
Abstract
The extracellular matrix (ECM) plays critical roles in cytoskeletal support, biomechanical transduction and biochemical signal transformation. Tumor-associated macrophage (TAM) function is regulated by matrix stiffness in solid tumors and is often associated with poor prognosis. ECM stiffness-induced mechanical cues can activate cell membrane mechanoreceptors and corresponding mechanotransducers in the cytoplasm, modulating the phenotype of TAMs. Currently, tuning TAM polarization through matrix stiffness-induced mechanical stimulation has received increasing attention, whereas its effect on TAM fate has rarely been summarized. A better understanding of the relationship between matrix stiffness and macrophage function will contribute to the development of new strategies for cancer therapy. In this review, we first introduced the overall relationship between macrophage polarization and matrix stiffness, analyzed the changes in mechanoreceptors and mechanotransducers mediated by matrix stiffness on macrophage function and tumor progression, and finally summarized the effects of targeting ECM stiffness on tumor prognosis to provide insight into this new field.
Similar content being viewed by others

Introduction
The extracellular matrix (ECM) is the basis of maintaining tissue structure and organ homeostasis [1]. It is also a vital component of the tumor microenvironment (TME), supporting the initiation, progression, and invasion of tumors [2]. ECM is mainly composed of collagen, fibrin, elastin, fibronectin, glass adhesin, glycoproteins and other matrix proteins, which determine the stiffness and elasticity of tumor tissue [3]. Tissue stiffness is dynamically regulated and remodeled by the synthesis and degradation of ECM proteins. Degradation of peripheral ECM components by matrix metalloproteinases (MMPs), cathepsin, and hyaluronidase is an important pathogenic mechanism of the dynamic regulation of ECM structure [4]. During tumor progression, ECM degradation releases a suite of growth factors and cytokines that induce tumor cell growth, angiogenesis and inflammation [5]. Moreover, ECM degradation is accompanied by the deposition of different tumor-specific ECMs, finally contributing to an increase in tissue density and rigidity[6]. Generally, the matrix stiffness of soft tissues, such as the brain, liver, colon and fat tissue, is typically less than 10 kPa; however, in diseased conditions, such as fibrosis and solid tumors, the mean stiffness value can exceed 20 kPa and 50 kPa, respectively [7]. Recently, accumulating evidences demonstrate that increased matrix stiffness is associated with poor clinical outcomes [8, 9], and ECM stiffness is widely recognized as a new hallmark in solid tumors [10, 11]. Therefore, targeting ECM stiffness is emerging as a potential option for cancer therapy.
Tumor-associated macrophages (TAMs) are the most prominent innate immune cells in the TME, accounting for approximately 50% of CD45+ cells in the tumor tissue, with heterogeneity and plasticity ranging from antitumor to protumor [12]. There are two main sources of TAMs: (I) monocytes produced by myeloid progenitors of bone marrow, including classical Ly6C+ monocytes and nonclassical Ly6C− monocytes. These monocytes leave the blood circulation and enter tumor tissue to differentiatiate into macrophages [13]. (II) Early embryonic source: yolk sac or fetal liver. Macrophages become tissue-resident macrophages, which are present in various healthy tissues and participate in cancer growth and metastasis [14]. After monocytes dissociate from the bone marrow, monocytes are recruited to the TME by chemokines such as CCL2, CCL5 and CXCL12, which are produced by cancer cells in the early stages of tumorigenesis [15]. At present, the most widely used traditional classification of TAMs is the dual classification, that is, the antitumor concomitant pro-inflammatory M1 phenotype (classical activation) and the protumor concomitant anti-inflammatory M2 phenotype (alternative activation) [16,17,18]. For instance, bacterial products such as lipopolysaccharides and pro-inflammatory factors such as IFN-γ can induce M1 macrophages to produce inflammatory factors and chemokines (including IL-1, IL-6, IL-12, TNF-α, CCL3, CCL5, CXCL8, CXCL9, and CXCL10), with the upregulation of iNOS as well as CD80, CD86, and MHCII [19]. While certain signaling pathways, such as STAT6 and PPARγ, along with parasitic infections, have the capacity to induce M2 macrophages to produce anti-inflammatory cytokines (including IL-4, IL-10, and IL-13), as well as express markers such as Arg1, TGF-β, CD163, CD206, VEGF, and MMPs, these signaling molecules accelerate TME remodeling, angiogenesis and tumor growth [20]. In addition, some researchers have added an additional subcategory to M2 TAMs, such as M2a, M2b, M2c and M2d, due to the complexity of TAMs [21]. Furthermore, single-cell sequencing analysis revealed that TAM subsets can express both M1- and M2-related genes with complicated phenotypes, which indicates a state of phenotypic transition between M1 and M2 TAMs [22]. In fact, M1 and M2 represent only the extremes of the more complex and continuum spectrum of macrophages activation states, and macrophages are an extremely plastic cellular population depending on tumor context and stage of disease [23].
In the early stage of tumorigenesis, TAMs are mainly the M1 phenotype, and with the development of the tumor, TAMs are mainly the M2 phenotype with a significant immunosuppressive function in the TME [24]. Besides the hypoxic microenvironment, tumor cells, and fibroblasts, matrix stiffness represents a significant factor driving macrophage polarization toward the M2 phenotype. Furthermore, accumulating evidence demonstrates that the ECM is not only the core component of tumor tissue but also has extensive and complex biochemical and biomechanical contact with TAMs in the cancer microenvironment, supporting the proliferation, progression, and invasion of cancer cells [25]. Given that matrix stiffness affects the phenotype and function of TAMs and is often associated with a poor clinical prognosis, a better understanding of the mechanisms underlying the regulatory network between matrix stiffness and macrophages is critical for targeted therapy [26].
Matrix stiffness or mechanical stimulation first senses mechanical changes through the mechanoreceptors on the plasma membrane and then transmits mechanical signals through mechanically related molecules in the cytoplasm, ultimately modulating nuclear transcription or the function of macrophages. Therefore, in this review, we discuss the mechanoreceptors and the corresponding key mechanical molecules of macrophages regulated by matrix stiffness or mechanical stimulation and summarize ECM stiffness-based targeted cancer therapy as well as the shortcomings and challenges in this field.
Matrix stiffness and macrophage polarization
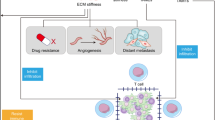
The excessive secretion and crosslinking of collagen proteins lead to an increase in matrix stiffness, which increases macrophage mechanosensing [27]. In turn, M2 macrophages activate fibroblasts to become myofibroblasts, which secrete large amounts of collagen to promote matrix deposition while also remodeling the ECM by regulating the balance of MMPs and their inhibitors [28]. Therefore, matrix stiffness and macrophages are mutually regulated in a feedforward manner, ultimately mediating tumor invasion and metastasis (Fig. 1).

The relationship between matrix stiffness and macrophage polarization in the TME. With the deposition of collagen and the increase in matrix stiffness in the TME, stiff ECM can polarize macrophages into tumor-promoting M2-type TAMs, which in turn also promotes the stiffness of the ECM
Matrix stiffness regulates macrophage polarization
A growing quantity of evidence suggests that enhanced matrix stiffness promotes macrophages to the M2 phenotype. For example, bone marrow-derived macrophages (BMDMs) cultured on stiff hydrogels upregulate the expression of CD206, IL-4 and TGF-β with less production of reactive oxygen species (ROS), while M2 macrophages can convert to the M1 phenotype when BMDMs are cultured on soft hydrogels[29]. In addition, human-derived macrophages exhibit an M2 phenotype in stiffer 3D matrices, as determined by the overexpression of IL-10 cytokines [30], similar to ECM deposition contributing to macrophage M2 polarization in metastatic breast cancer [31]. Likewise, single-cell RNA sequencing was used to assess the effects of matrix stiffness on intratumor heterogeneity between stiff and compliant mouse mammary tumors. The results showed a significantly higher proportion of M2-like macrophages in the stiffer TME [32]. Furthermore, BMDMs and TAMs isolated from murine tumors cultured in high collagen matrices mimicking tumor tissue showed similar expression of immunosuppressive genes and chemokines, which inhibited the chemotaxis and proliferation of cytotoxic CD8+ T cells in coculture assays [33].
Consistent with the above viewpoint, cancer-associated fibroblast (CAF)-induced ECM deposition and matrix stiffness increases can promote cancer progression with poor prognosis [27, 34]. It has been reported that CAFs are highly correlated with TAMs, accompanied by high expression of both CAF and TAM markers, such as α-SMA, FAP, and CD163, in patients with worse clinical prognosis [35, 36]. Furthermore, CAFs are able to facilitate monocyte migration into tumors and polarize into the M2 phenotype. For instance, CAF-derived M-CSF1, IL-6, and CCL2 in monocyte recruitment increased the M2/M1 TAM ratio in pancreatic cancer [37]. Similarly, CCL2, IL-8, IL-10, and TGF-β secreted by CAFs have also been shown to promote the recruitment of monocytes and their transformation into M2 macrophages [38, 39]. Furthermore, CAFs are the top secreting factors primarily to TAMs and engage in mutual paracrine interactions with TAMs, as verified by an experimental-mathematical approach in female breast cancer [40]. Together, these results suggest that matrix stiffness may promote macrophage M2 polarization in tumors.
