
Abstract
The hallmark of age-related neurodegenerative diseases is the appearance of cellular protein deposits and spreading of this pathology throughout the central nervous system. Growing evidence has shown the involvement and critical role of proteins with prion-like properties in the formation of these characteristic cellular aggregates. Prion-like domains of such proteins with their proposed function in the organization of membraneless organelles are prone for misfolding and promoting further aggregation. Spreading of these toxic aggregates between cells and across tissues can explain the progression of clinical phenotypes and pathology in a stereotypical manner, characteristic for almost every neurodegenerative disease. Here, we want to review the current evidence for the role of prion-like mechanisms in classical neurodegenerative diseases and ALS in particular. We will also discuss an intriguingly central role of the protein TDP-43 in the majority of cases of this devastating disease.

Similar content being viewed by others
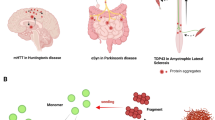

References
Acevedo-Morantes CY, Wille H (2014) The structure of human prions: from biology to structural models-considerations and pitfalls. Viruses 6:3875–3892. https://doi.org/10.3390/v6103875
Afroz T, Hock EM, Ernst P, Foglieni C, Jambeau M, Gilhespy LAB, Laferriere F, Maniecka Z, Plückthun A, Mittl P, Paganetti P, Allain FHT, Polymenidou M (2017) Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat Commun 8:45. https://doi.org/10.1038/s41467-017-00062-0
Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, Parhizkar S, Ward MA, Cavallini A, Jackson S, Bose S, Clavaguera F, Tolnay M, Lavenir I, Goedert M, Hutton ML, O’Neill MJ (2014) A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 127:667–683. https://doi.org/10.1007/s00401-014-1254-6
Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, Bilican B, Chaum E, Chandran S, Shaw CE, Eggan KC, Maniatis T, Taylor JP (2014) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81:536–543. https://doi.org/10.1016/j.neuron.2013.12.018
Alberti S, Halfmann R, King O, Kapila A, Lindquist S (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137:146–158. https://doi.org/10.1016/j.cell.2009.02.044
Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grofte M, Rask MB, Streicher W, Jungmichel S, Nielsen ML et al (2015) Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat commun 6:8088. https://doi.org/10.1038/ncomms9088
Anderson PM, Al-Chalabi A (2011) Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 7:603–615. https://doi.org/10.1038/nrneurol.2011.150
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kügler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593. https://doi.org/10.1038/nn.4132
Ayala YM, Pantano S, D’Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE (2005) Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 348:575–588
Ayers JI, Fromholt S, Koch M, DeBosier A, McMahon B, Xu G, Borchelt DR (2014) Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol 128:791–803. https://doi.org/10.1007/s00401-014-1342-7
Ayers JI, Fromholt SE, O’Neal VM, Diamond JH, Borchelt DR (2016) Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol 131:103–114. https://doi.org/10.1007/s00401-015-1514-0
Baker HF, Ridley RM, Duchen LW, Crow TJ, Bruton CJ (1994) Induction of beta (A4)-amyloid in primates by injection of Alzheimer’s disease brain homogenate. Comparison with transmission of spongiform encephalopathy. Mol Neurobiol 8:25–39
Banani SF, Rice AM, Peeples WB, Lin Y, Jain S, Parker R, Rosen MK (2016) Compositional control of phase-separated cellular bodies. Cell 166:651–663. https://doi.org/10.1016/j.cell.2016.06.010
Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, Kaitila I, McIntosh I, Francomano CA (1995) Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet 56:368–373
Bergh J, Zetterström P, Andersen PM, Brännström T, Graffmo KS, Jonsson PA, Lang L, Danielsson J, Oliveberg M, Marklund SL (2015) Structural and kinetic analysis of protein-aggregate strains in vivo using binary epitope mapping. Proc Natl Acad Sci USA 112:4489–4494. https://doi.org/10.1073/pnas.1419228112
Bétemps D, Verchère J, Brot S, Morignat E, Bousset L, Gaillard D, Lakhdar L, Melki R, Baron T (2014) Alpha-synuclein spreading in M83 mice brain revealed by detection of pathological alpha-synuclein by enhanced ELISA. Acta Neuropathol Commun 2:29. https://doi.org/10.1186/2051-5960-2-29
Bhandare VV, Ramaswamy A (2016) Identification of possible siRNA molecules for TDP43 mutants causing amyotrophic lateral sclerosis: in silico design and molecular dynamics study. Comput Biol Chem 61:97–108. https://doi.org/10.1016/j.compbiolchem.2016.01.001
Bidhendi EE, Bergh J, Zetterström P, Andersen PM, Marklund SL, Brannström T (2016) Two superoxide dismutase prion strains transmit amyotrophic lateral sclerosis-like disease. J Clin Invest 126:2249–2253. https://doi.org/10.1172/JCI84360
Boeynaems S, Bogaert E, Kovacs D et al (2017) Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol Cell 65(1044–1055):e5. https://doi.org/10.1016/j.molcel.2017.02.013
Bohnen NI, Warner MA, Kokmen E, Beard CM, Kurland LT (1994) Prior blood transfusions and Alzheimer’s disease. Neurology 44:1159–1160
Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M (2007) Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP × Tau transgenic mice. Am J Pathol 171:2012–2020
Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ (2015) Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol 129:221–237. https://doi.org/10.1007/s00401-014-1373-0
Borchelt DR, Wong PC, Becher MW, Pardo CA, Lee MK, Xu ZS, Thinakaran G, Jenkins NA, Copeland NG, Sisodia SS, Cleveland DW, Price DL, Hoffman PN (1998) Axonal transport of mutant superoxide dismutase 1 and focal axonal abnormalities in the proximal axons of transgenic mice. Neurobiol Dis 5:27–35
Borel F, Gernoux G, Cardozo B, Metterville JP, Toro Cabreja GC, Song L, Su Q, Gao GP, Elmallah MK, Brown RH Jr, Mueller C (2016) Therapeutic rAAVrh10 mediated SOD1 silencing in adult SOD1(G93A) mice and nonhuman primates. Hum Gene Ther 27:19–31. https://doi.org/10.1089/hum.2015.122
Bosque PJ, Prusiner SB (2000) Cultured cell sublines highly susceptible to prion infection. J Virol 74:4377–4386
Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Böckmann A, Meier BH, Melki R (2013) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4:2575. https://doi.org/10.1038/ncomms3575
Boylan K (2015) Familial amyotrophic lateral sclerosis. Neurol Clin 33:807–830. https://doi.org/10.1016/j.ncl.2015.07.001
Bozzoni V, Pansarasa O, Diamanti L, Nosari G, Cereda C, Ceroni M (2016) Amyotrophic lateral sclerosis and environmental factors. Funct Neurol 31:7–19
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Braak H, de Vos RA, Bohl J, Del Tredici K (2006) Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 396:67–72
Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K (2013) Amyotrophic lateral sclerosis—a model of corticofugal axonal spread. Nat Rev Neurol 9:708–714. https://doi.org/10.1038/nrneurol.2013.221
Brahic M, Bousset L, Bieri G, Melki R, Gitler AD (2016) Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon 1. Acta Neuropathol 131:539–548. https://doi.org/10.1007/s00401-016-1538-0
Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, Hyman AA (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324:1729–1732. https://doi.org/10.1126/science.1172046
Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, Suh E, Van Deerlin VM, Wood EM, Baek Y, Kwong L, Lee EB, Elman L, McCluskey L, Fang L, Feldengut S, Ludolph AC, Lee VMY, Braak H, Trojanowski JQ (2013) Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. https://doi.org/10.1002/ana.23937
Brown P, Preece M, Brandel JP, Sato T, McShane L, Zerr I, Fletcher A, Will RG, Pocchiari M, Cashman NR, d’Aignaux JH, Cervenáková L, Fradkin J, Schonberger LB, Collins SJ (2000) Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 55:1075–1081
Bruce ME, McConnell I, Fraser H, Dickinson AG (1991) The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol 72:595–603
Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H (1994) Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B 343:405–411
Buchan JR, Kolaitis RM, Taylor JP, Parker R (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153:1461–1474. https://doi.org/10.1016/j.cell.2013.05.037
Budini M, Romano V, Avendaño-Vázquez SE, Bembich S, Buratti E, Baralle FE (2012) Role of seolected mutations in the Q/N rich region of TDP-43 in EGFR-12xQ/N-induced aggregate formation. Brain Res 1462:139–150. https://doi.org/10.1016/j.brainres.2012.02.031
Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276:36337–36343
Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 280:37572–37584
Buratti E, De Conti L, Stuani C, Romano M, Baralle M, Baralle F (2010) Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J 277:2268–2281. https://doi.org/10.1111/j.1742-4658.2010.07643.x
Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11:11–24. https://doi.org/10.1038/nrneurol.2014.228
Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O’Neill MJ, Hutton ML, Citron M (2011) Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem 286:34457–34467. https://doi.org/10.1074/jbc.M111.229633
Chakrabortee S, Kayatekin C, Newby GA, Mendillo ML, Lancaster A, Lindquist S (2016) Luminidependens (LD) is an Arabidopsis protein with prion behavior. Proc Natl Acad Sci USA 113:6065–6070. https://doi.org/10.1073/pnas.1604478113
Chandler RL (1961) Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet 1:1378–1379
Chen-Plotkin AS, Lee VM, Trojanowski JQ (2010) TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol 6:211–220. https://doi.org/10.1038/nrneurol.2010.18
Chia R, Tattum MH, Jones S, Collinge J, Fisher EMC, Jackson GS (2010) Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS ONE 5:e10627. https://doi.org/10.1371/journal.pone.0010627
Chio A, Calvo A, Moglia C, Mazzini L, Mora G (2011) Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry 8:740–746. https://doi.org/10.1136/jnnp.2010.235952
Chu Y, Kordower JH (2010) Lewy body pathology in fetal grafts. Ann N Y Acad Sci 1184:55–67. https://doi.org/10.1111/j.1749-6632.2009.05229.x
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ et al (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347:1436–1441. https://doi.org/10.1126/science.aaa3650
Ciryam P, Tartaglia GG, Morimoto RI, Dobson CM, Vendruscolo M (2013) Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep 5:781–790. https://doi.org/10.1016/j.celrep.2013.09.043
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11:909–913. https://doi.org/10.1038/ncb1901
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, Ghetti B, Goedert M, Tolnay M (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 110:9535–9540. https://doi.org/10.1073/pnas.1301175110
Colby DW, Giles K, Legname G, Wille H, Baskakov IV, DeArmond SJ, Prusiner SB (2009) Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci USA 106:20417–20422. https://doi.org/10.1073/pnas.0910350106
Collinge J, Clarke AR (2007) A general model of prion strains and their pathogenicity. Science 318:930–936
Costessi L, Porro F, Iaconcig A, Muro AF (2014) TDP-43 regulates b-adducin (Add2) transcript stability. RNA Biol 11:1280–1290. https://doi.org/10.1080/15476286.2014.996081
Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, Liu AX, Ramos D, Jethava N, Hosangadi D et al (2011) A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci USA 108:20881–20890. https://doi.org/10.1073/pnas.1109434108
Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S et al (2012) Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 21:2899–2911. https://doi.org/10.1093/hmg/dds116
Cronier S, Laude H, Peyrin J-M (2004) Prions can infect primary cultured neurons and astrocytes and promote neuronal cell death. Proc Natl Acad Sci USA 101:12271–12276
Cronin S, Hardiman O, Traynor BJ (2007) Ethnic variation in the incidence of ALS: a systematic review. Neurology 68:1002–1007
Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY (1998) Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19:148–154
Cushman M, Johnson BS, King OD, Gitler AD, Shorter J (2010) Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci 123:1191–1201. https://doi.org/10.1242/jcs.051672
Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B (2009) Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha-synuclein pathology. J Neurochem 111:192–203. https://doi.org/10.1111/j.1471-4159.2009.06324.x
Daviglus ML, Plassman BL, Pirzada A, Bell CC, Bowen PE, Burke JR, Connolly ES Jr, Dunbar-Jacob JM, Granieri EC, McGarry K, Patel D, Trevisan M, Williams JW Jr (2011) Risk factors and preventive interventions for Alzheimer disease: state of the science. Arch Neurol 68:1185–1190. https://doi.org/10.1001/archneurol.2011.100
de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT (2012) Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73:685–697. https://doi.org/10.1016/j.neuron.2011.11.033
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477:211–215. https://doi.org/10.1038/nature10353
Deng H, Gao K, Jankovic J (2014) The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol 10:337–348. https://doi.org/10.1038/nrneurol.2014.78
Desplats P, Lee H-J, Bae E-J, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee S-J (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 106:13010–13015. https://doi.org/10.1073/pnas.0903691106
Dickson DW (2012) Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2:a009258. https://doi.org/10.1101/cshperspect.a009258
Dieriks BV, Park TIH, Fourie C, Faull RLM, Dragunow M, Curtis MA (2017) Α-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci Rep 7:42984. https://doi.org/10.1038/srep42984
