
Abstract
Despite the considerable progress made towards understanding ALS pathophysiology, several key features of ALS remain unexplained, from its aetiology to its epidemiological aspects. The glymphatic system, which has recently been recognised as a major clearance pathway for the brain, has received considerable attention in several neurological conditions, particularly Alzheimer’s disease. Its significance in ALS has, however, been little addressed. This perspective article therefore aims to assess the possibility of CSF contribution in ALS by considering various lines of evidence, including the abnormal composition of ALS-CSF, its toxicity and the evidence for impaired CSF dynamics in ALS patients. We also describe a potential role for CSF circulation in determining disease spread as well as the importance of CSF dynamics in ALS neurotherapeutics. We propose that a CSF model could potentially offer additional avenues to explore currently unexplained features of ALS, ultimately leading to new treatment options for people with ALS.
Similar content being viewed by others

Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive fatal neurodegenerative disorder characterised by the selective death of motor neurons. Although the underlying cause of ALS is unknown, recent discoveries in the genetics and molecular pathology of ALS have provided important new insights. These include the finding that monogenetic causes of ALS—accounting for approximately 10% of cases—are phenotypically and pathologically largely indistinguishable from sporadic ALS. Furthermore, over 97% of ALS cases and half of frontotemporal dementia (FTD) are pathologically defined by cytoplasmic mis-accumulation of insoluble TDP-43, leading to these disorders being classified as TDP-43 proteinopathies [135].
Although much remains to be established about ALS pathophysiology, multiple mechanisms are implicated including proteostasis, glutamate excitotoxicity, dysregulation of RNA metabolism, nuclear-cytoplasmic transport and autophagy [16, 36, 73]. Notwithstanding these advances in our mechanistic understanding of ALS, a number of key questions remain unanswered. These include the primary cause of the disease and the significance of ageing as a risk factor, as well as a male predilection [8, 107].
The glymphatic system has recently been recognised as an important clearance pathway for the brain, playing a major role in the regulation of brain metabolites, including glucose and lipids [86]. Notably, its involvement in protein homeostasis has led to it receiving considerable attention in neurodegenerative diseases, particularly Alzheimer’s disease. ALS, despite also being a proteinopathy, has received comparatively less attention in the context of glymphatic function.
Against this background we describe the various potential roles of cerebrospinal fluid (CSF) in ALS pathophysiology, including as a possible driver for the disease process. We start by providing a brief overview of CSF circulation, followed by a primer on the glymphatic system, including the different factors affecting its function (Box 1), both of which have been comprehensively described by excellent reviews [21, 86]. We then assess the possibility of CSF contribution in ALS by considering various lines of evidence.
Overview of CSF circulation
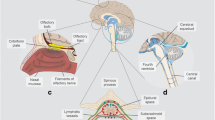
Surrounding most of the brain and spinal cord, CSF is believed to originate primarily from the choroid plexus, which is an extension of the ependymal lining of the brain ventricles. The majority of CSF is secreted into the two lateral ventricles, from where it converges into the third ventricle through the foramen of Monro, and subsequently flows into the fourth ventricle through the aqueduct of Sylvius. CSF then leaves the fourth ventricle and reaches the subarachnoid space via the foramen of Magendie and the two foramina of Luschka, with an indeterminate fraction of CSF also thought to flow into the central canal of the spinal cord.
CSF drainage however remains a topic of debate, with recent studies challenging the traditional view of reabsorption into dural venous sinuses by arachnoid granulations, which are outgrowths of the arachnoid mater [23, 163]. Alternative exit routes have been evidenced and include the olfactory route and meningeal lymphatics [12, 88, 109, 133, 151], although their relative contributions have not yet been established.
The total volume of CSF in an adult is widely estimated to be about 150 mL, with the larger majority of this volume distributed in the subarachnoid spaces and about 25 mL present in the brain ventricles. Renewal of CSF takes place three to four times in a single day. Whilst the primary role of CSF has long been considered to be the provision of buoyant support to the brain and protection against mechanical damage, another important function of CSF, which will be the focus of this review, is the clearance of metabolic waste products.
A primer on the glymphatic system
The earliest descriptions of perivascular spaces, or Virchow-Robin spaces, arose as early as the mid-1800s, when investigators, including Rudolf Virchow and Charles Robin, observed spaces surrounding blood vessels penetrating the brain parenchyma [202]. However, it was only in the next century, based on findings derived from dye injection experiments, that perivascular spaces first came to be functionally associated with fluid flow [202]. Today, perivascular spaces are widely regarded as an important site of exchange between CSF and interstitial fluid (ISF), underlying what is now known as the glymphatic system (Fig. 1).

Overview of the glymphatic system. A combination of various forces, including vascular pulsation and respiration, drives the influx of CSF from the subarachnoid space into the periarterial space, or Virchow-Robin space. CSF then moves into the interstitial space, with its entry being promoted by AQP4 channels lining the astrocyte endfeet. A convective flow drives ISF towards the perivenous spaces, carrying solutes, including metabolic waste products, along. As ISF moves from the extracellular space into the perivenous space, it can be drained from the CSF circulation through pathways such as the olfactory route. This highly organised system, enabling rapid CSF-ISF exchange, is known as the glymphatic system
Anatomically, these perivascular spaces are formed by pial arteries that perforate the brain parenchyma after traversing the subarachnoid space. As these pial arteries transition into penetrating arterioles, CSF from the subarachnoid space also extends into the brain parenchyma, bordering the blood vessels and creating CSF-filled spaces that are able to interact with the extracellular space [86, 217]. These perivascular spaces are themselves surrounded by a leptomeningeal layer, which, on one side, adheres to the blood vessel wall, and, on the other side, extends into the pia mater. Importantly, the outer wall of the perivascular space facing the brain parenchyma is lined by astrocyte endfeet expressing AQP4 channels [86]. It should, however, be noted at this point that the precise anatomy of perivascular spaces is still controversial, and much remains to be established about certain key aspects, including the exact fluid flow pathways and the interconnections between different compartments.
Whilst evidence for the role of perivascular spaces in solute transport from the brain interstitium had already emerged in the 1980s [162], the importance of the glymphatic system was only recognised relatively recently, following a landmark study by Iliff et al. who injected fluorescent tracers into mice and characterised the flow of CSF using two-photon microscopy [80]. CSF from the subarachnoid space is first forced into the perivascular space, driven by processes including vascular pulsation, respiration and sleep (Fig. 2). Following this, AQP4 channels lining the astrocytic endfeet are believed to promote the influx of CSF into the brain parenchyma by reducing resistance to inflow [80]. Although the role of AQP4 channels in CSF-ISF exchange has been challenged [181], recent findings from AQP4-knockout mice support the importance of AQP4 channels in glymphatic transport and amyloid-beta clearance [121]. Following CSF influx into the extracellular space, a convective flow transports interstitial solutes from the periarterial to the perivenous end. Once ISF reaches the perivenous space, it can subsequently be drained through one of the previously described outflow paths. This elaborate clearance system, also shown to be impacted by ageing (Box 1), has thus been termed the ‘glymphatic system’, given its similarity with the lymphatic system and its reliance on glial cells, in this case, astrocytes.
Despite past studies usually focussing on either the glymphatic system or the meningeal lymphatic system, increasing evidence supports the view that these two systems could be interdependent, with recent findings demonstrating a decrease in glymphatic influx and efflux of interstitial solutes following disruption of the meningeal lymphatic vasculature [47]. Similar to the glymphatic system, meningeal lymphatic function was found to be reduced in ageing, whilst also being linked to neurological disorders, such as Alzheimer’s disease and Parkinson’s disease [5, 47, 112, 220]. We refer readers to previous review articles for a more in-depth discussion of the meningeal lymphatic system [46, 108].
CSF toxicity in ALS
Although CSF from ALS patients is now known to be constitutionally abnormal [18, 27], with raised levels of proteins, including TDP-43 and neurofilaments [115, 210], its toxicity started being recognised early on, when it was shown to significantly reduce the survival of rat primary neuronal cultures [45]. Since then, numerous studies have been performed on various cell types, including NSC-34 cell lines, and, more recently, hESC-derived and iPSC-derived motor neurons, showing greater degeneration when the cells were exposed to CSF from ALS patients than to CSF from control patients [33, 136, 189, 197]. Interestingly, ALS-CSF was also shown in NSC-34 cells to result in TDP-43 mislocalisation to the cytoplasm, a feature that could be reversed by VEGF [174]. Nevertheless, the exact mechanism by which ALS-CSF induces neuronal degeneration remains to be established, although processes such as excitotoxicity and mitochondrial dysfunction have been suggested [175, 179].
The in vitro effect of ALS-CSF also extends to both astrocytes and microglia, demonstrating a non-cell autonomous component to CSF toxicity. Upon exposure to ALS-CSF, astrocytes undergo a change in morphology, accompanied by increased GFAP reactivity, further acquiring a neuroinflammatory profile [126]. Release of inflammatory markers is also common to microglia exposed to ALS-CSF [127]. Furthermore, rat primary motor neurons co-cultured with glia responded differently to ALS-CSF than when cultured alone [17], adding to the growing body of literature evidencing the role of glia in mediating neurotoxicity [214].
Supporting these in vitro findings, several studies have also reported various changes caused by ALS-CSF in animal models. Neurofilament phosphorylation was observed in rat motor neurons following injection of ALS-CSF into the spinal subarachnoid space [160]. Moreover, intrathecal injection of ALS-CSF led to changes in the Golgi complex [158], potentially affecting protein trafficking and causing endoplasmic reticular stress [196]. Histologically, close similarities were observed between tissue exposed to ALS-CSF and sporadic ALS cases [68]. ALS-CSF was also found to produce phenotypic changes, with rats subjected to intraventricular injections experiencing motor dysfunction [165]. Muscular atrophy was observed in a different study, possibly arising through motor neuron degeneration [173].
Accepting inherent limitations in patient studies, including sample heterogeneity, and that CSF concentration may not reflect cellular levels, these in vitro and in vivo studies nonetheless show that ALS-CSF is toxic to neurons and possesses pro-inflammatory properties [136]. The in vivo finding of toxicity distant to the site of CSF injection also raises a potential role for CSF in disease spread [68], a possibility supported by recent findings demonstrating the onset of motor and cognitive decline, as well as TDP-43 proteinopathy, following ALS-CSF infusion in mice [125].
Altered CSF dynamics in ALS patients
Evidence for the disruption of CSF flow in ALS includes findings from a phase-contrast electrocardiography-triggered MRI study, which revealed different CSF dynamics in ALS patients, with a delay in CSF flow upon systole and a higher maximum velocity [169]. Abnormal CSF dynamics in ALS patients is also supported by a more recent study demonstrating reduced CSF flow magnitude along with a greater pulse wave velocity, although the causes, as well as implications, of these findings remain to be established [168].

