancer types, including lung cancer, acute myeloid leukemia, and hepatocellular carcinoma [469,470,471]. (Table 2) Pirfenidone, an antifibrotic agent, selectively destabilizes Gli2 [472]. Imiquimod, an agonist of toll-like receptors 7 and 8, directly suppresses Hh signaling by stimulating PKA-mediated Gli phosphorylation [473].
Conclusion and future perspective
Since the first discovery of conserved key signaling pathways, such as Notch and Hh, in Drosophila, researchers have been persistently exploring their structure, regulatory mechanisms, physiology, and pathology and developing therapeutic strategies to target some of the signaling pathway components. This review summarizes the work that has been done in both the Notch and Hh signaling pathways and clarifies the scope for further basic research and the value of developing clinical applications for Notch and Hh signaling in cancer, where the activation of Hh is usually associated with tumor development. Inhibitors of its components (SHh, Smo, Gli) have been quite effective in some tumors (BCC) and some unsatisfactory. One of the most interesting aspects of NOTCH signaling is its context-dependent nature, which means that it possesses pro- or anticancer effects under different conditions. First, different types of ligands in the pathway have their own roles or different combinations of ligands and receptors, producing distinct effects. In addition, Notch function differs in diverse tumor types or even different subclones of the same tumor, which is perhaps related to Notch-dependent proximity secretion. Of course, the downstream gene expression of Notch contains both positive and negative regulators of tumor growth, perhaps also related to its double-edged sword effect.
NOTCH- and Hh-targeted therapies have been studied for decades but have failed to meet expectations. The main reasons for this include high pan-inhibition-induced organismal toxicity, low affinity for drug delivery systems, upregulation of bypass activation pathways, and the mechanism-driven group of individual drugs whose activity can easily be overestimated in preclinical models. The development of new drugs targeting molecules with higher specificity within the pathway and affinity antibody‒drug couples may be a countermeasure. New immunotherapeutic strategies, including DC pulse vaccines, CAR-T cells and DLL3-CAR-NK-cell therapies, are more attractive. Combination therapies with radiotherapy, chemotherapy, immunotherapy and other small molecule inhibitors can be considered. miRNAs interfere with both the Notch and Hh pathways and can be used as potential biomarkers and therapeutic approaches, although their delivery still poses technical challenges. As intradermal lysoviral therapy has entered the standard of care phase of oncology, viruses could be considered designed to respond to high Notch and Hh activity within the target cells. The antitumor effects of natural products of related pathways should not be underestimated [474, 475].
With the unveiling of findings on the mechanisms of embryonic development, a complex circuit formed between the two signaling pathways has been laid out. Notch and Hh signaling control each other in this feedback fashion. A tightly controlled loop prevents uncontrolled proliferation and incorrect patterns. It is reasonable to speculate whether there is a break in the control loop in tumor cell populations that proliferate out of control and whether the re-establishment of feedback can in turn gently promote tumor regression, all of which deserve further exploration. The field of cell signaling crosstalk is rapidly expanding, and one would expect them all to be part of the same conversation, taking a more coordinated and comprehensive view of the problem and solving it.
Availability of data and materials
Not applicable.
References
Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801. https://doi.org/10.1038/287795a0.
Zotter B, Dagan O, Brady J, Baloui H, Samanta J, Salzer JL. Gli1 Regulates the Postnatal Acquisition of Peripheral Nerve Architecture. J Neurosci. 2022;42(2):183–201. https://doi.org/10.1523/jneurosci.3096-20.2021.
Xie Y, Kuan AT, Wang W, Herbert ZT, Mosto O, Olukoya O, et al. Astrocyte-neuron crosstalk through Hedgehog signaling mediates cortical synapse development. Cell Rep. 2022;38(8):110416. https://doi.org/10.1016/j.celrep.2022.110416.
Wang X, Ma Z, Wu Y, Chen J, Peng X, Wang Y, et al. Expression pattern of Ptch2 in mouse embryonic maxillofacial development. Acta Histochem. 2022;124(1):151835. https://doi.org/10.1016/j.acthis.2021.151835.
Sivakumar S, Qi S, Cheng N, Sathe AA, Kanchwala M, Kumar A, et al. TP53 promotes lineage commitment of human embryonic stem cells through ciliogenesis and sonic hedgehog signaling. Cell Rep. 2022;38(7):110395. https://doi.org/10.1016/j.celrep.2022.110395.
Embalabala RJ, Brockman AA, Jurewicz AR, Kong JA, Ryan K, Guinto CD, et al. GLI3 Is Required for OLIG2+ Progeny Production in Adult Dorsal Neural Stem Cells. Cells. 2022;11(2):218. https://doi.org/10.3390/cells11020218.
Ohtsuka T, Kageyama R. Dual activation of Shh and Notch signaling induces dramatic enlargement of neocortical surface area. Neurosci Res. 2022;176:18–30. https://doi.org/10.1016/j.neures.2021.09.006.
Seppala M, Thivichon-Prince B, Xavier GM, Shaffie N, Sangani I, Birjandi AA, et al. Gas1 Regulates Patterning of the Murine and Human Dentitions through Sonic Hedgehog. J Dent Res. 2022;101(4):473–82. https://doi.org/10.1177/00220345211049403.
Reynolds JI, Vitek RA, Geiger PG, Johnson BP. Engineering Epithelial-Mesenchymal Microtissues to Study Cell-Cell Interactions in Development. Methods Mol Biol. 2022;2403:201–13. https://doi.org/10.1007/978-1-0716-1847-9_13.
Zhao C, Zhang Z, Qu X, Bai X, Liu X, Tao W, et al. Desert hedgehog mediates the proliferation of medaka spermatogonia through Smoothened signaling. Reproduction. 2022;163(4):209–18. https://doi.org/10.1530/rep-21-0468.
Yin W, Liontos A, Koepke J, Ghoul M, Mazzocchi L, Liu X, et al. An essential function for autocrine hedgehog signaling in epithelial proliferation and differentiation in the trachea. Development. 2022;149(3):dev199804. https://doi.org/10.1242/dev.199804.
Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–33. https://doi.org/10.1016/j.cell.2009.03.045.
Shi H, Wang H, Zhang C, Lu Y, Yao J, Chen Z, et al. Mutations in OSBPL2 cause hearing loss associated with primary cilia defects via sonic hedgehog signaling. JCI Insight. 2022;7(4):e149626. https://doi.org/10.1172/jci.insight.149626.
Xu R, Liu Y, Zhou Y, Lin W, Yuan Q, Zhou X, et al. Gnas Loss Causes Chondrocyte Fate Conversion in Cranial Suture Formation. J Dent Res. 2022:220345221075215. https://doi.org/10.1177/00220345221075215
Wu Y, Kurosaka H, Wang Q, Inubushi T, Nakatsugawa K, Kikuchi M, et al. Retinoic Acid Deficiency Underlies the Etiology of Midfacial Defects. J Dent Res. 2022:220345211062049. https://doi.org/10.1177/00220345211062049
Smith AE, Sigurbjörnsdóttir ES, Steingrímsson E, Sigurbjörnsdóttir S. Hedgehog signalling in bone and osteoarthritis: the role of Smoothened and cholesterol. Febs j. 2022. https://doi.org/10.1111/febs.16440.
Deng H, Xiao X, Chilufya MM, Qiao L, Lv Y, Guo Z, et al. Altered Expression of the Hedgehog Pathway Proteins BMP2, BMP4, SHH, and IHH Involved in Knee Cartilage Damage of Patients With Osteoarthritis and Kashin-Beck Disease. Cartilage. 2022;13(1):19476035221087704. https://doi.org/10.1177/19476035221087706.
Lei J, Chen S, Jing J, Guo T, Feng J, Ho TV, et al. Inhibiting Hh Signaling in Gli1(+) Osteogenic Progenitors Alleviates TMJOA. J Dent Res. 2022:220345211059079. https://doi.org/10.1177/00220345211059079
Rahi S, Mehan S. Understanding Abnormal SMO-SHH Signaling in Autism Spectrum Disorder: Potential Drug Target and Therapeutic Goals. Cell Mol Neurobiol. 2022;42(4):931–53. https://doi.org/10.1007/s10571-020-01010-1.
Wang H, Zang C, Liu XS, Aster JC. The role of Notch receptors in transcriptional regulation. J Cell Physiol. 2015;230(5):982–8. https://doi.org/10.1002/jcp.24872.
Le H, Kleinerman R, Lerman OZ, Brown D, Galiano R, Gurtner GC, et al. Hedgehog signaling is essential for normal wound healing. Wound Repair Regen. 2008;16(6):768–73. https://doi.org/10.1111/j.1524-475X.2008.00430.x.
Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432(7015):324–31. https://doi.org/10.1038/nature03100.
Zhang L, Fu X, Ni L, Liu C, Zheng Y, You H, et al. Hedgehog Signaling Controls Bone Homeostasis by Regulating Osteogenic/Adipogenic Fate of Skeletal Stem/Progenitor Cells in Mice. J Bone Miner Res. 2022;37(3):559–76. https://doi.org/10.1002/jbmr.4485.
Wu J, Wang R, Kan X, Zhang J, Sun W, Goltzman D, et al. A Sonic Hedgehog-Gli-Bmi1 signaling pathway plays a critical role in p27 deficiency induced bone anabolism. Int J Biol Sci. 2022;18(3):956–69. https://doi.org/10.7150/ijbs.65954.
Li Y, Wei Y, Li H, Che H, Miao D, Ma C, et al. Exogenous Parathyroid Hormone Alleviates Intervertebral Disc Degeneration through the Sonic Hedgehog Signalling Pathway Mediated by CREB. Oxid Med Cell Longev. 2022;2022:9955677. https://doi.org/10.1155/2022/9955677.
Yang WS, Shen YQ, Yang X, Li XH, Xu SH, Zhao LB, et al. MicroRNA Transcriptomics Analysis Identifies Dysregulated Hedgehog Signaling Pathway in a Mouse Model of Acute Intracerebral Hemorrhage Exposed to Hyperglycemia. J Stroke Cerebrovasc Dis. 2022;31(3):106281. https://doi.org/10.1016/j.jstrokecerebrovasdis.2021.106281.
Wang J, Ware K, Bedolla A, Allgire E, Turcato FC, Weed M, et al. Disruption of Sonic Hedgehog Signaling Accelerates Age-Related Neurogenesis Decline and Abolishes Stroke-Induced Neurogenesis and Leads to Increased Anxiety Behavior in Stroke Mice. Transl Stroke Res. 2022. https://doi.org/10.1007/s12975-022-00994-w.
Ghasemi H, Pegah A, Tayebinia H, Khazaei S, Feizi F, Mazaheri S, et al. The Overexpression of Sonic Hedgehog Associates with Collateral Development and Amelioration of Oxidative Stress in Stroke Patients. J Stroke Cerebrovasc Dis. 2022;31(5):106408. https://doi.org/10.1016/j.jstrokecerebrovasdis.2022.106408.
Tail M, Zhang H, Zheng G, Hatami M, Skutella T, Unterberg A, et al. The Sonic Hedgehog Pathway Modulates Survival, Proliferation, and Differentiation of Neural Progenitor Cells under Inflammatory Stress In Vitro. Cells. 2022;11(4):736. https://doi.org/10.3390/cells11040736.
Zhou JX, Jia LW, Liu WM, Miao CL, Liu S, Cao YJ, et al. Role of sonic hedgehog in maintaining a pool of proliferating stem cells in the human fetal epidermis. Hum Reprod. 2006;21(7):1698–704. https://doi.org/10.1093/humrep/del086.
Palla AR, Hilgendorf KI, Yang AV, Kerr JP, Hinken AC, Demeter J, et al. Primary cilia on muscle stem cells are critical to maintain regenerative capacity and are lost during aging. Nat Commun. 2022;13(1):1439. https://doi.org/10.1038/s41467-022-29150-6.
Uehara K, Koyanagi-Aoi M, Koide T, Itoh T, Aoi T. Epithelial-derived factors induce muscularis mucosa of human induced pluripotent stem cell-derived gastric organoids. Stem Cell Reports. 2022. https://doi.org/10.1016/j.stemcr.2022.02.002.
Chakrabarti J, Dua-Awereh M, Schumacher M, Engevik A, Hawkins J, Helmrath MA, et al. Sonic Hedgehog acts as a macrophage chemoattractant during regeneration of the gastric epithelium. NPJ Regen Med. 2022;7(1):3. https://doi.org/10.1038/s41536-021-00196-2.
Lama-Sherpa TD, Das S, Hinshaw DC, Kammerud SC, Song PN, Alsheikh HA, et al. Quantitative Longitudinal Imaging Reveals that Inhibiting Hedgehog Activity Alleviates the Hypoxic Tumor Landscape. Mol Cancer Res. 2022;20(1):150–60. https://doi.org/10.1158/1541-7786.Mcr-21-0257.
Haraguchi R, Kitazawa R, Kohara Y, Imai Y, Kitazawa S. Novel animal model of soft tissue tumor due to aberrant hedgehog signaling activation in pericyte lineage. Cell Tissue Res. 2022. https://doi.org/10.1007/s00441-022-03578-0.
Braune EB, Lendahl U. Notch – a goldilocks signaling pathway in disease and cancer therapy. Discov Med. 2016;21(115):189–96.
Majumder S, Crabtree JS, Golde TE, Minter LM, Osborne BA, Miele L. Targeting Notch in oncology: the path forward. Nat Rev Drug Discov. 2021;20(2):125–44. https://doi.org/10.1038/s41573-020-00091-3.
Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacol Ther. 2013;139(2):95–110. https://doi.org/10.1016/j.pharmthera.2013.02.003.
Ayaz F, Osborne BA. Non-canonical notch signaling in cancer and immunity. Front Oncol. 2014;4:345. https://doi.org/10.3389/fonc.2014.00345.
Jenkins D. Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal. 2009;21(7):1023–34. https://doi.org/10.1016/j.cellsig.2009.01.033.
Quatannens D, Verhoeven Y, Van Dam P, Lardon F, Prenen H, Roeyen G, et al. Targeting hedgehog signaling in pancreatic ductal adenocarcinoma. Pharmacol Ther. 2022;236:108107. https://doi.org/10.1016/j.pharmthera.2022.108107.
Nguyen NM, Cho J. Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance. Int J Mol Sci. 2022;23(3):1733. https://doi.org/10.3390/ijms23031733.
Rocha C, Prinos P. Post-transcriptional and Post-translational Modifications of Primary Cilia: How to Fine Tune Your Neuronal Antenna. Front Cell Neurosci. 2022;16:809917. https://doi.org/10.3389/fncel.2022.809917.
Aanstad P, Santos N, Corbit KC, Scherz PJ, le Trinh A, Salvenmoser W, et al. The extracellular domain of Smoothened regulates ciliary localization and is required for high-level Hh signaling. Curr Biol. 2009;19(12):1034–9. https://doi.org/10.1016/j.cub.2009.04.053.
Sigafoos AN, Paradise BD, Fernandez-Zapico ME. Hedgehog/GLI Signaling Pathway: Transduction, Regulation, and Implications for Disease. Cancers (Basel). 2021;13(14):3410. https://doi.org/10.3390/cancers13143410.