Ding X, Ma M, Teng J, Teng RKF, Zhou S, Yin J, Fonkem E, Huang JH, Wu E, Wang X (2015) Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 6:24178–24191
Domert J, Rao SB, Agholme L, Brorsson AC, Marcusson J, Hallbeck M, Nath S (2014) Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol Dis 65:82–92. https://doi.org/10.1016/j.nbd.2013.12.019
Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IR, Neumann M et al (2012) Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J 31:4258–4275. https://doi.org/10.1038/emboj.2012.261
Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M (2010) Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 330:980–982. https://doi.org/10.1126/science.1194516
Fakhoury M (2015) Role of immunity and inflammation in the pathophysiology of neurodegenerative diseases. Neurodegener Dis 15:63–69. https://doi.org/10.1159/000369933
Fallini C, Bassell GJ, Rossoll W (2012) The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet 21:3703–3718. https://doi.org/10.1093/hmg/dds205
Fang YS, Tsai KJ, Chang YJ, Kao P, Woods R, Kuo PH, Wu CC, Liao JY, Chou SC, Lin V, Jin LW, Yuan HS, Cheng IH, Tu PH, Chen YR (2014) Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat Commun 5:4824. https://doi.org/10.1038/ncomms5824
Fatima M, Tan R, Halliday GM, Kril JJ (2015) Spread of pathology in amyotrophic lateral sclerosis: assessment of phosphorylated TDP-43 along axonal pathways. Acta Neuropathol Commun 3:47. https://doi.org/10.1186/s40478-015-0226-y
Fehlinger A, Wolf H, Hossinger A, Duernberger Y, Pleschka C, Riemschoss K, Liu S, Bester R, Paulsen L, Priola SA, Groschup MH, Schätzl HM, Vorberg IM (2017) Prion strains depend on different endocytic routes for productive infection. Sci Rep 7:6923. https://doi.org/10.1038/s41598-017-07260-2
Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, Li D, Thal DR, Walther P, Ludolph AC, Danzer KM, Weishaupt JH (2015) TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol 211:897–911. https://doi.org/10.1083/jcb.201504057
Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G (2004) Cells release prions in association with exosomes. Proc Natl Acad Sci USA 101:9683–9688
Flores BN, Dulchavsky ME, Krans A, Sawaya MR, Paulson HL, Todd PK, Barmada SJ, Ivanova MI (2016) Distinct C9orf72-associated dipeptide repeat structures correlate with neuronal toxicity. PLoS ONE 11:e0165084. https://doi.org/10.1371/journal.pone.0165084
Fraser H (1982) Neuronal spread of scrapie agent and targeting of lesions within the retino-tectal pathway. Nature 295:149–150
Fraser H, Dickinson AG (1973) Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. J Comp Pathol 83:29–40
Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, Covert M, Melki R, Kirkegaard K, Brahic M (2012) Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann Neurol 72:517–524. https://doi.org/10.1002/ana.23747
Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284:12845–12852. https://doi.org/10.1074/jbc.M808759200
Fujimura-Kiyono C, Kimura F, Ishida S, Nakajima H, Hosokawa T, Sugino M, Hanafusa T (2011) Onset and spreading patterns of lower motor neuron involvments predict survival in sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 82:1244–1249. https://doi.org/10.1136/jnnp-2011-300141
Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N (2011) A seeding reaction recapitulates intracellular formation of sarkosyl-insoluble TAR DNA binding protein-43 inclusions. J Biol Chem 286:18664–18672. https://doi.org/10.1074/jbc.M111.231209
Gajdusek DC, Gibbs CJ, Alpers M (1966) Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature 209:794–796
Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, Patrick C, Ubhi K, Nuber S, Sacayon P, Zago W, Seubert P, Barbour R, Schenk D, Masliah E (2014) Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci 34:9441–9454. https://doi.org/10.1523/JNEUROSCI.5314-13.2014
Gargiulo-Monachelli GM, Janota F, Bettini M, Shoesmith CL, Strong MJ, Sica RE (2012) Regional spread pattern predicts survival in patients with sporadic amyotrophic lateral sclerosis. Eur J Neurol 19:834–841. https://doi.org/10.1111/j.1468-1331.2011.03616.x
Gems D, Partridge L (2013) Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol 75:621–644. https://doi.org/10.1146/annurev-physiol-030212-183712
Gendron TF, Rademakers R, Petrucelli L (2013) TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43. J Alzheimers Dis 33(Suppl 1):S35–S45
Gitler AD, Shorter J (2011) RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 5:179–187. https://doi.org/10.4161/pri.5.3.17230
Gitler AD, Tsuiji H (2016) There has been an awakening: emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res 1647:19–29. https://doi.org/10.1016/j.brainres.2016.04.004
Goldschmidt L, Teng PK, Riek R, Eisenberg D (2010) Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci USA 107:3487–3492. https://doi.org/10.1073/pnas.0915166107
González L, Martin S, Jeffrey M (2003) Distinct profiles of PrP(d) immunoreactivity in the brain of scrapie- and BSE-infected sheep: implications for differential cell targeting and PrP processing. J Gen Virol 84:1339–1350
Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J, Olivo-Marin JC, Männel D, Zurzolo C (2009) Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11:328–336. https://doi.org/10.1038/ncb1841
Grad LI, Cashman NR (2014) Prion-like activity of Cu/Zn superoxide dismutase: implications for amyotrophic lateral sclerosis. Prion 8:33–41
Grad LI, Guest WC, Yanai A, Pokrishevsky E, O’Neill MA, Gibbs E, Semenchenko V, Yousefi M, Wishart DS, Plotkin SS, Cashman NR (2011) Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci USA 108:16398–16403. https://doi.org/10.1073/pnas.1102645108
Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O’Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M, Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR (2014) Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci USA 111:3620–3625. https://doi.org/10.1073/pnas.1312245111
Guo JL (2011) Lee VMY (2011) Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 286:15317–15331. https://doi.org/10.1074/jbc.M110.209296
Guo L, Shorter J (2015) It’s raining liquids: RNA tunes viscoelasticity and dynamics of membraneless organelles. Mol Cell 60:189–192. https://doi.org/10.1016/j.molcel.2015.10.006
Guo W, Chen Y, Zhou X, Kar A, Ray P, Chen X, Rao EJ, Yang M, Ye H, Zhu L, Liu J, Xu M, Yang Y, Wang C, Zhang D, Bigio EH, Mesulam M, Shen Y, Xe Q, Fushimi K, Wu JY (2011) An ALS associated mutation affecting TDP43 enhances protein aggregation, fibril formation and neurotoxicity. Nat Struct Mol Biol 18:822–830. https://doi.org/10.1038/nsmb.2053
Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VMY (2013) Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. https://doi.org/10.1016/j.cell.2013.05.057
Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, Gathagan RJ, Iba M, McBride JD, Trojanowski JQ, Lee VM (2016) Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med 213:2635–2654
Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G et al (2012) Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149:768–779. https://doi.org/10.1016/j.cell.2012.04.016
Handwerger KE, Cordero JA, Gall JG (2005) Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structure. Mol Biol Cell 16:202–211
Hansen C, Angot E, Bergström A, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K (2011) α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Investig 121:715–725. https://doi.org/10.1172/JCI43366
Harrison AF, Shorter J (2017) RNA-binding proteins with prion-like domains in health and disease. Biochem J 474:1417–1438. https://doi.org/10.1042/BCJ20160499
He B, Zheng M, Liu Q, Shi Z, Long S, Lu X, Pei Z, Yuan TF, Su H, Yao X (2017) Injected amyloid beta in the olfactory bulb transfers to other brain regions via neural connections in mice. Mol Neurobiol. https://doi.org/10.1007/s12035-017-0446-1
Heilbronner G, Eisele YS, Langer F, Kaeser SA, Novotny R, Nagarathinam A, Aslund A, Hammarström P, Nilsson KPR, Jucker M (2013) Seeded strain-like transmission of β-amyloid morphotypes in APP transgenic mice. EMBO Rep 14:1017–1022. https://doi.org/10.1038/embor.2013.137
Hennig S, Kong G, Mannen T, Sadowska A, Kobelke S, Blythe A, Knott GJ, Iyer KS, Ho D, Newcombe EA et al (2015a) Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J Cell Biol 210:529–539. https://doi.org/10.1083/jcb.201504117
Hennig S, Kong G, Mannen T, Sadowska A, Kobelke S, Blythe A, Knott GJ, Iyer KS, Ho D, Newcombe EA, Hosoki K, Goshima N, Kawaguchi T, Hatters D, Trinkle-Mulcahy L, Hirose T, Bond CS, Fox AH (2015b) Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J Cell Biol 210:529–539. https://doi.org/10.1083/jcb.201504117
Herrera F, Tenreiro S, Miller-Fleming L, Outeiro TF (2011) Visualization of cell-to-cell transmission of mutant huntingtin oligomers. PLoS Curr. https://doi.org/10.1371/currents.rrn1210
Herva ME, Spillantini MG (2015) Parkinson’s disease as a member of Prion-like disorders. Virus Res 207:38–46. https://doi.org/10.1016/j.virusres.2014.10.016
Hesketh S, Thompsett AR, Brown DR (2012) Prion protein polymerisation triggered by manganese-generated prion protein seeds. J Neurochem 120:177–189. https://doi.org/10.1111/j.1471-4159.2011.07540.x
Higelin J, Demestre M, Putz S, Delling JP, Jacob C, Lutz AK, Bausinger J, Huber AK, Klingenstein M, Barbi G, Speit G, Huebers A, Weishaupt JH, Hermann A, Liebau S, Ludolph AC, Boeckers TM (2016) FUS mislocalization and vulnerability to DNA damage in ALS patients derived hiPSCs and aging motoneurons. Front Cell Neurosci 10:290. https://doi.org/10.3389/fncel.2016.00290
Hinrichs MH, Jalal A, Brenner B, Mandelkow E, Kumar S, Scholz T (2012) Tau protein diffuses along the microtubule lattice. J Biol Chem 287:38559–38568. https://doi.org/10.1074/jbc.M112.369785
Holmes BB, Diamond MI (2012) Amyotrophic lateral sclerosis and organ donation: is there risk of disease transmission? Ann Neurol 72:832–836. https://doi.org/10.1002/ana.23684
Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, Wang ZY, Roybon L, Melki R, Li JY (2014) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128:805–820. https://doi.org/10.1007/s00401-014-1343-6
Hommes OR, Leblond CP (1967) Mitotic division of neuroglia in the normal adult rat. J Comp Neurol 129:269–278
Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee J-M (2009) Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci USA 106:20324–20329. https://doi.org/10.1073/pnas.0911281106
Hyman AA, Weber CA, Julicher F (2014) Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol 30:39–58. https://doi.org/10.1146/annurev-cellbio-100913-013325
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VMY (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33:1024–1037. https://doi.org/10.1523/JNEUROSCI.2642-12.2013
Iguchi Y, Eid L, Parent M, Soucy G, Soucy G, Bareil C, Riku Y, Kawai K, Takagi S, Yoshida M, Katsuno M, Sobue G, Julien JP (2016) Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 139:3187–3201
Imran M, Mahmood S (2011a) An overview of human prion diseases. Virol J 8:559. https://doi.org/10.1186/1743-422X-8-559
Imran M, Mahmood S (2011b) An overview of animal prion diseases. Virol J 8:493. https://doi.org/10.1186/1743-422X-8-493
Ingre C, Roos PM, Piehl F, Kamel F, Fang F (2015) Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol 7:181–193. https://doi.org/10.2147/CLEP.S37505
Irwin DJ (2016) Tauopathies as clinicopathological entities. Parkinsonism Relat Disord 22:S29–S33. https://doi.org/10.1016/j.parkreldis.2015.09.020
Ishii T, Kawakami E, Endo K, Misawa H, Watabe K (2017) Formation and spreading of TDP-43 aggregates in cultured neuronal and glial cells demonstrated by time-lapse imaging. PLoS ONE 12:e0179375. https://doi.org/10.1371/journal.pone.0179375
Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, Geng J, Amin P, DeWitt JP, Mookhtiar AK, Florez M, Ouchida AT, Fan J, Pasparakis M, Kelliher MA, Ravits J, Yuan J (2016) RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353:603–608. https://doi.org/10.1126/science.aaf6803
Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R (2016) ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164:487–498. https://doi.org/10.1016/j.cell.2015.12.038
Japtok J, Lojewski X, Naumann M, Klingenstein M, Reinhardt P, Sterneckert J, Putz S, Demestre M, Boeckers TM, Ludolph AC, Liebau S, Storch A, Hermann A (2015) Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol Dis 82:420–429. https://doi.org/10.1016/j.nbd.2015.07.017
Jiang LL, Zhao J, Yin XF, He WT, Yang H, Che MX, Hu HY (2016) Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci Rep 6:23928. https://doi.org/10.1038/srep23928
Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD (2009) TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284:20329–20339. https://doi.org/10.1074/jbc.M109.010264
Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149:753–767. https://doi.org/10.1016/j.cell.2012.04.017
Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, Miller TM, Diamond MI (2016) Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 92:796–812. https://doi.org/10.1016/j.neuron.2016.09.055
Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, Atarashi R, Race B, Qing L, Gambetti P, Caughey B, Surewicz WK (2010) Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 285:14083–14087. https://doi.org/10.1074/jbc.C110.113464
Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU (2013a) Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82:323–355. https://doi.org/10.1146/annurev-biochem-060208-092442
Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A et al (2013b) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. https://doi.org/10.1038/nature11922
