
Abstract
Patients with advanced gastric cancer usually have a poor prognosis and limited therapeutic options. Overcoming this challenge requires novel targets and effective drugs. The Hedgehog (Hh) signaling pathway plays a crucial role in the development of the gastrointestinal tract and maintenance of the physiologic function of the stomach. Aberrantly activated Hh signaling is implicated in carcinogenesis as well as maintenance of cancer stem cells. Somatic mutations in the components of Hh signaling (PTCH1 and SMO) have been shown to be a major cause of basal cell carcinoma, and dozens of Hh inhibitors have been developed. To date, two inhibitors (GDC-0449 and LDE225) have been approved by the U.S. Food and Drug Administration to treat basal cell carcinoma and medulloblastoma. Here, we review the role of the Hh signaling in the carcinogenesis and progression of gastric cancer and summarize recent findings on Hh inhibitors in gastric cancer. Hedgehog signaling is often aberrantly activated and plays an important role during inflammation and carcinogenesis of gastric epithelial cells. Further study of the precise mechanisms of Hh signaling in this disease is needed for the validation of therapeutic targets and evaluation of the clinical utility of Hh inhibitors for gastric cancer.
Similar content being viewed by others


Background
Gastric cancer (GC) is highly heterogeneous, largely because of the complex molecular mechanisms of its carcinogenesis (e.g., genetic alterations, epigenetic changes, infection, and interactions with the microenvironment) [1,2,3]. Consequently, existing targeted therapies, such as HER2 antagonist trastuzumab and VEGFR2 antagonist ramucirumab, are effective only in a small percentage of patients with GC. Therefore, more research is needed to develop effective personalized therapy for this disease [4]. In this context, the Hedgehog (Hh) signaling pathway is important to explore.
The Hh signaling pathway is crucial in embryonic development and tissue homeostasis [5,6,7]. Over the past decades, increasing evidence has also implicated aberrant Hh signaling in the initiation and progression of many cancers, including prostate [8], breast [9], pancreatic [10], and hepatocellular [11] cancers as well as GC [12]. In basal cell carcinoma (BCC) [13] and medulloblastoma [14] in particular, the role of Hh signaling has been well established. On the basis of these results, Hh signaling inhibitors have been developed, and two have been approved by the U.S. Food and Drug Administration (FDA) to treat BCC and medulloblastoma [15, 16]. However, the molecular mechanisms of the Hh signaling in other tumors appear far more complex than those in BCC. In addition to the canonical Hh signaling cascade, the crosstalk between components of Hh and other pathways contributes to carcinogenesis [17]. Moreover, Hh signaling is needed for the maintenance of cancer stem cells (CSCs) [18,19,20]. Thus, a deeper understanding of the role of Hh signaling in CSCs will provide a rationale for development of a druggable target to block metastasis and overcome chemotherapy resistance in GC.
In this review, we will focus on the role of the Hh signaling pathway in GC and CSCs and discuss the therapeutic potential of Hh inhibitors for GC.
Hh signaling pathway overview
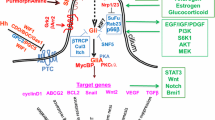
The central components of the Hh signaling pathway are three Hh ligands (Sonic hedgehog [SHH], Indian hedgehog [IHH], and Desert hedgehog [DHH]), the transmembrane receptor Patched1 (PTCH1), the G-protein-coupled receptor Smoothened (SMO), the negative regulator Suppressor-of-fused (SUFU), and the transcription factors GLI1, GLI2, and GLI3 [7]. In general, Hh signaling is activated through the binding of the Hh ligand to PTCH1, which acts as a negative regulator of SMO [21]. The inhibition of SMO is then released, which activates a cascade that leads to the translocation of the downstream GLI transcription factors (GLIs) to the nucleus. Subsequently, the activated GLIs induce expression of various Hh target genes, such as BCL2, SNAI1, FOXM1, cMYC, and CCND1 [22]. In contrast, in the absence of Hh ligands, PTCH1 is located in the primary cilium and suppresses SMO activation by preventing its localization and accumulation in the cilium (Fig. 1).

Hh signaling pathway transduction. a In the absence of Hh ligands, PTCH is located in the primary cilium and suppresses SMO activation by preventing SMO localization to and accumulation in the cilium. The GLI transcription factors are phosphorylated and processed into transcriptional repressors (GLI R) by several protein mediators (PKA, CK1, and GSK3β). GLI R translocates into the nucleus and inhibits transcription. b When an Hh ligand binds to PTCH, the inhibition of SMO is relieved, allowing dissociation of GLI transcription factors from KIF7 and SUFU. Transcriptional activators (GLI1/2 A) then enter the nucleus to induce expression of Hh target genes. Figure created with BioRender
GLIs play a central role in Hh signaling because abnormal activation of Hh signaling, whether due to mutations of pathway components or ligand-dependent or -independent mechanisms, triggers the downstream effector GLIs. Among the three GLIs, GLI1 acts exclusively as a transcription activator, while GLI2 and GLI3 act either as transcriptional activators or truncated transcriptional repressors [23]. Indeed, GLI1 was shown to activate the expression of genes involved in multiple cellular functions, including cell proliferation (cyclin D1/D2 and FOXM1) [24, 25], angiogenesis (VEGF family) [26], epithelial-mesenchymal transition (SNAI1) [27], and invasion (osteopontin and MMP) [28,29,30]. Furthermore, GLI1 exerts its function through crosstalk with several non-canonical Hh signaling, such as mTOR [31, 32], KRAS [33], TGFβ [34, 35], and WNT [36, 37]. Therefore, GLI1 is emerging as a promising target for blocking Hh signaling and treating cancer.
Broadly, the canonical mechanisms linking abnormal activation of the Hh signaling pathway to cancer can be ligand dependent or ligand independent. The best-known example of the ligand-independent mechanism is in BCC [13]. Somatic mutations in PTCH1 have been identified in more than 90% of sporadic BCC, and the dysfunctional PTCH1 leads to constitutively active SMO. While these tumors are insensitive to Hh ligands, small-molecule SMO inhibitors (e.g., cyclopamine) can effectively suppress these tumors’ growth. So far, BCC is the only tumor known to almost exclusively depend on mutation of Hh signaling pathway components. By contrast, mutations in PTCH1 and SMO are infrequent in other tumors and are rare in GC [38, 39].