Yang S, Zheng X. The Immunofluorescence-Based Detection of Hedgehog Pathway Components in Primary Cilia of Cultured Cells. Methods Mol Biol. 2022;2374:89–94. https://doi.org/10.1007/978-1-0716-1701-4_8.
Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17(11):722–35. https://doi.org/10.1038/nrm.2016.94.
Siebel C, Lendahl U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol Rev. 2017;97(4):1235–94. https://doi.org/10.1152/physrev.00005.2017.
Kovall RA, Gebelein B, Sprinzak D, Kopan R. The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev Cell. 2017;41(3):228–41. https://doi.org/10.1016/j.devcel.2017.04.001.
Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, Blacklow SC. Structural basis for autoinhibition of Notch. Nat Struct Mol Biol. 2007;14(4):295–300. https://doi.org/10.1038/nsmb1227.
Capaccione KM, Pine SR. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis. 2013;34(7):1420–30. https://doi.org/10.1093/carcin/bgt127.
Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369(1):20–7. https://doi.org/10.1016/j.canlet.2015.07.048.
Stephenson NL, Avis JM. Direct observation of proteolytic cleavage at the S2 site upon forced unfolding of the Notch negative regulatory region. Proc Natl Acad Sci U S A. 2012;109(41):E2757–65. https://doi.org/10.1073/pnas.1205788109.
Groot AJ, Vooijs MA. The role of Adams in Notch signaling. Adv Exp Med Biol. 2012;727:15–36. https://doi.org/10.1007/978-1-4614-0899-4_2.
Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, et al. Three-dimensional structure of human γ-secretase. Nature. 2014;512(7513):166–70. https://doi.org/10.1038/nature13567.
Gomez-Lamarca MJ, Falo-Sanjuan J, Stojnic R, Abdul Rehman S, Muresan L, Jones ML, et al. Activation of the Notch Signaling Pathway In Vivo Elicits Changes in CSL Nuclear Dynamics. Dev Cell. 2018;44(5):611-623.e7. https://doi.org/10.1016/j.devcel.2018.01.020.
Sprinzak D, Blacklow SC. Biophysics of Notch Signaling. Annu Rev Biophys. 2021;50:157–89. https://doi.org/10.1146/annurev-biophys-101920-082204.
Conner SD. Regulation of Notch Signaling Through Intracellular Transport. Int Rev Cell Mol Biol. 2016;323:107–27. https://doi.org/10.1016/bs.ircmb.2015.12.002.
Pamarthy S, Kulshrestha A, Katara GK, Beaman KD. The curious case of vacuolar ATPase: regulation of signaling pathways. Mol Cancer. 2018;17(1):41. https://doi.org/10.1186/s12943-018-0811-3.
Jin K, Wen Z, Wu B, Zhang H, Qiu J, Wang Y, et al. NOTCH-induced rerouting of endosomal trafficking disables regulatory T cells in vasculitis. J Clin Invest. 2021;131(1):e136042. https://doi.org/10.1172/jci136042.
Steinbuck MP, Arakcheeva K, Winandy S. Novel TCR-Mediated Mechanisms of Notch Activation and Signaling. J Immunol. 2018;200(3):997–1007. https://doi.org/10.4049/jimmunol.1700070.
Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol. 2013;13(6):427–37. https://doi.org/10.1038/nri3445.
Song LL, Peng Y, Yun J, Rizzo P, Chaturvedi V, Weijzen S, et al. Notch-1 associates with IKKalpha and regulates IKK activity in cervical cancer cells. Oncogene. 2008;27(44):5833–44. https://doi.org/10.1038/onc.2008.190.
Chen LJ, Zhang NN, Zhou CX, Yang ZX, Li YR, Zhang T, et al. Gm364 coordinates MIB2/DLL3/Notch2 to regulate female fertility through AKT activation. Cell Death Differ. 2022;29(2):366–80. https://doi.org/10.1038/s41418-021-00861-5.
Mangolini M, Götte F, Moore A, Ammon T, Oelsner M, Lutzny-Geier G, et al. Notch2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat Commun. 2018;9(1):3839. https://doi.org/10.1038/s41467-018-06069-5.
Kim W, Khan SK, Yang Y. Interacting network of Hippo, Wnt/β-catenin and Notch signaling represses liver tumor formation. BMB Rep. 2017;50(1):1–2. https://doi.org/10.5483/bmbrep.2017.50.1.196.
Katoh M, Katoh M. Transcriptional mechanisms of WNT5A based on NF-kappaB, Hedgehog, TGFbeta, and Notch signaling cascades. Int J Mol Med. 2009;23(6):763–9. https://doi.org/10.3892/ijmm_00000190.
Fernández-Majada V, Aguilera C, Villanueva A, Vilardell F, Robert-Moreno A, Aytés A, et al. Nuclear IKK activity leads to dysregulated notch-dependent gene expression in colorectal cancer. Proc Natl Acad Sci U S A. 2007;104(1):276–81. https://doi.org/10.1073/pnas.0606476104.
Zhu H, Bhaijee F, Ishaq N, Pepper DJ, Backus K, Brown AS, et al. Correlation of Notch1, pAKT and nuclear NF-κB expression in triple negative breast cancer. Am J Cancer Res. 2013;3(2):230–9.
Kuramoto T, Goto H, Mitsuhashi A, Tabata S, Ogawa H, Uehara H, et al. Dll4-Fc, an inhibitor of Dll4-notch signaling, suppresses liver metastasis of small cell lung cancer cells through the downregulation of the NF-κB activity. Mol Cancer Ther. 2012;11(12):2578–87. https://doi.org/10.1158/1535-7163.Mct-12-0640.
Kohlhaas V, Blakemore SJ, Al-Maarri M, Nickel N, Pal M, Roth A, et al. Active Akt signaling triggers CLL toward Richter transformation via overactivation of Notch1. Blood. 2021;137(5):646–60. https://doi.org/10.1182/blood.2020005734.
Li Y, Jin C, Bai H, Gao Y, Sun S, Chen L, et al. Human NOTCH4 is a key target of RUNX1 in megakaryocytic differentiation. Blood. 2018;131(2):191–201. https://doi.org/10.1182/blood-2017-04-780379.
Malik N, Yan H, Moshkovich N, Palangat M, Yang H, Sanchez V, et al. The transcription factor CBFB suppresses breast cancer through orchestrating translation and transcription. Nat Commun. 2019;10(1):2071. https://doi.org/10.1038/s41467-019-10102-6.
Gallo C, Fragliasso V, Donati B, Torricelli F, Tameni A, Piana S, et al. The bHLH transcription factor DEC1 promotes thyroid cancer aggressiveness by the interplay with NOTCH1. Cell Death Dis. 2018;9(9):871. https://doi.org/10.1038/s41419-018-0933-y.
Stahl M, Uemura K, Ge C, Shi S, Tashima Y, Stanley P. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J Biol Chem. 2008;283(20):13638–51. https://doi.org/10.1074/jbc.M802027200.
Yao D, Huang Y, Huang X, Wang W, Yan Q, Wei L, et al. Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood. 2011;117(21):5652–62. https://doi.org/10.1182/blood-2010-12-326074.
Takeuchi H, Haltiwanger RS. Role of glycosylation of Notch in development. Semin Cell Dev Biol. 2010;21(6):638–45. https://doi.org/10.1016/j.semcdb.2010.03.003.
Acar M, Jafar-Nejad H, Takeuchi H, Rajan A, Ibrani D, Rana NA, et al. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell. 2008;132(2):247–58. https://doi.org/10.1016/j.cell.2007.12.016.
Liu P, Verhaar AP, Peppelenbosch MP. Signaling Size: Ankyrin and SOCS Box-Containing ASB E3 Ligases in Action. Trends Biochem Sci. 2019;44(1):64–74. https://doi.org/10.1016/j.tibs.2018.10.003.
Guo Y, Zhang K, Cheng C, Ji Z, Wang X, Wang M, et al. Numb(-/low) Enriches a Castration-Resistant Prostate Cancer Cell Subpopulation Associated with Enhanced Notch and Hedgehog Signaling. Clin Cancer Res. 2017;23(21):6744–56. https://doi.org/10.1158/1078-0432.Ccr-17-0913.
Overstreet E, Fitch E, Fischer JA. Fat facets and Liquid facets promote Delta endocytosis and Delta signaling in the signaling cells. Development. 2004;131(21):5355–66. https://doi.org/10.1242/dev.01434.
Daskalaki A, Shalaby NA, Kux K, Tsoumpekos G, Tsibidis GD, Muskavitch MA, et al. Distinct intracellular motifs of Delta mediate its ubiquitylation and activation by Mindbomb1 and Neuralized. J Cell Biol. 2011;195(6):1017–31. https://doi.org/10.1083/jcb.201105166.
Le Borgne R, Remaud S, Hamel S, Schweisguth F. Two distinct E3 ubiquitin ligases have complementary functions in the regulation of delta and serrate signaling in Drosophila. PLoS Biol. 2005;3(4):e96. https://doi.org/10.1371/journal.pbio.0030096.
Baek C, Freem L, Goïame R, Sang H, Morin X, Tozer S. Mib1 prevents Notch Cis-inhibition to defer differentiation and preserve neuroepithelial integrity during neural delamination. PLoS Biol. 2018;16(4):e2004162. https://doi.org/10.1371/journal.pbio.2004162.
Sprinzak D, Lakhanpal A, Lebon L, Santat LA, Fontes ME, Anderson GA, et al. Cis-interactions between Notch and Delta generate mutually exclusive signalling states. Nature. 2010;465(7294):86–90. https://doi.org/10.1038/nature08959.
Guo J, Li P, Liu X, Li Y. NOTCH signaling pathway and non-coding RNAs in cancer. Pathol Res Pract. 2019;215(11):152620. https://doi.org/10.1016/j.prp.2019.152620.
Chandimali N, Huynh DL, Zhang JJ, Lee JC, Yu DY, Jeong DK, et al. MicroRNA-122 negatively associates with peroxiredoxin-II expression in human gefitinib-resistant lung cancer stem cells. Cancer Gene Ther. 2019;26(9–10):292–304. https://doi.org/10.1038/s41417-018-0050-1.
Antfolk D, Antila C, Kemppainen K, Landor SK, Sahlgren C. Decoding the PTM-switchboard of Notch. Biochim Biophys Acta Mol Cell Res. 2019;1866(12):118507. https://doi.org/10.1016/j.bbamcr.2019.07.002.
Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5(12):1026–33. https://doi.org/10.1038/nrd2086.
Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30(6):303–12. https://doi.org/10.1016/j.tips.2009.03.007.
Atwood SX, Chang AL, Oro AE. Hedgehog pathway inhibition and the race against tumor evolution. J Cell Biol. 2012;199(2):193–7. https://doi.org/10.1083/jcb.201207140.
Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14(7):416–29. https://doi.org/10.1038/nrm3598.
Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22(18):2454–72. https://doi.org/10.1101/gad.1693608.
Chen X, Tukachinsky H, Huang CH, Jao C, Chu YR, Tang HY, et al. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J Cell Biol. 2011;192(5):825–38. https://doi.org/10.1083/jcb.201008090.
Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem. 1998;273(22):14037–45. https://doi.org/10.1074/jbc.273.22.14037.
Tang X, Chen R, Mesias VSD, Wang T, Wang Y, Poljak K, et al. A SURF4-to-proteoglycan relay mechanism that mediates the sorting and secretion of a tagged variant of sonic hedgehog. Proc Natl Acad Sci U S A. 2022;119(11):e2113991119. https://doi.org/10.1073/pnas.2113991119.
Tukachinsky H, Kuzmickas RP, Jao CY, Liu J, Salic A. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2012;2(2):308–20. https://doi.org/10.1016/j.celrep.2012.07.010.
Creanga A, Glenn TD, Mann RK, Saunders AM, Talbot WS, Beachy PA. Scube/You activity mediates release of dually lipid-modified Hedgehog signal in soluble form. Genes Dev. 2012;26(12):1312–25. https://doi.org/10.1101/gad.191866.112.
Ehring K, Manikowski D, Goretzko J, Froese J, Gude F, Jakobs P, et al. Conserved cholesterol-related activities of Dispatched 1 drive Sonic hedgehog shedding from the cell membrane. J Cell Sci. 2022;135(5):jcs258672. https://doi.org/10.1242/jcs.258672.
Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010;29(4):469–81. https://doi.org/10.1038/onc.2009.392.
Gallet A, Ruel L, Staccini-Lavenant L, Thérond PP. Cholesterol modification is necessary for controlled planar long-range activity of Hedgehog in Drosophila epithelia. Development. 2006;133(3):407–18. https://doi.org/10.1242/dev.02212.
Eugster C, Panáková D, Mahmoud A, Eaton S. Lipoprotein-heparan sulfate interactions in the Hh pathway. Dev Cell. 2007;13(1):57–71. https://doi.org/10.1016/j.devcel.2007.04.019.
Panáková D, Sprong H, Marois E, Thiele C, Eaton S. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature. 2005;435(7038):58–65. https://doi.org/10.1038/nature03504.
Davis SJ. Release and transportation of Hedgehog molecules. Curr Opin Cell Biol. 2012;24(2):173–80. https://doi.org/10.1016/j.ceb.2012.02.001.
Holtz AM, Griffiths SC, Davis SJ, Bishop B, Siebold C, Allen BL. Secreted HHIP1 interacts with heparan sulfate and regulates Hedgehog ligand localization and function. J Cell Biol. 2015;209(5):739–57. https://doi.org/10.1083/jcb.201411024.
Wen J, Hadden MK. Affinity-based protein profiling identifies vitamin D3 as a heat shock protein 70 antagonist that regulates hedgehog transduction in murine basal cell carcinoma. Eur J Med Chem. 2022;228:114005. https://doi.org/10.1016/j.ejmech.2021.114005.
Blotta S, Jakubikova J, Calimeri T, Roccaro AM, Amodio N, Azab AK, et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood. 2012;120(25):5002–13. https://doi.org/10.1182/blood-2011-07-368142.
He M, Subramanian R, Bangs F, Omelchenko T, Liem KF Jr, Kapoor TM, et al. The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat Cell Biol. 2014;16(7):663–72. https://doi.org/10.1038/ncb2988.
Tang C, Wu X, Ren Q, Yao M, Xu S, Yan Z. Hedgehog signaling is controlled by Rac1 activity. Theranostics. 2022;12(3):1303–20. https://doi.org/10.7150/thno.67702.
Cai E, Zhang J, Ge X. Control of the Hedgehog pathway by compartmentalized PKA in the primary cilium. Sci China Life Sci. 2022;65(3):500–14. https://doi.org/10.1007/s11427-021-1975-9.
Bonifas JM, Pennypacker S, Chuang PT, McMahon AP, Williams M, Rosenthal A, et al. Activation of expression of hedgehog target genes in basal cell carcinomas. J Invest Dermatol. 2001;116(5):739–42. https://doi.org/10.1046/j.1523-1747.2001.01315.x.
Zhang Y, Zheng A, Xu R, Zhou F, Hao A, Yang H, et al. NR2F1-induced NR2F1-AS1 promotes esophageal squamous cell carcinoma progression via activating Hedgehog signaling pathway. Biochem Biophys Res Commun. 2019;519(3):497–504. https://doi.org/10.1016/j.bbrc.2019.09.015.
