
Abstract
Focal adhesion kinase (FAK), a nonreceptor cytoplasmic tyrosine kinase, is a vital participant in primary cellular functions, such as proliferation, survival, migration, and invasion. In addition, FAK regulates cancer stem cell activities and contributes to the formation of the tumor microenvironment (TME). Importantly, increased FAK expression and activity are strongly associated with unfavorable clinical outcomes and metastatic characteristics in numerous tumors. In vitro and in vivo studies have demonstrated that modulating FAK activity by application of FAK inhibitors alone or in combination treatment regimens could be effective for cancer therapy. Based on these findings, several agents targeting FAK have been exploited in diverse preclinical tumor models. This article briefly describes the structure and function of FAK, as well as research progress on FAK inhibitors in combination therapies. We also discuss the challenges and future directions regarding anti-FAK combination therapies.
Similar content being viewed by others
Background
Focal adhesion kinase is a tyrosine kinase composed of 1052 amino acids with a molecular weight of 125kD [1]. FAK is a crucial regulator of vital cellular processes, including cell adhesion [2], migration [3], proliferation [4], and survival [5]. Such processes have significant implications for the development and progression of cancer. Moreover, multiple studies have confirmed FAK upregulation in a diverse range of human malignancies, including colorectal, lung, ovarian, neck, bladder, breast, and esophageal cancers [6]. In addition, by promoting tumor angiogenesis, epithelial-mesenchymal transformation, cancer stemness, and immunomodulatory capacity [7,8,9], FAK significantly contributes to malignant progression. The available evidence suggests that FAK represents a promising target for cancer therapy. This article aims to provide an overview of the significant impact of FAK on both cancer cells and cancer-associated cells within tumors while also offering a comprehensive discussion of the recent advances made in developing therapeutic agents that target FAK and the combination of these agents with other approaches.
FAK structure and activation
Molecular structure of FAK
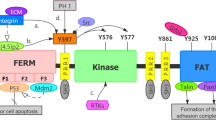
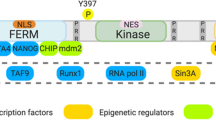
FAK is composed of three distinct domains: a four-point-one-ezrin-radixin-moesin (FERM) domain, a kinase domain, and a C-terminal focal adhesion-targeting (FAT) domain [10]. To target different sites on FAK, inhibitors specific to each domain have been developed (Fig. 1). The FERM domain is further divided into F1, F2, and F3 subdomains. F1 contains a nuclear export sequence, and F2 contains a nuclear localization sequence [11]. As the names indicate, these sequences are important for the nuclear transport of FAK. Over the past few years, research has emphasized the significance of nuclear FAK in the regulation of gene expression; it interacts with distinct E3 ligases to induce the degradation of transcription factors [12]. For example, the F1 and F2 lobes interact with p53, followed by the combination of the F3 lobe with murine double minute2 (Mdm2) and subsequent p53 ubiquitination and degradation, thus facilitating cancer cell proliferation and inhibiting apoptosis [13]. Nuclear FAK also alters expression of GATA4 and IL-33, suppressing the inflammatory response and inducing immune escape [14,15,16,17]. Aside from controlling FAK transportation from the cytoplasm to the nucleus, the FERM domain also has a crucial function in triggering the activation of FAK. For example, the link between the FERM domain and the kinase domain prevents FAK activation by blocking the autophosphorylation of Y397, which is the sole autophosphorylation site of FAK [18]. The FERM domain contains binding sites for numerous proteins, such as integrin [19], growth factor receptors [20, 21], and G-protein coupled estrogen receptors [22], and such interactions trigger downstream signaling cascades and induces FAK activation. The KAKTLRK sequence, which is in the F2 lobe, also participates in FAK activation [23].

Schematic diagram of the structural domain of FAK. FAK comprises three primary components, including a central kinase domain, a FERM domain on the N-terminal side, and a FAT domain on the C-terminal side. The kinase domain, which is crucial for catalytic activity, is flanked by three proline-rich regions that are responsible for protein‒protein interactions. The FAK phosphorylation sites and important binding proteins that regulate FAK activity and downstream signaling are highlighted in the diagram
The bilobal central kinase domain, which contains catalytic sites and ATP-binding sites, is highly homological with other tyrosine kinases, particularly proline-rich tyrosine kinase 2 (Pyk2), and serves as the major component for FAK enzymatic activity [24]. Nuclear export signal 2 (NES2), which is regarded as the only biologically active nuclear export signal (NES) exerting nuclear export activities, is located in the central kinase domain [25].