Factors affecting CSF dynamics in ALS patients. Several components of the glymphatic pathway are potentially disrupted in ALS patients. Ageing, for instance, is linked to reduced vascular pulsation, due to an increase in vessel wall stiffness. ALS patients also tend to suffer from poor sleep and impaired respiratory function, particularly towards later stages of the disease. Impairment of two major drivers of glymphatic influx, coupled to disturbed sleep, could underlie highly reduced glymphatic clearance in ALS patients. Other features of ALS that could impact on glymphatic function include abnormal AQP4 expression, raised norepinephrine levels, and vascular factors, such as hypertension and hypoperfusion. Whilst neuroimaging studies have demonstrated abnormal CSF dynamics in ALS patients, further studies may be required to specifically determine the influence of these different factors on glymphatic clearance in ALS patients

Possible roles for cerebrospinal fluid in ALS. Abnormal CSF dynamics could have important implications in ALS, with both glymphatic clearance and glymphatic influx possibly involved. a–d Reduced glymphatic clearance could result in the accumulation of various neurotoxic factors, notably that of major pathogenic proteins, including TDP-43 and SOD1. A rise in the levels of CSF components such as glutamate, inflammatory factors and other toxic metabolites could also favour an increasingly toxic interstitial environment (ISF: interstitial fluid). e Given the importance of glymphatic function in lipid transport, impaired clearance or influx may possibly affect the regulation of lipid metabolism. f Impaired glymphatic influx could also influence pharmacokinetics, particularly in the context of intrathecally administered drugs, and may therefore deserve investigation in ALS neurotherapeutics. g Lastly, various lines of evidence suggest that the CSF circulation could act as an important medium for the spread of the disease, with a possible link to proteostasis. Note Shape sizes are weighted by their relative significance
Additionally, with human neuroimaging studies revealing changes in CSF dynamics with age [166, 170] and recent evidence also suggesting reduced glymphatic clearance in older individuals [216], a major proportion of ALS patients, given the late onset of ALS, likely experience altered CSF flow and impaired glymphatic function. Sleep, which plays an important role in glymphatic clearance, is also known to be disrupted in ALS patients, who often have apnoea, or sleep poorly, owing to muscle cramps and fasciculations [28]. Although sleep has yet to be acknowledged as a risk factor for ALS, it remains strongly associated with disease severity [4, 28, 157]. Poor sleep, coupled to the severely affected respiratory function in ALS patients, could therefore significantly impair CSF dynamics and therefore glymphatic clearance.
The finding of increased norepinephrine concentration in CSF from ALS patients is also intriguing [22, 35]. Norepinephrine reduces the extracellular space volume and thus increases resistance to glymphatic influx into the brain parenchyma [208]. Of further interest is that whilst sleep quality and respiratory function are known to be negatively correlated with ALS severity [4], a higher norepinephrine level has also been linked to more severe symptoms in FTD [58].
Abnormal vascular changes in ALS
Vascular function, which is intimately connected to the glymphatic system, is known to be affected in ALS. Multiple studies employing single-photon emission computed tomography (SPECT) have demonstrated hypoperfusion in the brain of both ALS and FTD patients, with greater involvement of the frontal and temporal lobes [1, 82, 199]. Abnormal vascular changes in ALS and FTD have further been evidenced through arterial-spin coupling [53, 177]. In the SOD1 mouse model, blood-spinal cord barrier (BSCB) breakdown may precede hypoperfusion [215], a feature which has previously been linked to disease severity in ALS patients [2, 40].
Genetic association studies linking ALS risk with angiogenic genes, ANG and VEGF, provide further indirect evidence of the role of vascular factors in ALS [61, 66, 70, 147, 195]. VEGF has been heavily implicated in ALS pathophysiology, with several studies demonstrating the protective effect of VEGF therapy on neuronal death, both in vitro and in vivo [15, 98, 187, 198]. Lack of the VEGF hypoxia-response element in mice also results in motor neuron degeneration [145]. VEGF-associated neurodegeneration has been suggested to arise from ischaemia following reduced vascular perfusion, although this has yet to be established.
The link between hypertension and ALS is currently less clear. Multiple associations have been made between hypertension and ALS onset and survival [116, 129], but contradicting results have also been obtained by other studies [76, 94, 128]. This could possibly be explained by variations in methodology, and further studies are therefore required to confirm this association. Although one study found the use of angiotensin-converting enzyme inhibitors to be linked to a reduced risk of ALS [105], this was not replicated by a later study [63]. Nevertheless, the different vascular aspects of ALS could constitute a further impediment to proper CSF dynamics in ALS patients.
AQP4 channels and ALS
Overexpression of AQP4 in ALS is well documented in rodent models [19, 48, 137]. In addition to increased expression, reduced AQP4 polarisation with decreased endfeet localisation has also been observed in the SOD1 mouse model [48]. The exact implications of these changes in neuronal degeneration are currently unclear. However, given the various roles played by AQP4 channels, several downstream consequences have been suggested, including loss of blood-brain barrier (BBB) integrity, glutamate dysregulation and impaired potassium homeostasis [219]. The disrupted AQP4 polarisation could also contribute to impaired glymphatic function, commensurate with the features observed in aged mice [95].
Another intriguing finding is that reduced AQP4 expression in SOD1 mice triggered earlier disease onset and reduced survival [204]. One explanation suggested by the authors was that the absence of AQP4 channels led to reduced SOD1 clearance, favouring an increasingly neurotoxic extracellular environment. Notwithstanding this, further studies are required to confirm whether such changes in AQP4 expression and their possible implications in glymphatic clearance are features specific to SOD1 ALS.
The implications of abnormal CSF dynamics in ALS
Taken together, there are multiple and converging lines of evidence that suggest that the glymphatic system plays a role in ALS pathophysiology. We therefore outline several key processes in ALS that might be impacted by a dysregulated glymphatic system (Fig. 3).
Proteostasis
Accumulation of various proteins that may directly or indirectly be toxic in ALS is well-established (Box 2). These include mis-accumulated phosphorylated TDP-43, SOD1 aggregates and dipeptide repeats (DPRs) found in the most common inherited form of ALS, due to a repeat expansion mutation in C9ORF72. TDP-43 proteinopathy is known to be the most prevalent and has been implicated in many pathways, including mitochondrial dysfunction, autophagy dysregulation and impaired endocytosis [153].
The reasons for accumulation could reflect excess production and/or impaired clearance through autophagy and the ubiquitin-proteasome system (UPS). These are both involved in TDP-43 and SOD1 clearance [89, 101, 212], with another mechanism promoting TDP-43 clearance being the more recently discovered endolysosomal pathway [101, 106]. Thus, indirect evidence from rare inherited forms of ALS due to mutations in genes linked to autophagy or the UPS, such as VCP, OPTN and UBQLN2, argues for an important role for dysregulated clearance in ALS. Clear evidence supporting the overproduction of TDP-43 in ALS, however, remains to be established.
Given the established role of the glymphatic system in protein clearance, impairment of glymphatic function could also contribute to the accumulation of the different toxic proteins in ALS. The presence of TDP-43, SOD1 and DPRs in ALS-CSF is well-evidenced and points to a potential role for CSF in their regulation. It is therefore possible that ageing, coupled to the different possible sources of glymphatic disturbance already discussed, could result in an imbalance between production and clearance of these pathogenic proteins. Notably, ageing has further been argued to promote the deterioration of processes such as autophagy and the UPS [138], which could further exacerbate protein clearance in the diseased state. The numerous clearance mechanisms involved in amyloid-beta clearance in Alzheimer’s disease [190] also suggest that other major clearance pathways in ALS beyond intracellular mechanisms are yet to be defined, of which the glymphatic system would be one notable example.
Notwithstanding this, protein inclusions associated with ALS, including those enriched in TDP-43, SOD1 and DPRs, are predominantly intracellular, as opposed to amyloid-beta aggregates, found in Alzheimer’s disease, which also reside extracellularly. Thus, glymphatic clearance in the context of ALS could potentially follow from processes such as exosome secretion [34, 78] or necroptosis, in which intracellular contents are released from the dying cell into the interstitial space [83]. A possible interplay between intracellular mechanisms and extracellular processes could also be involved, with UPS clearance, for instance, being influenced by an altered extracellular environment, owing to impaired glymphatic function.
Whilst considerable progress has been made towards understanding ALS pathophysiology, its primary cause has remained a matter of debate, with genetic mutations accounting for only a minority of ALS cases. Hence, the significance of ageing in the impairment of clearance mechanisms could provide a promising avenue to explore in ALS.
Glutamate excitotoxicity
Glutamate excitotoxicity has long been viewed as an important pathophysiological mechanism in ALS and FTD. Riluzole, which inhibits glutamate release, is currently the only globally approved drug for ALS [84]. One reason for the particular susceptibility of motor neurons to AMPA-mediated glutamate excitotoxicity is thought to be their low buffering ability upon calcium influx [92]. This influx is promoted by permeability of the AMPA receptor, which can be affected by changes to its subunit composition. For instance, reduced expression of the impermeable GluR2 subunit results in an increased influx of calcium ions, potentially contributing to neurodegeneration in ALS [91, 194].
Glutamate excitotoxicity could also be caused by direct overstimulation of glutamate receptors, due to an increased level of extracellular glutamate [92]. Although increased synaptic release has been suggested to contribute to raised glutamate levels, strong evidence supports the involvement of the glutamate transporter EAAT2, with studies demonstrating reduced EAAT2 expression in different ALS models, including in SOD1 mice [64, 77, 164, 167, 203]. Intriguingly, a minor increase in glutamate levels has also been found to trigger neurodegeneration [29, 71, 103], even when raised to levels comparable to that of ALS-CSF [42].
Whilst the cause of increased glutamate levels could result from several processes, the role of CSF in glutamate regulation [6] could indicate a possible contribution in promoting potentially neurotoxic levels of glutamate in ALS [62, 185]. This could again be driven by the different aspects of ALS that affect CSF dynamics, including ageing, sleep disruption and vascular factors, amongst others. It should, however, be noted that CSF glutamate levels may not necessarily reflect extracellular levels, although increased CSF glutamate concentrations could likely affect exposed astrocytes.
Energy metabolism
Dysregulated energy metabolism is now a widely known feature of ALS, with patients generally suffering from weight loss, hypermetabolism and hyperlipidaemia [55]. Although the energy imbalance in ALS could stem from several processes, two important contributing factors are thought to be the higher basal energy expenditure in ALS patients [50, 65], and reduced nutrition due to dysphagia [96]. The causes of hyperlipidaemia, which could further affect energy metabolism, also remain controversial [55]. Along with increased apoE levels [97] and type 2 diabetes [85, 118], hyperlipidaemia has in fact been shown to be protective in ALS [51, 54]. Nevertheless, the link between energy metabolism and ALS pathogenesis remains unclear despite animal studies providing a number of suggestions [55] with therapeutic promise [119].
Hence, given the recently characterised role of the glymphatic system in lipid transport [159], an impairment in CSF circulation could potentially be a further underlying cause of the abnormal energy metabolism observed in ALS. BBB impermeability largely restricts influx of lipids and cholesterol, which are believed to be produced within the CNS by the choroid plexus and astrocytes [3, 31, 59, 209], with excess cholesterol being released into the blood circulation [24, 111]. Following synthesis, apoE is observed to distribute through the perivascular space in order to reach the brain [159]. We speculate that impaired lipid regulation by the glymphatic system, coupled to increased BBB permeability in ALS, may well disrupt energy metabolism. This is supported by numerous studies demonstrating an altered CSF metabolome in ALS patients [25, 26, 69, 207]. Again however, whether these changes could contribute to ALS pathogenesis is difficult to answer.