Yang S, Wu X, Daoutidou EI, Zhang Y, Shimell M, Chuang KH, et al. The NDNF-like factor Nord is a Hedgehog-induced extracellular BMP modulator that regulates Drosophila wing patterning and growth. Elife. 2022;11:e73357. https://doi.org/10.7554/eLife.73357.
Kinnebrew M, Johnson KA, Radhakrishnan A, Rohatgi R. Measuring and Manipulating Membrane Cholesterol for the Study of Hedgehog Signaling. Methods Mol Biol. 2022;2374:73–87. https://doi.org/10.1007/978-1-0716-1701-4_7.
Zhou M, Jiang J. Gli Phosphorylation Code in Hedgehog Signal Transduction. Front Cell Dev Biol. 2022;10:846927. https://doi.org/10.3389/fcell.2022.846927.
Han Y, Jiang J. Ci/Gli Phosphorylation by the Fused/Ulk Family Kinases. Methods Mol Biol. 2022;2374:213–29. https://doi.org/10.1007/978-1-0716-1701-4_19.
Persson M, Stamataki D, te Welscher P, Andersson E, Böse J, Rüther U, et al. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 2002;16(22):2865–78. https://doi.org/10.1101/gad.243402.
Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26(9):3365–77. https://doi.org/10.1128/mcb.26.9.3365-3377.2006.
Ogata R, Mori S, Ohmori H, Kishi S, Fujiwara-Tani R, Sasaki T, et al. Suppressive GLI2 fragment enhances liver metastasis in colorectal cancer. Oncotarget. 2022;13:122–35. https://doi.org/10.18632/oncotarget.28170.
Regl G, Neill GW, Eichberger T, Kasper M, Ikram MS, Koller J, et al. Human GLI2 and GLI1 are part of a positive feedback mechanism in Basal Cell Carcinoma. Oncogene. 2002;21(36):5529–39. https://doi.org/10.1038/sj.onc.1205748.
Lex RK, Zhou W, Ji Z, Falkenstein KN, Schuler KE, Windsor KE, et al. GLI transcriptional repression is inert prior to Hedgehog pathway activation. Nat Commun. 2022;13(1):808. https://doi.org/10.1038/s41467-022-28485-4.
Brennan D, Chen X, Cheng L, Mahoney M, Riobo NA. Noncanonical Hedgehog signaling. Vitam Horm. 2012;88:55–72. https://doi.org/10.1016/b978-0-12-394622-5.00003-1.
Sari IN, Phi LTH, Jun N, Wijaya YT, Lee S, Kwon HY. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells. 2018;7(11):208. https://doi.org/10.3390/cells7110208.
Hewavitharana T, Wedegaertner PB. Non-canonical signaling and localizations of heterotrimeric G proteins. Cell Signal. 2012;24(1):25–34. https://doi.org/10.1016/j.cellsig.2011.08.014.
Riobo NA, Saucy B, Dilizio C, Manning DR. Activation of heterotrimeric G proteins by Smoothened. Proc Natl Acad Sci U S A. 2006;103(33):12607–12. https://doi.org/10.1073/pnas.0600880103.
Akhshi T, Shannon R, Trimble WS. The complex web of canonical and non-canonical Hedgehog signaling. BioEssays. 2022;44(3):e2100183. https://doi.org/10.1002/bies.202100183.
Jäger T, Ocker M, Kiesslich T, Neureiter E, Neureiter D. Thoughts on investigational hedgehog pathway inhibitors for the treatment of cancer. Expert Opin Investig Drugs. 2017;26(2):133–6. https://doi.org/10.1080/13543784.2017.1274392.
Praktiknjo SD, Saad F, Maier D, Ip P, Hipfner DR. Activation of Smoothened in the Hedgehog pathway unexpectedly increases Gα(s)-dependent cAMP levels in Drosophila. J Biol Chem. 2018;293(35):13496–508. https://doi.org/10.1074/jbc.RA118.001953.
Polizio AH, Chinchilla P, Chen X, Kim S, Manning DR, Riobo NA. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem. 2011;286(22):19589–96. https://doi.org/10.1074/jbc.M110.197111.
Qu C, Liu Y, Kunkalla K, Singh RR, Blonska M, Lin X, et al. Trimeric G protein-CARMA1 axis links smoothened, the hedgehog receptor transducer, to NF-κB activation in diffuse large B-cell lymphoma. Blood. 2013;121(23):4718–28. https://doi.org/10.1182/blood-2012-12-470153.
Yam PT, Langlois SD, Morin S, Charron F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 2009;62(3):349–62. https://doi.org/10.1016/j.neuron.2009.03.022.
Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191(2):415–28. https://doi.org/10.1083/jcb.201004108.
de la Roche M, Asano Y, Griffiths GM. Origins of the cytolytic synapse. Nat Rev Immunol. 2016;16(7):421–32. https://doi.org/10.1038/nri.2016.54.
Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernández-Zapico ME, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23(1):24–36. https://doi.org/10.1101/gad.1753809.
Riobo NA, Haines GM, Emerson CP Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006;66(2):839–45. https://doi.org/10.1158/0008-5472.Can-05-2539.
Singh R, Dhanyamraju PK, Lauth M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget. 2017;8(1):833–45. https://doi.org/10.18632/oncotarget.13662.
Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 2012;21(3):374–87. https://doi.org/10.1016/j.ccr.2011.12.028.
Mizuarai S, Kawagishi A, Kotani H. Inhibition of p70S6K2 down-regulates Hedgehog/GLI pathway in non-small cell lung cancer cell lines. Mol Cancer. 2009;8:44. https://doi.org/10.1186/1476-4598-8-44.
Dennler S, André J, Alexaki I, Li A, Magnaldo T, ten Dijke P, et al. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007;67(14):6981–6. https://doi.org/10.1158/0008-5472.Can-07-0491.
Tang YA, Chen YF, Bao Y, Mahara S, Yatim S, Oguz G, et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci U S A. 2018;115(26):E5990-e5999. https://doi.org/10.1073/pnas.1801348115.
Gruber W, Hutzinger M, Elmer DP, Parigger T, Sternberg C, Cegielkowski L, et al. DYRK1B as therapeutic target in Hedgehog/GLI-dependent cancer cells with Smoothened inhibitor resistance. Oncotarget. 2016;7(6):7134–48. https://doi.org/10.18632/oncotarget.6910.
Beauchamp E, Bulut G, Abaan O, Chen K, Merchant A, Matsui W, et al. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem. 2009;284(14):9074–82. https://doi.org/10.1074/jbc.M806233200.
Deng W, Vanderbilt DB, Lin CC, Martin KH, Brundage KM, Ruppert JM. SOX9 inhibits β-TrCP-mediated protein degradation to promote nuclear GLI1 expression and cancer stem cell properties. J Cell Sci. 2015;128(6):1123–38. https://doi.org/10.1242/jcs.162164.
Han B, Qu Y, Jin Y, Yu Y, Deng N, Wawrowsky K, et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015;13(5):1046–58. https://doi.org/10.1016/j.celrep.2015.09.063.
Yoon JW, Gallant M, Lamm ML, Iannaccone S, Vieux KF, Proytcheva M, et al. Noncanonical regulation of the Hedgehog mediator GLI1 by c-MYC in Burkitt lymphoma. Mol Cancer Res. 2013;11(6):604–15. https://doi.org/10.1158/1541-7786.Mcr-12-0441.
Pietrobono S, Gagliardi S, Stecca B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front Genet. 2019;10:556. https://doi.org/10.3389/fgene.2019.00556.
Stecca B, Ruiz iAltaba A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. Embo J. 2009;28(6):663–76. https://doi.org/10.1038/emboj.2009.16.
Di Marcotullio L, Ferretti E, Greco A, De Smaele E, Po A, Sico MA, et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat Cell Biol. 2006;8(12):1415–23. https://doi.org/10.1038/ncb1510.
Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med. 2010;16(12):1429–33. https://doi.org/10.1038/nm.2251.
Antonucci L, Di Magno L, D’Amico D, Manni S, Serrao SM, Di Pastena F, et al. Mitogen-activated kinase kinase kinase 1 inhibits hedgehog signaling and medulloblastoma growth through GLI1 phosphorylation. Int J Oncol. 2019;54(2):505–14. https://doi.org/10.3892/ijo.2018.4638.
Lu J, Liu L, Zheng M, Li X, Wu A, Wu Q, et al. MEKK2 and MEKK3 suppress Hedgehog pathway-dependent medulloblastoma by inhibiting GLI1 function. Oncogene. 2018;37(28):3864–78. https://doi.org/10.1038/s41388-018-0249-5.
Zhao D, Cui Z. MicroRNA-361-3p regulates retinoblastoma cell proliferation and stemness by targeting hedgehog signaling. Exp Ther Med. 2019;17(2):1154–62. https://doi.org/10.3892/etm.2018.7062.
Ferretti E, De Smaele E, Miele E, Laneve P, Po A, Pelloni M, et al. Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. Embo j. 2008;27(19):2616–27. https://doi.org/10.1038/emboj.2008.172.
Sargazi ML, Jafarinejad-Farsangi S, Moazzam-Jazi M, Rostamzadeh F, Karam ZM. The crosstalk between long non-coding RNAs and the hedgehog signaling pathway in cancer. Med Oncol. 2022;39(9):127. https://doi.org/10.1007/s12032-022-01710-2.
Kosinski C, Li VS, Chan AS, Zhang J, Ho C, Tsui WY, et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc Natl Acad Sci U S A. 2007;104(39):15418–23. https://doi.org/10.1073/pnas.0707210104.
Jacobs CT, Huang P. Complex crosstalk of Notch and Hedgehog signalling during the development of the central nervous system. Cell Mol Life Sci. 2021;78(2):635–44. https://doi.org/10.1007/s00018-020-03627-3.
Crompton T, Outram SV, Hager-Theodorides AL. Sonic hedgehog signalling in T-cell development and activation. Nat Rev Immunol. 2007;7(9):726–35. https://doi.org/10.1038/nri2151.
Pelullo M, Zema S, Nardozza F, Checquolo S, Screpanti I, Bellavia D. Wnt, Notch, and TGF-β Pathways Impinge on Hedgehog Signaling Complexity: An Open Window on Cancer. Front Genet. 2019;10:711. https://doi.org/10.3389/fgene.2019.00711.
Chatterjee S, Sil PC. Targeting the crosstalks of Wnt pathway with Hedgehog and Notch for cancer therapy. Pharmacol Res. 2019;142:251–61. https://doi.org/10.1016/j.phrs.2019.02.027.
Yabut O, Pleasure SJ, Yoon K. A Notch above Sonic Hedgehog. Dev Cell. 2015;33(4):371–2. https://doi.org/10.1016/j.devcel.2015.05.001.
Patni AP, Harishankar MK, Joseph JP, Sreeshma B, Jayaraj R, Devi A. Comprehending the crosstalk between Notch, Wnt and Hedgehog signaling pathways in oral squamous cell carcinoma - clinical implications. Cell Oncol (Dordr). 2021;44(3):473–94. https://doi.org/10.1007/s13402-021-00591-3.
Wall DS, Mears AJ, McNeill B, Mazerolle C, Thurig S, Wang Y, et al. Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J Cell Biol. 2009;184(1):101–12. https://doi.org/10.1083/jcb.200805155.
Dave RK, Ellis T, Toumpas MC, Robson JP, Julian E, Adolphe C, et al. Sonic hedgehog and notch signaling can cooperate to regulate neurogenic divisions of neocortical progenitors. PLoS ONE. 2011;6(2):e14680. https://doi.org/10.1371/journal.pone.0014680.
Stasiulewicz M, Gray SD, Mastromina I, Silva JC, Björklund M, Seymour PA, et al. A conserved role for Notch signaling in priming the cellular response to Shh through ciliary localisation of the key Shh transducer Smo. Development. 2015;142(13):2291–303. https://doi.org/10.1242/dev.125237.
Jacobs CT, Huang P. Notch signalling maintains Hedgehog responsiveness via a Gli-dependent mechanism during spinal cord patterning in zebrafish. Elife. 2019;8:e49252. https://doi.org/10.7554/eLife.49252.
McGill MA, McGlade CJ. Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem. 2003;278(25):23196–203. https://doi.org/10.1074/jbc.M302827200.
Hayward P, Kalmar T, Arias AM. Wnt/Notch signalling and information processing during development. Development. 2008;135(3):411–24. https://doi.org/10.1242/dev.000505.
Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12(8):445–64. https://doi.org/10.1038/nrclinonc.2015.61.
Burns MA, Liao ZW, Yamagata N, Pouliot GP, Stevenson KE, Neuberg DS, et al. Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia. 2018;32(10):2126–37. https://doi.org/10.1038/s41375-018-0097-x.
Bakshi A, Chaudhary SC, Rana M, Elmets CA, Athar M. Basal cell carcinoma pathogenesis and therapy involving hedgehog signaling and beyond. Mol Carcinog. 2017;56(12):2543–57. https://doi.org/10.1002/mc.22690.
Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, et al. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33(3):416–21. https://doi.org/10.1038/ng1099.
Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22(3):373–88. https://doi.org/10.1016/j.ccr.2012.07.016.
Aster JC, Pear WS, Blacklow SC. The Varied Roles of Notch in Cancer. Annu Rev Pathol. 2017;12:245–75. https://doi.org/10.1146/annurev-pathol-052016-100127.
Reynolds TC, Smith SD, Sklar J. Analysis of DNA surrounding the breakpoints of chromosomal translocations involving the beta T cell receptor gene in human lymphoblastic neoplasms. Cell. 1987;50(1):107–17. https://doi.org/10.1016/0092-8674(87)90667-2.
Bonfiglio F, Bruscaggin A, Guidetti F, Terzi di Bergamo L, Faderl M, Spina V, et al. Genetic and phenotypic attributes of splenic marginal zone lymphoma. Blood. 2022;139(5):732–47. https://doi.org/10.1182/blood.2021012386.
Tardivon D, Antoszewski M, Zangger N, Nkosi M, Sordet-Dessimoz J, Hendriks R, et al. Notch signaling promotes disease initiation and progression in murine chronic lymphocytic leukemia. Blood. 2021;137(22):3079–92. https://doi.org/10.1182/blood.2020006701.
Callahan R, Smith GH. MMTV-induced mammary tumorigenesis: gene discovery, progression to malignancy and cellular pathways. Oncogene. 2000;19(8):992–1001. https://doi.org/10.1038/sj.onc.1203276.
Yavropoulou MP, Maladaki A, Yovos JG. The role of Notch and Hedgehog signaling pathways in pituitary development and pathogenesis of pituitary adenomas. Hormones (Athens). 2015;14(1):5–18. https://doi.org/10.1007/bf03401377.
Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45(7):791–8. https://doi.org/10.1038/ng.2643.
Yuan X, Wu H, Xu H, Han N, Chu Q, Yu S, et al. Meta-analysis reveals the correlation of Notch signaling with non-small cell lung cancer progression and prognosis. Sci Rep. 2015;5:10338. https://doi.org/10.1038/srep10338.