Macrophages promote extracellular matrix deposition
ECM stiffness can influence the TAM phenotype; in turn, TAMs can also promote tumor invasion and metastasis by remodeling the ECM composition and structure by instructing the degradation, deposition, crosslinking, and linearization of collagen fibers during tumor development [41]. A recent study showed that TAMs can promote fibrosis in pancreatic cancer by mannose receptor-mediated collagen internalization and subsequent lysosomal degradation-induced metabolic reprogramming [42]. Likewise, TAMs advance tumor progression by the synthesis and assembly of collagenous ECM, specifically collagen types I, VI, and XIV, resulting in the remodeling of its ECM composition and structure in a colorectal cancer mouse model [41]. Similarly, TAMs contribute to tumor progression via legumain-mediated remodeling of ECM deposition and angiogenesis in diffuse large B-cell lymphoma [43]. Interestingly, macrophages mediate the development of fibrosis via the secretion of growth factors and matricellular proteins within obese adipose tissue [44]. In addition, TAMs can fabricate an immunosuppressive network by secreting immunosuppressive factors, mediating the activation of CAFs, and finally leading to ECM deposition [45]. However, when macrophages were engineered with a designed chimeric antigen receptor (CAR), the infused CAR-147 macrophages reduced collagen deposition and promoted T-cell infiltration within HER2-4T1 tumors, which significantly inhibited tumor growth in a BALB/c mouse model [46]. Thus, TAMs have obvious plasticity when inhabiting different environments and can affect tumor progression by remodeling ECM stiffness.
Matrix stiffness regulates macrophage polarization through mechanoreceptors
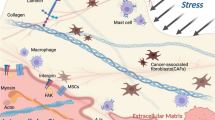
Matrix stiffness shapes cell morphology and function during adult homeostasis. In addition, it also signals to macrophages via mechanoreceptors to affect their polarization and function in the TME. Currently, Piezo1, transient receptor potential (TRP) ion channels, and integrins are the three main mechanoreceptors on macrophages regulated by matrix stiffness or mechanical stimulation, and the regulatory relationship between them is complex (Fig. 2).

Matrix stiffness regulates macrophage polarization via mechanoreceptors. In different studies, the mechanoreceptors Piezo1, TRP ion channels and integrins on macrophages regulated by matrix stiffness can polarize into M1- or M2-type macrophages
Piezo1
Piezo1, a non-selective Ca2+-permeable channel receptor that senses external mechanical forces, can be activated in response to different matrix stiffnesses as well as a variety of mechanical stimuli [47,48,49].
Macrophages sense and migrate to fibroblast-mediated ECM deposition [50], with subsequent activation of mechanoreceptors such as Piezo1. When BMDMs are mechanically stimulated, they first induce the activation of AP-1 through Piezo1, promoting the expression of endothelin-1 and the stabilization of HIF1α, which in turn promotes the expression of inflammatory cytokines such as IL1β, PTGS2 and CXCL10 [51]. Consistent with this perspective, matrix stiffness-activated ion channel Piezo1 induces macrophage M1 polarization by triggering the NFκB pathway with the upregulation of the pro-inflammatory cytokines of iNOS, TNFα, and IL-6 [52]. Similarly, the complex of Piezo1 and TLR4 together remodeled macrophage F-actin organization and consequently activated the CaMKII-Mst1/2-Rac axis to augment phagocytosis and killing ability [53]. In contrast, mechanical stretch can induce RAW264.7 macrophages toward M2 polarization and TGF-β1 release through the Piezo1 channel to enhance bone formation [54, 55]. In addition, using myeloid cell-specific knockout mice (Lyz2Cre; Piezo1fl/fl) and transplanting orthotopic KPC cell-derived tumors confirmed that Piezo1 deletion unleashes innate immunity against pancreatic ductal adenocarcinoma [56]. Hence, the effects of the mechanoreceptor Piezo1 on macrophages are complex and multiple, with the ability to regulate both M1 polarization and M2 polarization.
TRP
TRP ion channels are transmembrane ion channels that allow cations to pass through cell membranes non-selectively. In mammals, the TRP channel family consists of six subfamilies, TRPC, TRPV, TRPM, TRPML, TRPP and TRPA, which participate in the proliferation, invasion, metastasis and angiogenesis of cancer cells and are responsible for a variety of sensory responses, including mechanical force [57, 58]. While increased Ca2+ influx through TRP channels, including TRPM2, TRPM7 and TRPC1, is widely confirmed to contribute to macrophage polarization by inflammatory agonists [59,60,61], the role of matrix stiffness on macrophage function through TRP ion channels remains less explored.
A recent study showed that TRPV4 participates in matrix stiffness-induced macrophage M1 polarization in fibrotic skin tissue in vivo and in vitro, while TRPV4 knockdown reduces M1 markers such as IL-1b and Mcp1 [62]. In addition, cyclic mechanical stretch promotes resident CCR2− cardiac macrophage activation through a TRPV4-dependent pathway, representing a protective population that mediates adaptive cardiac remodeling and survival of the chronically failing heart [63]. Similarly, in pulmonary cystic fibrosis, matrix stiffness activates the phagocytic and bactericidal functions of alveolar macrophages by stimulating TRPC6 overexpression [64]. Conversely, the activation of TRP channels, such as TRPM8 and TRPM7, can promote macrophage M2 polarization with the stimulation of inflammatory factors [65, 66]. At present, although many studies have explored the relationship between TRP channels and macrophage function, only a few studies have explore the regulation of matrix stiffness or mechanical stimulation on the polarization and function of macrophages, and this field deserves further exploration.
Integrins
Integrins are heterodimeric proteins consisting of one α-chain and one β-chain. Each subunit contains three parts, including a long stalk that senses extracellular signals, a transmembrane helix, and a short intracellular tail that connects with cytoplasmic signaling and cytoskeletal proteins [67]. Integrins have been widely recognized as mechanoreceptor proteins that form a mechanical conduction network, which affects normal physiological behaviors such as cell migration, proliferation and differentiation as well as pathological processes such as cancer and tissue fibrosis [68, 69].
A recent study revealed that fibrotic ECM promotes alveolar macrophage M1 polarization via the integrin-NFκB signaling axis, and the activation effect of macrophages could be abrogated by integrin pan-inhibitors as well as NFκB inhibitors [70]. Consistent with the above results, macrophages cultured in stiffer poly(ethylene glycol) hydrogels show enhanced αV integrin staining with a classical activation phenotype (TNF-α, IL-1β, and IL-6) while cultured in lower stiffness hydrogels with reduced macrophage activation [71]. Similarly, the expression of integrin is reduced when macrophages are cultured on a soft matrix, and the phagocytosis function of macrophages is subsequently restricted [72]. In contrast with the above conclusions, matrix stiffness can also activate macrophages toward the M2 phenotype. To clarify whether matrix stiffness alters LOXL2 expression in macrophages within the TME, THP-1 cells cultured on 6 kPa, 10 kPa, and 16 kPa stiffness substrates were induced by phorbol 12-myristate 13-acetate (PMA) and subsequently treated with IL-4 and IL-13. The results showed that increased matrix stiffness remarkably strengthened macrophage M2 polarization and promoted LOXL2 expression by activating the integrin β5-FAK-MEK1/2-ERK1/2-HIF-1α pathway [73]. Similarly, activating αvβ3 integrin in macrophages by mechanical cues can enhance anti-inflammatory M2 macrophage polarization in a 3D macrophage-ECM hydrogel model [74]. In total, a comprehensive assessment, such as the detection of both M1- and M2- macrophage biomarkers in vivo and in vitro together with multiple detection methods, may provide a more definitive conclusion for the role of integrin in macrophage function.
Matrix stiffness regulates macrophage polarization through mechanotransducers
Mechanotransducers are key mechanically dependent molecules mediating mechanical signal transduction in the cytoplasm and nucleus. In the TME, YAP/TAZ, Rho/ROCK, FAK, and LOX are the main mechanotransducers that are mostly overexpressed in tumor tissues and regulate macrophage polarization and function stimulated by matrix stiffness and mechanical force (Fig. 3).

Matrix stiffness regulates macrophage polarization by mechanotransducers. When mechanoreceptors are activated by matrix stiffness, they can transmit mechanical signals to biological signals through different mechanotransducers, thus mediating the polarization of M1- or M2-macrophages by different pathways
YAP/TAZ
Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are downstream transducers of the Hippo pathway, engaging in cell proliferation and survival and playing vital roles in controlling organ growth, stem cell self-renewal and cell differentiation [75, 76]. In recent years, accumulating evidences demonstrate that mechanical force has widely been linked to the activity of the transcriptional coactivators YAP and TAZ, establishing a connection between extracellular biomechanics and gene regulation [77, 78].
Macrophage adhesion on soft hydrogel substrates reduces inflammation when compared to that of gels with stiffness, accompanied by decreased YAP expression and nuclear localization [79]. Similarly, macrophages cultured in stiff scaffolds upregulate YAP expression and induce inflammation and doxorubicin drug resistance in osteosarcoma [80]. In another study, low ECM protein expression and reduced YAP activation were accompanied by an increase in M1 macrophages, showing a favorable clinical prognosis in uterine sarcomas [81]. In stiff breast cancer tissue, YAP also induces macrophage M2 polarization and subsequently inhibits CD8+ T-cell activity and promotes tumor progression [82]. Furthermore, macrophage M2 polarization was increased in colorectal cancer by activating the Rho/Hippo/YAP signaling pathway [83]. Taken together, matrix stiffness can shape macrophage polarization and function through YAP/TAZ mechanotransduction molecules in the cancer microenvironment.
Rho/ROCK
The Ras homolog family (Rho) and Rho-associated coiled-coil containing protein serine/threonine kinase (ROCK) signaling pathways are involved in a variety of key biological processes including the regulation of the cytoskeleton and morphogenesis, which shape the TME [84, 85]. Generally, the Rho GTPase family can be divided into three classes: Rho (RhoA, RhoB, and RhoC), Rac (Rac1, Rac2, and Rac3), and cell division cycle 42 (Cdc42), while the ROCK family includes two members, ROCK1 and ROCK2 [86,87,88]. There is accumulating evidence that the Rho-ROCK pathway is engaged in ECM stiffness and composition, which ultimately promotes the growth, migration, and invasion of cancer cells [89].