The C-terminal region, which resembles a bundle composed of four helices, is primarily comprised of the FAT domain and has a significant role in controlling the activation of FAK. Through its interaction with proteins associated with focal adhesions such as paxillin and talin, the FAT domain prompts the recruitment and activation of FAK at the site of focal adhesions [26, 27]. This interaction is pivotal for FA assembly and turnover, which influences cell motility. In contrast, other studies have indicated that the FAT domain functions as an inhibitor of FAK activation by competing with the FERM domain for the binding of specific intracellular receptors and inducing dephosphorylation [28].
In addition to the three major domains, there are some special residues regulating FAK activation and function. FAK contains three proline-rich regions (PRRs), PRR1 localizes in the N-terminal domain, and PRR2/3 localizes in the C-terminal domain next to the FAT region. PRRs attach to proteins that contain Src homology 3 (SH3) domains, including small GTPases and p130Cas, to regulate kinase activity and support the cytoskeleton [29]. Additionally, there are at minimum six tyrosine residues that undergo phosphorylation throughout the entire region: Y397, Y407, Y576, Y577, Y861, and Y925. The autophosphorylation of Y397, which is situated at the N-terminus of the FERM structural domain, is vital for the activation of FAK [30]. Phosphorylated Y397 provides a binding site for Src-family kinases and other proteins containing the SH2 domain [31], which also interact with other tyrosine residues and are pivotal for FAK catalytic activity. Located in the activation loop of the kinase domain [32, 33], Y576 and Y577 positively regulate FAK kinase function, while another kinase domain-located tyrosine phosphorylation site, Y407, inhibits FAK activity. Additionally, Y861 and Y925 are in the C-terminal domains. Phosphorylated Y925 interacts with the SH2 domain of the adaptor protein GRB2 to trigger downstream Ras/MAPK signaling and induce integrin internalization and focal adhesion disassembly [34]. This FAK-GRB2-MAPK linkage was also demonstrated to be essential for tumor angiogenesis [35].
The mechanism and regulation of FAK activation
FAK is a vital mediator in the transmission of signals from the extracellular matrix (ECM) to the cell cytoplasm. It regulates pivotal cellular functions, including cell survival, proliferation, migration, and invasion (Fig. 2). In response to upstream stimuli, FAK initiates downstream signaling cascades, thereby prompting a series of events. Additionally, FAK is versatile as a signaling molecule because it can demonstrate kinase-dependent or kinase-independent activity [36]. Stimuli, integrin signaling activation, in particular, disrupts the inhibitory interaction between FERM and kinase domains, which leads to FAK dimerization and causes subsequent Y397 autophosphorylation [28]. The extensively phosphorylated Y397 has high affinity for the SH2 domain of kinases belonging to the Src family. When this interaction occurs, these kinases bind to and phosphorylate Y576 and Y577, which are positioned in the activation loop, to achieve complete activation of FAK and endow it with its enzymatic function [24].

FAK-mediated signaling cascades involved in tumor progression. FAK activation is multifaceted and can be mediated by various factors, such as integrins, receptor tyrosine kinases (RTKs), mechanical stimuli, cytokines, G-protein-coupled receptors (GPCRs), and a change in intracellular pH (H+). Upon phosphorylation, FAK may induce the activation of different transduction pathways, including RAS/RAF/ERK, JNK, YAP, and PI3K/AKT/mTOR signaling. This process can lead to the regulation of relevant oncogenes, which in turn supports cancer cell survival. FAK also exerts nuclear functions, acting as a scaffold for p53 and Mdm2 while also promoting the polyubiquitination and degradation of p53. In doing so, FAK again promotes resistance to apoptosis. As shown in the diagram, the highlighted red boxes indicate targets of interest for the development of combination therapy using FAK inhibitors
The activation of FAK is regulated by various internal and external factors, which mainly target the FERM domain to induce conformational changes, thus relieving the autoinhibitory structure between the FERM and the kinase domain and stimulating FAK activation. In addition to integrin, the uncovered binding partners of the FERM domain include extracellular matrix, phosphoinositide lipids, diacylglycerol kinase α, serine/threonine kinase PKCθ, and membrane-associated proteins such as tetraspanin transmembrane 4 L6 family member 5 (TM4LFM5) and EMP2 [24, 28, 37,38,39]. Intriguingly, glutathione peroxidase-1 can bind to FAK and prevent H2O2-induced oxidative inactivation of FAK [40]. Furthermore, growth factor receptors, including the Met receptor for hepatocyte growth factor, epidermal growth factor receptor, and platelet-derived growth factor receptor, enable conformational changes in the FERM domain by phosphorylating either the Y397 or Y197 sites [41, 42]. Other stimuli, such as elevated intracellular pH and increased matrix stiffness or forces, which occur during cancer progression, also trigger Y397 phosphorylation and FAK activation [43, 44]. Recently, it was revealed that FAK expression and activation are epigenetically regulated [45]. For example, microRNA miR-15b-5p inhibits FAK expression by binding to the 3′UTR of FAK mRNA. Moreover, intercellular adhesion molecule-1 suppresses miR-15b-5p activity and stimulates endothelial cell proliferation and migration [46]. The interaction between long noncoding RNA (lncRNA) MIR4435‑2HG and ganglioside synthesis enzyme ST8SIA1 induces the activation of FAK and downstream AKT/β‑catenin signaling, thus promoting prostate cancer cell viability [45]. Epidermal growth factor and IL-6, which are highly activated in glioblastoma, also initiate FAK activation [47].