Fre S, Pallavi SK, Huyghe M, Laé M, Janssen KP, Robine S, et al. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc Natl Acad Sci U S A. 2009;106(15):6309–14. https://doi.org/10.1073/pnas.0900427106.
Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, et al. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65(18):8530–7. https://doi.org/10.1158/0008-5472.Can-05-1069.
Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609-15.https://doi.org/10.1038/nature10166
Razumilava N, Gores GJ. Notch-driven carcinogenesis: the merging of hepatocellular cancer and cholangiocarcinoma into a common molecular liver cancer subtype. J Hepatol. 2013;58(6):1244–5. https://doi.org/10.1016/j.jhep.2013.01.017.
Chu Q, Orr BA, Semenkow S, Bar EE, Eberhart CG. Prolonged inhibition of glioblastoma xenograft initiation and clonogenic growth following in vivo Notch blockade. Clin Cancer Res. 2013;19(12):3224–33. https://doi.org/10.1158/1078-0432.Ccr-12-2119.
Tamagnone L, Zacchigna S, Rehman M. Taming the Notch Transcriptional Regulator for Cancer Therapy. Molecules. 2018;23(2):431. https://doi.org/10.3390/molecules23020431.
Thurston G, Kitajewski J. VEGF and Delta-Notch: interacting signalling pathways in tumour angiogenesis. Br J Cancer. 2008;99(8):1204–9. https://doi.org/10.1038/sj.bjc.6604484.
Hu J, Wang Y, Zhang Y, Yu Y, Chen H, Liu K, et al. Comprehensive genomic profiling of small cell lung cancer in Chinese patients and the implications for therapeutic potential. Cancer Med. 2019;8(9):4338–47. https://doi.org/10.1002/cam4.2199.
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–60. https://doi.org/10.1126/science.1208130.
Avila JL, Kissil JL. Notch signaling in pancreatic cancer: oncogene or tumor suppressor? Trends Mol Med. 2013;19(5):320–7. https://doi.org/10.1016/j.molmed.2013.03.003.
Lim JS, Ibaseta A, Fischer MM, Cancilla B, O’Young G, Cristea S, et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature. 2017;545(7654):360–4. https://doi.org/10.1038/nature22323.
Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C, et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med. 2011;208(10):1963–76. https://doi.org/10.1084/jem.20110198.
Ye YC, Zhao JL, Lu YT, Gao CC, Yang Y, Liang SQ, et al. NOTCH Signaling via WNT Regulates the Proliferation of Alternative, CCR2-Independent Tumor-Associated Macrophages in Hepatocellular Carcinoma. Cancer Res. 2019;79(16):4160–72. https://doi.org/10.1158/0008-5472.Can-18-1691.
Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31(3):306–10. https://doi.org/10.1038/ng916.
Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85(6):841–51. https://doi.org/10.1016/s0092-8674(00)81268-4.
Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–2. https://doi.org/10.1038/34201.
Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57(5):842–5.
Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013;19(11):1410–22. https://doi.org/10.1038/nm.3389.
Yuan L, Zhang H, Liu J, Malhotra A, Dey A, Yu B, et al. STAT3 is required for Smo-dependent signaling and mediates Smo-targeted treatment resistance and tumorigenesis in Shh medulloblastoma. Mol Oncol. 2022;16(4):1009–25. https://doi.org/10.1002/1878-0261.13097.
Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgård R, Undén AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. 2006;208(1):17–25. https://doi.org/10.1002/path.1882.
Chang J, Guo C, Li J, Liang Z, Wang Y, Yu A, et al. EN1 Regulates Cell Growth and Proliferation in Human Glioma Cells via Hedgehog Signaling. Int J Mol Sci. 2022;23(3):1123. https://doi.org/10.3390/ijms23031123.
Patel H, Joshi J, Raval A, Shah F. Identification of Natural Compounds to Inhibit Sonic Hedgehog Pathway in Oral Cancer. Anticancer Agents Med Chem. 2022;22(5):905–13. https://doi.org/10.2174/1871520621666210708100747.
Chakraborty B, Basu M, Mukhopadhyay D, Alam N, Ghosh S, Dutta S, et al. Differential promoter usages of PTCH1 and down regulation of HHIP are associated with HNSCC progression. Pathol Res Pract. 2022;232:153827. https://doi.org/10.1016/j.prp.2022.153827.
Tu YC, Yeh WC, Yu HH, Lee YC, Su BC. Hedgehog Suppresses Paclitaxel Sensitivity by Regulating Akt-Mediated Phosphorylation of Bax in EGFR Wild-Type Non-Small Cell Lung Cancer Cells. Front Pharmacol. 2022;13:815308. https://doi.org/10.3389/fphar.2022.815308.
Buyuk B, Jin S, Ye K. Epithelial-to-Mesenchymal Transition Signaling Pathways Responsible for Breast Cancer Metastasis. Cell Mol Bioeng. 2022;15(1):1–13. https://doi.org/10.1007/s12195-021-00694-9.
Zhang T, Zhou H, Wang K, Wang X, Wang M, Zhao W, et al. Role, molecular mechanism and the potential target of breast cancer stem cells in breast cancer development. Biomed Pharmacother. 2022;147:112616. https://doi.org/10.1016/j.biopha.2022.112616.
Szkandera J, Kiesslich T, Haybaeck J, Gerger A, Pichler M. Hedgehog signaling pathway in ovarian cancer. Int J Mol Sci. 2013;14(1):1179–96. https://doi.org/10.3390/ijms14011179.
Cui X, Shan T, Qiao L. Collagen type IV alpha 1 (COL4A1) silence hampers the invasion, migration and epithelial-mesenchymal transition (EMT) of gastric cancer cells through blocking Hedgehog signaling pathway. Bioengineered. 2022. https://doi.org/10.1080/21655979.2022.2053799.
Zhang J, Fan J, Zeng X, Nie M, Luan J, Wang Y, et al. Hedgehog signaling in gastrointestinal carcinogenesis and the gastrointestinal tumor microenvironment. Acta Pharm Sin B. 2021;11(3):609–20. https://doi.org/10.1016/j.apsb.2020.10.022.
Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425(6960):846–51. https://doi.org/10.1038/nature01972.
Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–6. https://doi.org/10.1038/nature02009.
van der Ploeg P, Uittenboogaard A, Bosch SL, van Diest PJ, Wesseling-Rozendaal YJW, van de Stolpe A, et al. Signal transduction pathway activity in high-grade serous carcinoma, its precursors and Fallopian tube epithelium. Gynecol Oncol. 2022. https://doi.org/10.1016/j.ygyno.2022.01.027.
Lin M, Zhu H, Shen Q, Sun LZ, Zhu X. GLI3 and androgen receptor are mutually dependent for their malignancy-promoting activity in ovarian and breast cancer cells. Cell Signal. 2022;92:110278. https://doi.org/10.1016/j.cellsig.2022.110278.
Zhao F, Yu XP, Zhao H, Song BB, Lyu AW, Zhang SH, et al. Expression and significance of GLI1 and Shh in the malignant transformation of ovarian endometriosis. Zhonghua Fu Chan Ke Za Zhi. 2022;57(2):125–32. https://doi.org/10.3760/cma.j.cn112141-20211219-00736.
Theunissen JW, de Sauvage FJ. Paracrine Hedgehog signaling in cancer. Cancer Res. 2009;69(15):6007–10. https://doi.org/10.1158/0008-5472.Can-09-0756.
Zeng S, Zhou F, Wang Y, Zhai Z, Xu L, Wang H, et al. Aberrant expression of the extracellular matrix component Biglycan regulated by Hedgehog signalling promotes colorectal cancer cell proliferation. Acta Biochim Biophys Sin (Shanghai). 2022;54(2):1–9. https://doi.org/10.3724/abbs.2021018.
Wang Y, Chen H, Jiao X, Wu L, Yang Y, Zhang J, et al. PTCH1 mutation promotes antitumor immunity and the response to immune checkpoint inhibitors in colorectal cancer patients. Cancer Immunol Immunother. 2022;71(1):111–20. https://doi.org/10.1007/s00262-021-02966-9.
Hegde GV, Peterson KJ, Emanuel K, Mittal AK, Joshi AD, Dickinson JD, et al. Hedgehog-induced survival of B-cell chronic lymphocytic leukemia cells in a stromal cell microenvironment: a potential new therapeutic target. Mol Cancer Res. 2008;6(12):1928–36. https://doi.org/10.1158/1541-7786.Mcr-08-0142.
Lv G, Wang Y, Ji C, Shi C, Li Y. SPRY1 promotes cell proliferation and inhibits apoptosis by activating Hedgehog pathway in acute myeloid leukemia. Hematology. 2022;27(1):1–10. https://doi.org/10.1080/16078454.2021.2010330.
Zhang Y, Cai R, Li H, Duan Y, Zhang Y, Jing W, et al. Construction of a target MSNs drugcarrier loaded with siRNA(GLI1) and siRNA(SMO) aim at hedgehog signal pathway and the pharmacodynamic study of drug-carriers in the treatment of leukemia stem cells. Drug Deliv Transl Res. 2022. https://doi.org/10.1007/s13346-020-00893-3.
Patmanathan SN, Tong BT, Teo JHJ, Ting YZJ, Tan NS, Sim SHK, et al. A PDZ Protein GIPC3 Positively Modulates Hedgehog Signaling and Melanoma Growth. J Invest Dermatol. 2022;142(1):179-188.e4. https://doi.org/10.1016/j.jid.2021.04.033.
Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A. 2007;104(14):5895–900. https://doi.org/10.1073/pnas.0700776104.
Zhang Z, Wang C, Liu T, Tang Z, Yan R, Zhang C, et al. miRNA-182-5p promotes human bladder cancer proliferation and migration through the FOXF2/SHH axis. Neoplasma. 2022. https://doi.org/10.4149/neo_2021_210903N1266.
Wang W, Qiu J, Qu P, Chen H, Lan J, Chen H, et al. Regulator of cullins-1 (ROC1) negatively regulates the Gli2 regulator SUFU to activate the hedgehog pathway in bladder cancer. Cancer Cell Int. 2021;21(1):75. https://doi.org/10.1186/s12935-021-01775-5.
Kotulak-Chrzaszcz A, Rybarczyk A, Klacz J, Matuszewski M, Kmiec Z, Wierzbicki PM. Expression levels of sonic hedgehog pathway genes and their targets are upregulated in early clear cell renal cell carcinoma. Int J Mol Med. 2022;49(5):58. https://doi.org/10.3892/ijmm.2022.5114.
Xia Y, Zhen L, Li H, Wang S, Chen S, Wang C, et al. MIRLET7BHG promotes hepatocellular carcinoma progression by activating hepatic stellate cells through exosomal SMO to trigger Hedgehog pathway. Cell Death Dis. 2021;12(4):326. https://doi.org/10.1038/s41419-021-03494-1.
O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16(12):3113–20. https://doi.org/10.1158/1078-0432.Ccr-09-2824.
Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol. 2019;234(6):8381–95. https://doi.org/10.1002/jcp.27740.
Senobari Z, Karimi G, Jamialahmadi K. Ellagitannins, promising pharmacological agents for the treatment of cancer stem cells. Phytother Res. 2022;36(1):231–42. https://doi.org/10.1002/ptr.7307.
Bhola NE, Jansen VM, Koch JP, Li H, Formisano L, Williams JA, et al. Treatment of Triple-Negative Breast Cancer with TORC1/2 Inhibitors Sustains a Drug-Resistant and Notch-Dependent Cancer Stem Cell Population. Cancer Res. 2016;76(2):440–52. https://doi.org/10.1158/0008-5472.Can-15-1640-t.
Mollen EWJ, Ient J, Tjan-Heijnen VCG, Boersma LJ, Miele L, Smidt ML, et al. Moving Breast Cancer Therapy up a Notch. Front Oncol. 2018;8:518. https://doi.org/10.3389/fonc.2018.00518.
Gelsomino L, Panza S, Giordano C, Barone I, Gu G, Spina E, et al. Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Lett. 2018;428:12–20. https://doi.org/10.1016/j.canlet.2018.04.023.
Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446–51. https://doi.org/10.1038/ng.2823.
Hirata N, Yamada S, Shoda T, Kurihara M, Sekino Y, Kanda Y. Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat Commun. 2014;5:4806. https://doi.org/10.1038/ncomms5806.
Man J, Yu X, Huang H, Zhou W, Xiang C, Huang H, et al. Hypoxic Induction of Vasorin Regulates Notch1 Turnover to Maintain Glioma Stem-like Cells. Cell Stem Cell. 2018;22(1):104-118.e6. https://doi.org/10.1016/j.stem.2017.10.005.
Onishi H, Nakamura K, Yanai K, Nagai S, Nakayama K, Oyama Y, et al. Cancer therapy that targets the Hedgehog signaling pathway considering the cancer microenvironment (Review). Oncol Rep. 2022;47(5):93. https://doi.org/10.3892/or.2022.8304.
Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers (Basel). 2015;7(3):1554–85. https://doi.org/10.3390/cancers7030851.
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz iAltaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17(2):165–72. https://doi.org/10.1016/j.cub.2006.11.033.
Batsaikhan BE, Yoshikawa K, Kurita N, Iwata T, Takasu C, Kashihara H, et al. Cyclopamine decreased the expression of Sonic Hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res. 2014;34(11):6339–44.
Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25(10):2524–33. https://doi.org/10.1634/stemcells.2007-0166.
Regan JL, Schumacher D, Staudte S, Steffen A, Haybaeck J, Keilholz U, et al. Non-Canonical Hedgehog Signaling Is a Positive Regulator of the WNT Pathway and Is Required for the Survival of Colon Cancer Stem Cells. Cell Rep. 2017;21(10):2813–28. https://doi.org/10.1016/j.celrep.2017.11.025.
Huang FT, Zhuan-Sun YX, Zhuang YY, Wei SL, Tang J, Chen WB, et al. Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int J Oncol. 2012;41(5):1707–14. https://doi.org/10.3892/ijo.2012.1597.
Chang WH, Lai AG. Aberrations in Notch-Hedgehog signalling reveal cancer stem cells harbouring conserved oncogenic properties associated with hypoxia and immunoevasion. Br J Cancer. 2019;121(8):666–78. https://doi.org/10.1038/s41416-019-0572-9.
LaFoya B, Munroe JA, Mia MM, Detweiler MA, Crow JJ, Wood T, et al. Notch: A multi-functional integrating system of microenvironmental signals. Dev Biol. 2016;418(2):227–41. https://doi.org/10.1016/j.ydbio.2016.08.023.
Díaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201(2):279–92. https://doi.org/10.1083/jcb.201209151.
Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010;12(10):943–53. https://doi.org/10.1038/ncb2103.
Mizukoshi K, Okazawa Y, Haeno H, Koyama Y, Sulidan K, Komiyama H, et al. Metastatic seeding of human colon cancer cell clusters expressing the hybrid epithelial/mesenchymal state. Int J Cancer. 2020;146(9):2547–62. https://doi.org/10.1002/ijc.32672.
Jordan NV, Bardia A, Wittner BS, Benes C, Ligorio M, Zheng Y, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537(7618):102–6. https://doi.org/10.1038/nature19328.