On the other hand, the ligand-dependent mechanism has been observed in some gastrointestinal adenocarcinomas, e.g., pancreatic cancer and colon cancer [40]. This mechanism tends to implicate interaction between tumor cells and stromal cells in the tumor microenvironment. Thus, three modes of ligand-dependent regulation of Hh signaling have been proposed: autocrine regulation, in which a tumor-derived ligand activates Hh signaling in tumor cells; paracrine regulation, in which a tumor-derived ligand activates Hh signaling in stromal cells; and reverse paracrine regulation, in which a stromal cell–derived ligand activates Hh signaling in tumor cells [41] (Fig. 2). Finally, beyond these canonical mechanisms, the crosstalk between components of Hh signaling and other pathways contributes to carcinogenesis and progression in GC [42].

Mechanisms of Hh signaling pathway in cancer. a Ligand-independent mechanism: somatic mutations in PTCH1 or SMO lead to constitutive activation of Hh signaling. b-d Ligand-dependent mechanisms: autocrine regulation (b), paracrine regulation (c), and reverse paracrine regulation (d). Figure created with BioRender

Targeting Hh signaling at different levels of the cascade. Hh ligand antibody, e.g., 5E1, blocks interaction of Hh ligand and receptor. SMO inhibitors, e.g., cyclopamine, directly bind to and inactivate SMO. GLI1 inhibitors, e.g., GANT61, interrupt transcriptional activity of GLI1 by directly binding to GLI1. BET inhibitor JQ1 represses Hh signaling by interfering with the regulation of BRD4 on GLI1/GLI2
Hh signaling in GC
Hh signaling has been implicated as a critical factor in gastric gland organogenesis and differentiation during embryonic development. Although all three Hh ligands activate identical signaling cascades by binding to PTCH1, the distribution of these ligands exhibits tissue specificity. SHH and IHH are most highly expressed in the gastrointestinal tract [43], while DHH expression is restricted to testes and the nervous system [44]. In the adult stomach, the Hh pathway not only regulates gastric epithelial cell differentiation and maturation [45, 46] but also is essential to the physiology of the stomach [47, 48]. Hh ligands (typically SHH in the stomach) secreted by the epithelial cells are recognized by receptors on stromal cells, which initiates the Hh signaling cascade in stromal cells and increases transcription of downstream target genes. In turn, these genes’ products are involved in maintaining the microenvironment of the epithelium. It is so-called “paracrine” regulation [49].
In precancerous lesions in the stomach, the balance of this “paracrine” regulation is broken, which results in metaplastic transformation and growth of the fibrous tissue, proceeding to carcinogenesis. Interestingly, in the chronic inflammatory setting, e.g., Helicobacter pylori infection, expression of SHH is downregulated in inflamed tissues [50, 51], mainly because of the loss of parietal cells and epithelium atrophy [52]. However, with gastric lesion progression, increasing expression of SHH is accompanied by epithelial regeneration and proliferation [53]. These observations underline the important role of SHH and Hh signaling in gastric epithelial repair and regeneration [54]. Furthermore, GC cells show not only elevated SHH expression but also increased PTCH1 receptor expression [55]. Thus, excess SHH stimulates Hh signaling and promotes GC cell proliferation and progression. In the latter case, besides “paracrine” regulation, “autocrine” regulation also contributes to the progression of cancer.
Previous studies demonstrated that the overactivity of Hh signaling is a common molecular event in GC and that this abnormal activity is blocked by Hh inhibitors (e.g., cyclopamine) and Hh antibodies [12, 56]. In addition, a number of studies showed that overexpression of SHH is associated with unfavorable clinical outcomes (e.g., advanced clinical stage, lymph node metastasis, and poor prognosis) in patients with GC [57]. Altogether, these results suggest that the Hh signaling pathway participates in cell migration and metastasis. Furthermore, the insulin-like growth factor/phosphoinositide 3-kinase (PI3K)/Akt pathway shows a reciprocal relationship with Hh-dependent tumor formation during GC cell migration. Yoo et al. reported that the Hh pathway promotes GC progression and metastases through activation of the PI3K/Akt pathway [58]. Akt, in turn, stabilizes full-length GLI2 through phosphorylation of S230, thereby amplifying the transcriptional output of Hh signaling [59]. This evidence not only confirms the role of Hh signaling in gastric carcinogenesis and progression but also raises the possibility of inhibition of Hh signaling for treatment of GC.
Hh signaling, CSCs, and drug resistance
Abundant evidence indicates that Hh signaling is involved in the maintenance of CSCs in many cancers [18,19,20]. Components of Hh signaling have been found to be specifically overexpressed in subpopulations of cancer cells with CSC properties. Moreover, these putative CSCs, such as those in pancreatic cancer (ALDH+ cells), colon cancer (CD133+ cells), breast cancer (CD44 + CD24− cells), and GC (CD44+ cells) are sensitive to Hh inhibitors [60,61,62,63]. For example, Yoon and colleagues found enrichment of CD44 along with increased levels of Hh pathway components and certain self-renewal marker proteins (SOX2, OCT4, and NANOG) in three GC cell lines [64]. In these GC lines, Hh inhibition with SMO shRNA or small-molecule inhibitors significantly suppressed spheroid formation and tumor growth. Furthermore, while CD44+ spheroid cells were highly resistant to chemotherapy (5-fluorouracil and cisplatin), this chemoresistance was reversed with Hh inhibition.
To date, the molecular characterization and functional relevance of CSCs in solid tumors are not well understood. Nevertheless, the close relationship between Hh signaling and CSCs raises the possibility of the combination of an Hh inhibitor and standard chemotherapy to improve antitumor efficacy. To achieve the goal, the precise molecular mechanisms of CSCs with Hh signaling need to be further investigated. Meanwhile, from a technical viewpoint, at least two challenges need to be resolved. First is the identification of reliable biomarkers to distinguish CSCs and to predict benefit from therapy. The baseline expression of Hh ligands in tumor tissue appears not to provide a positive association between clinical benefit and high activation of Hh signaling from treatment with Hh inhibition [65]. Instead, CSC-related biomarkers should be considered potential candidates for patient selection. Second is identifying which component of Hh signaling would be an ideal target. To date, different types of inhibitors have been developed that target multiple signal transduction elements of the Hh signaling cascade. However, most of these inhibitors have failed to show the desired results in clinical trials for GC.
Hh inhibitors and clinical potential in GC
Considering the critical role of the Hh pathway in carcinogenesis, targeting of Hh signaling has attracted substantial interest. So far, dozens of small-molecule inhibitors of Hh have been developed, and two (GDC-0449 and LDE225) have been approved by the FDA for treating BCC and medulloblastoma [15, 16], but none have been approved for GC. Here, we focus on the inhibitors that have been investigated in GC (Table 1, Fig. 3).