The functions of FAK on tumor cells
Tumor cell proliferation, apoptosis and survival
The involvement of FAK in tumor growth has been extensively investigated in human breast cancer. The PI3K/AKT/mTOR signaling pathway has been widely recognized as one of the most commonly disrupted pathways in cancer [48, 49], and is correlated with FAK-mediated tumor cell growth. The ablation of FAK reduced Wnt1-driven basal-like breast cancer growth and promotes apoptosis by downregulating AKT-mTOR signaling [50]. In addition, activation of FAK by insulin-like growth factor-1 (IGF-1) and its receptor system (IGF-1R) initiates the PI3K-AKT-YAP (yes-associated protein/yes-related protein) signaling cascade, which modulates the expression of genes targeted by YAP. This mechanism is implicated in the expansion of aggressive triple-negative breast cancer cells [51]. A similar result was observed in intrahepatic cholangiocarcinoma (iCCA) [52]. FAK activation, which is required for Y357 phosphorylation of YAP, strongly promotes AKT/YAP-driven mouse iCCA initiation [52].
FAK was also confirmed to stimulate cell cycle progression and promote cancer proliferation by targeting various cyclins and cyclin-dependent kinase (CDK) inhibitors [53]. Among them, cyclin D1, together with key CDK inhibitors p21 and p27, are the most extensively studied downstream targets of FAK, regulating the cell cycle transition from G1 to S phase. It was reported that knockdown of Mucin-like 1 (MUCL1) in HER2-amplified breast cancer resulted in FAK/Jun NH2-terminal kinase (JNK) signaling blockade and subsequent G1/S phase arrest, which was mediated by decreased cyclin D levels as well as increased p21 and p27 levels [54]. A mechanistic study revealed that integrin signaling through FAK activated the ERK pathway, which stimulated the transcriptional activation of cyclin D [55]. In addition to p21 and p27, it was also demonstrated that FAK ablation in glioblastoma repressed the expression of the autophagy cargo receptor p62/SQSTM-1, the inhibition of which post transcriptionally upregulated p27 expression to mediate G1 phase arrest and induced a cell senescence-like state [56]. Moreover, p62 synergized with its downstream target SKP2 to inhibit p21 and p27 activity. Nevertheless, contradictory results were observed in vascular smooth muscle cells: SKP2 was degraded by nuclear FAK to inhibit cell proliferation by promoting p21 and p27 expression [57]. It has been acknowledged that nuclear FAK acts as a scaffold for protein interactions and regulates specific gene transcription. In skin squamous cell carcinoma, nuclear FAK was reported to interact with runt-related transcription factor 1 (Runx1) and recruit Runx1 regulatory proteins such as sin3a to inhibit the transcription of insulin-like growth factor binding protein 3, which induces cell cycle arrest at the G1 phase by suppressing the expression of cyclins and CDKs and increasing p21 expression [58]. Likewise, the nuclear activation of FAK in colon cancer cells by fibrin resulted in a decline in p53, along with its subsequent targets, such as 14-3-3σ and p21, ultimately stimulating cell proliferation while repressing senescence [59].