Amantini C, Morelli MB, Nabissi M, Piva F, Marinelli O, Maggi F, et al. Expression Profiling of Circulating Tumor Cells in Pancreatic Ductal Adenocarcinoma Patients: Biomarkers Predicting Overall Survival. Front Oncol. 2019;9:874. https://doi.org/10.3389/fonc.2019.00874.
Sprouse ML, Welte T, Boral D, Liu HN, Yin W, Vishnoi M, et al. PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling. Int J Mol Sci. 2019;20(8):1916. https://doi.org/10.3390/ijms20081916.
Boareto M, Jolly MK, Goldman A, Pietilä M, Mani SA, Sengupta S, et al. Notch-Jagged signalling can give rise to clusters of cells exhibiting a hybrid epithelial/mesenchymal phenotype. J R Soc Interface. 2016;13(118):20151106. https://doi.org/10.1098/rsif.2015.1106.
Zhang J, Li N, Lu S, Chen Y, Shan L, Zhao X, et al. The role of Notch ligand Jagged1 in osteosarcoma proliferation, metastasis, and recurrence. J Orthop Surg Res. 2021;16(1):226. https://doi.org/10.1186/s13018-021-02372-y.
Zhang N, Ji J, Zhou D, Liu X, Zhang X, Liu Y, et al. The Interaction of the Senescent and Adjacent Breast Cancer Cells Promotes the Metastasis of Heterogeneous Breast Cancer Cells through Notch Signaling. Int J Mol Sci. 2021;22(2):849. https://doi.org/10.3390/ijms22020849.
Li LJ, Chang PM, Li CH, Chang YC, Lai TC, Su CY, et al. FAS receptor regulates NOTCH activity through ERK-JAG1 axis activation and controls oral cancer stemness ability and pulmonary metastasis. Cell Death Discov. 2022;8(1):101. https://doi.org/10.1038/s41420-022-00899-5.
Xie Q, Guo H, He P, Deng H, Gao Y, Dong N, et al. Tspan5 promotes epithelial-mesenchymal transition and tumour metastasis of hepatocellular carcinoma by activating Notch signalling. Mol Oncol. 2021;15(11):3184–202. https://doi.org/10.1002/1878-0261.12980.
Hu G, Ma J, Zhang J, Chen Y, Liu H, Huang Y, et al. Hypoxia-induced lncHILAR promotes renal cancer metastasis via ceRNA for the miR-613/206/ 1–1-3p/Jagged-1/Notch/CXCR4 signaling pathway. Mol Ther. 2021;29(10):2979–94. https://doi.org/10.1016/j.ymthe.2021.05.020.
Qiu L, Yang X, Wu J, Huang C, Miao Y, Fu Z. HIST2H2BF Potentiates the Propagation of Cancer Stem Cells via Notch Signaling to Promote Malignancy and Liver Metastasis in Colorectal Carcinoma. Front Oncol. 2021;11: 677646. https://doi.org/10.3389/fonc.2021.677646.
Zhang Y, Aodeng G, Liu P, Su W, Zhao H. Effects of HOX transcript antisense intergenic RNA on the metastasis, epithelial-mesenchymal transition, and Notch signaling pathway in tongue cancer. Transl Cancer Res. 2021;10(1):520–8. https://doi.org/10.21037/tcr-20-3452.
Kim BR, Na YJ, Kim JL, Jeong YA, Park SH, Jo MJ, et al. RUNX3 suppresses metastasis and stemness by inhibiting Hedgehog signaling in colorectal cancer. Cell Death Differ. 2020;27(2):676–94. https://doi.org/10.1038/s41418-019-0379-5.
Guo K, Wang P, Zhang L, Zhou Y, Dai X, Yan Y, et al. Transcription factor POU4F2 promotes colorectal cancer cell migration and invasion through hedgehog-mediated epithelial-mesenchymal transition. Cancer Sci. 2021;112(10):4176–86. https://doi.org/10.1111/cas.15089.
Chen B, Hu Z, Li B, Lin X, Luo Z, Hu Z. The expressions of Hedgehog and PI3K-AKT pathway components correlate with invasion and metastasis in hepatocellular carcinoma. Int J Clin Exp Pathol. 2019;12(6):2381–8.
Liu C, Qi M, Li L, Yuan Y, Wu X, Fu J. Natural cordycepin induces apoptosis and suppresses metastasis in breast cancer cells by inhibiting the Hedgehog pathway. Food Funct. 2020;11(3):2107–16. https://doi.org/10.1039/c9fo02879j.
Zeng C, Chen T, Zhang Y, Chen Q. Hedgehog signaling pathway regulates ovarian cancer invasion and migration via adhesion molecule CD24. J Cancer. 2017;8(5):786–92. https://doi.org/10.7150/jca.17712.
Li B, Huang W, Cao N, Lou G. Forkhead-box R2 promotes metastasis and growth by stimulating angiogenesis and activating hedgehog signaling pathway in ovarian cancer. J Cell Biochem. 2018;119(9):7780–9. https://doi.org/10.1002/jcb.27148.
Jia J, Martin TA, Ye L, Meng L, Xia N, Jiang WG, et al. Fibroblast activation protein-α promotes the growth and migration of lung cancer cells via the PI3K and sonic hedgehog pathways. Int J Mol Med. 2018;41(1):275–83. https://doi.org/10.3892/ijmm.2017.3224.
Zhao Z, Jia Q, Wu MS, Xie X, Wang Y, Song G, et al. Degalactotigonin, a Natural Compound from Solanum nigrum L., Inhibits Growth and Metastasis of Osteosarcoma through GSK3β Inactivation-Mediated Repression of the Hedgehog/Gli1 Pathway. Clin Cancer Res. 2018;24(1):130–44. https://doi.org/10.1158/1078-0432.Ccr-17-0692.
Chen J, Zhou X, Yang J, Sun Q, Liu Y, Li N, et al. Circ-GLI1 promotes metastasis in melanoma through interacting with p70S6K2 to activate Hedgehog/GLI1 and Wnt/β-catenin pathways and upregulate Cyr61. Cell Death Dis. 2020;11(7):596. https://doi.org/10.1038/s41419-020-02799-x.
Kumar V, Vashishta M, Kong L, Wu X, Lu JJ, Guha C, et al. The Role of Notch, Hedgehog, and Wnt Signaling Pathways in the Resistance of Tumors to Anticancer Therapies. Front Cell Dev Biol. 2021;9:650772. https://doi.org/10.3389/fcell.2021.650772.
Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299–309. https://doi.org/10.1038/s41586-019-1730-1.
Zhang CC, Yan Z, Zong Q, Fang DD, Painter C, Zhang Q, et al. Synergistic effect of the γ-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl Med. 2013;2(3):233–42. https://doi.org/10.5966/sctm.2012-0096.
Qiu S, Deng L, Bao Y, Jin K, Tu X, Li J, et al. Reversal of docetaxel resistance in prostate cancer by Notch signaling inhibition. Anticancer Drugs. 2018;29(9):871–9. https://doi.org/10.1097/cad.0000000000000659.
Rahman MT, Nakayama K, Rahman M, Katagiri H, Katagiri A, Ishibashi T, et al. Notch3 overexpression as potential therapeutic target in advanced stage chemoresistant ovarian cancer. Am J Clin Pathol. 2012;138(4):535–44. https://doi.org/10.1309/ajcpkdlrq8f3ewns.
Yao J, Qian C. Inhibition of Notch3 enhances sensitivity to gemcitabine in pancreatic cancer through an inactivation of PI3K/Akt-dependent pathway. Med Oncol. 2010;27(3):1017–22. https://doi.org/10.1007/s12032-009-9326-5.
Dai G, Deng S, Guo W, Yu L, Yang J, Zhou S, et al. Notch pathway inhibition using DAPT, a γ-secretase inhibitor (GSI), enhances the antitumor effect of cisplatin in resistant osteosarcoma. Mol Carcinog. 2019;58(1):3–18. https://doi.org/10.1002/mc.22873.
Melamed JR, Morgan JT, Ioele SA, Gleghorn JP, Sims-Mourtada J, Day ES. Investigating the role of Hedgehog/GLI1 signaling in glioblastoma cell response to temozolomide. Oncotarget. 2018;9(43):27000–15. https://doi.org/10.18632/oncotarget.25467.
Joshi I, Minter LM, Telfer J, Demarest RM, Capobianco AJ, Aster JC, et al. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood. 2009;113(8):1689–98. https://doi.org/10.1182/blood-2008-03-147967.
Tian D, Shi Y, Chen D, Liu Q, Fan F. The Wnt inhibitor LGK-974 enhances radiosensitivity of HepG2 cells by modulating Nrf2 signaling. Int J Oncol. 2017;51(2):545–54. https://doi.org/10.3892/ijo.2017.4042.
Chen Y, Bieber MM, Teng NN. Hedgehog signaling regulates drug sensitivity by targeting ABC transporters ABCB1 and ABCG2 in epithelial ovarian cancer. Mol Carcinog. 2014;53(8):625–34. https://doi.org/10.1002/mc.22015.
Wakabayashi N, Skoko JJ, Chartoumpekis DV, Kimura S, Slocum SL, Noda K, et al. Notch-Nrf2 axis: regulation of Nrf2 gene expression and cytoprotection by notch signaling. Mol Cell Biol. 2014;34(4):653–63. https://doi.org/10.1128/mcb.01408-13.
Nieszporek A, Skrzypek K, Adamek G, Majka M. Molecular mechanisms of epithelial to mesenchymal transition in tumor metastasis. Acta Biochim Pol. 2019;66(4):509–20. https://doi.org/10.18388/abp.2019_2899.
Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–29. https://doi.org/10.1038/nrclinonc.2017.44.
Liu T, Zhang Z, Wang C, Huang H, Li Y. BRD4 promotes the migration and invasion of bladder cancer cells through the Sonic hedgehog signaling pathway and enhances cisplatin resistance. Biochem Cell Biol. 2022:1–9. doi:https://doi.org/10.1139/bcb-2021-0552
Vermezovic J, Adamowicz M, Santarpia L, Rustighi A, Forcato M, Lucano C, et al. Notch is a direct negative regulator of the DNA-damage response. Nat Struct Mol Biol. 2015;22(5):417–24. https://doi.org/10.1038/nsmb.3013.
Adamowicz M, Vermezovic J, d’Adda di Fagagna F. NOTCH1 Inhibits Activation of ATM by Impairing the Formation of an ATM-FOXO3a-KAT5/Tip60 Complex. Cell Rep. 2016;16(8):2068–76. https://doi.org/10.1016/j.celrep.2016.07.038.
Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28(1):17–28. https://doi.org/10.1002/stem.261.
Gan GN, Eagles J, Keysar SB, Wang G, Glogowska MJ, Altunbas C, et al. Hedgehog signaling drives radioresistance and stroma-driven tumor repopulation in head and neck squamous cancers. Cancer Res. 2014;74(23):7024–36. https://doi.org/10.1158/0008-5472.Can-14-1346.
Hyuga T, Alcantara M, Kajioka D, Haraguchi R, Suzuki K, Miyagawa S, et al. Hedgehog Signaling for Urogenital Organogenesis and Prostate Cancer: An Implication for the Epithelial-Mesenchyme Interaction (EMI). Int J Mol Sci. 2019;21(1):58. https://doi.org/10.3390/ijms21010058.
Li D, Li T, Shang Z, Zhao L, Xu Q, Tan J, et al. Combined inhibition of Notch and FLT3 produces synergistic cytotoxic effects in FLT3/ITD(+) acute myeloid leukemia. Signal Transduct Target Ther. 2020;5(1):21. https://doi.org/10.1038/s41392-020-0108-z.
Bousquet Mur E, Bernardo S, Papon L, Mancini M, Fabbrizio E, Goussard M, et al. Notch inhibition overcomes resistance to tyrosine kinase inhibitors in EGFR-driven lung adenocarcinoma. J Clin Invest. 2020;130(2):612–24. https://doi.org/10.1172/jci126896.
Long GV, Eroglu Z, Infante J, Patel S, Daud A, Johnson DB, et al. Long-Term Outcomes in Patients With BRAF V600-Mutant Metastatic Melanoma Who Received Dabrafenib Combined With Trametinib. J Clin Oncol. 2018;36(7):667–73. https://doi.org/10.1200/jco.2017.74.1025.
Bora-Singhal N, Perumal D, Nguyen J, Chellappan S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non-Small Cell Lung Cancer. Neoplasia. 2015;17(7):538–51. https://doi.org/10.1016/j.neo.2015.07.001.
Chen H, Yang D, Wang Y, Tao H, Luo Y, Wu A, et al. Activation of the Hedgehog pathway mediates resistance to epidermal growth factor receptor inhibitors in non-small cell lung cancer. J Cancer. 2022;13(3):987–97. https://doi.org/10.7150/jca.63410.
Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. 2015;27(3):327–41. https://doi.org/10.1016/j.ccell.2015.02.001.
Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27(3):342–53. https://doi.org/10.1016/j.ccell.2015.02.002.
Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, Curran T, et al. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26(44):6442–7. https://doi.org/10.1038/sj.onc.1210467.
Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71(2):435–44. https://doi.org/10.1158/0008-5472.Can-10-2876.
Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2(51):51ra70. https://doi.org/10.1126/scitranslmed.3001599.
Whitson RJ, Lee A, Urman NM, Mirza A, Yao CY, Brown AS, et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat Med. 2018;24(3):271–81. https://doi.org/10.1038/nm.4476.
Zhao X, Pak E, Ornell KJ, Pazyra-Murphy MF, MacKenzie EL, Chadwick EJ, et al. A Transposon Screen Identifies Loss of Primary Cilia as a Mechanism of Resistance to SMO Inhibitors. Cancer Discov. 2017;7(12):1436–49. https://doi.org/10.1158/2159-8290.Cd-17-0281.
Kuijk LM, Verstege MI, Rekers NV, Bruijns SC, Hooijberg E, Roep BO, et al. Notch controls generation and function of human effector CD8+ T cells. Blood. 2013;121(14):2638–46. https://doi.org/10.1182/blood-2012-07-442962.
Zhang K, Hong X, Song Z, Xu Y, Li C, Wang G, et al. Identification of Deleterious NOTCH Mutation as Novel Predictor to Efficacious Immunotherapy in NSCLC. Clin Cancer Res. 2020;26(14):3649–61. https://doi.org/10.1158/1078-0432.Ccr-19-3976.
Oyama Y, Onishi H, Koga S, Murahashi M, Ichimiya S, Nakayama K, et al. Patched 1-interacting Peptide Represses Fibrosis in Pancreatic Cancer to Augment the Effectiveness of Immunotherapy. J Immunother. 2020;43(4):121–33. https://doi.org/10.1097/cji.0000000000000305.
Petty AJ, Li A, Wang X, Dai R, Heyman B, Hsu D, et al. Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J Clin Invest. 2019;129(12):5151–62. https://doi.org/10.1172/jci128644.
Jiang J, Ding Y, Chen Y, Lu J, Chen Y, Wu G, et al. Pan-cancer analyses reveal that increased Hedgehog activity correlates with tumor immunosuppression and resistance to immune checkpoint inhibitors. Cancer Med. 2022;11(3):847–63. https://doi.org/10.1002/cam4.4456.