Neuroinflammation
To date, several studies have helped to demonstrate the inflammatory profile of ALS-CSF, revealing raised levels of various immune components, including C3c, albumin and IgG in ALS-CSF [10, 11, 44, 67, 102]. Out of these, IgG, also present in post-mortem ALS samples, has been found to favour neurodegeneration, both in vitro and in vivo [49, 155, 156]. IgG further possesses pro-inflammatory properties, promoting microglia recruitment and upregulation of inflammatory cytokines [124, 141, 142]. Some of the other chemokines found to be elevated in ALS-CSF include IL-6, TNF-α and TGF-β [81, 130, 172], with the finding of raised TNF-α levels being particularly interesting, given its associations with glutamate excitotoxicity [43, 178, 218]. Whilst their direct involvement in ALS pathophysiology is still unknown, the presence of these different immune factors in ALS-CSF, some of which have been shown to be neurotoxic, could suggest an important role for CSF in neuroinflammation, with the neurotoxic environment possibly favoured by altered CSF dynamics and reduced clearance. This would in fact provide a potential explanation for the well-evidenced toxicity of ALS-CSF, which has, in turn, been suggested to possibly contribute to ALS pathophysiology [176]. Further studies are nevertheless required to confirm this association.
Oxidative stress
Given their high metabolic rate as well as their inability to divide, motor neurons could be particularly vulnerable to accumulation of toxic metabolites, including lactate, 4-hydroxynonenal (HNE) and peroxynitrite [176]. High lactic acid levels have been shown to induce neuronal degeneration and promote inflammation [72, 134], whilst HNE, a marker of lipid peroxidation, results in oxidative stress in cultured motor neuron hybrid cells at levels similar to those of ALS-CSF [183]. Peroxynitrite, although controversial, has also been suggested to promote oxidative stress [191]. It is therefore possible that CSF, which is known to regulate the level of metabolites, including that of lactate [75, 110], could potentially help to prevent the accumulation of waste products that would otherwise result in a highly neurotoxic extracellular environment.
CSF circulation and spread
The variable sites of disease onset and subsequent disease evolution have raised the question as to what determines ALS disease spread. This is unknown and debate has hitherto focussed on questions of anterograde or retrograde spread [41, 57]. Some general patterns can, however, be noted. Specifically, the disease is localised focally during early stages and spreads outwards in a contiguous pattern. The spread is more often directed caudally than rostrally. Additionally, the pattern of spread is heterogeneous in nature, making it difficult to predict [161].
More recently, a growing body of literature suggests that ALS spread could be explained by a prion-like mechanism. Notably, the mutant SOD1 protein, which has been demonstrated to possess high fibril-forming propensity under certain conditions, can promote fibrillation in wild-type SOD1 [39]. TDP-43 is also able to form aggregates in vitro, although the factors that predispose TDP-43 to aggregation are still incompletely understood [153]. Evidence further indicates that these properties could be shared by FUS [140] as well as DPRs [205]. These observations suggest a close similarity with other neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, which have also been characterised as proteinopathies.
Although the prion-like properties of the different ALS proteins are starting to gain acceptance within the scientific community, less is known about how intercellular spread could take place in vivo. Trans-synaptic transmission has been suggested as one possible mechanism [13, 32]. Another possibility is through exosome propagation. It has in fact been shown that SOD1-containing exosomes could be secreted into the extracellular space [192], with possible uptake by macropinocytosis [132]. There is evidence that TDP-43 could also spread via exosomes [60], although further studies are required to establish this. Another recently proposed model is that of corticofugal axonal spread, for which strong support arises from the particular vulnerability of neurons receiving connections from pyramidal cells of the neocortex [32].
A further pathway already described by others is that spread of the disease could occur via the CSF circulation [182]. The presence of misfolded proteins, including TDP-43 and SOD1, has already been demonstrated in ALS-CSF, and, whilst neurons could potentially directly ingest these proteins through phagocytic mechanisms [30], another plausible mechanism previously suggested would be via uptake of exosomes containing the misfolded proteins [90]. Indeed, CSF exosomes from Alzheimer’s disease and Parkinson’s disease patients have been shown to contain alpha-synuclein and misfolded tau respectively [188, 201]. Protein-containing exosomes are also known to be present in ALS-CSF [74] but whether they specifically contain TDP-43 or SOD1 remains to be established.
Hence, whilst we do not propose that spread of the disease occurs solely through CSF pathways, such a mechanism of spread could clearly complement other proposed models. It would also help to explain the non-contiguous pattern of spread and the multifocal pattern of initiation observed in some ALS cases [171], and even provide a potential link between ALS and FTD, by providing a direct route of spread from the brain to the spinal cord. Other arguments have also been made in support of spread via CSF; for instance, the proximity of vulnerable neurons to CSF and also the susceptibility of particular cranial nerves to the disease [182].
Towards a glymphatic system model in ALS
One major caveat in considering glymphatic involvement in ALS is that its function has so far only been demonstrated in the brain, with its contribution in the spinal cord still unclear. Indeed, spinal cord and brain anatomy differ considerably both in terms of parenchymal and leptomeningeal organisation, suggesting possible mechanistic differences in waste clearance. Whether clearance from the brain and clearance from the spinal cord are influenced by similar factors and could therefore be modulated via similar mechanisms are also not known. These distinctions may therefore need to be further explored, given their possible importance with regard to spinal cord involvement in ALS. Another point to consider is that most studies on the glymphatic system have been carried out in rodents, with definitive evidence for its existence in humans yet to be established. However, results have indicated a high possibility of this being the case, with some studies reporting a number of inter-species similarities, not only in terms of the associated anatomical structures, but also the factors affecting clearance, such as sleep [21, 56, 120, 163, 216].
Hence, the glymphatic system could play an important role in reconciling disease mechanism with epidemiology and other currently unexplained aspects of ALS. It would firstly support the existence of a threshold effect suggested by the significance of ageing as a risk factor. Secondly, it could help to explain the intriguing gender disparity in ALS, with the established protective influence of oestrogen in preserving vascular, and therefore glymphatic, function possibly accounting for the lower female incidence. This could further underlie the postmenopausal increase in female:male ratio observed by some studies [38, 117]. Adopting a glymphatic system model could not only suggest a primary, or at least contributory, cause for the disease, but also offer potential explanations for the clinical features of ALS, including its progression and heterogeneity.
Whether the intriguing difficulty in generating C9ORF72 mouse models that recapitulate the disease phenotype bears a connection to glymphatic function also deserves mention. Both CSF production and turnover rate are much greater in mice than in humans [87], and when coupled to the significantly higher basal heart rate in mice, have been suggested to result in improved waste clearance and therefore a reduction in the accumulation of toxic products [21]. This could possibly explain why C9ORF72 mouse models have failed to exhibit motor dysfunction. Interestingly, however, cognitive defects have been observed both in mutant TDP-43 and C9ORF72 mouse models [9, 20], although whether this can be attributed to the comparatively higher metabolic rate inherent to the mouse brain is unclear [21].
Understandably, many areas need to be addressed before acknowledging glymphatic dysfunction as a contributor to ALS pathophysiology. Despite the involvement of certain angiogenic genes, evidence for a clear genetic link between ALS and genes implicated in glymphatic function is still missing. Furthermore, definitive evidence for the significance of sleep [184] and hypertension as risk factors for ALS is also yet to be established. It is also unclear, according to this model, why ALS incidence would decrease above the age of about 70 [8, 107].
Glymphatic system and ALS neurotherapeutics
Whilst the glymphatic system has more often than not been associated with waste clearance from the brain, regulating glymphatic influx could also significantly impact ALS neurotherapeutics. With BBB impermeability acting as a major hurdle to drug delivery in ALS and other neurological disorders, intrathecal therapy has emerged as a promising treatment modality in recent years. Indeed, administering the drug directly into the cerebrospinal circulation has multiple advantages, such as allowing for more accurate monitoring of drug levels, thereby ensuring better pharmacokinetic results [193]. The obvious downside to intrathecal administration, however, is the risk of complications owing to its invasive nature.
Several intrathecal therapies are currently being trialled in ALS, including anti-sense oligonucleotides (ASOs), mesenchymal stem cells (MSCs) and growth factors, such as brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF) and vascular endothelial growth factor (VEGF) [193]. Promisingly, these have shown positive safety profiles so far, although evidence for their efficacy remains to be established [123, 143, 144, 186]. Based on this, further studies may be required to investigate the influence of CSF dynamics on the pharmacokinetics of intrathecal drugs. Key considerations include whether the intrathecally administered drug is appropriately taken up by the spinal cord, and, perhaps more importantly, whether it can penetrate the brain parenchyma. Pre-clinical studies in mice have indicated widespread transduction following intrathecal delivery [14, 146, 200]. This has, however, been more difficult to confirm in humans, with pharmacokinetic analysis being largely restricted to monitoring CSF drug levels [193].
To further emphasise the significance of achieving more efficient drug delivery, different variables, such as diffusion rate and anatomical constraints, have been suggested to limit drug access to deeper parts of the brain and the spinal cord [206]. Additionally, many of the factors impacting on CSF dynamics in ALS patients, including ageing and hypertension, could potentially affect drug distribution following intrathecal injection. However, studies performed in mice have shown that CSF influx into the brain parenchyma could be improved by administering hypertonic saline or mannitol, which both increase plasma osmolality, or ketamine-xylazine, a modulator of slow-wave activity [150, 208]. A more recent study further demonstrated that dexmedetomidine, an α2-adrenergic agonist, which also affects slow-wave oscillations, could enhance brain distribution following intrathecal delivery [104].
Conclusion
The glymphatic system has promise in potentially unifying many different aspects of ALS, and may therefore be viewed as a novel paradigm when considering ALS pathophysiology. We suggest that further avenues be explored to specifically examine features of disease onset and spread. Several lines of enquiry from pre-clinical to epidemiological studies are thus required to shed light on the extent of CSF contribution in ALS, with the hope of ultimately identifying new treatment options for people with ALS.
Availability of data and materials
Not applicable.