FAK plays a vital role in sustaining cancer cell survival and regulating cell apoptosis [60,61,62,63], anoikis [64,65,66,67,68], autophagy [68], and senescence [69]. The disruption of FAK-mediated signaling in breast cancer cells stimulated the Fas-associated death domain (FADD) and caspase-8 apoptotic pathways, which induced cell apoptosis and inhibited anchorage-independent survival [70]. Additionally, components of death receptor pathways, including the FAK-binding partner death domain kinase receptor-interacting protein (RIP), are involved in this process [61]. The antiapoptotic effect of FAK was further confirmed to be associated with FAK-dependent activation of the phosphatidylinositide 3′-OH-kinase-AKT survival pathway, concomitant with the subsequent stimulation of NF-kB and inhibitor-of-apoptosis proteins [71]. Moreover, nuclear FAK also plays an important role in regulating cancer cell apoptosis. Nuclear FAK interacts with the N-terminal transactivation domain of p53 through its N-terminal fragment; this attenuates p53 transcriptional activity and inhibits p53-mediated apoptosis to promote cell survival [72]. Anoikis, a distinct type of apoptosis that occurs in normal epithelial and endothelial cells, is also negatively regulated by FAK signaling In human breast cancer, the mechanism of FAK-induced cell resistance to anoikis was reported to be correlated with the increased activity of NF-kB, which is induced by the functional interaction between the N-terminal domain of FAK and TRAF2, a RING finger adaptor protein [65]. Notably, integrin endocytosis has gradually been uncovered to be critical for FAK activation depending on endosome antigen-1 and small GTPase Rab21, and FAK activation ultimately promotes anoikis resistance and anchorage-independent cell growth [66, 67].
Cell migration and invasion
Integrin aggregation in the ECM plays a crucial role in inducing FAK signaling, which is fundamental for cell motility and cytoskeletal reorganization. Chemotactic signals activate integrins, leading to FAK activation and the formation of focal adhesion complexes, which subsequently trigger the polymerization of actin filaments toward the cytoplasmic membrane [73, 74]. The FAK/Src complex and kinase activity lead to p130Cas phosphorylation, promoting the formation of Cas/Crk complexes, which significantly influence cell migration [75]. Additionally, MLCK-mediated focal adhesion disassembly and JNK-mediated paxillin phosphorylation promote cytoskeleton reorganization [76, 77]. FAK’s associations with PI3-kinase and/or Grb7 govern intracellular signaling pathways correlated with cellular mobility [78, 79]. Furthermore, integrin-mediated FAK signaling critically controls adhesion dynamics during cell migration. The formation of FAK/Src complexes at focal adhesion sites enhances ERK2 activity, resulting in the activation of Calpain 2 [80,81,82]. FAK’s effects on small GTPases impact cytoskeletal reorganization and adhesion stabilization [83]. RhoA, Rac1, and Cdc42, among small GTPases, play a crucial role in cytoskeletal reorganization and tumorigenesis [84, 85]. In this regard, RhoA influences cell‒cell or cell–ECM associations by inducing shifts within the cytoskeleton, while Rac1 initiates actin polymerization, enabling membrane folding, whereas Cdc42 initiates actin filament production in the generation of filopodia [86,87,88]. These investigations present compelling proof that the expression of FAK and the activation of FAK signaling pathways are mainly mediated by Rho GTPases, indicating the indispensable role of FAK in cytoskeletal reorganization.
Epithelial–mesenchymal transformation
Epithelial–mesenchymal transition (EMT) is a physiological process in normal embryonic development and tissue regeneration. However, aberrant reactivation of EMT is associated with malignant properties of tumor cells, including migration, invasiveness, increased tumor stemness, and resistance to chemotherapy and immunotherapy [88, 89]. Multiple studies have established the role of FAK in promoting EMT and increasing cell invasion and metastasis [90, 91]. EMT is regulated by FAK-mediated alterations in E-cadherin expression, a key molecule in the process [92,93,94]. Evidence provided by Gayrard et al. illustrates that SRC-FAK-mediated reconstruction of actomyosin results in the loosening of E-cadherin junctions without disrupting those involving β-associated proteins [94]. Avizienyte’s team confirmed the importance of FAK phosphorylation in the decrease in E-cadherin induced by Src in colon cancer cells [91]. The investigation carried out by Hauck’s team suggests that restraining FAK function restricts cell invasion stimulated by Src and obstructs the metastasis and invasion aimed by FAK-targeted drugs [95]. The downregulation of KIF26A clearly enhances EMT and decreases E-cadherin expression by augmenting the binding of c-MYC to the promoter section of FAK [96]. Slug expression, which balances EMT and cellular migration, is triggered by TGF-β1 in squamous cell carcinoma cells. However, when FAK inhibitors are administered, such an effect is alleviated [97]. These findings underline the crucial role of FAK in EMT, invasion, and metastasis. Nonetheless, additional research is required to clarify the downstream molecular mechanisms by which FAK regulates EMT. These mechanisms include E-cadherin-mediated cell‒cell adhesion, integrin-ECM-based adhesion, and the collaboration of both these mechanisms.