Hh ligand antibody
The anti-SHH monoclonal antibody 5E1 blocks Hh signaling by binding at the SHH pseudo-active site groove [78]. 5E1 has been used as a research tool to study Hh signaling under physiologic or pathologic circumstances. A previous study showed that mesenchymal stem cells are recruited from bone marrow and contribute to a tumor niche that promotes and sustains GC progression [79]. Blocking Hh signaling using 5E1 significantly inhibited the proliferative response of mesenchymal stem cells to the cytokine interferon-gamma [66]. These data support a synergic interaction of Hh signaling and cytokines during precancerous lesions.
SMO inhibitors
Cyclopamine is the first small-molecule inhibitor of Hh signaling, extracted from corn lilies, and it inactivates SMO by directly binding to its heptahelical bundle [80, 81]. Cyclopamine has been found to effectively inhibit Hh and tumor growth in vivo and in vitro [55, 69,70,71]. However, the off-target effects that accompany a high concentration of cyclopamine require careful evaluation to avoid false-positive data. Moreover, owing to its severe toxicity and low oral bioavailability, cyclopamine is not a suitable drug [82]. A recently developed derivative of cyclopamine, IPI-926 (also called saridegib), showed improved properties compared with cyclopamine [83]. In preclinical studies, IPI-926 was a reliable inhibitor of tumor growth and overcame chemoresistance in a number of cancers [74, 84, 85]. Based on these results, several phase I and phase II clinical trials in pancreatic cancer, head and neck cancer, and other cancers have been conducted. However, to date, detailed results remain to be disclosed.
GDC-0449 (vismodegib), which acts by binding to the extracellular domain of SMO and antagonizing Hh signaling, has shown promising anti-tumor activity in advanced BCC and became the first Hh signaling inhibitor approved by the FDA in 2012 for the treatment of metastases and locally advanced BCC [15, 16]. Although GDC-0449 significantly suppressed tumor proliferation and invasion in vivo and in vitro [72, 86, 87], no satisfactory results have been obtained from clinical trials in GC so far [65]. In a randomized phase II study (NCT00982592), GDC-0449 in combination with chemotherapy was investigated in advanced GC. There was no difference in response rate or survival time with the addition of GDC-0449 to chemotherapy. However, in the patients given chemotherapy combined with GDC-0449, those with higher CD44 expression tended to fare better [64]. In contrast, in the chemotherapy-only group, patient survival was not stratified by CD44. As mentioned above, an accurate biomarker is necessary to identify a subpopulation that could benefit from Hh inhibition.
Other SMO inhibitors, including LDE225 (sonidegib), PF-04449913 (glasdegib), itraconazole, LEQ506, BMS-833923, LY2940680, and vitamin D3, have been effective against a variety of human cancers [88,89,90], and some have reached clinical trials. These agents are being investigated as potential treatments for hematologic malignancies, medulloblastoma, and other solid tumors, but not for GC [91, 92].
GLI1 inhibitors
Since the GLI1 transcription factor is the final effector of the Hh signaling cascade, targeting GLI1 may yield more efficient antitumor activity than other targets have thus far. In 2007, two small-molecule GLI1 inhibitors (GANT58 and GANT61) were identified through cell-based screening [93]. GANT61 directly binds GLI1 protein between zinc fingers 2 and 3 and interrupts GLI1 binding to target DNA, thereby inhibiting transcription [94].
Yan and colleagues treated GC cell lines with cyclopamine and GANT61 and found that both inhibitors repressed cell growth [77]. Another study, by Xu et al., showed that GANT61 increased apoptosis of CD44+/Musashi-1+ GC cells [63]. The study also demonstrated a synergistic effect between the Hh signaling inhibitor and the chemotherapy drug doxorubicin. Thus, GANT61 shows therapeutic potential, and further in vivo study and clinical trials are required to investigate the antitumor activity of GANT61 in GC.
Besides direct GLI1 inhibitors, indirect GLI1 inhibitors have also recently emerged as candidates for suppressing Hh-dependent tumors. JQ1, the bromodomain and extra-terminal domain (BET) protein inhibitor, specifically impacts GLI1 transcription [73]. BET proteins act as a transcriptional regulator by binding to chromatin during mitosis, promoting cell cycle progression. BET proteins also form multiprotein complexes with the positive transcription elongation factor b, activate RNA polymerase II, and enhance gene expression [95]. Recently, Tang and colleagues demonstrated that BRD4, a member of the BET family, is the critical regulator of GLI1/GLI2 by occupying their promoters and modulating transcription [73]. Furthermore, JQ1 effectively suppresses Hh-dependent tumor growth by preventing BRD4 from binding to GLI1/GLI2. Even more encouragingly, JQ1 impaired GLI signaling in the setting of acquired resistance to SMO inhibitors. These results suggests a novel strategy for the treatment of Hh-dependent tumors using BET inhibition. Previous studies demonstrated that JQ1 efficiently suppressed proliferation and induced apoptosis of GC cells through BRD4 and downstream genes [96, 97]. However, no study on the effect of JQ1 on Hh/GLI1 signaling in GC has been reported. Interestingly, in certain GC cell lines with low expression of BRD4 or c-MYC (a BRD4 target protein), the antitumor activity of JQ1 was still remarkable [98]. This finding suggests that other molecular mechanisms are involved in this process.
Perspective
The Hh signaling pathway has long been thought to play a crucial role in embryonic development, tissue homeostasis, carcinogenesis, and maintenance of CSCs. Since the predominant role of Hh signaling in BCC has been verified, leading to the development and marketing of vismodegib, efforts are underway to exploit this pathway to treat other cancers. However, so far, the results from clinical trials targeting Hh signaling in a number of cancers, such as GC, colorectal cancer, ovarian cancer, and pancreatic cancer, have been inconsistent with expectations [64, 65, 99, 100]. The explanation for these results is likely that other genes or signaling pathways are also involved in carcinogenesis and progression.
The reasonable way to overcome this problem is the identification of precise predictive biomarkers and optimized combination strategies, such as an Hh signaling inhibitor combined with standard chemotherapy or other targeted therapy. In one study of Hh signaling in medulloblastoma, Shou and colleagues validated a five-gene signature that identifies Hh pathway activation and patients most likely to respond to therapy targeting the Hh signaling pathway [101]. Although the panel is not yet mature and needs to be improved [102], it still broadens our research strategy for identifying better predictive and prognostic markers. Furthermore, given the crucial role of Hh signaling in the maintenance of CSCs, CSC-related proteins could be used to select patients who are likely to benefit from an Hh inhibitor. Indeed, several proteins or panels have been proposed as CSC biomarkers in GC [103,104,105,106,107,108,109,110,111], such as ALDH1, CD24/CD44, CD54/CD44, EPCAM/CD44, LGR5, CD90, and CD133. Thoroughly understanding the mechanisms of CSCs and Hh signaling will provide rationales for more precise approach.