Kim WS, Kågedal K, Halliday GM (2014) Alpha-synuclein biology in Lewy body diseases. Alzheimers Res Ther 6:73. https://doi.org/10.1186/s13195-014-0073-2
Kimberlin RH, Hall SM, Walker CA (1983) Pathogenesis of mouse scrapie. Evidence for direct neural spread of infection to the CNS after injection of sciatic nerve. J Neurol Sci 61:315–325
King OD, Gitler AD, Shorter J (2012) The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 1462:61–80. https://doi.org/10.1016/j.brainres.2012.01.016
Kirkwood TB (2005) Understanding the odd science of aging. Cell 120:437–447
Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E (2008) Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9:505–518. https://doi.org/10.1038/nrn2417
Kokmen E, Beard CM, O’Brien PC, Kurland LT (1996) Epidemiology of dementia in Rochester, Minnesota. Mayo Clin Proc 71:275–282
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14:504–506. https://doi.org/10.1038/nm1747
Körner S, Kollewe K, Fahlbusch M, Zapf A, Dengler R, Krampfl K, Petri S (2011) Onset and spreading patterns of upper and lower motor neuron symptoms in amyotrophic lateral sclerosis. Muscle Nerve 43:636–642. https://doi.org/10.1002/mus.21936
Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208. https://doi.org/10.1126/science.1166066
Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL (2013) Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155:1049–1060. https://doi.org/10.1016/j.cell.2013.10.033
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C et al (2012) Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15:1488–1497. https://doi.org/10.1038/nn.3230
Lalmansingh AS, Urekar CJ, Reddi PP (2011) TDP-43 is a transcriptional repressor. The testis-specific mouse acrv1 gene is a TDP-43 target in vivo. J Biol Chem 286:10970–10982. https://doi.org/10.1074/jbc.M110.166587
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A et al (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167(774–788):e17. https://doi.org/10.1016/j.cell.2016.10.002
Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14:501–503. https://doi.org/10.1038/nm1746
Li YR, King OD, Shorter J, Gitler AD (2013) Stress granules as crucibles of ALS pathogenesis. J Cell Biol 201:361–372. https://doi.org/10.1083/jcb.201302044
Liangzhong L, Yuanyuan W, Yimei L, Jianxing S (2016) ALS-causing mutations significantly perturb the self-assembly and interaction with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol 14:e1002338. https://doi.org/10.1371/journal.pbio.1002338
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Lin Y, Mori E, Kato M, Xiang S, Wu L, Kwon I, McKnight SL (2016) Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167(789–802):e12. https://doi.org/10.1016/j.cell.2016.10.003
Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K (2012) Trans-synaptic spread of tau pathology in vivo. PLoS ONE 7:e31302. https://doi.org/10.1371/journal.pone.0031302
Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, Millul A, Benn E, Beghi E, EURALS (2010) Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry 81:385–390. https://doi.org/10.1136/jnnp.2009.183525
Ludwin SK (1984) Proliferation of mature oligodendrocytes after trauma to the central nervous system. Nature 308:274–275
Luk KC, Song C, Brien PO, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM (2009) Exogenous alpha-synuclein fibrils seed the formation of Lewy bodylike intracellular inclusions in cultured cells. Proc Natl Acad Sci USA 106:20051–20056. https://doi.org/10.1073/pnas.0908005106
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012a) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. https://doi.org/10.1126/science.1227157
Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VMY (2012b) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209:975–986. https://doi.org/10.1084/jem.20112457
Majcher V, Goode A, James V, Layfield R (2015) Autophagy receptor defects and ALS-FTLD. Mol Cell Neurosci 66:43–52. https://doi.org/10.1016/j.mcn.2015.01.002
Majumder P, Chu JF, Chatterjee B, Swamy KBS, Shen CKJ (2016) Co-regulation of mRNA translation by TDP-43 and Fragile X Syndrome protein FMRP. Acta Neuropathol 132:721–738
Malaspina A, Puentes F, Amor S (2015) Disease origin and progression in amyotrophic lateral sclerosis: an immunology perspective. Int Immunol 27:117–129. https://doi.org/10.1093/intimm/dxu099
March ZM, King OD, Shorter J (2016) Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res 1647:9–18. https://doi.org/10.1016/j.brainres.2016.02.037
Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465:223–226. https://doi.org/10.1038/nature08971
Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DMA, Hasegawa M (2013) Prion-like spreading of pathological a-synuclein in brain. Brain. https://doi.org/10.1093/brain/awt037
May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grässer FA, Mori K, Kremmer E, Banzhaf-Strathmann J, Mann M, Meissner F, Edbauer D (2014) C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128:485–503. https://doi.org/10.1007/s00401-014-1329-4
McCann H, Stevens CH, Cartwright H, Halliday GM (2014) α-Synucleinopathy phenotypes. Parkinsonism Relat Disord 20:S62–S67. https://doi.org/10.1016/S1353-8020(13)70017-8
McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Velde CV (2011) TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20:1400–1410. https://doi.org/10.1093/hmg/ddr021
Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret J-M, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M (2006) Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 313:1781–1784
Mizuno Y, Amari M, Takatama M, Aizawa H, Mihara B, Okamoto K (2006) Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J Neurol 249:13–18
Mohamed N-V, Herrou T, Plouffe V, Piperno N, Leclerc N (2013) Spreading of tau pathology in Alzheimer’s disease by cell-to-cell transmission. Eur J Neurosci 37:1939–1948. https://doi.org/10.1111/ejn.12229
Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163:123–133. https://doi.org/10.1016/j.cell.2015.09.015
Moloney DM, Slaney SF, Oldridge M, Wall SA, Sahlin P, Stenman G, Wilkie AO (1996) Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet 13:48–53
Morris JA, Gajdusek DC (1963) Encephalopathy in mice following inoculation of scrapie sheep brain. Nature 197:1084–1086
Mougenot AL, Nicot S, Bencsik A, Morignat E, Verchère J, Lakhdar L, Legastelois S, Baron T (2012) Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging 33:2225–2228. https://doi.org/10.1016/j.neurobiolaging.2011.06.022
Münch C, Brien JO, Bertolotti A (2011) Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci USA 108:3548–3553. https://doi.org/10.1073/pnas.1017275108
Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH et al (2015) ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88:678–690. https://doi.org/10.1016/j.neuron.2015.10.030