FAK in cancer stem cells
In various tumor types, FAK has been found to contribute to the activities of cancer stem cells (CSCs), particularly in breast cancer [98, 99]. Loss of FAK leads to a decrease in mammary cancer stem cells (MaCSCs) and suppresses their protumorigenic functions [100]. FAK facilitates the formation of a ternary complex with connexin and NANOG, which sustains CSC self-renewal and maintenance in triple-negative breast cancer [101]. By interrupting the interaction between FAK and endophilin A2, the stem-like population, gene signature, self-renewal, and tumorigenicity of mammary CSCs can be suppressed [102]. In triple-negative breast cancer (TNBC), inhibiting FAK genetically or with drugs decreases anchorage-independent spheroid cell growth, reduces chemotherapy-dependent CSC enrichment, and delays metastatic outgrowth [99, 103]. A recent study reported that inhibiting FAK, a protein found in head and neck squamous cell carcinoma (HNSCC), could significantly reduce the expression of stem cell markers, including Oct4, Sox2, and Nanog, leading to a decrease in cell self-renewal [104]. Additionally, FAK-mediated signaling pathways have been discovered to play a crucial role in regulating CSC properties in esophageal squamous cell carcinoma [105].
The effects of FAK on tumor-associated cells
Immune cells
Tumor-associated macrophage and regulatory T cells (Treg) are major inhibitory cells for anti-cancer immune response [106,107,108]. The importance of FAK expression in the regulation of the tumor environment has been emphasized in current research (Fig. 3). Enhancement of the expression of several chemokines occurs due to the elevation of FAK levels in tumors, which promotes TME remodeling by recruiting immunosuppressive cells and secreting cytokines [109]. FAK inhibitors have been shown to suppress leucocyte and macrophage infiltration and the growth of breast cancer [110, 111] and pancreatic ductal adenocarcinoma (PDAC) tumors in mouse models [112]. FAK signaling enhances the expression of homing signals such as CCL5, CCL7, CXCL10, and TGFβ2 [113], which play a crucial role in recruiting Tregs [114]. FAK contributes to the increased expression of IL-33 [115], an alarmin cytokine, produced by stromal and epithelial cells, which binds to ST2L on immune cells and enhances the transcription of chemokine genes such as CCL5 [116]. The increased production of CCL5 leads to the recruitment of Tregs and other immune cells, promoting immunosuppression. However, recruited Tregs promote tumor survival by depleting CD8+ cytotoxic cells [117]. CD28 is a costimulatory molecule on T cells that enhances T-cell activation and proliferation. Amplification of CD28+ T cells within the TME can enhance their antitumor effects and facilitate tumor cell elimination. FAK depletion can lead to tumor regression by increasing the number of CD28+ T cells in the TME [118]. FAK overexpression slows tumor growth and promotes natural killer cell infiltrations while FAK knockdown promotes tumor growth and suppresses natural killer cell infiltrations [119].