Conclusions
Hh signaling is often aberrantly activated and plays an important role in the inflammation and carcinogenesis of gastric epithelial cells. However, the clinical utility of Hh inhibitors for GC should be further evaluated through more well-designed clinical trials. In addition, given the complexity of Hh signaling and the heterogeneity of GC, the precise mechanisms of Hh signaling need to be studied further for the validation of therapeutic targets and ideal biomarkers.
Availability of data and materials
Not applicable.
Abbreviations
- BCC:
-
basal cell carcinoma
- BET:
-
bromodomain and extra-terminal domain
- CSC:
-
cancer stem cell
- DHH:
-
Desert hedgehog
- FDA:
-
U.S. Food and Drug Administration
- GC:
-
gastric cancer
- GLI:
-
glioma-associated oncogene
- Hh:
-
Hedgehog
- IHH:
-
Indian hedgehog
- PTCH1:
-
Patched1
- SHH:
-
Sonic hedgehog
- SMO:
-
Smoothened
References
Ajani JA, Lee J, Sano T, Janjigian YY, Fan D, Song S. Gastric adenocarcinoma. Nat Rev Dis Primers. 2017;3:17036. https://doi.org/10.1038/nrdp.2017.36.
Wadhwa R, Song S, Lee JS, Yao Y, Wei Q, Ajani JA. Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol. 2013;10(11):643–55. https://doi.org/10.1038/nrclinonc.2013.170.
Song S, Ajani JA. The role of microRNAs in cancers of the upper gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2013;10(2):109–18. https://doi.org/10.1038/nrgastro.2012.210.
Lee J, Bass AJ, Ajani JA. Gastric adenocarcinoma: an update on genomics, immune system modulations, and targeted therapy. Am Soc Clin Oncol Educ Book. 2016;36:104–11. https://doi.org/10.1200/EDBK_159091.
Warner JF, McClay DR. Perturbations to the hedgehog pathway in sea urchin embryos. Methods Mol Biol. 2014;1128:211–21. https://doi.org/10.1007/978-1-62703-974-1_14.
van den Brink GR. Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiol Rev. 2007;87(4):1343–75. https://doi.org/10.1152/physrev.00054.2006.
Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22(18):2454–72. https://doi.org/10.1101/gad.1693608.
Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707–12. https://doi.org/10.1038/nature02962.
Riobo-Del Galdo NA, Lara Montero Á, Wertheimer EV. Role of Hedgehog Signaling in Breast Cancer: Pathogenesis and Therapeutics. Cells. 2019;8(4). https://doi.org/10.3390/cells8040375.
Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del Castillo C, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–6. https://doi.org/10.1038/nature02009.
Huang S, He J, Zhang X, Bian Y, Yang L, Xie G, Zhang K, Tang W, Stelter AA, Wang Q, Zhang H, Xie J. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis. 2006;27(7):1334–40. https://doi.org/10.1093/carcin/bgi378.
Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. Widespread requirement for hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425(6960):846–51. https://doi.org/10.1038/nature01972.
Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH Jr, de Sauvage FJ. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–2. https://doi.org/10.1038/34201.
Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, James CD. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57(5):842–5.
Rubin LL, de Sauvage FJ. Targeting the hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5(12):1026–33. https://doi.org/10.1038/nrd2086.
Khatra H, Bose C, Sinha S. Discovery of hedgehog antagonists for Cancer therapy. Curr Med Chem. 2017;24(19):2033–58. https://doi.org/10.2174/0929867324666170316115500.
Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev. 2014;40(6):750–9. https://doi.org/10.1016/j.ctrv.2014.02.003.
Briscoe J, Thérond PP. The mechanisms of hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013 Jul;14(7):416–29. https://doi.org/10.1038/nrm3598.
Ramos EK, Hoffmann AD, Gerson SL, Liu H. New opportunities and challenges to defeat Cancer stem cells. Trends Cancer. 2017;3(11):780–96. https://doi.org/10.1016/j.trecan.2017.08.007.
Sari IN, Phi LTH, Jun N, Wijaya YT, Lee S, Kwon HY. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells. 2018;7(11). https://doi.org/10.3390/cells7110208.
Huang P, Zheng S, Wierbowski BM, Kim Y, Nedelcu D, Aravena L, Liu J, Kruse AC, Salic A. Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell. 2018;174(2):312–324.e16. https://doi.org/10.1016/j.cell.2018.04.029.
Infante P, Alfonsi R, Botta B, Mori M, Di Marcotullio L. Targeting GLI factors to inhibit the hedgehog pathway. Trends Pharmacol Sci. 2015;36(8):547–58. https://doi.org/10.1016/j.tips.2015.05.006.
Wu F, Zhang Y, Sun B, McMahon AP, Wang Y. Hedgehog signaling: from basic biology to Cancer therapy. Cell Chem Biol. 2017;24(3):252–80. https://doi.org/10.1016/j.chembiol.2017.02.010.
Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20(23):9055–67.
Teh MT, Wong ST, Neill GW, Ghali LR, Philpott MP, Quinn AG. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002;62(16):4773–80.
Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake Pepinsky R, Shapiro R, Taylor FR, Baker DP, Asahara T, Isner JM. The morphogen sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat Med. 2001;7(6):706–11. https://doi.org/10.1038/89083.
Li X, Deng W, Nail CD, Bailey SK, Kraus MH, Ruppert JM, Lobo-Ruppert SM. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene. 2006;25(4):609–21. https://doi.org/10.1038/sj.onc.1209077.
Das S, Harris LG, Metge BJ, Liu S, Riker AI, Samant RS, Shevde LA. The hedgehog pathway transcription factor GLI1 promotes malignant behavior of cancer cells by up-regulating osteopontin. J Biol Chem. 2009;284(34):22888–97. https://doi.org/10.1074/jbc.M109.021949.
Kwon YJ, Hurst DR, Steg AD, Yuan K, Vaidya KS, Welch DR, Frost AR. Gli1 enhances migration and invasion via up-regulation of MMP-11 and promotes metastasis in ERα negative breast cancer cell lines. Clin Exp Metastasis. 2011;28(5):437–49. https://doi.org/10.1007/s10585-011-9382-z.