Abbreviations
- ADAR:
-
Adenosine deaminases acting on RNA
- ALS:
-
Amyotrophic lateral sclerosis
- ALS-CSF:
-
CSF from ALS patients
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ANG:
-
Angiogenin
- apoE:
-
Apolipoprotein E
- AQP4:
-
Aquaporin 4
- ASO:
-
Anti-sense oligonucleotide
- BBB:
-
Blood-brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- BSCB:
-
Blood-spinal cord barrier
- C9ORF72:
-
Chromosome 9 open reading frame 72
- CNS:
-
Central nervous system
- CNTF:
-
Ciliary neurotrophic factor
- CSF:
-
Cerebrospinal fluid
- DPR:
-
Dipeptide repeat
- EAAT2:
-
Excitatory amino acid transporter 2
- FTD:
-
Frontotemporal dementia
- FUS:
-
Fused-in-sarcoma
- hESC:
-
Human embryonic stem cell
- HNE:
-
4‐hydroxynonenal
- IgG:
-
Immunoglobin G
- IL-6:
-
Interleukin-6
- iPSC:
-
Induced pluripotent stem cell
- ISF:
-
Interstitial fluid
- MSC:
-
Mesenchymal stem cell
- MREG:
-
Magnetic resonance encephalography
- NSC-34:
-
Mouse spinal cord-neuroblastoma hybrid cell line
- OPTN:
-
Optineurin
- SOD1:
-
Superoxide dismutase 1
- SPECT:
-
Single-photon emission computed tomography
- TDP-43:
-
TAR DNA binding protein 43
- TGF-β:
-
Transforming growth factor beta
- TNF-α:
-
Tumour necrosis factor alpha
- UBQLN2:
-
Ubiquilin-2
- UPS:
-
Ubiquitin-proteasome system
- VCP:
-
Valosin containing protein
- VEGF:
-
Vascular endothelial growth factor
References
Abe K, Fujimura H, Toyooka K, Hazama T, Hirono N, Yorifuji S, Yanagihara T (1993) Single-photon emission computed tomographic investigation of patients with motor-neuron disease. Neurology 43:1569–1573. https://doi.org/10.1212/Wnl.43.8.1569
Abe K, Fujimura H, Toyooka K, Sakoda S, Yorifuji S, Yanagihara T (1997) Cognitive function in amyotrophic lateral sclerosis. J Neurol Sci 148:95–100. https://doi.org/10.1016/s0022-510x(96)05338-5
Achariyar TM, Li BM, Peng WG, Verghese PB, Shi Y, McConnell E, Benraiss A, Kasper T, Song W, Takano T et al (2016) Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol Neurodegener 11:74. https://doi.org/10.1186/s13024-016-0138-8
Ahmed RM, Newcombe RE, Piper AJ, Lewis SJ, Yee BJ, Kiernan MC, Grunstein RR (2016) Sleep disorders and respiratory function in amyotrophic lateral sclerosis. Sleep Med Rev 26:33–42. https://doi.org/10.1016/j.smrv.2015.05.007
Ahn JH, Cho H, Kim JH, Kim SH, Ham JS, Park I, Suh SH, Hong SP, Song JH, Hong YK et al (2019) Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 572:62–66. https://doi.org/10.1038/s41586-019-1419-5
Akanuma S, Sakurai T, Tachikawa M, Kubo Y, Hosoya K (2015) Transporter-mediated L-glutamate elimination from cerebrospinal fluid: possible involvement of excitatory amino acid transporters expressed in ependymal cells and choroid plexus epithelial cells. Fluids Barriers CNS 12:11. https://doi.org/10.1186/s12987-015-0006-x
Albeck MJ, Skak C, Nielsen PR, Olsen KS, Borgesen SE, Gjerris F (1998) Age dependency of resistance to cerebrospinal fluid outflow. J Neurosurg 89:275–278. https://doi.org/10.3171/jns.1998.89.2.0275
Alonso A, Logroscino G, Jick SS, Hernan MA (2009) Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur J Neurol 16:745–751
Alrafiah AR (2018) From mouse models to human disease: an approach for amyotrophic lateral sclerosis. In Vivo 32:983–998. https://doi.org/10.21873/invivo.11339
Annunziata P, Volpi N (1985) High levels of C3c in the cerebrospinal fluid from amyotrophic lateral sclerosis patients. Acta Neurol Scand 72:61–64. https://doi.org/10.1111/j.1600-0404.1985.tb01548.x
Apostolski S, Nikolic J, Bugarski-Prokopljevic C, Miletic V, Pavlovic S, Filipovic S (1991) Serum and CSF immunological findings in ALS. Acta Neurol Scand 83:96–98. https://doi.org/10.1111/j.1600-0404.1991.tb04656.x
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H, Alitalo K (2015) A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 212:991–999. https://doi.org/10.1084/jem.20142290
Ayers JI, Cashman NR (2018) Prion-like mechanisms in amyotrophic lateral sclerosis. Handb Clin Neurol 153:337–354. https://doi.org/10.1016/B978-0-444-63945-5.00018-0
Ayers JI, Fromholt S, Sinyavskaya O, Siemienski Z, Rosario AM, Li A, Crosby KW, Cruz PE, DiNunno NM, Janus C et al (2015) Widespread and efficient transduction of spinal cord and brain following neonatal AAV injection and potential disease modifying effect in ALS mice. Mol Therapy 23:53–62. https://doi.org/10.1038/mt.2014.180
Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND (2004) VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 429:413–417. https://doi.org/10.1038/nature02544
Balendra R, Isaacs AM (2018) C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol 14:544–558. https://doi.org/10.1038/s41582-018-0047-2
Barber SC, Wood-Allum CA, Sargsyan SA, Walsh T, Cox LE, Monk PN, Shaw PJ (2011) Contrasting effects of cerebrospinal fluid from motor neuron disease patients on the survival of primary motor neurons cultured with or without glia. Amyotroph Lateral Scler 12:257–263. https://doi.org/10.3109/17482968.2011.560672
Barschke P, Oeckl P, Steinacker P, Ludolph A, Otto M (2017) Proteomic studies in the discovery of cerebrospinal fluid biomarkers for amyotrophic lateral sclerosis. Exp Rev Proteom 14:769–777. https://doi.org/10.1080/14789450.2017.1365602
Bataveljic D, Nikolic L, Milosevic M, Todorovic N, Andjus PR (2012) Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1G93A rat model. Glia 60:1991–2003. https://doi.org/10.1002/glia.22414
Batra R, Lee CW (2017) Mouse models of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis/frontotemporal dementia. Front Cell Neurosci 11:196. https://doi.org/10.3389/fncel.2017.00196
Benveniste H, Liu X, Koundal S, Sanggaard S, Lee H, Wardlaw J (2019) The glymphatic system and waste clearance with brain aging: a review. Gerontology 65:106–119. https://doi.org/10.1159/000490349
Bertel O, Malessa S, Sluga E, Hornykiewicz O (1991) Amyotrophic-lateral-sclerosis - changes of noradrenergic and serotonergic transmitter systems in the spinal-cord. Brain Res 566:54–60. https://doi.org/10.1016/0006-8993(91)91680-Y
Biceroglu H, Albayram S, Ogullar S, Hasiloglu ZI, Selcuk H, Yuksel O, Karaaslan B, Yildiz C, Kiris A (2012) Direct venous spinal reabsorption of cerebrospinal fluid: a new concept with serial magnetic resonance cisternography in rabbits. J Neurosurg Spine 16:394–401. https://doi.org/10.3171/2011.12.SPINE11108
Bjorkhem I, Lutjohann D, Diczfalusy U, Stahle L, Ahlborg G, Wahren J (1998) Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res 39:1594–1600
Blasco H, Corcia P, Moreau C, Veau S, Fournier C, Vourc’h P, Emond P, Gordon P, Pradat PF, Praline J et al (2010) H-1-NMR-based metabolomic profiling of CSF in early amyotrophic lateral sclerosis. PLoS ONE 5:e13223. https://doi.org/10.1371/journal.pone.0013223
Blasco H, Corcia P, Pradat PF, Bocca C, Gordon PH, Veyat-Durebex C, Mavel S, Nadal-Desbarats L, Moreau C, Devos D et al (2013) Metabolomics in cerebrospinal fluid of patients with amyotrophic lateral sclerosis: an untargeted approach via high-resolution mass spectrometry. J Proteome Res 12:3746–3754. https://doi.org/10.1021/pr400376e
Blasco H, Veyrat-Durebex C, Bocca C, Patin F, Vourc’h P, Kouassi Nzoughet J, Lenaers G, Andres CR, Simard G, Corcia P et al (2017) Lipidomics reveals cerebrospinal-fluid signatures of ALS. Sci Rep 7:17652. https://doi.org/10.1038/s41598-017-17389-9
Boentert M (2019) Sleep disturbances in patients with amyotrophic lateral sclerosis: current perspectives. Nat Sci Sleep 11:97–111. https://doi.org/10.2147/Nss.S183504
Bonini D, Filippini A, La Via L, Fiorentini C, Fumagalli F, Colombi M, Barbon A (2015) Chronic glutamate treatment selectively modulates AMPA RNA editing and ADAR expression and activity in primary cortical neurons. RNA Biol 12:43–53. https://doi.org/10.1080/15476286.2015.1008365
Bowen S, Ateh DD, Deinhardt K, Bird MM, Price KM, Baker CS, Robson JC, Swash M, Shamsuddin W, Kawar S et al (2007) The phagocytic capacity of neurones. Eur J Neurosci 25:2947–2955. https://doi.org/10.1111/j.1460-9568.2007.05554.x
Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM (1985) Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest 76:1501–1513. https://doi.org/10.1172/JCI112130
Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K (2013) Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat Rev Neurol 9:708–714. https://doi.org/10.1038/nrneurol.2013.221
Brauer S, Gunther R, Sterneckert J, Glass H, Hermann A (2020) Human spinal motor neurons are particularly vulnerable to cerebrospinal fluid of amyotrophic lateral sclerosis patients. Int J Mol Sci 21(10):3564. https://doi.org/10.3390/ijms21103564
Brauer S, Zimyanin V, Hermann A (2018) Prion-like properties of disease-relevant proteins in amyotrophic lateral sclerosis. J Neural Transm (Vienna) 125:591–613. https://doi.org/10.1007/s00702-018-1851-y
Brooks BR, Zielger MG, Lake CR, Wood JH, Enna SJ, Engel WK (1980) Cerebrospinal-fluid norepinephrine and free gamma-aminobutyric acid in amyotrophic lateral sclerosis. Brain Res Bull 5:765–768. https://doi.org/10.1016/0361-9230(80)90126-4
Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377:162–172. https://doi.org/10.1056/NEJMra1603471
Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, Heverin M, Jordan N, Kenna K, Lynch C et al (2012) Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol 11:232–240. https://doi.org/10.1016/S1474-4422(12)70014-5
Chancellor AM, Hendry A, Caird FI, Warlow CP, Weir AI (1993) Motor neuron disease: a disease of old age. Scott Med J 38:178–182. https://doi.org/10.1177/003693309303800606
Chattopadhyay M, Durazo A, Sohn SH, Strong CD, Gralla EB, Whitelegge JP, Valentine JS (2008) Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc Natl Acad Sci USA 105:18663–18668. https://doi.org/10.1073/pnas.0807058105
Chio A, Pagani M, Agosta F, Calvo A, Cistaro A, Filippi M (2014) Neuroimaging in amyotrophic lateral sclerosis: insights into structural and functional changes. Lancet Neurol 13:1228–1240. https://doi.org/10.1016/S1474-4422(14)70167-X
Chou SM, Norris FH (1993) Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve 16:864–869. https://doi.org/10.1002/mus.880160810
Cid C, Alvarez-Cermeno JC, Regidor I, Salinas M, Alcazar A (2003) Low concentrations of glutamate induce apoptosis in cultured neurons: implications for amyotrophic lateral sclerosis. J Neurol Sci 206:91–95. https://doi.org/10.1016/s0022-510x(02)00339-8