Studies from Tu et al. suggest that mechanical stretch promotes RAW264.7 macrophage polarization and inflammatory secretion via the activation of the RhoA-ROCK-NF-κB pathway [90]. Moreover, to explore the effect of different polyacrylamide gel stiffnesses on macrophage polarization state and function, soft (11 kPa), medium (88 kPa), and stiff (323 kPa) gels were constructed, and the results showed that the substrate stiffness-mediated Rho-ROCK pathway plays an important role in directing macrophage behavior [91]. Similarly, pirfenidone treatment can disrupt the polarization and mechanical activation of macrophages by suppressing ROCK2 protein expression (92). In addition, monocyte and macrophage migration into tumor tissues requires the rearrangement of their actin cytoskeleton and is confirmed to be mediated by ROCK, as the ROCK inhibitor Y-27632 contributes to decreased macrophage infiltration in breast tumor tissue [93]. Likewise, examining the influence of ECM (composition, architecture, and stiffness) on the 3D migration of human macrophages demonstrated that macrophages migrate into tissues using either the protease-dependent mesenchymal migration mode or the Rho-ROCK-mediated amoeboid migration mode [94]. Therefore, ECM stiffness can widely affect macrophage behavior via the Rho-ROCK pathway.
FAK
Focal adhesion kinase (FAK) is a cytoplasmic non-receptor protein tyrosine kinase belonging to the protein tyrosine kinase superfamily. FAK plays crucial roles in cell signal transduction, receives signals from integrins, growth factors and mechanical stimulation, activates intracellular PI3K/Akt, Ras/MAPK and RAS/RAF/MEK pathways, and is related to tumorigenesis and migration [95, 96].
Stiffer polydimethylsiloxane substrates accelerated BMDM and RAW264.7 macrophage osteoclast differentiation by activating the cytoskeleton-associated adhesion molecules fibronectin and integrin αvβ3 and subsequently the biochemical signaling cascades of FAK, PKC, and RhoA [97]. In addition, matrix stiffness-mediated HIF-1α overexpression promotes THP-1-derived macrophage M2 polarization by activating the integrin β5-FAK-MEK1/2-ERK1/2 pathway [73]. Furthermore, the excessive secretion of S100A7 protein stimulated by stiff esophageal squamous carcinoma promotes macrophage M2 polarization and angiogenesis through the activation of the p-FAK and p-ErK pathways [98]. However, by using a Flexcell Tension system, RAW264.7 macrophages promoted M1 polarization-related gene expression and cytokine release after mechanical stretch, and mechanically stretch-preconditioned RAW264.7 cells showed a tumoricidal effect on melanoma in vitro and in vivo [99]. In addition, increased extracellular pressure during infection or inflammation promotes THP-1-derived macrophage phagocytosis by inhibiting FAK expression [100]. These results suggest that FAK can sense stiffness to regulate macrophage function.
LOX
Lysyl oxidase (LOX) family members, including LOX, LOXL1, LOXL2, LOXL3, and LOXL4, are extracellular copper-dependent enzymes that play a crucial role in ECM crosslinking, which is relevant to fibrosis and oncogenesis [101]. The crosslinking of ECM components, especially collagens and elastin, is strongly engaged in collagen deposition and matrix stiffness [102].
Macrophage-secreted oncostatin M induces LOXL2 expression and ECM collagen remodeling, which promotes primary and metastatic tumor progression and decreases overall survival (OS) in KRasG12D-driven pancreatic ductal adenocarcinoma [103]. Furthermore, macrophages drive stromal cell-dependent collagen crosslinking and stiffening promoting breast cancer aggression due to the high expression of LOX [104]. Moreover, LOXL4 promotes macrophage infiltration and matrix deposition, followed by the activation of an immunosuppressive M2 phenotype and programmed death ligand 1 (PD-L1) expression, which further suppress the function of CD8+ T cells in hepatocellular carcinoma [105]. During breast tumorigenesis, the EZH2-miR-29b/miR-30d-LOXL4 signaling pathway is activated accompanied by the infiltration of macrophages [106]. In addition, epigenetic regulation of LOX plays an important role in tumor progression; for instance, LOX derived from M2-like macrophages promoted breast cancer cell migration and collagen crosslinking, and this phenomenon was suppressed by an H3K27 demethylase inhibitor [107]. These results suggest that LOX-induced matrix stiffness is involved in M2 macrophage polarization.
Drugs targeting matrix stiffness for tumor therapy
Since matrix stiffness can affect macrophage function and promote tumor progression in the TME, targeting matrix stiffness can promote macrophages toward an antitumor phenotype and may be a new strategy for cancer treatment [108]. In addition, targeting matrix stiffness can also anchor other stromal cells and delay tumor progression in macrophage-dependent and macrophage-independent ways [109]. At present, three main strategies can be used to interfere with the effect of matrix stiffness on tumor progression: [1] reducing matrix protein production, [2] degrading matrix protein and crosslinking, [3] targeting mechanoreceptors and mechanotransducers stimulated by matrix stiffness (Fig. 4, Table 1).

Drugs targeting matrix stiffness for tumor therapy
Reducing matrix protein production
CAFs and TGF-β are the main factors leading to the production of matrix proteins in the TME. To reduce matrix protein production and matrix stiffness, targeting CAFs and TGF-β has achieved great attractions in preclinical and clinical studies.
Targeting CAFs
CAFs actively participate in tumor progression through complicated interactions with other types of stromal cells and produce ECM components that contribute to the remodeling of the tumor stroma [39, 110]. An array of CAF biomarkers have been identified, including but not limited to αSMA, FSP1, FAP, PDGFRα, PDGFRβ, CLEC3B, Desmin, DDR2 and Vimentin, and many therapeutic approaches targeting CAFs have been well reviewed [111,112,113]. CAFs have long been considered an attractive therapeutic target with the majority of studies showing tumor-promoting roles of CAFs [114, 115].
At present, erdafitinib, a pan-FGFR inhibitor, has achieved expedited approval from the US Food and Drug Administration (FDA) for locally advanced or metastatic urothelial carcinoma in patients with FGFR alterations due to the positive objective tumor response in a phase 2 clinical trial [116]. However, erdafitinib has obvious adverse events in clinical trials with poor tolerability, and the antitumor effect of erdafitinib should be further anticipated in subsequent studies [116, 117]. In contrast, in some preclinical studies, targeting the elimination of CAFs even promotes tumor progression and results in poor prognosis [118, 119]. Moreover, targeting CAFs-related signaling pathways, such as SHH–SMO signaling or hyaluronic acid, even shortened patient survival in clinical trials [120,121,122]. Recently, single-cell analysis techniques revealed distinct CAF subpopulations compared to conventional CAF subpopulations [123, 124]. Therefore, we should systemically define the functional roles of CAFs and CAF subpopulations in future studies to comprehensively develop accurate diagnostic and therapeutic approaches based on specific CAF subpopulations.
Targeting TGF-β
TGF-β is a subtype of the TGF-β family that plays a crucial role in fibrosis and solid tumors [125]. TGF-β promotion of tumor progression is multifaceted, including altering the polarization of macrophages, damaging the activities of tumor-infiltrating lymphocytes (TILs), and inducing regulatory T (Treg) cell differentiation in the TME [126]. Moreover, TGF-β hinders TIL penetration by increasing peritumoral collagen production [127]. In addition, TGF-β-induced phosphorylation of Smad2/3 also promotes the deposition of α-SMA, MMP1, and collagen type I, which increase matrix stiffness by the overexpression of LOXL1 [128].
At present, several drugs have shown promising results in clinical trials. For example, fresolimumab is a neutralizing antibody targeting all human isoforms of TGF-β and has demonstrated longer median OS in metastatic breast cancer when combined with local radiotherapy [129]. In addition, fresolimumab treatment decreases biomarkers of thrombospondin-1 and cartilage oligomeric protein and improves clinical symptoms in systemic sclerosis patients [130]. Galunisertib is a TGF-β receptor kinase inhibitor that selectively blocks TGF-β signaling and results in the improvement of OS in pancreatic cancer patients when combined with gemcitabine therapy [131]. Similarly, the combination of galunisertib and sorafenib demonstrated acceptable safety and prolonged OS in patients with advanced hepatocellular carcinoma [132]. Other drugs, such as SHR-1701 (bifunctional anti-PD-L1/TGF-βRII agent) and ACE-536 (TGF-β superfamily ligand trap), have reached phase 3 clinical trials (CTR20210880, NCT04064060, NCT04717414) with promising potential. Although TGF-β pathways contribute to the pathological processes of fibrosis and tumor progression, the physiological functions and side effects of TGF-β cannot be ignored, and the dosing and drug delivery systems of TGF-β-based therapies need further study in the future.
Matrix protein degrading and crosslinking
To reduce the matrix stiffness of the cancer environment, degrading matrix proteins and crosslinking is another good strategy in addition to the elimination of matrix protein production. Therefore, targeting MMPs and LOX has been conducted in clinical trials for cancer therapy.
Targeting MMPs
MMPs are zinc-dependent endopeptidase enzymes that can degrade various ECM proteins [133]. The initial clinical trials were designed based on the theory that ECM provides a biological barrier preventing endothelial cell migration or tumor cell invasion, and activated MMPs can degrade ECM, helping cancer cells break through the matrix barrier and promoting the invasion and metastasis of malignant tumors [134, 135]. Due to the high expression of MMPs in metastatic tissues, targeting MMP may be a viable solution. However, phase 3 clinical trials of inhibitors (Prinomastat, Tanomastat, Marimastat) targeting MMPs have all ended in failure [136,137,138]. Thus, to achieve better cancer therapy targeting MMPs, a comprehensive understanding of the roles of MMPs in cancer progression must be evaluated in depth.
Targeting LOX
The LOX family of enzymes is responsible for matrix remodeling by covalent crosslinking of collagen and elastin at primary and metastatic tumor sites [139]. Due to the high expression of LOX in most types of human cancers, LOX family members have emerged as potential clinical targets for cancer therapy [140].