Ohta H, Aoyagi K, Fukaya M, Danjoh I, Ohta A, Isohata N, Saeki N, Taniguchi H, Sakamoto H, Shimoda T, Tani T, Yoshida T, Sasaki H. Cross talk between hedgehog and epithelial-mesenchymal transition pathways in gastric pit cells and in diffuse-type gastric cancers. Br J Cancer. 2009;100(2):389–98. https://doi.org/10.1038/sj.bjc.6604846.
Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, Li CW, Hsu JL, Miller SA, Wang X, Lee DF, Hsu JM, Huo L, Labaff AM, Liu D, Huang TH, Lai CC, Tsai FJ, Chang WC, Chen CH, Wu TT, Buttar NS, Wang KK, Wu Y, Wang H, Ajani J, Hung MC. The crosstalk of mTOR/S6K1 and hedgehog pathways. Cancer Cell. 2012;21(3):374–87. https://doi.org/10.1016/j.ccr.2011.12.028.
Yao Y, Zhou D, Shi D, Zhang H, Zhan S, Shao X, Sun K, Sun L, Wu G, Tian K, Zhu X, He S. GLI1 overexpression promotes gastric cancer cell proliferation and migration and induces drug resistance by combining with the AKT-mTOR pathway. Biomed Pharmacother. 2019;111:993–1004. https://doi.org/10.1016/j.biopha.2019.01.018.
Wu M, Ingram L, Tolosa EJ, Vera RE, Li Q, Kim S, Ma Y, Spyropoulos DD, Beharry Z, Huang J, Fernandez-Zapico ME, Cai H. Gli transcription factors mediate the oncogenic transformation of prostate basal cells induced by a Kras-androgen receptor Axis. J Biol Chem. 2016;291(49):25749–60.
Luo K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb Perspect Biol. 2017;9(1). https://doi.org/10.1101/cshperspect.a022137.
Tang YA, Chen YF, Bao Y, Mahara S, Yatim SMJM, Oguz G, Lee PL, Feng M, Cai Y, Tan EY, Fong SS, Yang ZH, Lan P, Wu XJ, Yu Q. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci U S A. 2018;115(26):E5990–9. https://doi.org/10.1073/pnas.1801348115.
Yanai K, Nakamura M, Akiyoshi T, Nagai S, Wada J, Koga K, Noshiro H, Nagai E, Tsuneyoshi M, Tanaka M, Katano M. Crosstalk of hedgehog and Wnt pathways in gastric cancer. Cancer Lett. 2008;263(1):145–56. https://doi.org/10.1016/j.canlet.2007.12.030.
Chatterjee S, Sil PC. Targeting the crosstalks of Wnt pathway with hedgehog and notch for cancer therapy. Pharmacol Res. 2019 Apr;142:251–61. https://doi.org/10.1016/j.phrs.2019.02.027.
Wang XD, Inzunza H, Chang H, Qi Z, Hu B, Malone D, Cogswell J. Mutations in the hedgehog pathway genes SMO and PTCH1 in human gastric tumors. PLoS One. 2013;8(1):e54415. https://doi.org/10.1371/journal.pone.0054415.
Holbrook JD, Parker JS, Gallagher KT, Halsey WS, Hughes AM, Weigman VJ, Lebowitz PF, Kumar R. Deep sequencing of gastric carcinoma reveals somatic mutations relevant to personalized medicine. J Transl Med. 2011;9:119. https://doi.org/10.1186/1479-5876-9-119.
Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, Nannini-Pepe M, Kotkow K, Marsters JC, Rubin LL, de Sauvage FJ. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455(7211):406–10. https://doi.org/10.1038/nature07275.
Saqui-Salces M, Merchant JL. Hedgehog signaling and gastrointestinal cancer. Biochim Biophys Acta. 2010;1803(7):786–95. https://doi.org/10.1016/j.bbamcr.2010.03.008.
Merchant JL, Saqui-Salces M. Inhibition of hedgehog signaling in the gastrointestinal tract: targeting the cancer microenvironment. Cancer Treat Rev. 2014;40(1):12–21. https://doi.org/10.1016/j.ctrv.2013.08.003.
Ramalho-Santos M, Melton DA, McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development. 2000;127(12):2763–72.
Mäkelä JA, Saario V, Bourguiba-Hachemi S, Nurmio M, Jahnukainen K, Parvinen M, Toppari J. Hedgehog signalling promotes germ cell survival in the rat testis. Reproduction. 2011;142(5):711–21. https://doi.org/10.1530/REP-11-0110.
van den Brink GR, Hardwick JC, Tytgat GN, Brink MA, Ten Kate FJ, Van Deventer SJ, Peppelenbosch MP. Sonic hedgehog regulates gastric gland morphogenesis in man and mouse. Gastroenterology. 2001;121(2):317–28. https://doi.org/10.1053/gast.2001.26261.
van den Brink GR, Hardwick JC, Nielsen C, Xu C, ten Kate FJ, Glickman J, van Deventer SJ, Roberts DJ, Peppelenbosch MP. Sonic hedgehog expression correlates with fundic gland differentiation in the adult gastrointestinal tract. Gut. 2002;51(5):628–33. https://doi.org/10.1136/gut.51.5.628.
Merchant JL. Hedgehog signalling in gut development, physiology and cancer. J Physiol. 2012;590(3):421–32. https://doi.org/10.1113/jphysiol.2011.220681.
Merchant JL, Ding L. Hedgehog signaling links chronic inflammation to gastric Cancer precursor lesions. Cell Mol Gastroenterol Hepatol. 2017;3(2):201–10. https://doi.org/10.1016/j.jcmgh.2017.01.004.
Kolterud A, Grosse AS, Zacharias WJ, Walton KD, Kretovich KE, Madison BB, Waghray M, Ferris JE, Hu C, Merchant JL, Dlugosz AA, Kottmann AH, Gumucio DL. Paracrine hedgehog signaling in stomach and intestine: new roles for hedgehog in gastrointestinal patterning. Gastroenterology. 2009;137(2):618–28. https://doi.org/10.1053/j.gastro.2009.05.002.
Suzuki H, Minegishi Y, Nomoto Y, Ota T, Masaoka T, van den Brink GR, Hibi T. Down-regulation of a morphogen (sonic hedgehog) gradient in the gastric epithelium of helicobacter pylori-infected Mongolian gerbils. J Pathol. 2005;206(2):186–97. https://doi.org/10.1002/path.1763.
Stasikowska-Kanicka O, Wągrowska-Danilewicz M, Białek I, Danilewicz M. The immunoexpression of Shh, Smo and Gli2 in helicobacter pylori positive and negative gastric biopsies. Pol J Pathol. 2012;63(1):25–30.