Nagy M, Fenton WA, Li D, Furtak K, Horwich AL (2016) Extended survival of misfolded G85R SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc Natl Acad Sci USA 113:5424–5428. https://doi.org/10.1073/pnas.1604885113
Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, He Z, Zhang B, Gathagan RJ, Trojanowski JQ, Lee VMY (2017) Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci 37:11406–11423. https://doi.org/10.1523/JNEUROSCI.1230-17.2017
Nath S, Agholme L, Kurudenkandy FR, Granseth B, Marcusson J, Hallbeck M (2012) Spreading of neurodegenerative pathology via neuron-to-neuron transmission of β-amyloid. J Neurosci 32:8767–8777. https://doi.org/10.1523/JNEUROSCI.0615-12.2012
Naumann M, Pal A, Goswami A, Lojewski X, Japtok J, Vehlow A, Naujock M, Günther R, Jin M, Stanslowski N, Reinhardt P, Sterneckert J, Frickenhaus M, Pan-Montojo F, Storkebaum E, Poser I, Freischmidt A, Weishaupt JH, Holzmann K, Troost D, Ludolph AC, Boeckers TM, Liebau S, Petri S, Cordes N, Hyman A, Wegner F, Grill S, Weis J (2018) Storch A and Hermann A impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and aggregation formation. Nat Commun. https://doi.org/10.1038/s41467-017-02299-1
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Nishida N, Harris DA, Vilette D, Laude H, Frobert Y, Grassi J, Casanova D, Milhavet O, Lehmann S (2000) Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wildtype mouse prion protein. J Virol 74:320–325
Nishimura AL, Shum C, Scotter EL, Abdelgany A, Sardone V, Wright J, Lee YB, Chen HJ, Bilican B, Carrasco M, Maniatis T, Chandran S, Rogelj B, Gallo JM, Shaw CE (2014) Allele-specific knockdown of ALS-associated mutant TDP-43 in neural stem cells derived from induced pluripotent stem cells. PLoS ONE 10:e0142287. https://doi.org/10.1371/journal.pone.0142287
Nomura T, Watanabe S, Kaneko K, Yamanaka K, Nukina N, Furukawa Y (2014) Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem 289:1192–1202. https://doi.org/10.1074/jbc.M113.516492
Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M (2010) Seeded aggregation and toxicity of alpha-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem 285:34885–34898. https://doi.org/10.1074/jbc.M110.148460
Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, Yoshida M, Murayama S, Mann DMA, Akiyama H, Hasegawa M (2013) Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 4:124–134. https://doi.org/10.1016/j.celrep.2013.06.007
Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD et al (2015) Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell 57:936–947. https://doi.org/10.1016/j.molcel.2015.01.013
Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, Tayler K, Wiltgen B, Hatami A, Rönicke R, Reymann K, Hutter-Paier B, Alexandru A, Jagla W, Graubner S, Glabe CG, Demuth HU, Bloom GS (2012) Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485:651–655. https://doi.org/10.1038/nature11060
Ogawa M, Furukawa Y (2014) A seeded propagation of Cu, Zn-superoxide dismutase aggregates in amyotrophic lateral sclerosis. Front Cell Neurosci 8:83. https://doi.org/10.3389/fncel.2014.00083
Okamoto K, Fujita Y, Hoshino E, Tamura Y, Fukuda T, Hasegawa M, Takatama M (2015) An autopsy case of familial amyotrophic lateral sclerosis with a TARDBP Q343R mutation. Neuropathology 35:462–468. https://doi.org/10.1111/neup.12209
Oldfield CJ, Dunker AK (2014) Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem 83:553–584. https://doi.org/10.1146/annurev-biochem-072711-164947
O’Meara ES, Kukull WA, Schellenberg GD, Bowen JD, McCormick WC, Teri L, Pfanschmidt M, Thompson JD, Larson EB (1997) Alzheimer’s disease and history of blood transfusion by apolipoprotein-E genotype. Neuroepidemiology 16:86–93
Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE (1993) Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA 90:10962–10966
Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O’Sullivan GA, Pal A, Said J, Marsico G, Verbavatz JM, Rodrigo-Angulo M, Gille G, Funk RH, Reichmann H (2012) Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep 2:898. https://doi.org/10.1038/srep00898
Parrón T, Requena M, Hernández AF, Alarcón R (2014) Environmental exposure to pesticides and cancer risk in multiple human organ systems. Toxicol Lett 230:157–165. https://doi.org/10.1016/j.toxlet.2013.11.009
Parry BR, Surovtsev IV, Cabeen MT, O’Hern CS, Dufresne ER, Jacobs-Wagner C (2014) The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 156:183–194. https://doi.org/10.1016/j.cell.2013.11.028
Passoni M, De Conti L, Baralle FE, Buratti E (2012) UG repeats/TDP-43 interactions near 5´ splice sites exert unpredictable effects on splicing modulation. J Mol Biol 415:46–60. https://doi.org/10.1016/j.jmb.2011.11.003
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM et al (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162:1066–1077. https://doi.org/10.1016/j.cell.2015.07.047
Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, Hyman AA (2017) ATP as a biological hydrotrope. Science 356:753–756. https://doi.org/10.1126/science.aaf6846
Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, Sandoval IM, Fleming S, Dirr E, Polinski NK, Trojanowski JQ, Lee VM, Sortwell CE (2015) Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82:185–199. https://doi.org/10.1016/j.nbd.2015.06.003
Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344. https://doi.org/10.1038/nature14547.c
Pesiridis GS, Lee VMY, Trojanowski JQ (2009) Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 18:R156–R162. https://doi.org/10.1093/hmg/ddp303
Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J et al (2015a) Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 88:902–909. https://doi.org/10.1016/j.neuron.2015.11.018
Peters OM, Ghasemi M, Brown RH (2015b) Emerging mechanisms of molecular pathology in ALS. J Clin Invest 125:1767–1779. https://doi.org/10.1172/JCI71601
Petkova AT, Leapman RD, Guo Z, Yau W-M, Mattson MP, Tycko R (2005) Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 307:262–265
Pirooznia SK, Dawson V, Dawson TM (2014) Motor neuron death in ALS—programmed by astrocytes? Neuron 81:961–963. https://doi.org/10.1016/j.neuron.2014.02.024
Polymenidou M, Cleveland DW (2011) The seeds of neurodegeneration: prion-like spreading in ALS. Cell 147:498–508. https://doi.org/10.1016/j.cell.2011.10.011
Polymenidou M, Cleveland DW (2012) Prion-like spread of protein aggregates in neurodegeneration. J Exp Med 209:889–893. https://doi.org/10.1084/jem.20120741
Prpar Mihevc S, Baralle M, Buratti E, Rogelj B (2016) TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci Rep 6:33996. https://doi.org/10.1038/srep33996
Prudlo J, König J, Schuster C, Kasper E, Büttner A, Teipel S, Neumann M (2016) TDP-43 pathology and cognition in ALS: a prospective clinicopathologic correlation study. Neurology 87:1019–1023. https://doi.org/10.1212/WNL.0000000000003062
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144
Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, Geschwind DH, Glidden DV, Halliday GM, Middleton LT, Gentleman SM, Grinberg LT, Giles K (2015) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci USA 112:E5308–E5317. https://doi.org/10.1073/pnas.1514475112
Ramaswami M, Taylor JP, Parker R (2013) Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 154:727–736. https://doi.org/10.1016/j.cell.2013.07.038
Rattan SI (2008) Increased molecular damage and heterogeneity as the basis of aging. Biol Chem 389:267–272. https://doi.org/10.1515/BC.2008.030
Ravits JM, La Spada AR (2009) ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 73:805–811. https://doi.org/10.1212/WNL.0b013e3181b6bbbd
Ravits J, Paul P, Jorg C (2007) Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 68:1571–1575
Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L, Koolen M, Nagata T, Papadimitriou D, Nagy P, Mitsumoto H, Kariya S, Wichterle H, Henderson CE, Przedborski S (2014) Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81:1001–1008. https://doi.org/10.1016/j.neuron.2014.01.011
Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, Fariñas I, Obeso JA, Bezard E, Vila M (2014) Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75:351–362. https://doi.org/10.1002/ana.24066
Reitz C, Mayeux R (2014) Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol 15(88):640–651. https://doi.org/10.1016/j.bcp.2013.12.024
Ren P, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol 11:219–225. https://doi.org/10.1038/ncb1830
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010
Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P (2016a) Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213:1759–1778. https://doi.org/10.1084/jem.20160368
Rey NL, Wesson DW, Brundin P (2016b) The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol Dis S0969–9961(16):30294–30297. https://doi.org/10.1016/j.nbd.2016.12.013
Romano M, Feiguin F, Buratti E (2016) TBPH/TDP-43 modulates translation of Drosophila futsch mRNA through an UG-rich sequence within its 5´UTR. Brain Res 1647:50–56. https://doi.org/10.1016/j.brainres.2016.02.022
Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, Le Merrer M, Munnich A (1996) Mutations of the fibroblast growth factor receptor-3 gene in achondroplasia. Horm Res 45:108–110
Rubenstein R, Hui D, Race R, Ju W, Scalici C, Papini M, Kascak R, Carp R (1994) Replication of scrapie strains in vitro and their influence on neuronal functions. AnnNY Acad Sci 724:331–337
Saleem U, Ejaz S, Ashraf M, Omer MO, Altaf I, Batool Z, Fatima R, Afzal M (2014) Mutagenic and cytotoxic potential of Endosulfan and Lambda-cyhalothrin—in vitro study describing individual and combined effects of pesticides. J Environ Sci (China) 26:1471–1479. https://doi.org/10.1016/j.jes.2014.05.013
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. https://doi.org/10.1016/j.neuron.2014.04.047
Sassa Y, Kataoka N, Inoshima Y, Ishiguro N (2010) Anti-PrP antibodies detected at terminal stage of prion-affected mouse. Cell Immunol 263:212–218. https://doi.org/10.1016/j.cellimm.2010.03.018
Scotter EL, Chen HJ, Shaw CE (2015) TDP-43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurotherapeutics 12:352–363. https://doi.org/10.1007/s13311-015-0338-x
Sephton CF, Good SK, Atkin S, Dewey CM, Mayer P III, Herz J, Yu G (2010) TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem 285:6826–6834. https://doi.org/10.1074/jbc.M109.061846
Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537:50–56. https://doi.org/10.1038/nature19323
Shimonaka S, Nonaka T, Suzuki G, Hisanaga S, Hasegawa M (2016) Templated aggregation of TAR DNA-binding protein of 43 kDa (TDP-43) by seeding with TDP-43 peptide fibrils. J Biol Chem 291:8896–8907. https://doi.org/10.1074/jbc.M115.713552
Smethurst P, Sidle KCL, Hardy J (2015) Review: Prion-like mechanisms of transactive response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis (ALS). Neuropathol Appl Neurobiol 41:578–597. https://doi.org/10.1111/nan.12206
Smethurst P, Newcombe J, Troakes C, Simone R, Chen Y, Patani R, Sidle K (2016) In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol Dis 96:236–247. https://doi.org/10.1016/j.nbd.2016.08.007
Smith R, Myers K, Ravits J, Bowser R (2015) Amyotrophic lateral sclerosis: is the spinal fluid pathway involved in seeding and spread? Med Hypotheses 85:576–583. https://doi.org/10.1016/j.mehy.2015.07.014
Song HL, Shim S, Kim DH, Won SH, Joo S, Kim S, Jeon NL, Yoon SY (2014) β-Amyloid is transmitted via neuronal connections along axonal membranes. Ann Neurol 75:88–97. https://doi.org/10.1002/ana.24029
Spencer B, Valera E, Rockenstein E, Overk C, Mante M, Adame A, Zago W, Seubert P, Barbour R, Schenk D, Games D, Rissman RA, Masliah E (2017) Anti-α-synuclein immunotherapy reduces α-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun 5:7. https://doi.org/10.1186/s40478-016-0410-8
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–227. https://doi.org/10.1016/j.cell.2011.11.031
Tardivel M, Bégard S, Bousset L, Dujardin S, Coens A, Melki R, Buée L, Colin M (2016) Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun 4:117
Tashiro T, Sun X, Tsuda M, Komiya Y (1996) Differential axonal transport of soluble and insoluble tau in the rat sciatic nerve. J Neurochem 67:1566–1574
Teloni F, Altmeyer M (2015) Readers of poly(ADP-ribose): designed to be fit for purpose. Nucleic Acids Res 44:993–1006. https://doi.org/10.1093/nar/gkv1383
Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM (2014) Α-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep 7:2054–2065. https://doi.org/10.1016/j.celrep.2014.05.033
Tripathi VB, Baskaran P, Shawb CE, Guthrie S (2014) Tar DNA-binding protein-43 (TDP-43) regulates axon growth in vitro and in vivo. Neurobiol Dis 65:25–34. https://doi.org/10.1016/j.nbd.2014.01.004
Tsuji H, Arai T, Kametani F, Nonaka T, Yamashita M, Suzukake M, Hosokawa M, Yoshida M, Hatsuta H, Takao M, Saito Y, Murayama S, Akiyama H, Hasegawa M, Mann DM, Tamaoka A (2012) Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 135:3380–3391. https://doi.org/10.1093/brain/aws230
Turner MR, Brockington A, Scaber J, Hollinger H, Marsden R, Shaw PJ, Talbot K (2010) Pattern of spread and prognosis in lower limb-onset ALS. Amyotroph Lateral Scler 11:369–373. https://doi.org/10.3109/17482960903420140
Uenal H, Rosenbohm A, Kufeldt J, Weydt P, Goder K, Ludolph A, Rothenbacher D, Nagel G, ALS registry Study Group (2014) Incidence and geographical variation of amyotrophic lateral sclerosis (ALS) in Southern Germany–completeness of the ALS registry Swabia. PLoS ONE 9:e93932. https://doi.org/10.1371/journal.pone.0093932
Ulusoy A, Rusconi R, Pérez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, Di Monte DA (2013) Caudorostral brain spreading of α-synuclein through vagal connections. EMBO Mol Med 5:1119–1127. https://doi.org/10.1002/emmm.201302475