Until now, an array of LOX family inhibitors, such as BAPN, PXS compounds, tetrathiomolybdate, CCT365623, PAT-1251, AB0023 and simtuzumab, have been developed for tumor therapy. BAPN is the first pan-inhibitor of the LOX family with the features of non-specificity and irreversibility [141]. Although BAPN showed an antitumor effect by suppressing LOX activity in many preclinical studies [142,143,144], no clinical trials have been conducted for BAPN due to the lack of suitable chemical modification sites as well as the non-tumorigenic toxicity and teratogenic effect [139, 145, 146]. PXS compounds are a new generation of oral pan-LOX inhibitors, including PXS-S1A, PXS-S2A, PXS-S1C, PXS-5153A, and PXS-5505. Among these, PXS-5505 is being tested in clinical trials for hepatocellular carcinoma (NCT05109052) and thrombocythemia myelofibrosis (NCT04676529). In addition, AB0023, a neutralizing antibody against LOXL2, can inhibit the activity of LOXL2, reduce activated fibroblasts, and inhibit tumor progression [147]. Similarly, simtuzumab is a humanized monoclonal antibody of AB0023, and has been engaged in phase 2 clinical trials for pancreatic adenocarcinoma and metastatic KRAS mutant colorectal adenocarcinoma, whereas, both studies ended in failure [148, 149]. A possible explanation is that although simtuzumab can prevent further collagen crosslinking, it cannot reverse the crosslinks already present in the ECM. Thus, simtuzumab may be effective in the early stage of malignancy.
Targeting mechanoreceptors and mechanotransducers
To interfere with matrix stiffness-induced signaling, mechanoreceptors and mechanotransducers may be alternative targets in addition to the elimination of CAFs and TGF-β as well as regulators of collagen deposition and crosslinking. Therefore, targeting Piezo1, the TRP family, integrins, FAK, Rho/ROCK, and YAP/TAZ has gained great interest in recent years.
Targeting Piezo1
Piezo1 is a cationic mechanical receptor that is widely regulated by matrix stiffness or mechanical stimulation and is involved in the progression of tumors [150]. Most studies report that Piezo1 is highly expressed in different types of tumors and is associated with poor prognosis [151, 152]. For example, Piezo1 activates integrin-FAK signaling, regulates ECM and reinforces tissue stiffness to promote glioma aggression, while deleting Piezo1 inhibits tumor aggression and prolongs survival [153]. At present, only a few Piezo1 inhibitors (GsMTx4, ruthenium red, Dooku1, gadolinium) have been reported in preclinical studies and no agents are engaged in clinical trials [56, 154,155,156]. The role of Piezo1 inhibitors in tumors is still in the initial stage, and with more in-depth research in the future, the development of drugs targeting Piezo1 may be of great value.
Targeting the TRP family
TRP family ion channels are other types of mechanoreceptors that are widely studied in the sensation of coldness, heat, pain, taste and vision as well as mechanic stimulation. Among them, TRPV, TRPM, and TRPC subfamily members are mostly studied and associated with malignant growth and progression [157], and most of them are overexpressed in different kinds of cancers such as breast cancer, pancreatic cancer, glioblastoma, ovarian cancer, lung cancer, and colon cancer [158,159,160]. In contrast, some studies have found that TRPV4 activation in endothelial cells can inhibit tumor angiogenesis, growth and metastasis by suppressing the expression of VEGFR2, p-ERK, and MMP-9 [161]. In addition, activation of TRPV4 also inhibits the progression of glioma, melanoma, and breast cancer [162,163,164]. At present, most of these results are evaluated in preclinical studies, and a few studies have been conducted in clinical trials when treated with tumors.
D-3263 hydrochloride is an enteric-coated, orally bioavailable TRPM8 agonist and is being tested in a phase 1 clinical trial in patients with advanced solid tumors [NCT00839631]. Similarly, SOR-C13 is a high-affinity antagonist of TRPV6 and was evaluated in a phase 1 clinical trial in advanced solid tumors, suggesting antitumor activity because of stable disease survival [165]. Delta[9]-Tetrahydrocannabinol and nabiximols, nonspecific agonists of TRPV2, were tested in a phase 1 clinical trial in recurrent glioblastoma and both showed antitumor activity [166, 167]. Together, future studies should focus on the exact relationship between the TRP family and tumors to lay a firm theoretical foundation for the design of specific targeted drugs.
Targeting integrins
The integrin family consists of 24 transmembrane glycoprotein members, and a large proportion of integrin subfamilies are overexpressed during cancer progression [168, 169]. Integrin-mediated sensing, stiffening and remodeling of ECM stiffness are crucially important for cancer progression, supporting invasion and drug resistance [168]. Therefore, targeting integrins may reverse the effects of ECM stiffness on tumorigenesis.
Currently, approximately 90 kinds of integrin-based therapeutic agents or imaging drugs have been used in clinical studies, among which 16 drug types, including cilengitide, antiangiotide, etaracizumab, volociximab, intetumumab, abituzumab, BGC-0222, ProAgio, CEND-1, HYD-PEP-06, 7HP-349, ATN-161, SGN-B6A, ABBV-382, OPC-415, and MT-1002, have been reported to target integrin subfamilies of αv, α5β1, αvβ3, αLβ2, α4β1, β6, α3β1, β1, and β7 in clinical trials for cancer treatment [170]. For preclinical research, targeting integrins is quite mature with remarkable antitumor effects [171,172,173]. However, most previous clinical trials targeting integrins are disappointing, even in the phase 3 clinical trial of cilengitide for treating glioblastoma [174,175,176]. Despite the disappointing outcome of targeting integrins in clinical studies, integrins play an important role in tumorigenesis, and further mechanistic research should be conducted between integrins and tumors.
Targeting Rho/ROCK
The Rho-ROCK signaling pathway participates in a variety of key biological processes, including cytoskeleton remodeling, ECM stiffness and cancer progression [85]. In most solid tumors, such as pancreatic cancer, breast cancer, renal cancer, urothelial cancer and osteosarcoma, the Rho-ROCK signal is overexpressed and activated and is often associated with shortened OS [177,178,179]. However, the research of Rho-ROCK signaling in malignant tumors is still insufficient and lacks major breakthroughs. Although some Rho/ROCK inhibitors, such as fasudil (HA-1077), KD025, RKI983, AT13148, verosudil (AR-12286), ripasudil (K-115), have been studied in clinical studies for other diseases, currently, only a few Rho-ROCK inhibitors have been conducted in clinical trials for cancer therapy. For instance, AT13148, an oral dual ROCK-AKT inhibitor, failed in a phase 1 clinical trial in patients with solid tumors due to the narrow therapeutic index and pharmacokinetic profile [180]. Therefore, future works should further clarify the roles of Rho-ROCK in tumors.
Targeting FAK
FAK is a nonreceptor tyrosine kinase that is upregulated in a wide variety of solid cancers (such as head, neck, oral, thyroid, lung, breast, bladder, colorectal, prostatic, hepatocellular carcinomas and ovarian cancer) [181]. Therefore, FAK can be considered a potential target for cancer therapy.
To date, a large number of inhibitors have been designed based on the crystallographic structures of FAK kinase domains such as defactinib, CEP-37440, conteltinib (CT-707), VS-4718, AMP-945, GSK-2256098 and BI-853520, which were all engaged in phase 1/2 clinical trials for cancer therapy [182]. Currently, most clinical studies are in the early stage, and its antitumor effect needs to be verified in subsequent studies. In addition, other attractive FAK inhibitors, such as TAE226, PF-573228 and Y15, were tested in preclinical studies with significant antitumor effects [183,184,185]. At present, although no compounds have been launched on the market, there is great interest in the development of novel FAK inhibitors, which represent a new emerging therapeutic method for cancer treatment.
Targeting YAP/TAZ
The YAP/TAZ pathway can be activated by mechanical cues such as matrix stiffness and cell stretch, and has been shown to promote tumor growth and invasion in multiple human cancers [186]. Although the majority of studies reveal that YAP/TAZ activation is associated with tumor-promoting effects [187,188,189,190], accumulating evidence also demonstrates that YAP/TAZ play vital roles in tumor-suppressive effects in human cancers.
YAP inhibits breast cancer growth by disrupting a TEAD-ERα signaling axis [191, 192]. In addition, YAP inhibits HIF-2α and renal cell carcinoma progression by disrupting the HIF-2α/TEAD signaling complex [193]. Furthermore, YAP1 and WWTR1 expression inhibits the tumor progression of Merkel cell carcinoma [194]. One possible reason for the different conclusions mentioned above may be that TEAD complexes target different enhancers in YAP-low and YAP-high expression cancers [195]. Currently, due to the special structural properties of YAP and TAZ, it is not easy to directly target YAP/TAZ proteins. Therefore, targeting the YAP/TAZ-TEAD complex is a better strategy. Verteporfin, an inhibitor of YAP1, has been shown to inhibit the interaction between YAP1 and TEAD [196], and a phase 1/2 study of visudyne (liposomal verteporfin) in patients with recurrent high-grade EGFR-mutated glioblastoma is recruiting [NCT04590664]. IAG933 and BPI-460372, two kinds of TEAD inhibitors disturbing the YAP-TEAD association, are in the recruitment stage of phase 1 clinical trials for the treatment of patients with advanced mesothelioma and other solid tumors [NCT04857372, NCT05789602]. Other targeting YAP/TAZ-TEAD inhibitors, such as flufenamic acid, TED-347, VGLL4-mimicking peptide, have been used in preclinical studies to evaluate their antitumor effects. Taken together, the effect of YAP/TAZ on various cancers is complex, and inhibitors targeting the YAP/TAZ-TEAD complex need to be further evaluated in clinical studies.