Allen F, Maillard I. Therapeutic Targeting of Notch Signaling: From Cancer to Inflammatory Disorders. Front Cell Dev Biol. 2021;9: 649205. https://doi.org/10.3389/fcell.2021.649205.
Jaiswal A, Murakami K, Elia A, Shibahara Y, Done SJ, Wood SA, et al. Therapeutic inhibition of USP9x-mediated Notch signaling in triple-negative breast cancer. Proc Natl Acad Sci U S A. 2021;118(38):e2101592118. https://doi.org/10.1073/pnas.2101592118.
Shen Q, Cohen B, Zheng W, Rahbar R, Martin B, Murakami K, et al. Notch Shapes the Innate Immunophenotype in Breast Cancer. Cancer Discov. 2017;7(11):1320–35. https://doi.org/10.1158/2159-8290.Cd-17-0037.
Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016;76(11):3156–65. https://doi.org/10.1158/0008-5472.Can-15-2528.
Grazioli P, Orlando A, Giordano N, Noce C, Peruzzi G, Abdollahzadeh B, et al. Notch-Signaling Deregulation Induces Myeloid-Derived Suppressor Cells in T-Cell Acute Lymphoblastic Leukemia. Front Immunol. 2022;13:809261. https://doi.org/10.3389/fimmu.2022.809261.
Tsukumo SI, Yasutomo K. Regulation of CD8(+) T Cells and Antitumor Immunity by Notch Signaling. Front Immunol. 2018;9:101. https://doi.org/10.3389/fimmu.2018.00101.
Maekawa Y, Minato Y, Ishifune C, Kurihara T, Kitamura A, Kojima H, et al. Notch2 integrates signaling by the transcription factors RBP-J and CREB1 to promote T cell cytotoxicity. Nat Immunol. 2008;9(10):1140–7. https://doi.org/10.1038/ni.1649.
Sugimoto K, Maekawa Y, Kitamura A, Nishida J, Koyanagi A, Yagita H, et al. Notch2 signaling is required for potent antitumor immunity in vivo. J Immunol. 2010;184(9):4673–8. https://doi.org/10.4049/jimmunol.0903661.
Kondo T, Morita R, Okuzono Y, Nakatsukasa H, Sekiya T, Chikuma S, et al. Notch-mediated conversion of activated T cells into stem cell memory-like T cells for adoptive immunotherapy. Nat Commun. 2017;8:15338. https://doi.org/10.1038/ncomms15338.
Meng J, Jiang YZ, Zhao S, Tao Y, Zhang T, Wang X, et al. Tumor-derived Jagged1 promotes cancer progression through immune evasion. Cell Rep. 2022;38(10):110492. https://doi.org/10.1016/j.celrep.2022.110492.
Cui Y, Li Q, Li W, Wang Y, Lv F, Shi X, et al. NOTCH3 is a Prognostic Factor and Is Correlated With Immune Tolerance in Gastric Cancer. Front Oncol. 2020;10:574937. https://doi.org/10.3389/fonc.2020.574937.
Sierra RA, Thevenot P, Raber PL, Cui Y, Parsons C, Ochoa AC, et al. Rescue of notch-1 signaling in antigen-specific CD8+ T cells overcomes tumor-induced T-cell suppression and enhances immunotherapy in cancer. Cancer Immunol Res. 2014;2(8):800–11. https://doi.org/10.1158/2326-6066.Cir-14-0021.
Onishi H, Morisaki T, Kiyota A, Koya N, Tanaka H, Umebayashi M, et al. The Hedgehog inhibitor cyclopamine impairs the benefits of immunotherapy with activated T and NK lymphocytes derived from patients with advanced cancer. Cancer Immunol Immunother. 2013;62(6):1029–39. https://doi.org/10.1007/s00262-013-1419-5.
Yánez DC, Lau CI, Chawda MM, Ross S, Furmanski AL, Crompton T. Hedgehog signaling promotes T(H)2 differentiation in naive human CD4 T cells. J Allergy Clin Immunol. 2019;144(5):1419-1423.e1. https://doi.org/10.1016/j.jaci.2019.07.011.
Merchant JL, Ding L. Hedgehog Signaling Links Chronic Inflammation to Gastric Cancer Precursor Lesions. Cell Mol Gastroenterol Hepatol. 2017;3(2):201–10. https://doi.org/10.1016/j.jcmgh.2017.01.004.
Onishi H, Morisaki T, Kiyota A, Koya N, Tanaka H, Umebayashi M, et al. The Hedgehog inhibitor suppresses the function of monocyte-derived dendritic cells from patients with advanced cancer under hypoxia. Biochem Biophys Res Commun. 2013;436(1):53–9. https://doi.org/10.1016/j.bbrc.2013.05.057.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–86. https://doi.org/10.1038/s41568-019-0238-1.
D’Assoro AB, Leon-Ferre R, Braune EB, Lendahl U. Roles of Notch Signaling in the Tumor Microenvironment. Int J Mol Sci. 2022;23(11):6241. https://doi.org/10.3390/ijms23116241.
Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol. 2014;25:61–8. https://doi.org/10.1016/j.semcancer.2014.02.006.
Farnie G, Clarke RB, Spence K, Pinnock N, Brennan K, Anderson NG, et al. Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J Natl Cancer Inst. 2007;99(8):616–27. https://doi.org/10.1093/jnci/djk133.
Farnie G, Willan PM, Clarke RB, Bundred NJ. Combined inhibition of ErbB1/2 and Notch receptors effectively targets breast ductal carcinoma in situ (DCIS) stem/progenitor cell activity regardless of ErbB2 status. PLoS ONE. 2013;8(2):e56840. https://doi.org/10.1371/journal.pone.0056840.
Strell C, Paulsson J, Jin SB, Tobin NP, Mezheyeuski A, Roswall P, et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J Natl Cancer Inst. 2019;111(9):983–95. https://doi.org/10.1093/jnci/djy234.
Xue B, Chuang CH, Prosser HM, Fuziwara CS, Chan C, Sahasrabudhe N, et al. miR-200 deficiency promotes lung cancer metastasis by activating Notch signaling in cancer-associated fibroblasts. Genes Dev. 2021;35(15–16):1109–22. https://doi.org/10.1101/gad.347344.120.
Yang Y, Ahn YH, Gibbons DL, Zang Y, Lin W, Thilaganathan N, et al. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest. 2011;121(4):1373–85. https://doi.org/10.1172/jci42579.
Kim B, Seo Y, Kwon JH, Shin Y, Kim S, Park SJ, et al. IL-6 and IL-8, secreted by myofibroblasts in the tumor microenvironment, activate HES1 to expand the cancer stem cell population in early colorectal tumor. Mol Carcinog. 2021;60(3):188–200. https://doi.org/10.1002/mc.23283.
Li L, Zhao F, Lu J, Li T, Yang H, Wu C, et al. Notch-1 signaling promotes the malignant features of human breast cancer through NF-κB activation. PLoS ONE. 2014;9(4):e95912. https://doi.org/10.1371/journal.pone.0095912.
Shimizu M, Cohen B, Goldvasser P, Berman H, Virtanen C, Reedijk M. Plasminogen activator uPA is a direct transcriptional target of the JAG1-Notch receptor signaling pathway in breast cancer. Cancer Res. 2011;71(1):277–86. https://doi.org/10.1158/0008-5472.Can-10-2523.
Guimaraes VSN, Vidal MTA, de Faro Valverde L, de Oliveira MG, de Oliveira Siquara da Rocha L, Coelho PLC, et al. Hedgehog pathway activation in oral squamous cell carcinoma: cancer-associated fibroblasts exhibit nuclear GLI-1 localization. J Mol Histol. 2020;51(6):675–84. https://doi.org/10.1007/s10735-020-09913-5.
Yoshida GJ. Regulation of heterogeneous cancer-associated fibroblasts: the molecular pathology of activated signaling pathways. J Exp Clin Cancer Res. 2020;39(1):112. https://doi.org/10.1186/s13046-020-01611-0.
Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–34. https://doi.org/10.1016/j.ccr.2014.04.005.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–47. https://doi.org/10.1016/j.ccr.2014.04.021.
Steele NG, Biffi G, Kemp SB, Zhang Y, Drouillard D, Syu L, et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin Cancer Res. 2021;27(7):2023–37. https://doi.org/10.1158/1078-0432.Ccr-20-3715.
Zhang X, Wang Y, Wang X, Zou B, Mei J, Peng X, et al. Extracellular vesicles-encapsulated microRNA-10a-5p shed from cancer-associated fibroblast facilitates cervical squamous cell carcinoma cell angiogenesis and tumorigenicity via Hedgehog signaling pathway. Cancer Gene Ther. 2021;28(5):529–42. https://doi.org/10.1038/s41417-020-00238-9.
Pitulescu ME, Schmidt I, Giaimo BD, Antoine T, Berkenfeld F, Ferrante F, et al. Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol. 2017;19(8):915–27. https://doi.org/10.1038/ncb3555.
Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444(7122):1083–7. https://doi.org/10.1038/nature05313.
Kangsamaksin T, Murtomaki A, Kofler NM, Cuervo H, Chaudhri RA, Tattersall IW, et al. NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov. 2015;5(2):182–97. https://doi.org/10.1158/2159-8290.Cd-14-0650.
Zhuo H, Zhou D, Wang Y, Mo H, Yu Y, Liu Y. Sonic hedgehog selectively promotes lymphangiogenesis after kidney injury through noncanonical pathway. Am J Physiol Renal Physiol. 2019;317(4):F1022-f1033. https://doi.org/10.1152/ajprenal.00077.2019.
Hui Z, Wang S, Li J, Wang J, Zhang Z. Compound Tongluo Decoction inhibits endoplasmic reticulum stress-induced ferroptosis and promoted angiogenesis by activating the Sonic Hedgehog pathway in cerebral infarction. J Ethnopharmacol. 2022;283:114634. https://doi.org/10.1016/j.jep.2021.114634.
Feng J, Wang C, Liu T, Li J, Wu L, Yu Q, et al. Procyanidin B2 inhibits the activation of hepatic stellate cells and angiogenesis via the Hedgehog pathway during liver fibrosis. J Cell Mol Med. 2019;23(9):6479–93. https://doi.org/10.1111/jcmm.14543.
Gampala S, Yang JY. Hedgehog Pathway Inhibitors against Tumor Microenvironment. Cells. 2021;10(11):3135. https://doi.org/10.3390/cells10113135.
Li Y, Liu Y, Wang G, Wang Y, Guo L. Cooperation of Indian Hedgehog and Vascular Endothelial Growth Factor in Tumor Angiogenesis and Growth in Human Hepatocellular Carcinomas, an Immunohistochemical Study. Appl Immunohistochem Mol Morphol. 2019;27(6):436–40. https://doi.org/10.1097/pai.0000000000000654.
Bausch D, Fritz S, Bolm L, Wellner UF, Fernandez-Del-Castillo C, Warshaw AL, et al. Hedgehog signaling promotes angiogenesis directly and indirectly in pancreatic cancer. Angiogenesis. 2020;23(3):479–92. https://doi.org/10.1007/s10456-020-09725-x.
Huang Y, Fang J, Lu W, Wang Z, Wang Q, Hou Y, et al. A Systems Pharmacology Approach Uncovers Wogonoside as an Angiogenesis Inhibitor of Triple-Negative Breast Cancer by Targeting Hedgehog Signaling. Cell Chem Biol. 2019;26(8):1143-1158.e6. https://doi.org/10.1016/j.chembiol.2019.05.004.
Zhdanovskaya N, Firrincieli M, Lazzari S, Pace E, Scribani Rossi P, Felli MP, et al. Targeting Notch to Maximize Chemotherapeutic Benefits: Rationale, Advanced Strategies, and Future Perspectives. Cancers (Basel). 2021;13(20):5106. https://doi.org/10.3390/cancers13205106.
Aggarwal V, Tuli HS, Varol M, Tuorkey M, Sak K, Parashar NC, et al. NOTCH signaling: Journey of an evolutionarily conserved pathway in driving tumor progression and its modulation as a therapeutic target. Crit Rev Oncol Hematol. 2021;164:103403. https://doi.org/10.1016/j.critrevonc.2021.103403.
Moore G, Annett S, McClements L, Robson T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells. 2020;9(6):1503. https://doi.org/10.3390/cells9061503.
Haapasalo A, Kovacs DM. The Many Substrates of Presenilin/γ-Secretase. J Alzheimers Dis. 2011;25(1):3–28. https://doi.org/10.3233/jad-2011-101065.
Imbimbo BP, Panza F, Frisardi V, Solfrizzi V, D’Onofrio G, Logroscino G, et al. Therapeutic intervention for Alzheimer’s disease with γ-secretase inhibitors: still a viable option? Expert Opin Investig Drugs. 2011;20(3):325–41. https://doi.org/10.1517/13543784.2011.550572.
Pine SR. Rethinking Gamma-secretase Inhibitors for Treatment of Non-small-Cell Lung Cancer: Is Notch the Target? Clin Cancer Res. 2018;24(24):6136–41. https://doi.org/10.1158/1078-0432.Ccr-18-1635.
Das A, Narayanam MK, Paul S, Mukhnerjee P, Ghosh S, Dastidar DG, et al. A novel triazole, NMK-T-057, induces autophagic cell death in breast cancer cells by inhibiting γ-secretase-mediated activation of Notch signaling. J Biol Chem. 2019;294(17):6733–50. https://doi.org/10.1074/jbc.RA119.007671.
Gilbert CA, Daou MC, Moser RP, Ross AH. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res. 2010;70(17):6870–9. https://doi.org/10.1158/0008-5472.Can-10-1378.
Plentz R, Park JS, Rhim AD, Abravanel D, Hezel AF, Sharma SV, et al. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2009;136(5):1741-9.e6. https://doi.org/10.1053/j.gastro.2009.01.008.
Akiyoshi T, Nakamura M, Yanai K, Nagai S, Wada J, Koga K, et al. Gamma-secretase inhibitors enhance taxane-induced mitotic arrest and apoptosis in colon cancer cells. Gastroenterology. 2008;134(1):131–44. https://doi.org/10.1053/j.gastro.2007.10.008.
Feng Z, Xu W, Zhang C, Liu M, Wen H. Inhibition of gamma-secretase in Notch1 signaling pathway as a novel treatment for ovarian cancer. Oncotarget. 2017;8(5):8215–25. https://doi.org/10.18632/oncotarget.14152.
Riccio O, van Gijn ME, Bezdek AC, Pellegrinet L, van Es JH, Zimber-Strobl U, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27 Kip1 and p57 Kip2. EMBO Rep. 2008;9(4):377–83. https://doi.org/10.1038/embor.2008.7.
Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, et al. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82(1):341–58. https://doi.org/10.1093/toxsci/kfh254.
Cui D, Dai J, Keller JM, Mizokami A, Xia S, Keller ET. Notch Pathway Inhibition Using PF-03084014, a gamma-Secretase Inhibitor (GSI), Enhances the Antitumor Effect of Docetaxel in Prostate Cancer. Clin Cancer Res. 2015;21(20):4619–29. https://doi.org/10.1158/1078-0432.CCR-15-0242.