Waghray M, Zavros Y, Saqui-Salces M, El-Zaatari M, Alamelumangapuram CB, Todisco A, Eaton KA, Merchant JL. Interleukin-1beta promotes gastric atrophy through suppression of sonic hedgehog. Gastroenterology. 2010;138(2):562–72. https://doi.org/10.1053/j.gastro.2009.10.043.
Shiotani A, Murao T, Uedo N, Iishi H, Yamanaka Y, Kamada T, Kusunoki H, Inoue K, Haruma K. Eradication of H. pylori did not improve abnormal sonic hedgehog expression in the high risk group for gastric cancer. Dig Dis Sci. 2012;57(3):643–9. https://doi.org/10.1007/s10620-011-1916-3.
Wessler S, Krisch LM, Elmer DP, Aberger F. From inflammation to gastric cancer - the importance of Hedgehog/GLI signaling in Helicobacter pylori-induced chronic inflammatory and neoplastic diseases. Cell Commun Signal. 2017;15(1):15. https://doi.org/10.1186/s12964-017-0171-4.
Fukaya M, Isohata N, Ohta H, Aoyagi K, Ochiya T, Saeki N, Yanagihara K, Nakanishi Y, Taniguchi H, Sakamoto H, Shimoda T, Nimura Y, Yoshida T, Sasaki H. Hedgehog signal activation in gastric pit cell and in diffuse-type gastric cancer. Gastroenterology. 2006;131(1):14–29. https://doi.org/10.1053/j.gastro.2006.05.008.
Abdel-Rahman O. Hedgehog pathway aberrations and gastric cancer; evaluation of prognostic impact and exploration of therapeutic potentials. Tumour Biol. 2015;36(3):1367–74. https://doi.org/10.1007/s13277-015-3216-6.
Ma X, Chen K, Huang S, Zhang X, Adegboyega PA, Evers BM, Zhang H, Xie J. Frequent activation of the hedgehog pathway in advanced gastric adenocarcinomas. Carcinogenesis. 2005;26(10):1698–705. https://doi.org/10.1093/carcin/bgi130.
Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, Kim JS, Oh SC. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71(22):7061–70. https://doi.org/10.1158/0008-5472.CAN-11-1338.
Shi Y, Chen J, Karner CM, Long F. Hedgehog signaling activates a positive feedback mechanism involving insulin-like growth factors to induce osteoblast differentiation. Proc Natl Acad Sci U S A. 2015;112(15):4678–83. https://doi.org/10.1073/pnas.1502301112.
Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, Koorstra JB, Habbe N, Karikari C, Mullendore M, Gabrielson KL, Sharma R, Matsui W, Maitra A. An orally bioavailable small-molecule inhibitor of hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008;7(9):2725–35. https://doi.org/10.1158/1535-7163.MCT-08-0573.
Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, Ruiz i Altaba A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009;1(6–7):338–51. https://doi.org/10.1002/emmm.200900039.
Kurebayashi J, Koike Y, Ohta Y, Saitoh W, Yamashita T, Kanomata N, Moriya T. Anti-cancer stem cell activity of a hedgehog inhibitor GANT61 in estrogen receptor-positive breast cancer cells. Cancer Sci. 2017;108(5):918–30. https://doi.org/10.1111/cas.13205.
Xu M, Gong A, Yang H, George SK, Jiao Z, Huang H, Jiang X, Zhang Y. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44(+)/Musashi-1(+) gastric cancer stem cells. Cancer Lett. 2015;369(1):124–33. https://doi.org/10.1016/j.canlet.2015.08.005.
Yoon C, Park DJ, Schmidt B, Thomas NJ, Lee HJ, Kim TS, Janjigian YY, Cohen DJ, Yoon SS. CD44 expression denotes a subpopulation of gastric cancer cells in which hedgehog signaling promotes chemotherapy resistance. Clin Cancer Res. 2014;20(15):3974–88. https://doi.org/10.1158/1078-0432.CCR-14-0011.
Berlin J, Bendell JC, Hart LL, Firdaus I, Gore I, Hermann RC, Mulcahy MF, Zalupski MM, Mackey HM, Yauch RL, Graham RA, Bray GL, Low JA. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin Cancer Res. 2013;19(1):258–67. https://doi.org/10.1158/1078-0432.CCR-12-1800.
Donnelly JM, Chawla A, Houghton J, Zavros Y. Sonic hedgehog mediates the proliferation and recruitment of transformed mesenchymal stem cells to the stomach. PLoS One. 2013;8(9):e75225. https://doi.org/10.1371/journal.pone.0075225.
Xiao C, Ogle SA, Schumacher MA, Schilling N, Tokhunts RA, Orr-Asman MA, Miller ML, Robbins DJ, Hollande F, Zavros Y. Hedgehog signaling regulates E-cadherin expression for the maintenance of the actin cytoskeleton and tight junctions. Am J Physiol Gastrointest Liver Physiol. 2010;299(6):G1252–65. https://doi.org/10.1152/ajpgi.00512.2009.
Song Z, Yue W, Wei B, Wang N, Li T, Guan L, Shi S, Zeng Q, Pei X, Chen L. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS One. 2011;6(3):e17687. https://doi.org/10.1371/journal.pone.0017687.
Kameda C, Nakamura M, Tanaka H, Yamasaki A, Kubo M, Tanaka M, Onishi H, Katano M. Oestrogen receptor-alpha contributes to the regulation of the hedgehog signalling pathway in ERalpha-positive gastric cancer. Br J Cancer. 2010;102(4):738–47. https://doi.org/10.1038/sj.bjc.6605517.
Yanai K, Nagai S, Wada J, Yamanaka N, Nakamura M, Torata N, Noshiro H, Tsuneyoshi M, Tanaka M, Katano M. Hedgehog signaling pathway is a possible therapeutic target for gastric cancer. J Surg Oncol. 2007;95(1):55–62. https://doi.org/10.1002/jso.20606.
Na YJ, Lee DH, Kim JL, Kim BR, Park SH, Jo MJ, Jeong S, Kim HJ, Lee SY, Jeong YA, Oh SC. Cyclopamine sensitizes TRAIL-resistant gastric cancer cells to TRAIL-induced apoptosis via endoplasmic reticulum stress-mediated increase of death receptor 5 and survivin degradation. Int J Biochem Cell Biol. 2017;89:147–56. https://doi.org/10.1016/j.biocel.2017.06.010.
Wu C, Cheng J, Hu S, Deng R, Muangu YW, Shi L, Wu K, Zhang P, Chang W, Wang G, Tao K. Reduced proliferation and increased apoptosis of the SGC-7901 gastric cancer cell line on exposure to GDC-0449. Mol Med Rep. 2016;13(2):1434–40. https://doi.org/10.3892/mmr.2015.4677.