Conclusions and perspectives
Exploring phenotype, function and mechanism links between ECM stiffness and macrophages in the TME will improve the current knowledge of cancer diagnosis, prognosis, and treatment strategies. By using multiple immunohistochemistry, spatial metabolomics, single-cell analysis techniques, and organoids culture together with flow cytometry analysis for tumors of different rigidities, we may capture more comprehensive and realistic conclusions of TAM status in vivo and may provide correct guidance for targeting therapy based on ECM stiffness. It is important to note that matrix stiffness not only directly affects TAM function but also indirectly regulates the TAM phenotype through other types of cells, especially tumor cells and CAFs [197]. When discussing the effect and mechanism of matrix stiffness on the regulation of macrophages, different kinds of biological materials and scaffolds, such as nanoparticles, fibers, hydrogels, and 3D printing with diverse components, have been used to simulate ECM stiffness and often contribute to inconsistencies in experimental results in vitro and in vivo. This phenomenon may be attributed to the large difference between simulated ECM and natural ECM as well as the different materials used in the experiments. At present, more in-depth research is needed to determine the impact of ECM stiffness on TAM function.
Macrophages have traditionally been divided into the pro-inflammatory M1 phenotype and the anti-inflammatory M2 phenotype. However, macrophage classification is complex in reality, and sometimes it is difficult to evaluate the macrophage phenotype based only on common M1- and M2 biomarkers when both types of biomarkers show a consistent trend of change.Currently, no studies have systematically analyze this situation and defined the classification of macrophages when discussing their phenotype and function based on these biomarkers. How to assess the weighted value of both kinds of biomarkers, whether negative feedback regulation leads to consistent trends or only macrophage biomarkers combined with functional detection, is the gold standard to determine macrophage phenotype. Future work requires more robust evidence to unify the consensus of macrophage classification.
ECM stiffness is widely considered an adverse factor that promotes cancer progression by remodeling stromal cells such as TAMs and CAFs. Therefore, targeting ECM stiffness or mechanical cues is an attractive alternative option for cancer therapy. Therefore, reducing matrix protein production by eliminating CAFs and TGF-β, degrading matrix proteins and crosslinking by blocking the enzyme's action of MMPs and LOX, or targeting mechanoreceptors such as Piezo1, TRP, integrins, and mechanotransducers such as Rho/ROCK, FAK, YAP/TAZ, is gaining increasing attention in preclinical and clinical research for cancer therapy. Unfortunately, no drugs (except for erdafitinib) are approved for the above targets of cancer therapy on the market. It seems that one possible reason for this situation is that degradation of matrix proteins can reduce matrix stiffness, which is beneficial to cancer treatment. However, a stiff matrix is also a physical barrier that prevents the invasion and metastasis of cancer cells. Understanding and balancing these contradictory effects is a key problem that urgently needs to be addressed. Taken together, ECM is an extremely complex system, and in the future, the abovementioned core issues need to be well addressed to provide an optimal strategy for cancer therapy.
Availability of data and materials
All data generated or analyzed are included in this published article.
Abbreviations
- ECM:
-
Extracellular matrix
- TAMs:
-
Tumor-associated macrophages
- TME:
-
Tumor microenvironment
- MMPs:
-
Matrix metalloproteinases
- BMDMs:
-
Bone marrow-derived macrophages
- CAFs:
-
Cancer-associated fibroblasts
- CAR:
-
Chimeric antigen receptor
- TRP:
-
Transient receptor potential
- PMA:
-
Phorbol 12-myristate 13-acetate
- YAP:
-
Yes-associated protein
- TAZ:
-
Transcriptional coactivator with PDZ-binding motif
- Rho:
-
Ras homolog family
- ROCK:
-
Rho-associated coiled-coil containing protein serine/threonine kinase
- Cdc42:
-
Cell division cycle 42
- FAK:
-
Focal adhesion kinase
- LOX:
-
Lysyl oxidase
- OS:
-
Overall survival
- PD-L1:
-
Programmed death ligand 1
- FDA:
-
Food and Drug Administration
- TILs:
-
Tumor-infiltrating lymphocytes
- Treg:
-
Regulatory T
- CT-707:
-
Conteltinib
- Visudyne:
-
Liposomal Verteporfin
References
Saraswathibhatla A, Indana D, Chaudhuri O. Cell-extracellular matrix mechanotransduction in 3D. Nat Rev Mol Cell Biol. 2023;24:495–516.
Peltanova B, Raudenska M, Masarik M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol Cancer. 2019;18:63.
Karamanos NK, Piperigkou Z, Passi A, Gotte M, Rousselle P, Vlodavsky I. Extracellular matrix-based cancer targeting. Trends Mol Med. 2021;27:1000–13.
Cox TR. The matrix in cancer. Nat Rev Cancer. 2021;21:217–38.
Jiang Y, Zhang H, Wang J, Liu Y, Luo T, Hua H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol. 2022;15:34.
He Y, Liu T, Dai S, Xu Z, Wang L, Luo F. Tumor-associated extracellular matrix: how to be a potential aide to anti-tumor immunotherapy? Front Cell Dev Biol. 2021;9: 739161.
Du H, Bartleson JM, Butenko S, Alonso V, Liu WF, Winer DA, et al. Tuning immunity through tissue mechanotransduction. Nat Rev Immunol. 2023;23:174–88.
Tadeo I, Berbegall AP, Navarro S, Castel V, Noguera R. A stiff extracellular matrix is associated with malignancy in peripheral neuroblastic tumors. Pediatric Blood Cancer. 2017. https://doi.org/10.1002/pbc.26449.
Pankova D, Jiang Y, Chatzifrangkeskou M, Vendrell I, Buzzelli J, Ryan A, et al. RASSF1A controls tissue stiffness and cancer stem-like cells in lung adenocarcinoma. EMBO J. 2019;38: e100532.
Liverani C, Mercatali L, Cristofolini L, Giordano E, Minardi S, Porta GD, et al. Investigating the mechanobiology of cancer cell-ECM interaction through collagen-based 3D scaffolds. Cell Mol Bioeng. 2017;10:223–34.
Liu XQ, Chen XT, Liu ZZ, Gu SS, He LJ, Wang KP, et al. Biomimetic matrix stiffness modulates hepatocellular carcinoma malignant phenotypes and macrophage polarization through multiple modes of mechanical feedbacks. ACS Biomater Sci Eng. 2020;6:3994–4004.
Robinson A, Han CZ, Glass CK, Pollard JW. Monocyte regulation in homeostasis and malignancy. Trends Immunol. 2021;42:104–19.
Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol. 2022;23:1148–56.
Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44:439–49.
Mantovani A, Allavena P, Sozzani S, Vecchi A, Locati M, Sica A. Chemokines in the recruitment and shaping of the leukocyte infiltrate of tumors. Semin Cancer Biol. 2004;14:155–60.
Chen S, Saeed A, Liu Q, Jiang Q, Xu H, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. 2023;8:207.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20.
Cassetta L, Pollard JW. A timeline of tumour-associated macrophage biology. Nat Rev Cancer. 2023;23:238–57.
Wang N, Wang S, Wang X, Zheng Y, Yang B, Zhang J, et al. Research trends in pharmacological modulation of tumor-associated macrophages. Clin Transl Med. 2021;11: e288.
Petty AJ, Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy. 2017;9:289–302.
Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262:153–66.
Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184(792–809): e23.
Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. 2020;15:123–47.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51.
Wei J, Yao J, Yan M, Xie Y, Liu P, Mao Y, et al. The role of matrix stiffness in cancer stromal cell fate and targeting therapeutic strategies. Acta Biomater. 2022;150:34–47.
Zinger A, Koren L, Adir O, Poley M, Alyan M, Yaari Z, et al. Collagenase nanoparticles enhance the penetration of drugs into pancreatic tumors. ACS Nano. 2019;13:11008–21.
Zhang JY, Zhu WW, Wang MY, Zhai RD, Wang Q, Shen WL, et al. Cancer-associated fibroblasts promote oral squamous cell carcinoma progression through LOX-mediated matrix stiffness. J Transl Med. 2021;19:513.
Simoes FC, Cahill TJ, Kenyon A, Gavriouchkina D, Vieira JM, Sun X, et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat Commun. 2020;11:600.
Chen M, Zhang Y, Zhou P, Liu X, Zhao H, Zhou X, et al. Substrate stiffness modulates bone marrow-derived macrophage polarization through NF-kappaB signaling pathway. Bioactive materials. 2020;5:880–90.
Friedemann M, Kalbitzer L, Franz S, Moeller S, Schnabelrauch M, Simon JC, et al. Instructing human macrophage polarization by stiffness and glycosaminoglycan functionalization in 3D collagen networks. Adv Healthc Mater. 2017. https://doi.org/10.1002/adhm.201600967.
Wang Y, Li Y, Zhong J, Li M, Zhou Y, Lin Q, et al. Tumor-derived Cav-1 promotes pre-metastatic niche formation and lung metastasis in breast cancer. Theranostics. 2023;13:1684–97.
Taufalele PV, Wang W, Simmons AJ, Southard-Smith AN, Chen B, Greenlee JD, et al. Matrix stiffness enhances cancer-macrophage interactions and M2-like macrophage accumulation in the breast tumor microenvironment. Acta Biomater. 2023;163:365–77.
Larsen AMH, Kuczek DE, Kalvisa A, Siersbaek MS, Thorseth ML, Johansen AZ, et al. Collagen density modulates the immunosuppressive functions of macrophages. J Immunol. 2020;205:1461–72.
Lu Y, Jin Z, Hou J, Wu X, Yu Z, Yao L, et al. Calponin 1 increases cancer-associated fibroblasts-mediated matrix stiffness to promote chemoresistance in gastric cancer. Matrix Biol. 2023;115:1–15.
Herrera M, Herrera A, Dominguez G, Silva J, Garcia V, Garcia JM, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013;104:437–44.
Fujii N, Shomori K, Shiomi T, Nakabayashi M, Takeda C, Ryoke K, et al. Cancer-associated fibroblasts and CD163-positive macrophages in oral squamous cell carcinoma: their clinicopathological and prognostic significance. J Oral Pathol Med. 2012;41:444–51.
Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Can Res. 2013;73:3007–18.
Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17:559–72.
Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:131.
Mayer S, Milo T, Isaacson A, Halperin C, Miyara S, Stein Y, et al. The tumor microenvironment shows a hierarchy of cell-cell interactions dominated by fibroblasts. Nat Commun. 2023;14:5810.
Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M, et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med. 2016;213:2315–31.
LaRue MM, Parker S, Puccini J, Cammer M, Kimmelman AC, Bar-Sagi D. Metabolic reprogramming of tumor-associated macrophages by collagen turnover promotes fibrosis in pancreatic cancer. Proc Natl Acad Sci USA. 2022;119: e2119168119.
Shen L, Li H, Shi Y, Wang D, Gong J, Xun J, et al. M2 tumour-associated macrophages contribute to tumour progression via legumain remodelling the extracellular matrix in diffuse large B cell lymphoma. Sci Rep. 2016;6:30347.
Kuziel G, Moore BN, Arendt LM. Obesity and fibrosis: setting the stage for breast cancer. Cancers. 2023;15(11):2929.
Tariq M, Zhang J, Liang G, Ding L, He Q, Yang B. Macrophage polarization: anti-cancer strategies to target tumor-associated macrophage in breast cancer. J Cell Biochem. 2017;118:2484–501.
Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer. 2019;121:837–45.
Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60.
Hill RZ, Loud MC, Dubin AE, Peet B, Patapoutian A. PIEZO1 transduces mechanical itch in mice. Nature. 2022;607:104–10.
Lin YC, Guo YR, Miyagi A, Levring J, MacKinnon R, Scheuring S. Force-induced conformational changes in PIEZO1. Nature. 2019;573:230–4.
Hoffmann EJ, Ponik SM. Biomechanical contributions to macrophage activation in the tumor microenvironment. Front Oncol. 2020;10:787.
Solis AG, Bielecki P, Steach HR, Sharma L, Harman CCD, Yun S, et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature. 2019;573:69–74.
Atcha H, Jairaman A, Holt JR, Meli VS, Nagalla RR, Veerasubramanian PK, et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat Commun. 2021;12:3256.
Geng J, Shi Y, Zhang J, Yang B, Wang P, Yuan W, et al. TLR4 signalling via Piezo1 engages and enhances the macrophage mediated host response during bacterial infection. Nat Commun. 2021;12:3519.
Cai G, Lu Y, Zhong W, Wang T, Li Y, Ruan X, et al. Piezo1-mediated M2 macrophage mechanotransduction enhances bone formation through secretion and activation of transforming growth factor-beta1. Cell Prolif. 2023;56: e13440.
Deng R, Li C, Wang X, Chang L, Ni S, Zhang W, et al. Periosteal CD68(+) F4/80(+) Macrophages Are Mechanosensitive for Cortical Bone Formation by Secretion and Activation of TGF-beta1. Advanced science. 2022;9: e2103343.
Aykut B, Chen R, Kim JI, Wu D, Shadaloey SAA, Abengozar R, et al. Targeting Piezo1 unleashes innate immunity against cancer and infectious disease. Science immunology. 2020;5(50):eabb5168. https://doi.org/10.1126/sciimmunol.abb5168.
Koivisto AP, Belvisi MG, Gaudet R, Szallasi A. Advances in TRP channel drug discovery: from target validation to clinical studies. Nat Rev Drug Discov. 2022;21:41–59.
Zhang M, Ma Y, Ye X, Zhang N, Pan L, Wang B. TRP (transient receptor potential) ion channel family: structures, biological functions and therapeutic interventions for diseases. Signal Transduct Target Ther. 2023;8:261.
Kashio M, Sokabe T, Shintaku K, Uematsu T, Fukuta N, Kobayashi N, et al. Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc Natl Acad Sci USA. 2012;109:6745–50.
Schappe MS, Szteyn K, Stremska ME, Mendu SK, Downs TK, Seegren PV, et al. Chanzyme TRPM7 Mediates the Ca(2+) Influx Essential for Lipopolysaccharide-Induced Toll-Like Receptor 4 Endocytosis and Macrophage Activation. Immunity. 2018;48(59–74): e5.
Chauhan A, Sun Y, Sukumaran P, Quenum Zangbede FO, Jondle CN, Sharma A, et al. M1 macrophage polarization is dependent on TRPC1-mediated calcium entry. iScience. 2018;8:85–102.
Dutta B, Goswami R, Rahaman SO. TRPV4 plays a role in matrix stiffness-induced macrophage polarization. Front Immunol. 2020;11: 570195.
Wong NR, Mohan J, Kopecky BJ, Guo S, Du L, Leid J, et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity. 2021;54(2072–88): e7.
Riazanski V, Gabdoulkhakova AG, Boynton LS, Eguchi RR, Deriy LV, Hogarth DK, et al. TRPC6 channel translocation into phagosomal membrane augments phagosomal function. Proc Natl Acad Sci USA. 2015;112:E6486–95.
Khalil M, Babes A, Lakra R, Forsch S, Reeh PW, Wirtz S, et al. Transient receptor potential melastatin 8 ion channel in macrophages modulates colitis through a balance-shift in TNF-alpha and interleukin-10 production. Mucosal Immunol. 2016;9:1500–13.
Schilling T, Miralles F, Eder C. TRPM7 regulates proliferation and polarisation of macrophages. J Cell Sci. 2014;127:4561–6.
Huse M. Mechanical forces in the immune system. Nat Rev Immunol. 2017;17:679–90.
Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15:802–12.
Sun Z, Guo SS, Fassler R. Integrin-mediated mechanotransduction. J Cell Biol. 2016;215:445–56.
Zhang Y, Zhu L, Hong J, Chen C. Extracellular matrix of early pulmonary fibrosis modifies the polarization of alveolar macrophage. Int Immunopharmacol. 2022;111: 109179.
Blakney AK, Swartzlander MD, Bryant SJ. The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. J Biomed Mater Res Part A. 2012;100:1375–86.
Hu Y, Li H, Wang W, Sun F, Wu C, Chen W, et al. Molecular force imaging reveals that integrin-dependent mechanical checkpoint regulates fcgamma-receptor-mediated phagocytosis in macrophages. Nano Lett. 2023;23:5562–72.
Xing X, Wang Y, Zhang X, Gao X, Li M, Wu S, et al. Matrix stiffness-mediated effects on macrophages polarization and their LOXL2 expression. FEBS J. 2021;288:3465–77.
Wang H, Morales RT, Cui X, Huang J, Qian W, Tong J, et al. A photoresponsive hyaluronan hydrogel nanocomposite for dynamic macrophage immunomodulation. Adv Healthcare Mater. 2019;8: e1801234.
Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–312.
Tremblay AM, Camargo FD. Hippo signaling in mammalian stem cells. Semin Cell Dev Biol. 2012;23:818–26.
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–83.
Dupont S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp Cell Res. 2016;343:42–53.
Meli VS, Atcha H, Veerasubramanian PK, Nagalla RR, Luu TU, Chen EY, et al. YAP-mediated mechanotransduction tunes the macrophage inflammatory response. Sci Adv. 2020;6(49):eabb8471. https://doi.org/10.1126/sciadv.abb8471.
Chim LK, Williams IL, Bashor CJ, Mikos AG. Tumor-associated macrophages induce inflammation and drug resistance in a mechanically tunable engineered model of osteosarcoma. Biomaterials. 2023;296: 122076.
Gultekin O, Gonzalez-Molina J, Hardell E, Moyano-Galceran L, Mitsios N, Mulder J, et al. FOXP3+ T cells in uterine sarcomas are associated with favorable prognosis, low extracellular matrix expression and reduced YAP activation. NPJ Precis Oncol. 2021;5:97.
Wang C, Shen N, Guo Q, Tan X, He S. YAP/STAT3 inhibited CD8(+) T cells activity in the breast cancer immune microenvironment by inducing M2 polarization of tumor-associated macrophages. Cancer Med. 2023;12:16295–309.
Ji D, Jia J, Cui X, Li Z, Wu A. FAP promotes metastasis and chemoresistance via regulating YAP1 and macrophages in mucinous colorectal adenocarcinoma. iScience. 2023;26:106600.
Chin VT, Nagrial AM, Chou A, Biankin AV, Gill AJ, Timpson P, et al. Rho-associated kinase signalling and the cancer microenvironment: novel biological implications and therapeutic opportunities. Expert Rev Mol Med. 2015;17: e17.
Porazinski S, Parkin A, Pajic M. Rho-ROCK signaling in normal physiology and as a key player in shaping the tumor microenvironment. Adv Exp Med Biol. 2020;1223:99–127.
Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35.
Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79.
Wei L, Surma M, Shi S, Lambert-Cheatham N, Shi J. Novel insights into the Roles of Rho kinase in cancer. Arch Immunol Ther Exp. 2016;64:259–78.
Johan MZ, Samuel MS. Rho-ROCK signaling regulates tumor-microenvironment interactions. Biochem Soc Trans. 2019;47:101–8.
Tu PC, Pan YL, Liang ZQ, Yang GL, Wu CJ, Zeng L, et al. Mechanical stretch promotes macrophage polarization and inflammation via the RhoA-ROCK/NF-kappaB Pathway. Biomed Res Int. 2022;2022:6871269.
Sridharan R, Cavanagh B, Cameron AR, Kelly DJ, O’Brien FJ. Material stiffness influences the polarization state, function and migration mode of macrophages. Acta Biomater. 2019;89:47–59.
Xu Y, Ying L, Lang JK, Hinz B, Zhao R. Modeling mechanical activation of macrophages during pulmonary fibrogenesis for targeted anti-fibrosis therapy. bioRxiv. 2023. https://doi.org/10.1101/2023.07.19.549794.
Guiet R, Van Goethem E, Cougoule C, Balor S, Valette A, Al Saati T, et al. The process of macrophage migration promotes matrix metalloproteinase-independent invasion by tumor cells. J Immunol. 2011;187:3806–14.
Van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, Le Cabec V. Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. J Immunol. 2010;184:1049–61.
Dawson JC, Serrels A, Stupack DG, Schlaepfer DD, Frame MC. Targeting FAK in anticancer combination therapies. Nat Rev Cancer. 2021;21:313–24.