Clark IA, Vissel B (2016) Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents. J Neuroinflammation 13:236. https://doi.org/10.1186/s12974-016-0708-2
Costa J, Streich L, Pinto S, Pronto-Laborinho A, Nimtz M, Conradt HS, de Carvalho M (2019) Exploring cerebrospinal fluid IgG N-glycosylation as potential biomarker for amyotrophic lateral sclerosis. Mol Neurobiol 56:5729–5739. https://doi.org/10.1007/s12035-019-1482-9
Couratier P, Hugon J, Sindou P, Vallat JM, Dumas M (1993) Cell-culture evidence for neuronal degeneration in amyotrophic-lateral-sclerosis being linked to glutamate ampa kainate receptors. Lancet 341:265–268. https://doi.org/10.1016/0140-6736(93)92615-Z
Da Mesquita S, Fu Z, Kipnis J (2018) The meningeal lymphatic system: a new player in neurophysiology. Neuron 100:375–388. https://doi.org/10.1016/j.neuron.2018.09.022
Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, Contarino C, Onengut-Gumuscu S, Farber E, Raper D et al (2018) Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 560:185–191. https://doi.org/10.1038/s41586-018-0368-8
Dai J, Lin W, Zheng M, Liu Q, He B, Luo C, Lu X, Pei Z, Su H, Yao X (2017) Alterations in AQP4 expression and polarization in the course of motor neuron degeneration in SOD1G93A mice. Mol Med Rep 16:1739–1746. https://doi.org/10.3892/mmr.2017.6786
Demestre M, Pullen A, Orrell RW, Orth M (2005) ALS-IgG-induced selective motor neurone apoptosis in rat mixed primary spinal cord cultures. J Neurochem 94:268–275. https://doi.org/10.1111/j.1471-4159.2005.03184.x
Desport JC, Torny F, Lacoste M, Preux PM, Couratier P (2005) Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener Dis 2:202–207. https://doi.org/10.1159/000089626
Dorst J, Kuhnlein P, Hendrich C, Kassubek J, Sperfeld AD, Ludolph AC (2011) Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J Neurol 258:613–617. https://doi.org/10.1007/s00415-010-5805-z
Dreha-Kulaczewski S, Joseph AA, Merboldt KD, Ludwig HC, Gartner J, Frahm J (2015) Inspiration is the major regulator of human CSF flow. J Neurosci 35:2485–2491. https://doi.org/10.1523/jneurosci.3246-14.2015
Du AT, Jahng GH, Hayasaka S, Kramer JH, Rosen HJ, Gorno-Tempini ML, Rankin KP, Miller BL, Weiner MW, Schuff N (2006) Hypoperfusion in frontotemporal dementia and Alzheimer disease by arterial spin labeling MRI. Neurology 67:1215–1220. https://doi.org/10.1212/01.wnl.0000238163.71349.78
Dupuis L, Corcia P, Fergani A, De Aguilar JLG, Bonnefont-Rousselot D, Bittar R, Seilhean D, Hauw JJ, Lacomblez L, Loeffler JP et al (2008) Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70:1004–1009. https://doi.org/10.1212/01.wnl.0000285080.70324.27
Dupuis L, Pradat PF, Ludolph AC, Loeffler JP (2011) Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol 10:75–82. https://doi.org/10.1016/S1474-4422(10)70224-6
Eide PK, Ringstad G (2019) Delayed clearance of cerebrospinal fluid tracer from entorhinal cortex in idiopathic normal pressure hydrocephalus: a glymphatic magnetic resonance imaging study. J Cerebr Blood F Met 39:1355–1368. https://doi.org/10.1177/0271678x18760974
Eisen A, Kim S, Pant B (1992) Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve 15:219–224. https://doi.org/10.1002/mus.880150215
Engelborghs S, Vloeberghs E, Le Bastard N, Van Buggenhout M, Marien P, Somers N, Nagels G, Pickut BA, De Deyn PP (2008) The dopaminergic neurotransmitter system is associated with aggression and agitation in frontotemporal dementia. Neurochem Int 52:1052–1060. https://doi.org/10.1016/j.neuint.2007.10.018
Fagan AM, Holtzman DM, Munson G, Mathur T, Schneider D, Chang LK, Getz GS, Reardon CA, Lukens J, Shah JA et al (1999) Unique lipoproteins secreted by primary astrocytes from wild type, apoE (-/-), and human apoE transgenic mice. J Biol Chem 274:30001–30007. https://doi.org/10.1074/jbc.274.42.30001
Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, Li D, Thal DR, Walther P, Ludolph AC et al (2015) TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol 211:897–911. https://doi.org/10.1083/jcb.201504057
Fernandez-Santiago R, Hoenig S, Lichtner P, Sperfeld AD, Sharma M, Berg D, Weichenrieder O, Illig T, Eger K, Meyer T et al (2009) Identification of novel Angiogenin (ANG) gene missense variants in German patients with amyotrophic lateral sclerosis. J Neurol 256:1337–1342. https://doi.org/10.1007/s00415-009-5124-4
Fiszman ML, Ricart KC, Latini A, Rodriguez G, Sica RE (2010) In vitro neurotoxic properties and excitatory aminoacids concentration in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Relationship with the degree of certainty of disease diagnoses. Acta Neurol Scand 121:120–126. https://doi.org/10.1111/j.1600-0404.2009.01200.x
Franchi C, Bianchi E, Pupillo E, Poloni M, Nobili A, Fortino I, Bortolotti A, Merlino L, Beghi E (2016) Angiotensin-converting enzyme inhibitors and motor neuron disease: an unconfirmed association. Amyotroph Lat Scl Fr 17:385–388. https://doi.org/10.3109/21678421.2016.1143515
Fray AE, Ince PG, Banner SJ, Milton ID, Usher PA, Cookson MR, Shaw PJ (1998) The expression of the glial glutamate transporter protein EAAT2 in motor neuron disease: an immunohistochemical study. Eur J Neurosci 10:2481–2489. https://doi.org/10.1046/j.1460-9568.1998.00273.x
Funalot B, Desport JC, Sturtz F, Camu W, Couratier P (2009) High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler 10:113–117. https://doi.org/10.1080/17482960802295192
Gellera C, Colombrita C, Ticozzi N, Castellotti B, Bragato C, Ratti A, Taroni F, Silani V (2008) Identification of new ANG gene mutations in a large cohort of Italian patients with amyotrophic lateral sclerosis. Neurogenetics 9:33–40. https://doi.org/10.1007/s10048-007-0111-3
Gille B, De Schaepdryver M, Dedeene L, Goossens J, Claeys KG, Van Den Bosch L, Tournoy J, Van Damme P, Poesen K (2019) Inflammatory markers in cerebrospinal fluid: independent prognostic biomarkers in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry. https://doi.org/10.1136/jnnp-2018-319586
Gomez-Pinedo U, Galan L, Yanez M, Matias-Guiu J, Valencia C, Guerrero-Sola A, Lopez-Sosa F, Brin JR, Benito-Martin MS, Leon-Espinosa G et al (2018) Histological changes in the rat brain and spinal cord following prolonged intracerebroventricular infusion of cerebrospinal fluid from amyotrophic lateral sclerosis patients are similar to those caused by the disease. Neurologia 33:211–223. https://doi.org/10.1016/j.nrl.2016.07.002
Gray E, Larkin JR, Claridge TDW, Talbot K, Sibson NR, Turner MR (2015) The longitudinal cerebrospinal fluid metabolomic profile of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotempor Degenerat 16:456–463. https://doi.org/10.3109/21678421.2015.1053490
Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J et al (2006) ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 38:411–413. https://doi.org/10.1038/ng1742
Ha JS, Lee CS, Maeng JS, Kwon KS, Park SS (2009) Chronic glutamate toxicity in mouse cortical neuron culture. Brain Res 1273:138–143. https://doi.org/10.1016/j.brainres.2009.03.050
Haas R, Cucchi D, Smith J, Pucino V, Macdougall CE, Mauro C (2016) Intermediates of metabolism: from bystanders to signalling molecules. Trends Biochem Sci 41:460–471. https://doi.org/10.1016/j.tibs.2016.02.003
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3:17085. https://doi.org/10.1038/nrdp.2017.85
Hayashi N, Doi H, Kurata Y, Kagawa H, Atobe Y, Funakoshi K, Tada M, Katsumoto A, Tanaka K, Kunii M et al (2019) Proteomic analysis of exosome-enriched fractions derived from cerebrospinal fluid of amyotrophic lateral sclerosis patients. Neurosci Res. https://doi.org/10.1016/j.neures.2019.10.010
Hladky SB, Barrand MA (2019) Metabolite clearance during wakefulness and sleep. Handb Exp Pharmacol 253:385–423. https://doi.org/10.1007/164_2017_37
Hollinger SK, Okosun IS, Mitchell CS (2016) Antecedent disease and amyotrophic lateral sclerosis: what is protecting whom? Front Neurol 7:47. https://doi.org/10.3389/fneur.2016.00047
Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G et al (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA 99:1604–1609. https://doi.org/10.1073/pnas.032539299
Iguchi Y, Eid L, Parent M, Soucy G, Bareil C, Riku Y, Kawai K, Takagi S, Yoshida M, Katsuno M et al (2016) Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 139:3187–3201. https://doi.org/10.1093/brain/aww237
Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M (2013) Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 33:18190–18199. https://doi.org/10.1523/JNEUROSCI.1592-13.2013
Iliff JJ, Wang MH, Liao YH, Plogg BA, Peng WG, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA et al (2012) A paravascular pathway facilitates CSF Flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4:147ra111. https://doi.org/10.1126/scitranslmed.3003748
Ilzecka J, Stelmasiak Z, Dobosz B (2002) Transforming growth factor-Beta 1 (tgf-Beta 1) in patients with amyotrophic lateral sclerosis. Cytokine 20:239–243. https://doi.org/10.1006/cyto.2002.2005
Ishikawa T, Morita M, Nakano I (2007) Constant blood flow reduction in premotor frontal lobe regions in ALS with dementia - a SPECT study with 3D-SSP. Acta Neurol Scand 116:340–344. https://doi.org/10.1111/j.1600-0404.2007.00876.x
Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L et al (2016) RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353:603–608. https://doi.org/10.1126/science.aaf6803
Jayaprakash K, Glasmacher SA, Pang B, Beswick E, Mehta AR, Dakin R, Newton J, Chandran S, Pal S, Colville S et al (2020) Riluzole prescribing, uptake and treatment discontinuation in people with amyotrophic lateral sclerosis in Scotland. J Neurol 267:2459–2461. https://doi.org/10.1007/s00415-020-09919-9
Jawaid A, Salamone AR, Strutt AM, Murthy SB, Wheaton M, McDowell EJ, Simpson E, Appel SH, York MK, Schulz PE (2010) ALS disease onset may occur later in patients with pre-morbid diabetes mellitus. Eur J Neurol 17:733–739. https://doi.org/10.1111/j.1468-1331.2009.02923.x
Jessen NA, Munk AS, Lundgaard I, Nedergaard M (2015) The glymphatic system: a beginner’s guide. Neurochem Res 40:2583–2599. https://doi.org/10.1007/s11064-015-1581-6
Johanson CE, Duncan JA 3rd, Klinge PM, Brinker T, Stopa EG, Silverberg GD (2008) Multiplicity of cerebrospinal fluid functions: new challenges in health and disease. Cerebrospinal Fluid Res 5:10. https://doi.org/10.1186/1743-8454-5-10