Tang Y, Gholamin S, Schubert S, Willardson MI, Lee A, Bandopadhayay P, Bergthold G, Masoud S, Nguyen B, Vue N, Balansay B, Yu F, Oh S, Woo P, Chen S, Ponnuswami A, Monje M, Atwood SX, Whitson RJ, Mitra S, Cheshier SH, Qi J, Beroukhim R, Tang JY, Wechsler-Reya R, Oro AE, Link BA, Bradner JE, Cho YJ. Epigenetic targeting of hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20(7):732–40. https://doi.org/10.1038/nm.3613.
Ma H, Tian Y, Yu X. Targeting Smoothened Sensitizes Gastric Cancer to Chemotherapy in Experimental Models. Med Sci Monit. 2017;23:1493–500. https://doi.org/10.12659/MSM.903012.
Hu Q, Hou YC, Huang J, Fang JY, Xiong H. Itraconazole induces apoptosis and cell cycle arrest via inhibiting hedgehog signaling in gastric cancer cells. J Exp Clin Cancer Res. 2017;36(1):50. https://doi.org/10.1186/s13046-017-0526-0.
Baek S, Lee YS, Shim HE, Yoon S, Baek SY, Kim BS, Oh SO. Vitamin D3 regulates cell viability in gastric cancer and cholangiocarcinoma. Anat Cell Biol. 2011;44(3):204–9. https://doi.org/10.5115/acb.2011.44.3.204.
Yan R, Peng X, Yuan X, Huang D, Chen J, Lu Q, Lv N, Luo S. Suppression of growth and migration by blocking the hedgehog signaling pathway in gastric cancer cells. Cell Oncol (Dordr). 2013;36(5):421–35. https://doi.org/10.1007/s13402-013-0149-1.
Maun HR, Wen X, Lingel A, de Sauvage FJ, Lazarus RA, Scales SJ, Hymowitz SG. Hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. J Biol Chem. 2010;285(34):26570–80. https://doi.org/10.1074/jbc.M110.112284.
Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, Friedman R, Varro A, Tycko B, Wang TC. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–72. https://doi.org/10.1016/j.ccr.2011.01.020.
Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280(5369):1603–7. https://doi.org/10.1126/science.280.5369.1603.
Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of hedgehog signaling by direct binding of cyclopamine to smoothened. Genes Dev. 2002;16(21):2743–8. https://doi.org/10.1101/gad.1025302.
Lipinski RJ, Hutson PR, Hannam PW, Nydza RJ, Washington IM, Moore RW, Girdaukas GG, Peterson RE, Bushman W. Dose- and route-dependent teratogenicity, toxicity, and pharmacokinetic profiles of the hedgehog signaling antagonist cyclopamine in the mouse. Toxicol Sci. 2008;104(1):189–97. https://doi.org/10.1093/toxsci/kfn076.
Tremblay MR, Lescarbeau A, Grogan MJ, Tan E, Lin G, Austad BC, Yu LC, Behnke ML, Nair SJ, Hagel M, White K, Conley J, Manna JD, Alvarez-Diez TM, Hoyt J, Woodward CN, Sydor JR, Pink M, MacDougall J, Campbell MJ, Cushing J, Ferguson J, Curtis MS, McGovern K, Read MA, Palombella VJ, Adams J, Castro AC. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J Med Chem. 2009;52(14):4400–18. https://doi.org/10.1021/jm900305z.
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Rückert F, Grützmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–61. https://doi.org/10.1126/science.1171362.
Keysar SB, Le PN, Anderson RT, Morton JJ, Bowles DW, Paylor JJ, Vogler BW, Thorburn J, Fernandez P, Glogowska MJ, Takimoto SM, Sehrt DB, Gan GN, Eagles-Soukup JR, Serracino H, Hirsch FR, Lucia MS, Thorburn A, Song JI, Wang XJ, Jimeno A. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer Res. 2013;73(11):3381–92. https://doi.org/10.1158/0008-5472.CAN-12-4047.
Lee SJ, Do IG, Lee J, Kim KM, Jang J, Sohn I, Kang WK. Gastric cancer (GC) patients with hedgehog pathway activation: PTCH1 and GLI2 as independent prognostic factors. Target Oncol. 2013;8(4):271–80. https://doi.org/10.1007/s11523-013-0253-1.
Magistri P, Battistelli C, Strippoli R, Petrucciani N, Pellinen T, Rossi L, Mangogna L, Aurello P, D'Angelo F, Tripodi M, Ramacciato G, Nigri G. SMO inhibition modulates cellular plasticity and invasiveness in colorectal Cancer. Front Pharmacol. 2018;8:956. https://doi.org/10.3389/fphar.2017.00956.
Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the hedgehog pathway in cancer. Nat Med. 2013;19(11):1410–22. https://doi.org/10.1038/nm.3389.
Akyala AI, Peppelenbosch MP. Gastric cancer and Hedgehog signaling pathway: emerging new paradigms. Genes Cancer. 2018;9(1–2):1–10. https://doi.org/10.18632/genesandcancer.168.
Merchant AA, Matsui W. Targeting hedgehog--a cancer stem cell pathway. Clin Cancer Res. 2010;16(12):3130–40. https://doi.org/10.1158/1078-0432.CCR-09-2846.
Burness CB, Scott LJ. Sonidegib: a review in locally advanced basal cell carcinoma. Target Oncol. 2016;11(2):239–46. https://doi.org/10.1007/s11523-016-0418-9.
Pan S, Wu X, Jiang J, Gao W, Wan Y, Cheng D, Han D, Liu J, Englund NP, Wang Y, Peukert S, Miller-Moslin K, Yuan J, Guo R, Matsumoto M, Vattay A, Jiang Y, Tsao J, Sun F, Pferdekamper AC, Dodd S, Tuntland T, Maniara W, Kelleher JF 3rd, Yao YM, Warmuth M, Williams J, Dorsch M. Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med Chem Lett. 2010;1(3):130–4. https://doi.org/10.1021/ml1000307.
Lauth M, Bergström A, Shimokawa T, Toftgård R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104(20):8455–60. https://doi.org/10.1073/pnas.0609699104.
Agyeman A, Jha BK, Mazumdar T, Houghton JA. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget. 2014;5(12):4492–503. https://doi.org/10.18632/oncotarget.2046.
Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19(4):523–34. https://doi.org/10.1016/j.molcel.2005.06.027.
Ba M, Long H, Yan Z, Wang S, Wu Y, Tu Y, Gong Y, Cui S. BRD4 promotes gastric cancer progression through the transcriptional and epigenetic regulation of c-MYC. J Cell Biochem. 2018;119(1):973–82. https://doi.org/10.1002/jcb.26264.