Das A, Narayanam MK, Paul S, Mukhnerjee P, Ghosh S, Dastidar DG, et al. A novel triazole, NMK-T-057, induces autophagic cell death in breast cancer cells by inhibiting gamma-secretase-mediated activation of Notch signaling. J Biol Chem. 2019;294(17):6733–50. https://doi.org/10.1074/jbc.RA119.007671.
Han B, Liu SH, Guo WD, Zhang B, Wang JP, Cao YK, et al. Notch1 downregulation combined with interleukin-24 inhibits invasion and migration of hepatocellular carcinoma cells. World J Gastroenterol. 2015;21(33):9727–35. https://doi.org/10.3748/wjg.v21.i33.9727.
Pine SR. Rethinking Gamma-secretase Inhibitors for Treatment of Non-small-Cell Lung Cancer: Is Notch the Target? Clin Cancer Res. 2018;24(24):6136–41. https://doi.org/10.1158/1078-0432.Ccr-18-1635.
Aung KL, El-Khoueiry AB, Gelmon K, Tran B, Bajaj G, He B, et al. A multi-arm phase I dose escalating study of an oral NOTCH inhibitor BMS-986115 in patients with advanced solid tumours. Invest New Drugs. 2018;36(6):1026–36. https://doi.org/10.1007/s10637-018-0597-6.
Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol. 2012;30(19):2348–53. https://doi.org/10.1200/jco.2011.36.8282.
Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol. 2011;29(26):3529–34. https://doi.org/10.1200/jco.2011.35.7806.
Diaz-Padilla I, Wilson MK, Clarke BA, Hirte HW, Welch SA, Mackay HJ, et al. A phase II study of single-agent RO4929097, a gamma-secretase inhibitor of Notch signaling, in patients with recurrent platinum-resistant epithelial ovarian cancer: A study of the Princess Margaret, Chicago and California phase II consortia. Gynecol Oncol. 2015;137(2):216–22. https://doi.org/10.1016/j.ygyno.2015.03.005.
Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y, et al. Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer. 2015;121(3):432–40. https://doi.org/10.1002/cncr.29055.
Peereboom DM, Ye X, Mikkelsen T, Lesser GJ, Lieberman FS, Robins HI, et al. A Phase II and Pharmacodynamic Trial of RO4929097 for Patients With Recurrent/Progressive Glioblastoma. Neurosurgery. 2021;88(2):246–51. https://doi.org/10.1093/neuros/nyaa412.
Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer. 2012;48(7):997–1003. https://doi.org/10.1016/j.ejca.2012.02.056.
Kummar S, O’Sullivan Coyne G, Do KT, Turkbey B, Meltzer PS, Polley E, et al. Clinical Activity of the γ-Secretase Inhibitor PF-03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis). J Clin Oncol. 2017;35(14):1561–9. https://doi.org/10.1200/jco.2016.71.1994.
Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L. γ-Secretase inhibitors and modulators. Biochim Biophys Acta. 2013;1828(12):2898–907. https://doi.org/10.1016/j.bbamem.2013.06.005.
Habets RA, de Bock CE, Serneels L, Lodewijckx I, Verbeke D, Nittner D, et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci Transl Med. 2019;11(494):eaau6246. https://doi.org/10.1126/scitranslmed.aau6246.
Murciano-Goroff YR, Taylor BS, Hyman DM, Schram AM. Toward a More Precise Future for Oncology. Cancer Cell. 2020;37(4):431–42. https://doi.org/10.1016/j.ccell.2020.03.014.
Hann CL, Burns TF, Dowlati A, Morgensztern D, Ward PJ, Koch MM, et al. A Phase 1 Study Evaluating Rovalpituzumab Tesirine in Frontline Treatment of Patients With Extensive-Stage SCLC. J Thorac Oncol. 2021;16(9):1582–8. https://doi.org/10.1016/j.jtho.2021.06.022.
Zheng H, Bae Y, Kasimir-Bauer S, Tang R, Chen J, Ren G, et al. Therapeutic Antibody Targeting Tumor- and Osteoblastic Niche-Derived Jagged1 Sensitizes Bone Metastasis to Chemotherapy. Cancer Cell. 2017;32(6):731-747.e6. https://doi.org/10.1016/j.ccell.2017.11.002.
Santos MA, Sarmento LM, Rebelo M, Doce AA, Maillard I, Dumortier A, et al. Notch1 engagement by Delta-like-1 promotes differentiation of B lymphocytes to antibody-secreting cells. Proc Natl Acad Sci U S A. 2007;104(39):15454–9. https://doi.org/10.1073/pnas.0702891104.
Owen DH, Giffin MJ, Bailis JM, Smit MD, Carbone DP, He K. DLL3: an emerging target in small cell lung cancer. J Hematol Oncol. 2019;12(1):61. https://doi.org/10.1186/s13045-019-0745-2.
Blackhall F, Jao K, Greillier L, Cho BC, Penkov K, Reguart N, et al. Efficacy and Safety of Rovalpituzumab Tesirine Compared With Topotecan as Second-Line Therapy in DLL3-High SCLC: Results From the Phase 3 TAHOE Study. J Thorac Oncol. 2021;16(9):1547–58. https://doi.org/10.1016/j.jtho.2021.02.009.
Morgensztern D, Besse B, Greillier L, Santana-Davila R, Ready N, Hann CL, et al. Efficacy and Safety of Rovalpituzumab Tesirine in Third-Line and Beyond Patients with DLL3-Expressing, Relapsed/Refractory Small-Cell Lung Cancer: Results From the Phase II TRINITY Study. Clin Cancer Res. 2019;25(23):6958–66. https://doi.org/10.1158/1078-0432.Ccr-19-1133.
Spino M, Kurz SC, Chiriboga L, Serrano J, Zeck B, Sen N, et al. Cell Surface Notch Ligand DLL3 is a Therapeutic Target in Isocitrate Dehydrogenase-mutant Glioma. Clin Cancer Res. 2019;25(4):1261–71. https://doi.org/10.1158/1078-0432.Ccr-18-2312.
Liu SK, Bham SA, Fokas E, Beech J, Im J, Cho S, et al. Delta-like ligand 4-notch blockade and tumor radiation response. J Natl Cancer Inst. 2011;103(23):1778–98. https://doi.org/10.1093/jnci/djr419.
Chiorean EG, LoRusso P, Strother RM, Diamond JR, Younger A, Messersmith WA, et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin Cancer Res. 2015;21(12):2695–703. https://doi.org/10.1158/1078-0432.Ccr-14-2797.
Smith DC, Eisenberg PD, Manikhas G, Chugh R, Gubens MA, Stagg RJ, et al. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin Cancer Res. 2014;20(24):6295–303. https://doi.org/10.1158/1078-0432.Ccr-14-1373.
Li Y, Hickson JA, Ambrosi DJ, Haasch DL, Foster-Duke KD, Eaton LJ, et al. ABT-165, a Dual Variable Domain Immunoglobulin (DVD-Ig) Targeting DLL4 and VEGF, Demonstrates Superior Efficacy and Favorable Safety Profiles in Preclinical Models. Mol Cancer Ther. 2018;17(5):1039–50. https://doi.org/10.1158/1535-7163.Mct-17-0800.
Jimeno A, Moore KN, Gordon M, Chugh R, Diamond JR, Aljumaily R, et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Invest New Drugs. 2019;37(3):461–72. https://doi.org/10.1007/s10637-018-0665-y.
Liao W, Li G, You Y, Wan H, Wu Q, Wang C, et al. Antitumor activity of Notch-1 inhibition in human colorectal carcinoma cells. Oncol Rep. 2018;39(3):1063–71. https://doi.org/10.3892/or.2017.6176.
Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2006;6(5):347–59. https://doi.org/10.1038/nrc1880.
Ferrarotto R, Eckhardt G, Patnaik A, LoRusso P, Faoro L, Heymach JV, et al. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol. 2018;29(7):1561–8. https://doi.org/10.1093/annonc/mdy171.
Mazur PK, Einwächter H, Lee M, Sipos B, Nakhai H, Rad R, et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2010;107(30):13438–43. https://doi.org/10.1073/pnas.1002423107.
Lin L, Mernaugh R, Yi F, Blum D, Carbone DP, Dang TP. Targeting specific regions of the Notch3 ligand-binding domain induces apoptosis and inhibits tumor growth in lung cancer. Cancer Res. 2010;70(2):632–8. https://doi.org/10.1158/0008-5472.Can-09-3293.
Hu ZI, Bendell JC, Bullock A, LoConte NK, Hatoum H, Ritch P, et al. A randomized phase II trial of nab-paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019;8(11):5148–57. https://doi.org/10.1002/cam4.2425.
Smith DC, Chugh R, Patnaik A, Papadopoulos KP, Wang M, Kapoun AM, et al. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Invest New Drugs. 2019;37(4):722–30. https://doi.org/10.1007/s10637-018-0714-6.
Rosen LS, Wesolowski R, Baffa R, Liao KH, Hua SY, Gibson BL, et al. A phase I, dose-escalation study of PF-06650808, an anti-Notch3 antibody-drug conjugate, in patients with breast cancer and other advanced solid tumors. Invest New Drugs. 2020;38(1):120–30. https://doi.org/10.1007/s10637-019-00754-y.
Moss ML, Minond D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediators Inflamm. 2017;2017:9673537. https://doi.org/10.1155/2017/9673537.
Li DD, Zhao CH, Ding HW, Wu Q, Ren TS, Wang J, et al. A novel inhibitor of ADAM17 sensitizes colorectal cancer cells to 5-Fluorouracil by reversing Notch and epithelial-mesenchymal transition in vitro and in vivo. Cell Prolif. 2018;51(5):e12480. https://doi.org/10.1111/cpr.12480.
Astudillo L, Da Silva TG, Wang Z, Han X, Jin K, VanWye J, et al. The Small Molecule IMR-1 Inhibits the Notch Transcriptional Activation Complex to Suppress Tumorigenesis. Cancer Res. 2016;76(12):3593–603. https://doi.org/10.1158/0008-5472.Can-16-0061.
Platonova N, Parravicini C, Sensi C, Paoli A, Colombo M, Neri A, et al. Identification of small molecules uncoupling the Notch: Jagged interaction through an integrated high-throughput screening. PLoS ONE. 2017;12(11):e0182640. https://doi.org/10.1371/journal.pone.0182640.
Lehal R, Zaric J, Vigolo M, Urech C, Frismantas V, Zangger N, et al. Pharmacological disruption of the Notch transcription factor complex. Proc Natl Acad Sci U S A. 2020;117(28):16292–301. https://doi.org/10.1073/pnas.1922606117.
Zito PM, Nassereddin A, Scharf R. Vismodegib. StatPearls. StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.; 2022. https://pubmed.ncbi.nlm.nih.gov/30020732/.
Villani A, Potestio L, Fabbrocini G, Scalvenzi M. New Emerging Treatment Options for Advanced Basal Cell Carcinoma and Squamous Cell Carcinoma. Adv Ther. 2022;39(3):1164–78. https://doi.org/10.1007/s12325-022-02044-1.
Jain R, Dubey SK, Singhvi G. The Hedgehog pathway and its inhibitors: Emerging therapeutic approaches for basal cell carcinoma. Drug Discov Today. 2022;27(4):1176–83. https://doi.org/10.1016/j.drudis.2021.12.005.
Sandhiya S, Melvin G, Kumar SS, Dkhar SA. The dawn of hedgehog inhibitors: Vismodegib. J Pharmacol Pharmacother. 2013;4(1):4–7. https://doi.org/10.4103/0976-500X.107628.
Niyaz M, Khan MS, Mudassar S. Hedgehog Signaling: An Achilles’ Heel in Cancer. Transl Oncol. 2019;12(10):1334–44. https://doi.org/10.1016/j.tranon.2019.07.004.
Mahindroo N, Punchihewa C, Fujii N. Hedgehog-Gli Signaling Pathway Inhibitors as Anticancer Agents. J Med Chem. 2009;52(13):3829–45. https://doi.org/10.1021/jm801420y.
Coon V, Laukert T, Pedone CA, Laterra J, Kim KJ, Fults DW. Molecular therapy targeting Sonic hedgehog and hepatocyte growth factor signaling in a mouse model of medulloblastoma. Mol Cancer Ther. 2010;9(9):2627–36. https://doi.org/10.1158/1535-7163.MCT-10-0486.
Chaudary N, Pintilie M, Hedley D, Hill RP, Milosevic M, Mackay H. Hedgehog inhibition enhances efficacy of radiation and cisplatin in orthotopic cervical cancer xenografts. Br J Cancer. 2017;116(1):50–7. https://doi.org/10.1038/bjc.2016.383.
Petrova E, Rios-Esteves J, Ouerfelli O, Glickman JF, Resh MD. Inhibitors of Hedgehog acyltransferase block Sonic Hedgehog signaling. Nat Chem Biol. 2013;9(4):247–9. https://doi.org/10.1038/nchembio.1184.
Matevossian A, Resh MD. Hedgehog Acyltransferase as a target in estrogen receptor positive, HER2 amplified, and tamoxifen resistant breast cancer cells. Mol Cancer. 2015;14:72. https://doi.org/10.1186/s12943-015-0345-x.
Petrova E, Matevossian A, Resh MD. Hedgehog acyltransferase as a target in pancreatic ductal adenocarcinoma. Oncogene. 2015;34(2):263–8. https://doi.org/10.1038/onc.2013.575.
Wang F, Huang X, Sun Y, Li Z, Sun R, Zhao T, et al. Sulforaphane regulates the proliferation of leukemia stem-like cells via Sonic Hedgehog signaling pathway. Eur J Pharmacol. 2022;919:174824. https://doi.org/10.1016/j.ejphar.2022.174824.
Yun T, Wang J, Yang J, Huang W, Lai L, Tan W, et al. Discovery of Small Molecule Inhibitors Targeting the Sonic Hedgehog. Front Chem. 2020;8:498. https://doi.org/10.3389/fchem.2020.00498.
Espinosa-Bustos C, Mella J, Soto-Delgado J, Salas CO. State of the art of Smo antagonists for cancer therapy: advances in the target receptor and new ligand structures. Future Med Chem. 2019;11(6):617–38. https://doi.org/10.4155/fmc-2018-0497.
Axelson M, Liu K, Jiang X, He K, Wang J, Zhao H, et al. U.S. Food and Drug Administration approval: vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin Cancer Res. 2013;19(9):2289–93. https://doi.org/10.1158/1078-0432.CCR-12-1956.
Aditya S, Rattan A. Vismodegib: A smoothened inhibitor for the treatment of advanced basal cell carcinoma. Indian Dermatol Online J. 2013;4(4):365–8. https://doi.org/10.4103/2229-5178.120685.
Zhang Y, Vagiannis D, Budagaga Y, Sabet Z, Hanke I, Rozkoš T, et al. Sonidegib potentiates the cancer cells’ sensitivity to cytostatic agents by functional inhibition of ABCB1 and ABCG2 in vitro and ex vivo. Biochem Pharmacol. 2022;199:115009. https://doi.org/10.1016/j.bcp.2022.115009.