Johnston M, Zakharov A, Papaiconomou C, Salmasi G, Armstrong D (2004) Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cerebrospinal Fluid Res 1:2. https://doi.org/10.1186/1743-8454-1-2
Kabuta T, Suzuki Y, Wada K (2006) Degradation of amyotrophic lateral sclerosis-linked mutant Cu, Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem 281:30524–30533. https://doi.org/10.1074/jbc.M603337200
Kanouchi T, Ohkubo T, Yokota T (2012) Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion-like propagation? J Neurol Neurosur Ps 83:739–745. https://doi.org/10.1136/jnnp-2011-301826
Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S (2004) Glutamate receptors: RNA editing and death of motor neurons. Nature 427:801. https://doi.org/10.1038/427801a
King AE, Woodhouse A, Kirkcaldie MT, Vickers JC (2016) Excitotoxicity in ALS: overstimulation, or overreaction? Exp Neurol 275(Pt 1):162–171. https://doi.org/10.1016/j.expneurol.2015.09.019
Kiviniemi V, Wang X, Korhonen V, Keinanen T, Tuovinen T, Autio J, LeVan P, Keilholz S, Zang YF, Hennig J et al (2016) Ultra-fast magnetic resonance encephalography of physiological brain activity—glymphatic pulsation mechanisms? J Cereb Blood Flow Metab 36:1033–1045. https://doi.org/10.1177/0271678x15622047
Korner S, Kollewe K, Ilsemann J, Muller-Heine A, Dengler R, Krampfl K, Petri S (2013) Prevalence and prognostic impact of comorbidities in amyotrophic lateral sclerosis. Eur J Neurol 20:E647–E652. https://doi.org/10.1111/ene.12015
Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA et al (2014) Impairment of paravascular clearance pathways in the aging brain. Ann Neurol 76:845–861. https://doi.org/10.1002/ana.24271
Kuhnlein P, Gdynia HJ, Sperfeld AD, Lindner-Pfleghar B, Ludolph AC, Prosiegel M, Riecker A (2008) Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat Clin Pract Neurol 4:366–374. https://doi.org/10.1038/ncpneuro0853
Lacomblez L, Doppler V, Beucler I, Costes G, Salachas F, Raisonnier A, Le Forestier N, Pradat PF, Bruckert E, Meininger V (2002) APOE: a potential marker of disease progression in ALS. Neurology 58:1112–1114. https://doi.org/10.1212/Wnl.58.7.1112
Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I et al (2003) VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet 34:383–394. https://doi.org/10.1038/ng1211
Lee H, Xie L, Yu M, Kang H, Feng T, Deane R, Logan J, Nedergaard M, Benveniste H (2015) The effect of body posture on brain glymphatic transport. J Neurosci 35:11034–11044. https://doi.org/10.1523/JNEUROSCI.1625-15.2015
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A et al (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167(774–788):e717. https://doi.org/10.1016/j.cell.2016.10.002
Leibiger C, Deisel J, Aufschnaiter A, Ambros S, Tereshchenko M, Verheijen BM, Buttner S, Braun RJ (2018) Endolysosomal pathway activity protects cells from neurotoxic TDP-43. Microb Cell 5:212–214. https://doi.org/10.15698/mic2018.04.627
Leonardi A, Abbruzzese G, Arata L, Cocito L, Vische M (1984) Cerebrospinal fluid (CSF) findings in amyotrophic lateral sclerosis. J Neurol 231:75–78. https://doi.org/10.1007/bf00313720
Lewerenz J, Maher P (2015) Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence? Front Neurosci-Switz 9:469. https://doi.org/10.3389/fnins.2015.00469
Lilius TO, Blomqvist K, Hauglund NL, Liu G, Staeger FF, Baerentzen S, Du T, Ahlstrom F, Backman JT, Kalso EA et al (2019) Dexmedetomidine enhances glymphatic brain delivery of intrathecally administered drugs. J Control Release 304:29–38. https://doi.org/10.1016/j.jconrel.2019.05.005
Lin FC, Tsai CP, Lee JKW, Wu MT, Lee CTC (2015) Angiotensin-converting enzyme inhibitors and amyotrophic lateral sclerosis risk: a total population-based case-control study. Jama Neurol 72:40–48. https://doi.org/10.1001/jamaneurol.2014.3367
Liu G, Coyne AN, Pei F, Vaughan S, Chaung M, Zarnescu DC, Buchan JR (2017) Endocytosis regulates TDP-43 toxicity and turnover. Nat Commun 8:2092. https://doi.org/10.1038/s41467-017-02017-x
Logroscino G, Traynor BJ, Hardiman O, Chio A, Mitchell D, Swingler RJ, Millul A, Benn E, Beghi E (2010) Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry 81:385–390. https://doi.org/10.1136/jnnp.2009.183525
Louveau A, Plog BA, Antila S, Alitalo K, Nedergaard M, Kipnis J (2017) Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J Clin Invest 127:3210–3219. https://doi.org/10.1172/JCI90603
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS et al (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341. https://doi.org/10.1038/nature14432
Lundgaard I, Lu ML, Yang E, Peng W, Mestre H, Hitomi E, Deane R, Nedergaard M (2017) Glymphatic clearance controls state-dependent changes in brain lactate concentration. J Cereb Blood Flow Metab 37:2112–2124. https://doi.org/10.1177/0271678X16661202
Lutjohann D, Breuer O, Ahlborg G, Nennesmo I, Siden A, Diczfalusy U, Bjorkhem I (1996) Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci USA 93:9799–9804. https://doi.org/10.1073/pnas.93.18.9799
Ma QL, Ineichen BV, Detmar M, Proulx ST (2017) Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun 8:1434. https://doi.org/10.1038/s41467-017-01484-6
Macedo AC, Balouch S, Tabet N (2017) Is sleep disruption a risk factor for Alzheimer’s disease? J Alzheimers Dis 58:993–1002. https://doi.org/10.3233/jad-161287
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11:323–330. https://doi.org/10.1016/S1474-4422(12)70043-1
Majumder V, Gregory JM, Barria MA, Green A, Pal S (2018) TDP-43 as a potential biomarker for amyotrophic lateral sclerosis: a systematic review and meta-analysis. BMC Neurol 18:90. https://doi.org/10.1186/s12883-018-1091-7
Mandrioli J, Ferri L, Fasano A, Zucchi E, Fini N, Moglia C, Lunetta C, Marinou K, Ticozzi N, Ferrante GD et al (2018) Cardiovascular diseases may play a negative role in the prognosis of amyotrophic lateral sclerosis. Eur J Neurol 25:861–868. https://doi.org/10.1111/ene.13620
Manjaly ZR, Scott KM, Abhinav K, Wijesekera L, Ganesalingam J, Goldstein LH, Janssen A, Dougherty A, Willey E, Stanton BR et al (2010) The sex ratio in amyotrophic lateral sclerosis: a population based study. Amyotroph Lateral Scler 11:439–442. https://doi.org/10.3109/17482961003610853
Mariosa D, Kamel F, Bellocco R, Ye W, Fang F (2015) Association between diabetes and amyotrophic lateral sclerosis in Sweden. Eur J Neurol 22:1436–1442. https://doi.org/10.1111/ene.12632
Mehta AR, Walters R, Waldron FM, Pal S, Selvaraj BT, Macleod MR, Hardingham GE, Chandran S, Gregory JM (2019) Targeting mitochondrial dysfunction in amyotrophic lateral sclerosis: a systematic review and meta-analysis. Brain Commun 1:fcz009. https://doi.org/10.1093/braincomms/fcz009
Meng Y, Abrahao A, Heyn CC, Bethune AJ, Huang Y, Pople CB, Aubert I, Hamani C, Zinman L, Hynynen K et al (2019) Glymphatics visualization after focused ultrasound-induced blood-brain barrier opening in humans. Ann Neurol 86:975–980. https://doi.org/10.1002/ana.25604
Mestre H, Hablitz LM, Xavier ALR, Feng WX, Zou WY, Pu TL, Monai H, Murlidharan G, Rivera RMC, Simon MJ et al (2018) Aquaporin-4-dependent glymphatic solute transport in the rodent brain. Elife 7:e40070. https://doi.org/10.7554/eLife.40070
Mestre H, Tithof J, Du T, Song W, Peng W, Sweeney AM, Olveda G, Thomas JH, Nedergaard M, Kelley DH (2018) Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Commun 9:4878. https://doi.org/10.1038/s41467-018-07318-3
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K et al (2013) An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 12:435–442. https://doi.org/10.1016/S1474-4422(13)70061-9
Milosevic M, Milicevic K, Bozic I, Lavrnja I, Stevanovic I, Bijelic D, Dubaic M, Zivkovic I, Stevic Z, Giniatullin R et al (2017) Immunoglobulins G from sera of amyotrophic lateral sclerosis patients induce oxidative stress and upregulation of antioxidative system in BV-2 microglial cell line. Front Immunol 8:1619. https://doi.org/10.3389/fimmu.2017.01619
Mishra PS, Boutej H, Soucy G, Bareil C, Kumar S, Picher-Martel V, Dupre N, Kriz J, Julien JP (2020) Transmission of ALS pathogenesis by the cerebrospinal fluid. Acta Neuropathol Commun 8:65. https://doi.org/10.1186/s40478-020-00943-4
Mishra PS, Dhull DK, Nalini A, Vijayalakshmi K, Sathyaprabha TN, Alladi PA, Raju TR (2016) Astroglia acquires a toxic neuroinflammatory role in response to the cerebrospinal fluid from amyotrophic lateral sclerosis patients. J Neuroinflammation 13:212. https://doi.org/10.1186/s12974-016-0698-0
Mishra PS, Vijayalakshmi K, Nalini A, Sathyaprabha TN, Kramer BW, Alladi PA, Raju TR (2017) Etiogenic factors present in the cerebrospinal fluid from amyotrophic lateral sclerosis patients induce predominantly pro-inflammatory responses in microglia. J Neuroinflam 14:251. https://doi.org/10.1186/s12974-017-1028-x
Moglia C, Calvo A, Canosa A, Bertuzzo D, Cugnasco P, Solero L, Grassano M, Bersano E, Cammarosano S, Manera U et al (2017) Influence of arterial hypertension, type 2 diabetes and cardiovascular risk factors on ALS outcome: a population-based study. Amyotroph Lateral Scler Frontotempor Degenerat 18:590–597. https://doi.org/10.1080/21678421.2017.1336560
Moreau C, Brunaud-Danel V, Dallongeville J, Duhamel A, Laurier-Grymonprez L, de Reuck J, Wiart AC, Perez T, Richard F, Amouyel P et al (2012) Modifying effect of arterial hypertension on amyotrophic lateral sclerosis. Amyotroph Lateral Scler 13:194–201. https://doi.org/10.3109/17482968.2011.610110
Moreau C, Devos D, Brunaud-Danel V, Defebvre L, Perez T, Destee A, Tonnel AB, Lassalle P, Just N (2005) Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia? Neurology 65:1958–1960. https://doi.org/10.1212/01.wnl.0000188907.97339.76
Mortensen KN, Sanggaard S, Mestre H, Lee H, Kostrikov S, Xavier ALR, Gjedde A, Benveniste H, Nedergaard M (2019) Impaired glymphatic transport in spontaneously hypertensive rats. J Neurosci 39:6365–6377. https://doi.org/10.1523/JNEUROSCI.1974-18.2019
Munch C, O’Brien J, Bertolotti A (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci USA 108:3548–3553. https://doi.org/10.1073/pnas.1017275108
Murtha LA, Yang Q, Parsons MW, Levi CR, Beard DJ, Spratt NJ, McLeod DD (2014) Cerebrospinal fluid is drained primarily via the spinal canal and olfactory route in young and aged spontaneously hypertensive rats. Fluids Barriers CNS 11:12. https://doi.org/10.1186/2045-8118-11-12
Nedergaard M, Goldman SA, Desai S, Pulsinelli WA (1991) Acid-induced death in neurons and glia. J Neurosci 11:2489–2497