Dong X, Hu X, Chen J, Hu D, Chen LF. BRD4 regulates cellular senescence in gastric cancer cells via E2F/miR-106b/p21 axis. Cell Death Dis. 2018;9(2):203. https://doi.org/10.1038/s41419-017-0181-6.
Montenegro RC, Clark PG, Howarth A, Wan X, Ceroni A, Siejka P, Nunez-Alonso GA, Monteiro O, Rogers C, Gamble V, Burbano R, Brennan PE, Tallant C, Ebner D, Fedorov O, O'Neill E, Knapp S, Dixon D, Müller S. BET inhibition as a new strategy for the treatment of gastric cancer. Oncotarget. 2016;7(28):43997–4012. https://doi.org/10.18632/oncotarget.9766.
Kaye SB, Fehrenbacher L, Holloway R, Amit A, Karlan B, Slomovitz B, Sabbatini P, Fu L, Yauch RL, Chang I, Reddy JC. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin Cancer Res. 2012;18(23):6509–18. https://doi.org/10.1158/1078-0432.CCR-12-1796.
Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L, Cohen D, Wade J, Sleckman B, Lenz HJ, Stiff P, Kumar P, Xu P, Henderson L, Takebe N, Salgia R, Wang X, Stadler WM, de Sauvage FJ, Kindler HL. Randomized phase Ib/II study of gemcitabine plus placebo or Vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic Cancer. J Clin Oncol. 2015;33(36):4284–92. https://doi.org/10.1200/JCO.2015.62.8719.
Shou Y, Robinson DM, Amakye DD, Rose KL, Cho YJ, Ligon KL, Sharp T, Haider AS, Bandaru R, Ando Y, Geoerger B, Doz F, Ashley DM, Hargrave DR, Casanova M, Tawbi HA, Rodon J, Thomas AL, Mita AC, MacDonald TJ, Kieran MW. A five-gene hedgehog signature developed as a patient preselection tool for hedgehog inhibitor therapy in medulloblastoma. Clin Cancer Res. 2015;21(3):585–93. https://doi.org/10.1158/1078-0432.CCR-13-1711.
Kieran MW, Chisholm J, Casanova M, Brandes AA, Aerts I, Bouffet E, Bailey S, Leary S, MacDonald TJ, Mechinaud F, Cohen KJ, Riccardi R, Mason W, Hargrave D, Kalambakas S, Deshpande P, Tai F, Hurh E, Geoerger B. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-Oncology. 2017;19(11):1542–52. https://doi.org/10.1093/neuonc/nox109.
Katsuno Y, Ehata S, Yashiro M, Yanagihara K, Hirakawa K, Miyazono K. Coordinated expression of REG4 and aldehyde dehydrogenase 1 regulating tumourigenic capacity of diffuse-type gastric carcinoma-initiating cells is inhibited by TGF-β. J Pathol. 2012;228(3):391–404. https://doi.org/10.1002/path.4020.
Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, Gordon SA, Shimada Y, Wang TC. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells. 2009;27(5):1006–20. https://doi.org/10.1002/stem.30.
Chen T, Yang K, Yu J, Meng W, Yuan D, Bi F, Liu F, Liu J, Dai B, Chen X, Wang F, Zeng F, Xu H, Hu J, Mo X. Identification and expansion of cancer stem cells in tumor tissues and peripheral blood derived from gastric adenocarcinoma patients. Cell Res. 2012;22(1):248–58. https://doi.org/10.1038/cr.2011.109.
Jiang J, Zhang Y, Chuai S, Wang Z, Zheng D, Xu F, Zhang Y, Li C, Liang Y, Chen Z. Trastuzumab (herceptin) targets gastric cancer stem cells characterized by CD90 phenotype. Oncogene. 2012;31(6):671–82. https://doi.org/10.1038/onc.2011.282.
Han ME, Jeon TY, Hwang SH, Lee YS, Kim HJ, Shim HE, Yoon S, Baek SY, Kim BS, Kang CD, Oh SO. Cancer spheres from gastric cancer patients provide an ideal model system for cancer stem cell research. Cell Mol Life Sci. 2011;68(21):3589–605. https://doi.org/10.1007/s00018-011-0672-z.
Jiang Y, He Y, Li H, Li HN, Zhang L, Hu W, Sun YM, Chen FL, Jin XM. Expressions of putative cancer stem cell markers ABCB1, ABCG2, and CD133 are correlated with the degree of differentiation of gastric cancer. Gastric Cancer. 2012;15(4):440–50. https://doi.org/10.1007/s10120-012-0140-y.
Hashimoto K, Aoyagi K, Isobe T, Kouhuji K, Shirouzu K. Expression of CD133 in the cytoplasm is associated with cancer progression and poor prognosis in gastric cancer. Gastric Cancer. 2014;17(1):97–106. https://doi.org/10.1007/s10120-013-0255-9.
Li XB, Yang G, Zhu L, Tang YL, Zhang C, Ju Z, Yang X, Teng Y. Gastric Lgr5(+) stem cells are the cellular origin of invasive intestinal-type gastric cancer in mice. Cell Res. 2016;26(7):838–49. https://doi.org/10.1038/cr.2016.47.
Nguyen PH, Giraud J, Chambonnier L, Dubus P, Wittkop L, Belleannée G, Collet D, Soubeyran I, Evrard S, Rousseau B, Senant-Dugot N, Mégraud F, Mazurier F, Varon C. Characterization of biomarkers of tumorigenic and Chemoresistant Cancer stem cells in human gastric carcinoma. Clin Cancer Res. 2017;23(6):1586–97. https://doi.org/10.1158/1078-0432.CCR-15-2157.
Acknowledgements
We appreciate Sarah Bronson, the scientific editor from Department of Scientific publications of MDACC for her excellent edition on English of this manuscript.
Ethical approval and consent to participate
Not applicable.
Funding
This work was supported by an MD Anderson Institutional Research Grant (3–0026317 to S. Song) and grants from the U.S. Department of Defense (CA160433; to S. Song) and National Institutes of Health (CA129906, CA138671, CA172741 to J.A. Ajani).
Author information
Authors and Affiliations
Contributions
ZW, SS, and JA contributed to conception and determined the final version. YX and SS drafted the manuscript and designed the figures. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, Y., Song, S., Wang, Z. et al. The role of hedgehog signaling in gastric cancer: molecular mechanisms, clinical potential, and perspective. Cell Commun Signal 17, 157 (2019). https://doi.org/10.1186/s12964-019-0479-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-019-0479-3