Utton MA, Connell J, Asuni AA, van Slegtenhorst M, Hutton M, de Silva R, Lees AJ, Miller CC, Anderton BH (2002) The slow axonal transport of the microtubule-associated protein tau and the transport rates of different isoforms and mutants in cultured neurons. J Neurosci 22:6394–6400
van Zundert B, Brown RH Jr (2017) Silencing strategies for therapy of SOD1-mediated ALS. Neurosci Lett 636:32–39. https://doi.org/10.1016/j.neulet.2016.07.059
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211. https://doi.org/10.1126/science.1165942
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714. https://doi.org/10.1038/nrm2970
Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM-Y (2011) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. https://doi.org/10.1016/j.neuron.2011.08.033
Vorberg I, Raines A, Story B, Priola SA (2004) Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J Infect Dis 189:431–439
Wächter N, Storch A, Hermann A (2015) Human TDP-43 and FUS selectively affect motor neuron maturation and survival in a murine cell model of ALS by non-cell-autonomous mechanisms. Amyotroph Lateral Scler Frontotemporal Degener 16:431–441. https://doi.org/10.3109/21678421.2015.1055275
Walker AK, Spiller KJ, Ge G, Zheng A, Xu Y, Zhou M, Tripathy K, Kwong LK, Trojanowski JQ, Lee VM (2015) Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol 130:643–660. https://doi.org/10.1007/s00401-015-1460-x
Wang HY, Wang IF, Bose J, Shen CK (2004) Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 83:130–139
Watts JC, Condello C, Stöhr J, Oehler A, Lee J, DeArmond SJ, Lannfelt L, Ingelsson M, Giles K, Prusiner SB (2014) Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc Natl Acad Sci USA 111:10323–10328. https://doi.org/10.1073/pnas.1408900111
Weber SC, Brangwynne CP (2015) Inverse size scaling of the nucleolus by a concentration-dependent phase transition. Curr Biol 25:641–646. https://doi.org/10.1016/j.cub.2015.01.012
Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, Pasinelli P, Trotti D (2017) Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep 17:645–652. https://doi.org/10.1016/j.celrep.2016.09.032
Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T (2007) Prions of fungi: inherited structures and biological roles. Nat Rev Microbiol 5:611–618
Wilkin DJ, Szabo JK, Cameron R, Henderson S, Bellus GA, Mack ML, Kaitila I, Loughlin J, Munnich A, Sykes B, Bonaventure J, Francomano CA (1998) Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. Am J Hum Genet 63:711–716
Wiltzius JJ, Landau M, Nelson R, Sawaya MR, Apostol MI, Goldschmidt L, Soriaga AB, Cascio D, Rajashankar K, Eisenberg D (2009) Molecular mechanisms for protein-encoded inheritance. Nat Struct Mol Biol 16:973–978. https://doi.org/10.1038/nsmb.1643
Wolozin B (2012) Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener 7:56. https://doi.org/10.1186/1750-1326-7-56
Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Margittai M, Kayed R, Zurzolo C, Di Paolo G, Duff KE (2013) Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288:1856–1870. https://doi.org/10.1074/jbc.M112.394528
Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM (2013) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80:402–414. https://doi.org/10.1016/j.neuron.2013.07.046
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. https://doi.org/10.1038/nature14973
Zhao W, Beers DR, Appel SH (2013) Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol 8:888–899. https://doi.org/10.1007/s11481-013-9489-x
Zhou Q, Lehmer C, Michaelsen M, Mori K, Alterauge D, Baumjohann D, Schludi MH, Greiling J, Farny D, Flatley A, Feederle R, May S, Schreiber F, Arzberger T, Kuhm C, Klopstock T, Hermann A, Haass C, Edbauer D (2017) Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins. EMBO Mol Med 9:687–702. https://doi.org/10.15252/emmm.201607054
Zhu S, Victoria GS, Marzo L, Ghosh R, Zurzolo C (2015) Prion aggregates transfer through tunneling nanotubes in endocytic vesicles. Prion 9:125–135. https://doi.org/10.1080/19336896.2015.1025189
Acknowledgements
This work was supported, in part, the NOMIS foundation and the Helmholtz Virtual Institute “RNA dysmetabolism in ALS and FTD (VH-VI-510)” to A.H.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
How to treat prions
Potential therapeutic strategies to target neurodegenerative diseases with prion-like mechanisms.
Neutralizing antibodies
In AD, several antibodies targeting β-amyloid have been developed. Recently, aducanumab, an antibody targeting aggregated β-amyloid, was shown to reduce amyloid load in PET imaging in human patients (Sevigny et al. 2016).
The potential effectiveness of targeted antibody therapy has also been demonstrated in mouse models for tau (Chai et al. 2011; Yanamandra et al. 2013) and α-synuclein (Games et al. 2014; Spencer et al. 2017; Tran et al. 2014). Zhou et al. summarized that the immunotherapy with anti-GA antibodies could be a possible treatment for patients with poly-GA pathology in C9orf72 disease (Zhou et al. 2017).
Resolution of the aggregates
Protein disaggregases to counter aberrant phase transition via diverse proteins like Hsp110, Hsp70, Hsp40 and further have been reviewed by Harrison and Shorter (Harrison and Shorter 2017). Addressing ALS, overexpression of chaperone Hsp 110 increased the median survival in a transgenetic G85R SOD1YFP mouse model (Nagy et al. 2016).
Silencing mechanisms
Silencing of ALS-causing genes is of debate, however—at least in cases of TDP43 and FUS—reduced expression of the protein itself could be harmful (Bhandare and Ramaswamy 2016). In addition, there is the possibility of allele-specific knockdown of mutant TDP-43 via siRNA (Nishimura et al. 2014). Furthermore, the silencing of SOD1 via microRNA delayed disease and death in an SOD1(G93A) mice (Borel et al. 2016) and is currently in phase 2 studies. Possible silencing mechanisms for SOD1 in ALS like siRNA, microRNA, and anti-sense oligonucleotides are reviewed by van Sundert et al. (van Zundert and Brown 2017).
Inhibiting necroptosis
By blocking necroptosis, a possible mechanism of aggregate release could be eliminated, and furthermore, this could block the toxicity of the glia component in ALS demonstrated by Re and colleagues (Re et al. 2014).
Blocking exosomes
Investigation of blocking the release via exosoms with, for example, neutral-sphingomyelinase-2-inhibitor showed different results. Blocking exosomes in a tau mouse model led to decreased tau propagation in the brains (Asai et al. 2015). Whereas blocking exosoms in TDP-43 ALS caused increasing cytoplasmic aggregates in vitro and symptom exacerbation in vivo in a TDP-43 mouse model (Smethurst et al. 2016).
Rights and permissions
About this article

Cite this article
Bräuer, S., Zimyanin, V. & Hermann, A. Prion-like properties of disease-relevant proteins in amyotrophic lateral sclerosis. J Neural Transm 125, 591–613 (2018). https://doi.org/10.1007/s00702-018-1851-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-018-1851-y