Tan X, Yan Y, Song B, Zhu S, Mei Q, Wu K. Focal adhesion kinase: from biological functions to therapeutic strategies. Exp Hematol Oncol. 2023;12:83.
Wang Q, Xie J, Zhou C, Lai W. Substrate stiffness regulates the differentiation profile and functions of osteoclasts via cytoskeletal arrangement. Cell Prolif. 2022;55: e13172.
Lu Z, Zheng S, Liu C, Wang X, Zhang G, Wang F, et al. S100A7 as a potential diagnostic and prognostic biomarker of esophageal squamous cell carcinoma promotes M2 macrophage infiltration and angiogenesis. Clin Transl Med. 2021;11: e459.
Shan S, Fang B, Zhang Y, Wang C, Zhou J, Niu C, et al. Mechanical stretch promotes tumoricidal M1 polarization via the FAK/NF-kappaB signaling pathway. FASEB J. 2019;33:13254–66.
Shiratsuchi H, Basson MD. Extracellular pressure stimulates macrophage phagocytosis by inhibiting a pathway involving FAK and ERK. Am J Physiol Cell Physiol. 2004;286:C1358–66.
Chen W, Yang A, Jia J, Popov YV, Schuppan D, You H. Lysyl Oxidase (LOX) family members: rationale and their potential as therapeutic targets for liver fibrosis. Hepatology. 2020;72:729–41.
Wei X, Lou H, Zhou D, Jia Y, Li H, Huang Q, et al. TAGLN mediated stiffness-regulated ovarian cancer progression via RhoA/ROCK pathway. J Exp Clin Cancer Res. 2021;40:292.
Alonso-Nocelo M, Ruiz-Canas L, Sancho P, Gorgulu K, Alcala S, Pedrero C, et al. Macrophages direct cancer cells through a LOXL2-mediated metastatic cascade in pancreatic ductal adenocarcinoma. Gut. 2023;72:345–59.
Maller O, Drain AP, Barrett AS, Borgquist S, Ruffell B, Zakharevich I, et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nat Mater. 2021;20:548–59.
Tan HY, Wang N, Zhang C, Chan YT, Yuen MF, Feng Y. Lysyl oxidase-like 4 fosters an immunosuppressive microenvironment during hepatocarcinogenesis. Hepatology. 2021;73:2326–41.
Yin H, Wang Y, Wu Y, Zhang X, Zhang X, Liu J, et al. EZH2-mediated epigenetic silencing of miR-29/miR-30 targets LOXL4 and contributes to tumorigenesis, metastasis, and immune microenvironment remodeling in breast cancer. Theranostics. 2020;10:8494–512.
Takemoto R, Kamiya T, Hara H, Adachi T. Lysyl oxidase expression is regulated by the H3K27 demethylase Jmjd3 in tumor-associated M2-like macrophages. J Clin Biochem Nutr. 2020;66:110–5.
Liang D, Liu L, Zhao Y, Luo Z, He Y, Li Y, et al. Targeting extracellular matrix through phytochemicals: a promising approach of multi-step actions on the treatment and prevention of cancer. Front Pharmacol. 2023;14:1186712.
Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15:366–81.
Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282–301.
Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol. 2021;18:792–804.
Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discovery. 2019;18:99–115.
Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020;17:487–505.
Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8:3932–48.
Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P, et al. NOX4 inhibition potentiates immunotherapy by overcoming cancer-associated fibroblast-mediated CD8 T-cell exclusion from tumors. Can Res. 2020;80:1846–60.
Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381:338–48.
Liow E, Howard N, Jung CH, Pope B, Campbell BK, Nguyen A, et al. Phase 2 study of neoadjuvant FGFR inhibition and androgen deprivation therapy prior to prostatectomy. Clin Genitourin Cancer. 2022;20:452–8.
Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–34.
McAndrews KM, Vazquez-Arreguin K, Kwak C, Sugimoto H, Zheng X, Li B, et al. alphaSMA(+) fibroblasts suppress Lgr5(+) cancer stem cells and restrain colorectal cancer progression. Oncogene. 2021;40:4440–52.
Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. 2015;33:4284–92.
Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, et al. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J Clin Oncol. 2019;37:1062–9.
Van Cutsem E, Tempero MA, Sigal D, Oh DY, Fazio N, Macarulla T, et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol. 2020;38:3185–94.
Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–23.
Luo H, Xia X, Huang LB, An H, Cao M, Kim GD, et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat Commun. 2022;13:6619.
Colak S, Ten Dijke P. Targeting TGF-beta signaling in cancer. Trends in cancer. 2017;3:56–71.
Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. 2019;50:924–40.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–8.
Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-beta in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. 2019;8(11):1419. https://doi.org/10.3390/cells8111419.
Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, et al. Focal irradiation and systemic TGFbeta blockade in metastatic breast cancer. Clin Cancer Res. 2018;24:2493–504.
Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Investig. 2015;125:2795–807.
Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. TGFbeta receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer. Cancer Chemother Pharmacol. 2019;83:975–91.
Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, Merle P, et al. A phase 2 study of galunisertib (TGF-beta1 receptor Type I Inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin Transl Gastroenterol. 2019;10: e00056.
Li M, Yan T, Cai Y, Wei Y, Xie Q. Expression of matrix metalloproteinases and their association with clinical characteristics of solid tumors. Gene. 2023;850: 146927.
Sagara A, Miura S, Kobinata A, Naganawa R, Yaginuma S, Saito S, et al. COL8A1 enhances the invasion/metastasis in MDA-MB-231 cells via the induction of IL1B and MMP1 expression. Biochem Biophys Res Commun. 2023;642:145–53.
Zhao Z, Yang W, Kong R, Zhang Y, Li L, Song Z, et al. circEIF3I facilitates the recruitment of SMAD3 to early endosomes to promote TGF-beta signalling pathway-mediated activation of MMPs in pancreatic cancer. Mol Cancer. 2023;22:152.
Bissett D, O’Byrne KJ, von Pawel J, Gatzemeier U, Price A, Nicolson M, et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J Clin Oncol. 2005;23:842–9.
Hirte H, Vergote IB, Jeffrey JR, Grimshaw RN, Coppieters S, Schwartz B, et al. A phase III randomized trial of BAY 12–9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: a National Cancer Institute of Canada Clinical Trials Group Study. Gynecol Oncol. 2006;102:300–8.
Sparano JA, Bernardo P, Stephenson P, Gradishar WJ, Ingle JN, Zucker S, et al. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J Clin Oncol. 2004;22:4683–90.
Wang W, Wang X, Yao F, Huang C. Lysyl oxidase family proteins: prospective therapeutic targets in cancer. Int J Mol Sci. 2022;20:12270. https://doi.org/10.3390/ijms232012270.
Setargew YFI, Wyllie K, Grant RD, Chitty JL, Cox TR. Targeting lysyl oxidase family meditated matrix cross-linking as an anti-stromal therapy in solid tumours. Cancers. 2021;13(3):491.
Hajdu I, Kardos J, Major B, Fabo G, Lorincz Z, Cseh S, et al. Inhibition of the LOX enzyme family members with old and new ligands. Selectivity analysis revisited. Bioorg Med Chem Lett. 2018;28(18):3113–8. https://doi.org/10.1016/j.bmcl.2018.07.001.
Zhu S, Zhang T, Gao H, Jin G, Yang J, He X, et al. Combination therapy of lox inhibitor and stimuli-responsive drug for mechanochemically synergistic breast cancer treatment. Adv Healthcare Mater. 2023;12: e2300103.
Chen Y, Li P, Chen X, Yan R, Zhang Y, Wang M, et al. Endoplasmic reticulum-mitochondrial calcium transport contributes to soft extracellular matrix-triggered mitochondrial dynamics and mitophagy in breast carcinoma cells. Acta Biomater. 2023;169:192–208.
Zhang C, Xu M, Zeng Z, Wei X, He S, Huang J, et al. A polymeric extracellular matrix nanoremodeler for activatable cancer photo-immunotherapy. Angew Chem. 2023;62: e202217339.
Gansner JM, Mendelsohn BA, Hultman KA, Johnson SL, Gitlin JD. Essential role of lysyl oxidases in notochord development. Dev Biol. 2007;307:202–13.
van Boxtel AL, Kamstra JH, Fluitsma DM, Legler J. Dithiocarbamates are teratogenic to developing zebrafish through inhibition of lysyl oxidase activity. Toxicol Appl Pharmacol. 2010;244:156–61.
Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16:1009–17.
Benson AB 3rd, Wainberg ZA, Hecht JR, Vyushkov D, Dong H, Bendell J, et al. A phase II randomized, double-blind, placebo-controlled study of simtuzumab or placebo in combination with gemcitabine for the first-line treatment of pancreatic adenocarcinoma. Oncologist. 2017;22:241-e15.
Hecht JR, Benson AB 3rd, Vyushkov D, Yang Y, Bendell J, Verma U. A phase II, randomized, double-blind, placebo-controlled study of simtuzumab in combination with folfiri for the second-line treatment of metastatic kras mutant colorectal adenocarcinoma. Oncologist. 2017;22:243-e23.
Zhuang C, Gould JE, Enninful A, Shao S, Mak M. Biophysical and mechanobiological considerations for T-cell-based immunotherapy. Trends Pharmacol Sci. 2023;44:366–78.
Karska J, Kowalski S, Saczko J, Moisescu MG, Kulbacka J. Mechanosensitive ion channels and their role in cancer cells. Membranes. 2023;13(2):167. https://doi.org/10.3390/membranes13020167.
Zhu Z, Li W, Gong M, Wang L, Yue Y, Qian W, et al. Piezo1 act as a potential oncogene in pancreatic cancer progression. Life Sci. 2022;310: 121035.
Chen X, Wanggou S, Bodalia A, Zhu M, Dong W, Fan JJ, et al. A feedforward mechanism mediated by mechanosensitive ion channel PIEZO1 and tissue mechanics promotes glioma aggression. Neuron. 2018;100(799–815): e7.