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. https://doi.org/10.1126/science.1134108
Ng Kee Kwong KC, Gregory JM, Pal S, Chandran S, Mehta AR, Cerebrospinal fluid cytotoxicity in amyotrophic lateral sclerosis: a systematic review of in vitro studies, Brain Communications, fcaa121. https://doi.org/10.1093/braincomms/fcaa12
Nicaise C, Soyfoo MS, Authelet M, De Decker R, Bataveljic D, Delporte C, Pochet R (2009) Aquaporin-4 overexpression in rat ALS model. Anat Rec (Hoboken) 292:207–213. https://doi.org/10.1002/ar.20838
Niccoli T, Partridge L, Isaacs AM (2017) Ageing as a risk factor for ALS/FTD. Hum Mol Genet 26:R105–R113. https://doi.org/10.1093/hmg/ddx247
Nolan M, Talbot K, Ansorge O (2016) Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun 4:99. https://doi.org/10.1186/s40478-016-0358-8
Nomura T, Watanabe S, Kaneko K, Yamanaka K, Nukina N, Furukawa Y (2014) Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem 289:1192–1202. https://doi.org/10.1074/jbc.M113.516492
Obal I, Jakab JS, Siklos L, Engelhardt JI (2001) Recruitment of activated microglia cells in the spinal cord of mice by ALS IgG. NeuroReport 12:2449–2452. https://doi.org/10.1097/00001756-200108080-00032
Obal I, Klausz G, Mandi Y, Deli M, Siklos L, Engelhardt JI (2016) Intraperitoneally administered IgG from patients with amyotrophic lateral sclerosis or from an immune-mediated goat model increase the levels of TNF-alpha, IL-6, and IL-10 in the spinal cord and serum of mice. J Neuroinflamm 13:121. https://doi.org/10.1186/s12974-016-0586-7
Oh KW, Moon C, Kim HY, Oh SI, Park J, Lee JH, Chang IY, Kim KS, Kim SH (2015) Phase I trial of repeated intrathecal autologous bone marrow-derived mesenchymal stromal cells in amyotrophic lateral sclerosis. Stem Cells Transl Med 4:590–597. https://doi.org/10.5966/sctm.2014-0212
Oh KW, Noh MY, Kwon MS, Kim HY, Oh SI, Park J, Kim HJ, Ki CS, Kim SH (2018) Repeated intrathecal mesenchymal stem cells for amyotrophic lateral sclerosis. Ann Neurol 84:361–373. https://doi.org/10.1002/ana.25302
Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S et al (2001) Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet 28:131–138. https://doi.org/10.1038/88842
Patel P, Kriz J, Gravel M, Soucy G, Bareil C, Gravel C, Julien JP (2014) Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol Therapy 22:498–510. https://doi.org/10.1038/mt.2013.239
Paubel A, Violette J, Amy M, Praline J, Meininger V, Camu W, Corcia P, Andres CR, Vourc’h P, French Amyotrophic Lateral Sclerosis Study G (2008) Mutations of the ANG gene in French patients with sporadic amyotrophic lateral sclerosis. Arch Neurol 65:1333–1336. https://doi.org/10.1001/archneur.65.10.1333
Pfefferbaum A, Mathalon DH, Sullivan EV, Rawles JM, Zipursky RB, Lim KO (1994) A quantitative magnetic resonance imaging study of changes in brain morphology from infancy to late adulthood. Arch Neurol 51:874–887. https://doi.org/10.1001/archneur.1994.00540210046012
Philips T, Rothstein JD (2014) Glial cells in amyotrophic lateral sclerosis. Exp Neurol 262:111–120. https://doi.org/10.1016/j.expneurol.2014.05.015
Plog BA, Mestre H, Olveda GE, Sweeney AM, Kenney HM, Cove A, Dholakia KY, Tithof J, Nevins TD, Lundgaard I et al (2018) Transcranial optical imaging reveals a pathway for optimizing the delivery of immunotherapeutics to the brain. JCI Insight. https://doi.org/10.1172/jci.insight.126138
Plog BA, Nedergaard M (2018) The glymphatic system in central nervous system health and disease: past, present, and future. Annu Rev Pathol Mech 13:379–394. https://doi.org/10.1146/annurev-pathol-051217-111018
Pokrishevsky E, Grad L, Cashman N (2015) Misfolded wild-type SOD1 induced by pathological FUS or TDP-43 transmits intercellularly and is propagation-competent. Prion 9:S7–S8
Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK (2019) Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci 12:25. https://doi.org/10.3389/fnmol.2019.00025
Preston JE (2001) Ageing choroid plexus-cerebrospinal fluid system. Microsc Res Techn 52:31–37. https://doi.org/10.1002/1097-0029(20010101)52:1%3c31:AID-JEMT5%3e3.0.CO;2-T
Pullen AH, Demestre M, Howard RS, Orrell RW (2004) Passive transfer of purified IgG from patients with amyotrophic lateral sclerosis to mice results in degeneration of motor neurons accompanied by Ca2 + enhancement. Acta Neuropathol 107:35–46. https://doi.org/10.1007/s00401-003-0777-z
Pullen AH, Humphreys P (2000) Ultrastructural analysis of spinal motoneurones from mice treated with IgG from ALS patients, healthy individuals, or disease controls. J Neurol Sci 180:35–45. https://doi.org/10.1016/s0022-510x(00)00427-5
Quaranta VN, Carratu P, Damiani MF, Dragonieri S, Capozzolo A, Cassano A, Resta O (2017) The prognostic role of obstructive sleep apnea at the onset of amyotrophic lateral sclerosis. Neurodegener Dis 17:14–21. https://doi.org/10.1159/000447560
Ramamohan PY, Gourie-Devi M, Nalini A, Shobha K, Ramamohan Y, Joshi P, Raju TR (2007) Cerebrospinal fluid from amyotrophic lateral sclerosis patients causes fragmentation of the Golgi apparatus in the neonatal rat spinal cord. Amyotroph Lateral Scler 8:79–82. https://doi.org/10.1080/08037060601145489
Rangroo Thrane V, Thrane AS, Plog BA, Thiyagarajan M, Iliff JJ, Deane R, Nagelhus EA, Nedergaard M (2013) Paravascular microcirculation facilitates rapid lipid transport and astrocyte signaling in the brain. Sci Rep 3:2582. https://doi.org/10.1038/srep02582
Rao MS, Devi MG, Nalini A, Shahani N, Raju TR (1995) Neurofilament phosphorylation is increased in ventral horn neurons of neonatal rat spinal cord exposed to cerebrospinal fluid from patients with amyotrophic lateral sclerosis. Neurodegeneration 4:397–401. https://doi.org/10.1006/neur.1995.0048
Ravits JM, La Spada AR (2009) ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 73:805–811. https://doi.org/10.1212/WNL.0b013e3181b6bbbd
Rennels ML, Gregory TF, Blaumanis OR, Fujimoto K, Grady PA (1985) Evidence for a paravascular fluid circulation in the mammalian central nervous-system, provided by the rapid distribution of tracer protein throughout the brain from the subarachnoid space. Brain Res 326:47–63. https://doi.org/10.1016/0006-8993(85)91383-6
Ringstad G, Vatnehol SAS, Eide PK (2017) Glymphatic MRI in idiopathic normal pressure hydrocephalus. Brain 140:2691–2705. https://doi.org/10.1093/brain/awx191
Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84. https://doi.org/10.1002/ana.410380114
Sankaranarayani R, Nalini A, Laxmi TR, Raju TR (2010) Altered neuronal activities in the motor cortex with impaired motor performance in adult rats observed after infusion of cerebrospinal fluid from amyotrophic lateral sclerosis patients. Behav Brain Res 206:109–119. https://doi.org/10.1016/j.bbr.2009.09.009
Sartoretti T, Wyss M, Sartoretti E, Reischauer C, Hainc N, Graf N, Binkert C, Najafi A, Sartoretti-Schefer S (2019) Sex and age dependencies of aqueductal cerebrospinal fluid dynamics parameters in healthy subjects. Front Aging Neurosci 11:199. https://doi.org/10.3389/fnagi.2019.00199
Sasaki S, Komori T, Iwata M (2000) Excitatory amino acid transporter 1 and 2 immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol 100:138–144. https://doi.org/10.1007/s004019900159
Sass LR, Khani M, Romm J, Schmid Daners M, McCain K, Freeman T, Carter GT, Weeks DL, Petersen B, Aldred J et al (2020) Non-invasive MRI quantification of cerebrospinal fluid dynamics in amyotrophic lateral sclerosis patients. Fluids Barriers CNS 17:4. https://doi.org/10.1186/s12987-019-0164-3
Sato K, Morimoto N, Matsuura T, Ohta Y, Tsunoda M, Ikeda Y, Abe K (2012) CSF flow dynamics in motor neuron disease. Neurol Res 34:512–517. https://doi.org/10.1179/1743132812Y.0000000043
Schmid Daners M, Knobloch V, Soellinger M, Boesiger P, Seifert B, Guzzella L, Kurtcuoglu V (2012) Age-specific characteristics and coupling of cerebral arterial inflow and cerebrospinal fluid dynamics. PLoS ONE 7:e37502. https://doi.org/10.1371/journal.pone.0037502
Sekiguchi T, Kanouchi T, Shibuya K, Noto Y, Yagi Y, Inaba A, Abe K, Misawa S, Orimo S, Kobayashi T et al (2014) Spreading of amyotrophic lateral sclerosis lesions–multifocal hits and local propagation? J Neurol Neurosurg Psychiatry 85:85–91. https://doi.org/10.1136/jnnp-2013-305617
Sekizawa T, Openshaw H, Ohbo K, Sugamura K, Itoyama Y, Niland JC (1998) Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases. J Neurol Sci 154:194–199. https://doi.org/10.1016/s0022-510x(97)00228-1
Shanmukha S, Narayanappa G, Nalini A, Alladi PA, Raju TR (2018) Sporadic amyotrophic lateral sclerosis (SALS)—skeletal muscle response to cerebrospinal fluid from SALS patients in a rat model. Dis Model Mech. https://doi.org/10.1242/dmm.031997
Shantanu S, Vijayalakshmi K, Shruthi S, Sagar BKC, Sathyaprabha TN, Nalini A, Raju TR, Alladi PA (2017) VEGF alleviates ALS-CSF induced cytoplasmic accumulations of TDP-43 and FUS/TLS in NSC-34 cells. J Chem Neuroanat 81:48–52. https://doi.org/10.1016/j.jchemneu.2017.01.007
Sharma A, Varghese AM, Vijaylakshmi K, Sumitha R, Prasanna VK, Shruthi S, Sagar BKC, Datta KK, Gowda H, Nalini A et al (2016) Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces mitochondrial and lysosomal dysfunction. Neurochem Res 41:965–984. https://doi.org/10.1007/s11064-015-1779-7
Shaw PJ (2002) Toxicity of CSF in motor neurone disease: a potential route to neuroprotection. Brain 125:693–694. https://doi.org/10.1093/brain/awf138
Shen D, Hou B, Xu Y, Cui B, Peng P, Li X, Tai H, Zhang K, Liu S, Fu H et al (2018) Brain structural and perfusion signature of amyotrophic lateral sclerosis with varying levels of cognitive deficit. Front Neurol 9:364. https://doi.org/10.3389/fneur.2018.00364
Shim HG, Jang SS, Kim SH, Hwang EM, Min JO, Kim HY, Kim YS, Ryu C, Chung G, Kim Y et al (2018) TNF-alpha increases the intrinsic excitability of cerebellar Purkinje cells through elevating glutamate release in Bergmann Glia. Sci Rep UK 8:11589. https://doi.org/10.1038/s41598-018-29786-9
Shobha K, Vijayalakshmi K, Alladi PA, Nalini A, Sathyaprabha TN, Raju TR (2007) Altered in vitro and in vivo expression of glial glutamate transporter-1 following exposure to cerebrospinal fluid of amyotrophic lateral sclerosis patients. J Neurol Sci 254:9–16. https://doi.org/10.1016/j.jns.2006.12.004
Shokri-Kojori E, Wang GJ, Wiers CE, Demiral SB, Guo M, Kim SW, Lindgren E, Ramirez V, Zehra A, Freeman C et al (2018) beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci USA 115:4483–4488. https://doi.org/10.1073/pnas.1721694115