Gounder MM, Rosenbaum E, Wu N, Dickson MA, Sheikh TN, D’Angelo SP, et al. A Phase Ib/II Randomized Study of RO4929097, a Gamma Secretase or Notch Inhibitor with or without Vismodegib, a Hedgehog Inhibitor. Advanced Sarcoma Clin Cancer Res. 2022. https://doi.org/10.1158/1078-0432.Ccr-21-3874.
Schnetzler G, Cuberos M, Bucher R, Stassen K, Mingrino R. Vismodegib (ERIVEDGE) pregnancy prevention programme: assessment of risk awareness. J Dermatolog Treat. 2022;33(1):466–72. https://doi.org/10.1080/09546634.2020.1770166.
Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel). 2016;8(2):22. https://doi.org/10.3390/cancers8020022.
Kim R, Ji JH, Kim JH, Hong JY, Lim HY, Kang WK, et al. Safety and anti-tumor effects of vismodegib in patients with refractory advanced gastric cancer: A single-arm, phase-II trial. J Cancer. 2022;13(4):1097–102. https://doi.org/10.7150/jca.67050.
Dummer R, Ascierto PA, Basset-Seguin N, Dréno B, Garbe C, Gutzmer R, et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: a joint expert opinion. J Eur Acad Dermatol Venereol. 2020;34(9):1944–56. https://doi.org/10.1111/jdv.16230.
LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17(8):2502–11. https://doi.org/10.1158/1078-0432.CCR-10-2745.
Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the Hedgehog Pathway in Advanced Basal-Cell Carcinoma. N Engl J Med. 2009;361(12):1164–72. https://doi.org/10.1056/NEJMoa0905360.
Bertrand N, Guerreschi P, Basset-Seguin N, Saiag P, Dupuy A, Dalac-Rat S, et al. Vismodegib in neoadjuvant treatment of locally advanced basal cell carcinoma: First results of a multicenter, open-label, phase 2 trial (VISMONEO study): Neoadjuvant Vismodegib in Locally Advanced Basal Cell Carcinoma. EClinicalMedicine. 2021;35:100844. https://doi.org/10.1016/j.eclinm.2021.100844.
Dréno B, Kunstfeld R, Hauschild A, Fosko S, Zloty D, Labeille B, et al. Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): a randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017;18(3):404–12. https://doi.org/10.1016/s1470-2045(17)30072-4.
Basset-Seguin N, Hauschild A, Grob J-J, Kunstfeld R, Dréno B, Mortier L, et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): a pre-planned interim analysis of an international, open-label trial. Lancet Oncol. 2015;16(6):729–36. https://doi.org/10.1016/s1470-2045(15)70198-1.
De Jesus-Acosta A, Sugar EA, O’Dwyer PJ, Ramanathan RK, Von Hoff DD, Rasheed Z, et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br J Cancer. 2020;122(4):498–505. https://doi.org/10.1038/s41416-019-0683-3.
Frappaz D, Barritault M, Montané L, Laigle-Donadey F, Chinot O, Le Rhun E, et al. MEVITEM-a phase I/II trial of vismodegib + temozolomide vs temozolomide in patients with recurrent/refractory medulloblastoma with Sonic Hedgehog pathway activation. Neuro Oncol. 2021;23(11):1949–60. https://doi.org/10.1093/neuonc/noab087.
Carr RM, Duma N, McCleary-Wheeler AL, Almada LL, Marks DL, Graham RP, et al. Targeting of the Hedgehog/GLI and mTOR pathways in advanced pancreatic cancer, a phase 1 trial of Vismodegib and Sirolimus combination. Pancreatology. 2020;20(6):1115–22. https://doi.org/10.1016/j.pan.2020.06.015.
Tang JY, Ally MS, Chanana AM, Mackay-Wiggan JM, Aszterbaum M, Lindgren JA, et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17(12):1720–31. https://doi.org/10.1016/s1470-2045(16)30566-6.
Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Brief Report: Treatment of Medulloblastoma with Hedgehog Pathway Inhibitor GDC-0449. N Engl J Med. 2009;361(12):1173–8. https://doi.org/10.1056/NEJMoa0902903.
Metcalfe C, de Sauvage FJ. Hedgehog fights back: mechanisms of acquired resistance against Smoothened antagonists. Cancer Res. 2011;71(15):5057–61. https://doi.org/10.1158/0008-5472.CAN-11-0923.
Casey D, Demko S, Shord S, Zhao H, Chen HY, He K, et al. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin Cancer Res. 2017;23(10):2377–81. https://doi.org/10.1158/1078-0432.Ccr-16-2051.
Burness CB. Sonidegib: First Global Approval. Drugs. 2015;75(13):1559–66. https://doi.org/10.1007/s40265-015-0458-y.
Brancaccio G, Pea F, Moscarella E, Argenziano G. Sonidegib for the Treatment of Advanced Basal Cell Carcinoma. Front Oncol. 2020;10:582866. https://doi.org/10.3389/fonc.2020.582866.
Migden MR, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015;16(6):716–28. https://doi.org/10.1016/s1470-2045(15)70100-2.
Lear JT, Migden MR, Lewis KD, Chang ALS, Guminski A, Gutzmer R, et al. Long-term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30-month analysis of the randomized phase 2 BOLT study. J Eur Acad Dermatol Venereol. 2018;32(3):372–81. https://doi.org/10.1111/jdv.14542.
Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J, et al. A Phase I, Multicenter, Open-Label, First-in-Human, Dose-Escalation Study of the Oral Smoothened Inhibitor Sonidegib (LDE225) in Patients with Advanced Solid Tumors. Clin Cancer Res. 2014;20(7):1900–9. https://doi.org/10.1158/1078-0432.Ccr-13-1710.
Lear JT, Hauschild A, Stockfleth E, Squittieri N, Basset-Seguin N, Dummer R. Efficacy and Safety of Sonidegib in Adult Patients with Nevoid Basal Cell Carcinoma Syndrome (Gorlin Syndrome): Results from a Phase 2, Double-Blind. Randomized Trial Clin Cosmet Investig Dermatol. 2020;13:117–21. https://doi.org/10.2147/CCID.S233097.
Jain S, Song R, Xie J. Sonidegib: mechanism of action, pharmacology, and clinical utility for advanced basal cell carcinomas. Onco Targets Ther. 2017;10:1645–53. https://doi.org/10.2147/ott.S130910.
Villani A, Fabbrocini G, Costa C, Scalvenzi M. Sonidegib: Safety and Efficacy in Treatment of Advanced Basal Cell Carcinoma. Dermatol Ther (Heidelb). 2020;10(3):401–12. https://doi.org/10.1007/s13555-020-00378-8.
Herms F, Baroudjian B, Delyon J, Laly P, Tetu P, Lebbe C, et al. Sonidegib in the Treatment of Locally Advanced Basal Cell Carcinoma: a Retrospective Study. Acta Derm Venereol. 2022. https://doi.org/10.2340/actadv.v102.1995.
Danial C, Sarin KY, Oro AE, Chang AL. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin Cancer Res. 2016;22(6):1325–9. https://doi.org/10.1158/1078-0432.CCR-15-1588.
Chai JY, Sugumar V, Alshanon AF, Wong WF, Fung SY, Looi CY. Defining the Role of GLI/Hedgehog Signaling in Chemoresistance: Implications in Therapeutic Approaches. Cancers (Basel). 2021;13(19):4746. https://doi.org/10.3390/cancers13194746.
Munchhof MJ, Li Q, Shavnya A, Borzillo GV, Boyden TL, Jones CS, et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med Chem Lett. 2012;3(2):106–11. https://doi.org/10.1021/ml2002423.
Norsworthy KJ, By K, Subramaniam S, Zhuang L, Del Valle PL, Przepiorka D, et al. FDA Approval Summary: Glasdegib for Newly Diagnosed Acute Myeloid Leukemia. Clin Cancer Res. 2019;25(20):6021–5. https://doi.org/10.1158/1078-0432.CCR-19-0365.
Hoy SM. Glasdegib: First Global Approval. Drugs. 2019;79(2):207–13. https://doi.org/10.1007/s40265-018-1047-7.
Sarkaria SM, Heaney ML. Glasdegib in newly diagnosed acute myeloid leukemia. Expert Rev Anticancer Ther. 2021;21(6):573–81. https://doi.org/10.1080/14737140.2021.1891885.
Martinelli G, Oehler VG, Papayannidis C, Courtney R, Shaik MN, Zhang X, et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study. The Lancet Haematology. 2015;2(8):e339–46. https://doi.org/10.1016/s2352-3026(15)00096-4.
Wagner AJ, Messersmith WA, Shaik MN, Li S, Zheng X, McLachlan KR, et al. A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21(5):1044–51. https://doi.org/10.1158/1078-0432.Ccr-14-1116.
Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin Cancer Res. 2018;24(10):2294–303. https://doi.org/10.1158/1078-0432.Ccr-17-2824.
Jin G, Sivaraman A, Lee K. Development of taladegib as a sonic hedgehog signaling pathway inhibitor. Arch Pharm Res. 2017;40(12):1390–3. https://doi.org/10.1007/s12272-017-0987-x.
Ohashi T, Oguro Y, Tanaka T, Shiokawa Z, Tanaka Y, Shibata S, et al. Discovery of the investigational drug TAK-441, a pyrrolo[3,2-c]pyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: modification of the core skeleton for improved solubility. Bioorg Med Chem. 2012;20(18):5507–17. https://doi.org/10.1016/j.bmc.2012.07.034.
Peukert S, He F, Dai M, Zhang R, Sun Y, Miller-Moslin K, et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem. 2013;8(8):1261–5. https://doi.org/10.1002/cmdc.201300217.
Zhang X, Tian Y, Yang Y, Hao J. Development of anticancer agents targeting the Hedgehog signaling. Cell Mol Life Sci. 2017;74(15):2773–82. https://doi.org/10.1007/s00018-017-2497-x.
Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27(3):315–6. https://doi.org/10.1016/j.ccell.2015.02.009.
Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci USA. 2007;104(20):8455–60. https://doi.org/10.1073/pnas.0609699104.
Garcia N, Ulin M, Ali M, Al-Hendy A, Carvalho KC, Yang Q. Evaluation of Hedgehog Pathway Inhibitors as a Therapeutic Option for Uterine Leiomyosarcoma Using the Xenograft Model. Reprod Sci. 2022;29(3):781–90. https://doi.org/10.1007/s43032-021-00731-y.
Lauth M, Bergström A, Shimokawa T, Toftgård R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104(20):8455–60. https://doi.org/10.1073/pnas.0609699104.
Huang L, Walter V, Hayes DN, Onaitis M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin Cancer Res. 2014;20(6):1566–75. https://doi.org/10.1158/1078-0432.Ccr-13-2195.
Harada K, Ohashi R, Naito K, Kanki K. Hedgehog Signal Inhibitor GANT61 Inhibits the Malignant Behavior of Undifferentiated Hepatocellular Carcinoma Cells by Targeting Non-Canonical GLI Signaling. Int J Mol Sci. 2020;21(9):3126. https://doi.org/10.3390/ijms21093126.
Bacelar Sacramento de Araújo T, de Oliveira Siquara da Rocha L, Torres Andion Vidal M, Cerqueira Coelho PL, Galvão Dos Reis M, Solano de Freitas Souza B, et al. GANT61 Reduces Hedgehog Molecule (GLI1) Expression and Promotes Apoptosis in Metastatic Oral Squamous Cell Carcinoma Cells. Int J Mol Sci. 2020;21(17):6076. https://doi.org/10.3390/ijms21176076.
Carballo GB, Ribeiro JH, Lopes GPF, Ferrer VP, Dezonne RS, Pereira CM, et al. GANT-61 Induces Autophagy and Apoptosis in Glioblastoma Cells despite their heterogeneity. Cell Mol Neurobiol. 2021;41(6):1227–44. https://doi.org/10.1007/s10571-020-00891-6.
Li J, Cai J, Zhao S, Yao K, Sun Y, Li Y, et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J Exp Clin Cancer Res. 2016;35(1):184. https://doi.org/10.1186/s13046-016-0463-3.
Konings K, Vandevoorde C, Belmans N, Vermeesen R, Baselet B, Walleghem MV, et al. The Combination of Particle Irradiation With the Hedgehog Inhibitor GANT61 Differently Modulates the Radiosensitivity and Migration of Cancer Cells Compared to X-Ray Irradiation. Front Oncol. 2019;9:391. https://doi.org/10.3389/fonc.2019.00391.
Zhang XQ, Tian Y, Yang YL, Hao JJ. Development of anticancer agents targeting the Hedgehog signaling. Cell Mol Life Sci. 2017;74(15):2773–82. https://doi.org/10.1007/s00018-017-2497-x.
Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci USA. 2010;107(30):13432–7. https://doi.org/10.1073/pnas.1006822107.
Wang ZY, Chen Z. Differentiation and apoptosis induction therapy in acute promyelocytic leukaemia. Lancet Oncol. 2000;1:101–6. https://doi.org/10.1016/s1470-2045(00)00017-6.
Kim J, Lee JJ, Kim J, Gardner D, Beachy PA. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A. 2010;107(30):13432–7. https://doi.org/10.1073/pnas.1006822107.
Ally MS, Ransohoff K, Sarin K, Atwood SX, Rezaee M, Bailey-Healy I, et al. Effects of Combined Treatment With Arsenic Trioxide and Itraconazole in Patients With Refractory Metastatic Basal Cell Carcinoma. JAMA Dermatol. 2016;152(4):452–6. https://doi.org/10.1001/jamadermatol.2015.5473.
Didiasova M, Singh R, Wilhelm J, Kwapiszewska G, Wujak L, Zakrzewicz D, et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. Faseb j. 2017;31(5):1916–28. https://doi.org/10.1096/fj.201600892RR.
Wolff F, Loipetzberger A, Gruber W, Esterbauer H, Aberger F, Frischauf AM. Imiquimod directly inhibits Hedgehog signalling by stimulating adenosine receptor/protein kinase A-mediated GLI phosphorylation. Oncogene. 2013;32(50):5574–81. https://doi.org/10.1038/onc.2013.343.
Wu Z, Zou B, Zhang X, Peng X. Eupatilin regulates proliferation and cell cycle of cervical cancer by regulating hedgehog signalling pathway. Cell Biochem Funct. 2020;38(4):428–35. https://doi.org/10.1002/cbf.3493.
Luo J, Wang J, Yang J, Huang W, Liu J, Tan W, et al. Saikosaponin B1 and Saikosaponin D inhibit tumor growth in medulloblastoma allograft mice via inhibiting the Hedgehog signaling pathway. J Nat Med. 2022. https://doi.org/10.1007/s11418-022-01603-8.
Acknowledgements
We appreciate BioRender.com which gave us materials for making figures.
Funding
None.
Author information
Authors and Affiliations
Contributions
Ruolan Xia reviewed the literature and wrote the manuscript. Maosen Xu wrote the manuscript. Xuelei Ma and Jing Yang designed this study. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xia, R., Xu, M., Yang, J. et al. The role of Hedgehog and Notch signaling pathway in cancer. Mol Biomed 3, 44 (2022). https://doi.org/10.1186/s43556-022-00099-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-022-00099-8