Shahidullah M, Rosales JL, Delamere N. Activation of Piezo1 Increases Na, K-ATPase-Mediated Ion Transport in Mouse Lens. Int Mol Sci. 2022;23(21):12870.
Dela Paz NG, Frangos JA. Yoda1-induced phosphorylation of Akt and ERK1/2 does not require Piezo1 activation. Biochem Biophys Res Commun. 2018;497:220–5.
Hatem A, Poussereau G, Gachenot M, Peres L, Bouyer G, Egee S. Dual action of Dooku1 on PIEZO1 channel in human red blood cells. Front Physiol. 2023;14:1222983.
Siveen KS, Nizamuddin PB, Uddin S, Al-Thani M, Frenneaux MP, Janahi IA, et al. TRPV2: a cancer biomarker and potential therapeutic target. Dis Markers. 2020;2020:8892312.
Yang SL, Cao Q, Zhou KC, Feng YJ, Wang YZ. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene. 2009;28:1320–8.
Chen SJ, Hoffman NE, Shanmughapriya S, Bao L, Keefer K, Conrad K, et al. A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia-inducible factor (HIF)-1/2alpha. J Biol Chem. 2014;289:36284–302.
Zhong T, Zhang W, Guo H, Pan X, Chen X, He Q, et al. The regulatory and modulatory roles of TRP family channels in malignant tumors and relevant therapeutic strategies. Acta Pharmaceutica Sinica B. 2022;12:1761–80.
Kanugula AK, Adapala RK, Jamaiyar A, Lenkey N, Guarino BD, Liedtke W, et al. Endothelial TRPV4 channels prevent tumor growth and metastasis via modulation of tumor angiogenesis and vascular integrity. Angiogenesis. 2021;24:647–56.
Peters AA, Jamaludin SYN, Yapa K, Chalmers S, Wiegmans AP, Lim HF, et al. Oncosis and apoptosis induction by activation of an overexpressed ion channel in breast cancer cells. Oncogene. 2017;36:6490–500.
Olivan-Viguera A, Garcia-Otin AL, Lozano-Gerona J, Abarca-Lachen E, Garcia-Malinis AJ, Hamilton KL, et al. Pharmacological activation of TRPV4 produces immediate cell damage and induction of apoptosis in human melanoma cells and HaCaT keratinocytes. PLoS ONE. 2018;13: e0190307.
Huang T, Xu T, Wang Y, Zhou Y, Yu D, Wang Z, et al. Cannabidiol inhibits human glioma by induction of lethal mitophagy through activating TRPV4. Autophagy. 2021;17:3592–606.
Fu S, Hirte H, Welch S, Ilenchuk TT, Lutes T, Rice C, et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2017;35:324–33.
Guzman M, Duarte MJ, Blazquez C, Ravina J, Rosa MC, Galve-Roperh I, et al. A pilot clinical study of Delta9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006;95:197–203.
Twelves C, Sabel M, Checketts D, Miller S, Tayo B, Jove M, et al. A phase 1b randomised, placebo-controlled trial of nabiximols cannabinoid oromucosal spray with temozolomide in patients with recurrent glioblastoma. Br J Cancer. 2021;124:1379–87.
Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18:533–48.
Xiong J, Yan L, Zou C, Wang K, Chen M, Xu B, et al. Integrins regulate stemness in solid tumor: an emerging therapeutic target. J Hematol Oncol. 2021;14:177.
Pang X, He X, Qiu Z, Zhang H, Xie R, Liu Z, et al. Targeting integrin pathways: mechanisms and advances in therapy. Signal Transduct Target Ther. 2023;8:1.
Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature. 2014;511:319–25.
Hosen N, Matsunaga Y, Hasegawa K, Matsuno H, Nakamura Y, Makita M, et al. The activated conformation of integrin beta(7) is a novel multiple myeloma-specific target for CAR T cell therapy. Nat Med. 2017;23:1436–43.
de Hurtado Mendoza T, Mose ES, Botta GP, Braun GB, Kotamraju VR, French RP, et al. Tumor-penetrating therapy for beta5 integrin-rich pancreas cancer. Nature Commun. 2021;12:1541.
Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1100–8.
Hussain M, Le Moulec S, Gimmi C, Bruns R, Straub J, Miller K, et al. Differential effect on bone lesions of targeting integrins: randomized phase II trial of abituzumab in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2016;22:3192–200.
Hersey P, Sosman J, O’Day S, Richards J, Bedikian A, Gonzalez R, et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or − dacarbazine in patients with stage IV metastatic melanoma. Cancer. 2010;116:1526–34.
Kamai T, Tsujii T, Arai K, Takagi K, Asami H, Ito Y, et al. Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin Cancer Res. 2003;9:2632–41.
Patel RA, Forinash KD, Pireddu R, Sun Y, Sun N, Martin MP, et al. RKI-1447 is a potent inhibitor of the Rho-associated ROCK kinases with anti-invasive and antitumor activities in breast cancer. Can Res. 2012;72:5025–34.
Rath N, Morton JP, Julian L, Helbig L, Kadir S, McGhee EJ, et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol Med. 2017;9:198–218.
McLeod R, Kumar R, Papadatos-Pastos D, Mateo J, Brown JS, Garces AHI, et al. First-in-human study of AT13148, a dual ROCK-AKT inhibitor in patients with solid Tumors. Clin Cancer Res. 2020;26:4777–84.
Wu X, Wang J, Liang Q, Tong R, Huang J, Yang X, et al. Recent progress on FAK inhibitors with dual targeting capabilities for cancer treatment. Biomed Pharmacother. 2022;151:113116.
Spallarossa A, Tasso B, Russo E, Villa C, Brullo C. The development of fak inhibitors: a five-year update. Int J Mol Sci. 2022;23(12):6381. https://doi.org/10.3390/ijms23126381.
Zhang H, Schaefer A, Wang Y, Hodge RG, Blake DR, Diehl JN, et al. Gain-of-function RHOA mutations promote focal adhesion kinase activation and dependency in diffuse gastric cancer. Cancer Discov. 2020;10:288–305.
Romito I, Porru M, Braghini MR, Pompili L, Panera N, Crudele A, et al. Focal adhesion kinase inhibitor TAE226 combined with Sorafenib slows down hepatocellular carcinoma by multiple epigenetic effects. J Exp Clin Cancer Res. 2021;40:364.
Zhang H, Shao H, Golubovskaya VM, Chen H, Cance W, Adjei AA, et al. Efficacy of focal adhesion kinase inhibition in non-small cell lung cancer with oncogenically activated MAPK pathways. Br J Cancer. 2016;115:203–11.
Mokhtari RB, Ashayeri N, Baghaie L, Sambi M, Satari K, Baluch N, et al. The hippo pathway effectors YAP/TAZ-TEAD oncoproteins as emerging therapeutic targets in the tumor microenvironment. Cancers. 2023;15(23):5497.
Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218–27.
Wu BK, Mei SC, Chen EH, Zheng Y, Pan D. YAP induces an oncogenic transcriptional program through TET1-mediated epigenetic remodeling in liver growth and tumorigenesis. Nat Genet. 2022;54:1202–13.
Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci USA. 2012;109:E2441-50.
Song S, Honjo S, Jin J, Chang SS, Scott AW, Chen Q, et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin Cancer Res. 2015;21:2580–90.
Zhu C, Li L, Zhang Z, Bi M, Wang H, Su W, et al. A non-canonical role of YAP/TEAD is required for activation of estrogen-regulated enhancers in breast cancer. Mol Cell. 2019;75(791–806): e8.
Li X, Zhuo S, Zhuang T, Cho YS, Wu G, Liu Y, et al. YAP inhibits ERalpha and ER(+) breast cancer growth by disrupting a TEAD-ERalpha signaling axis. Nat Commun. 2022;13:3075.
Li X, Cho YS, Zhu J, Zhuo S, Jiang J. The Hippo pathway effector YAP inhibits HIF2 signaling and ccRCC tumor growth. Cell discovery. 2022;8:103.
Frost TC, Gartin AK, Liu M, Cheng J, Dharaneeswaran H, Keskin DB, et al. YAP1 and WWTR1 expression inversely correlates with neuroendocrine markers in Merkel cell carcinoma. J Clin Invest. 2023;133(5):e157171. https://doi.org/10.1172/JCI157171.
Pearson JD, Huang K, Pacal M, McCurdy SR, Lu S, Aubry A, et al. Binary pan-cancer classes with distinct vulnerabilities defined by pro- or anti-cancer YAP/TEAD activity. Cancer Cell. 2021;39(1115–34): e12.
Nagashima S, Bao Y, Hata Y. The hippo pathway as drug targets in cancer therapy and regenerative medicine. Curr Drug Targets. 2017;18:447–54.
Zhang S, Regan K, Najera J, Grinstaff MW, Datta M, Nia HT. The peritumor microenvironment: physics and immunity. Trends in cancer. 2023;9:609–23.
Acknowledgements
We would like to thank BioRender (www. biorender.com) for assistance in creating figures.
Funding
This study was supported by the National Key Research and Development Program of China (2021YFC2009105, 2022YFC2704100), the National Natural Science Foundation of China (82001514, 82303671), the Major Science and Technology Foundation of Hubei Provincial (2023BCA002), Fundamental Research Funds for the Central Universities (2042023kf0084), and the Program of Excellent Doctor of Zhongnan Hospital of Wuhan University (ZNYB2023005).
Author information
Authors and Affiliations
Contributions
JX and RX structured and wrote the manuscript. JZ, QZ and ML were responsible for designing and drawing figures and tables. MW played a role in the design of the study, manuscript writing, and figure preparation. FL and WZ raised the conceptualization and supervised this work. All the authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xiong, J., Xiao, R., Zhao, J. et al. Matrix stiffness affects tumor-associated macrophage functional polarization and its potential in tumor therapy. J Transl Med 22, 85 (2024). https://doi.org/10.1186/s12967-023-04810-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04810-3