Smith AJ, Yao XM, Dix JA, Jin BJ, Verkman AS (2017) Test of the ‘glymphatic’ hypothesis demonstrates diffusive and aquaporin-4-independent solute transport in rodent brain parenchyma. Elife. https://doi.org/10.7554/elife.27679
Smith R, Myers K, Ravits J, Bowser R (2015) Amyotrophic lateral sclerosis: is the spinal fluid pathway involved in seeding and spread? Med Hypotheses 85:576–583. https://doi.org/10.1016/j.mehy.2015.07.014
Smith RG, Henry YK, Mattson MP, Appel SH (1998) Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann Neurol 44:696–699. https://doi.org/10.1002/ana.410440419
Sorensen TT, Horvath-Puho E, Norgaard M, Ehrenstein V, Henderson VW (2019) Risk of amyotrophic lateral sclerosis and other motor neuron disease among men with benign prostatic hyperplasia: a population-based cohort study. BMJ Open. https://doi.org/10.1136/bmjopen-2019-030015
Spreux-Varoquaux O, Bensimon G, Lacomblez L, Salachas F, Pradat PF, Le Forestier N, Marouan A, Dib M, Meininger V (2002) Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J Neurol Sci 193:73–78. https://doi.org/10.1016/s0022-510x(01)00661-x
Staff NP, Madigan NN, Morris J, Jentoft M, Sorenson EJ, Butler G, Gastineau D, Dietz A, Windebank AJ (2016) Safety of intrathecal autologous adipose-derived mesenchymal stromal cells in patients with ALS. Neurology 87:2230–2234. https://doi.org/10.1212/WNL.0000000000003359
Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M et al (2005) Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci 8:85–92. https://doi.org/10.1038/nn1360
Stuendl A, Kunadt M, Kruse N, Bartels C, Moebius W, Danzer KM, Mollenhauer B, Schneider A (2016) Induction of alpha-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 139:481–494. https://doi.org/10.1093/brain/awv346
Sumitha R, Manjunatha VM, Sabitha RK, Alladi PA, Nalini A, Rao LT, Chandrasekhar Sagar BK, Steinbusch HWM, Kramer BW, Sathyaprabha TN et al (2019) Cerebrospinal fluid from patients with sporadic amyotrophic lateral sclerosis induces degeneration of motor neurons derived from human embryonic stem cells. Mol Neurobiol 56:1014–1034. https://doi.org/10.1007/s12035-018-1149-y
Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV et al (2015) Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 11:457–470. https://doi.org/10.1038/nrneurol.2015.119
Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C (1999) Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amyotrophic lateral sclerosis. Ann Neurol 46:129–131. https://doi.org/10.1002/1531-8249(199907)46:1%3c129:aid-ana21%3e3.0.co;2-y
Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP (2006) Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci 9:108–118. https://doi.org/10.1038/nn1603
Van Damme P, Robberecht W (2014) Developments in treatments for amyotrophic lateral sclerosis via intracerebroventricular or intrathecal delivery. Exp Opin Inv Drug 23:955–963. https://doi.org/10.1517/13543784.2014.912275
Van den Bosch L, Van Damme P, Bogaert E, Robberecht W (2006) The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. BBA Mol Basis Dis 1762:1068–1082. https://doi.org/10.1016/j.bbadis.2006.05.002
van Es MA, Schelhaas HJ, van Vught PW, Ticozzi N, Andersen PM, Groen EJ, Schulte C, Blauw HM, Koppers M, Diekstra FP et al (2011) Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann Neurol 70:964–973. https://doi.org/10.1002/ana.22611
Vijayalakshmi K, Alladi PA, Ghosh S, Prasanna VK, Sagar BC, Nalini A, Sathyaprabha TN, Raju TR (2011) Evidence of endoplasmic reticular stress in the spinal motor neurons exposed to CSF from sporadic amyotrophic lateral sclerosis patients. Neurobiol Dis 41:695–705. https://doi.org/10.1016/j.nbd.2010.12.005
Vijayalakshmi K, Alladi PA, Sathyaprabha TN, Subramaniam JR, Nalini A, Raju TR (2009) Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces degeneration of a cultured motor neuron cell line. Brain Res 1263:122–133. https://doi.org/10.1016/j.brainres.2009.01.041
Vijayalakshmi K, Ostwal P, Sumitha R, Shruthi S, Varghese AM, Mishra P, Manohari SG, Sagar BC, Sathyaprabha TN, Nalini A et al (2015) Role of VEGF and VEGFR2 receptor in reversal of ALS-CSF induced degeneration of NSC-34 motor neuron cell line. Mol Neurobiol 51:995–1007. https://doi.org/10.1007/s12035-014-8757-y
Waldemar G, Vorstrup S, Jensen TS, Johnsen A, Boysen G (1992) Focal reductions of cerebral blood flow in amyotrophic lateral sclerosis: a [99mTc]-d, l-HMPAO SPECT study. J Neurol Sci 107:19–28. https://doi.org/10.1016/0022-510x(92)90204-x
Wang H, Yang B, Qiu L, Yang C, Kramer J, Su Q, Guo Y, Brown RH Jr, Gao G, Xu Z (2014) Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum Mol Genet 23:668–681. https://doi.org/10.1093/hmg/ddt454
Wang Y, Balaji V, Kaniyappan S, Kruger L, Irsen S, Tepper K, Chandupatla R, Maetzler W, Schneider A, Mandelkow E et al (2017) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 12:5. https://doi.org/10.1186/s13024-016-0143-y
Wardlaw JM, Benveniste H, Nedergaard M, Zlokovic BV, Mestre H, Lee H, Doubal FN, Brown R, Ramirez J, MacIntosh BJ et al (2020) Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol. https://doi.org/10.1038/s41582-020-0312-z
Warita H, Manabe Y, Murakami T, Shiote M, Shiro Y, Hayashi T, Nagano I, Shoji M, Abe K (2002) Tardive decrease of astrocytic glutamate transporter protein in transgenic mice with ALS-linked mutant SOD1. Neurol Res 24:577–581. https://doi.org/10.1179/016164102101200384
Watanabe-Matsumoto S, Moriwaki Y, Okuda T, Ohara S, Yamanaka K, Abe Y, Yasui M, Misawa H (2018) Dissociation of blood-brain barrier disruption and disease manifestation in an aquaporin-4-deficient mouse model of amyotrophic lateral sclerosis. Neurosci Res 133:48–57. https://doi.org/10.1016/j.neures.2017.11.001
Westergard T, Jensen BK, Wen XM, Cai JL, Kropf E, Iacovitti L, Pasinelli P, Trotti D (2016) Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep 17:645–652. https://doi.org/10.1016/j.celrep.2016.09.032
Wolak DJ, Thorne RG (2013) Diffusion of macromolecules in the brain: implications for drug delivery. Mol Pharm 10:1492–1504. https://doi.org/10.1021/mp300495e
Wuolikainen A, Moritz T, Marklund SL, Antti H, Andersen PM (2011) Disease-related changes in the cerebrospinal fluid metabolome in amyotrophic lateral sclerosis detected by GC/TOFMS. PLoS ONE. https://doi.org/10.1371/journal.pone.0017947
Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ et al (2013) Sleep drives metabolite clearance from the adult brain. Science 342:373–377. https://doi.org/10.1126/science.1241224
Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y (2006) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci 26:4985–4994. https://doi.org/10.1523/JNEUROSCI.5476-05.2006
Xu ZW, Henderson RD, David M, McCombe PA (2016) Neurofilaments as biomarkers for amyotrophic lateral sclerosis: a systematic review and meta-analysis. PLoS ONE. https://doi.org/10.1371/journal.pone.0164625
Yamada S, Miyazaki M, Yamashita Y, Ouyang C, Yui M, Nakahashi M, Shimizu S, Aoki I, Morohoshi Y, McComb JG (2013) Influence of respiration on cerebrospinal fluid movement using magnetic resonance spin labeling. Fluids Barriers CNS 10:36. https://doi.org/10.1186/2045-8118-10-36
Yung C, Sha D, Li L, Chin LS (2016) Parkin protects against misfolded SOD1 Toxicity By Promoting Its Aggresome Formation And Autophagic Clearance. Mol Neurobiol 53:6270–6287. https://doi.org/10.1007/s12035-015-9537-z
Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L et al (2014) Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128:505–524. https://doi.org/10.1007/s00401-014-1336-5
Zhao C, Devlin AC, Chouhan AK, Selvaraj BT, Stavrou M, Burr K, Brivio V, He X, Mehta AR, Story D et al (2019) Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia. https://doi.org/10.1002/glia.23761
Zhong Z, Deane R, Ali Z, Parisi M, Shapovalov Y, O’Banion MK, Stojanovic K, Sagare A, Boillee S, Cleveland DW et al (2008) ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci 11:420–422. https://doi.org/10.1038/nn2073
Zhou Y, Cai J, Zhang W, Gong X, Yan S, Zhang K, Luo Z, Sun J, Jiang Q, Lou M (2020) Impairment of the glymphatic pathway and putative meningeal lymphatic vessels in the aging human. Ann Neurol 87:357–369. https://doi.org/10.1002/ana.25670
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12:723–738. https://doi.org/10.1038/nrn3114
Zou JY, Crews FT (2005) TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res 1034:11–24. https://doi.org/10.1016/j.brainres.2004.11.014
Zou S, Lan YL, Wang HJ, Zhang B, Sun YG (2019) The potential roles of aquaporin 4 in amyotrophic lateral sclerosis. Neurol Sci 40:1541–1549. https://doi.org/10.1007/s10072-019-03877-5
Zou W, Pu T, Feng W, Lu M, Zheng Y, Du R, Xiao M, Hu G (2019) Blocking meningeal lymphatic drainage aggravates Parkinson’s disease-like pathology in mice overexpressing mutated alpha-synuclein. Transl Neurodegener 8:7. https://doi.org/10.1186/s40035-019-0147-y
Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP (2017) Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosur Psychiatry 88:540–549. https://doi.org/10.1136/jnnp-2016-315018
Acknowledgements
The figures in this manuscript were created via BioRender.com.
Funding
K.C.N.K.K. is an SSR National Scholar of Mauritius and acknowledges the Government of Mauritius for funding. A.R.M. is a Lady Edith Wolfson Clinical Fellow and is jointly funded by the Medical Research Council and the Motor Neurone Disease Association (MR/R001162/1). He also acknowledges support from the Rowling Scholars scheme, administered by the Anne Rowling Regenerative Neurology Clinic, University of Edinburgh, UK. The Chandran Laboratory is supported by the Euan MacDonald Centre, and the UK Dementia Research Institute partner funders: the Medical Research Council, Alzheimer’s Research UK and the Alzheimer’s Society.
Author information
Authors and Affiliations
Contributions
K.C.N.K.K. and A.R.M. drafted the manuscript, with critical input from M.N. and S.C. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Ng Kee Kwong, K., Mehta, A.R., Nedergaard, M. et al. Defining novel functions for cerebrospinal fluid in ALS pathophysiology. acta neuropathol commun 8, 140 (2020). https://doi.org/10.1186/s40478-020-01018-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-020-01018-0