The role of FAK in the tumor microenvironment. FAK not only maintains cancer malignancy but also influences remodeling of the immune microenvironment. For example, FAK can recruit immune cells, cancer-associated fibroblasts, and endothelial cells and even remodel the extracellular matrix. Preclinical studies support the importance of FAK inhibitors in combination with other immunotherapies, and relevant clinical trials are in progress, demonstrating the importance of FAK in oncology treatment
Cancer-associated fibroblasts (CAFs)
In the TME, CAFs are key stromal cells that play a critical role in tumor cell initiation, survival, proliferation, and metastasis through the secretion of various cytokines, growth factors, hormones, and ECM proteins [120]. For example, increased lumican expression in gastric CAFs promotes FAK activation via β1 integrin, promoting the invasion of gastric cancer cells [121]. PDAC cells activate CAFs and promote cancer stemness through increased expression of type I collagen via β1 integrin-FAK signaling [122]. Inhibition of FAK reduces CAF recruitment and TME fibrosis [123]. This reduces the stemness of PDAC cells [124] and suppresses breast cancer metastasis while increasing the levels of tumor suppressor microRNAs in exosomes [125]. Emerging evidence highlights the pivotal role of CAFs in governing tumor metabolic processes via FAK-regulated pathways. Notably, breast and pancreatic cancer patients exhibiting diminished FAK expression experience a significant decline in overall survival. Furthermore, experimental studies using mouse models have demonstrated that the depletion of FAK in CAFs actively promotes tumor growth. Mechanistically, this phenomenon can be attributed to the activation of protein kinase A within CAFs, resulting from the deficiency of FAK. Consequently, this activation leads to a pronounced enhancement of glycolysis in tumor cells [126]. The remodeling of the TME is facilitated by FAK, which functions as a crucial regulator in the TME. Therefore, a comprehensive understanding of the impact of FAK on tumor progression and TME remodeling could reveal new opportunities for cancer therapy.
Endothelial cells (ECs)
Integrins and growth factor receptors mediate the signals involved in angiogenesis. FAK is activated by integrin-mediated cell adhesion and associates with several proteins that contain the SH2 structural domain, including Src, Grb7, the p85 subunit of PI3K, and phospholipase C-g [127]. FAK interacts with epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), and platelet-derived growth factor receptor (PDGFR) via its N-terminus, and VEGF induces FAK phosphorylation through VEGFR activation, promoting angiogenesis [128]. When platelet-derived growth factor (PDGF) binds to PDGFR, it phosphorylates FAK, which activates endothelial cells, stromal cells, and CEPs, leading to matrix metalloproteinase-mediated breakdown of the ECM and angiogenesis [129]. FAK inhibition is a promising anticancer treatment strategy to hinder cell migration, invasion, proliferation, and angiogenesis. Inhibition of the phosphorylation of vascular endothelial growth factor receptor 2 (VEGFR2), Src, and FAK through Sema3A led to substantial reductions in tumor growth and angiogenesis in tongue SSC-9 cells, illustrating the potential of this approach for cancer therapy [63]. TAE226, a potent FAK inhibitor, effectively suppressed the growth of OSCC xenografts and angiogenesis in mice [130]. Moreover, FRNK, a negative inhibitor of FAK, was revealed to impede FAK phosphorylation, thereby reducing EGF-induced MMP-9 expression and ultimately hindering the invasion of follicular thyroid cancer cells [131]. These collective findings affirm the pivotal role of FAK in inducing cell invasion and angiogenesis and its potential as an attractive target for antiangiogenic therapy in cancer treatment.
Targeting FAK in combination therapies
FAK is considered a potential target for effective cancer therapy, and its key role in various types of cancer has been well established. Recently, FAK inhibitors have gained attention as novel and promising combination therapy partners (Fig. 3). Presented here is a summary of preclinical (Table 1) and clinical trials (Table 2) concerning FAK inhibitors. The primary focus is on combination trials incorporating FAK as a therapeutic target (Table 3) to assess its efficacy and its contribution to combination therapy.
Combination with immunotherapies
Antibodies to immune check point inhibitor that enhance the host immunologic activity against tumors have become standard of care in the treatment of many malignancies [132]. However, only a small percentage of patients have meaningful responses to these treatments. Searching for new pathways and molecules to improve responses and application of immune checkpoint inhibition therapy attracts great attention [133,134,135,136]. In TNBC, PD-L1 expression is elevated and significantly correlated with FAK mRNA expression, highlighting the functional relationship between immune checkpoints and FAK [137, 138]. Anti-PD-L1 antibody atezolizumab augments the suppressive impact of FAK inhibitors on cell invasion and migration through the restraint of FAK phosphorylation [138]. Cytokine-induced killer (CIK) cells are used as a treatment approach in adoptive cellular immunotherapy and are highly regarded as a promising candidate for cancer immunotherapy [139]. FAK knockdown/inhibition increased the sensitivity of TNBC cells to CIK cells in coculture system by enhancing CIK-mediated cell death. FAK knockdown also decreased PD-L1 mRNA and protein expression in TNBC cells [140].
Combining the FAK inhibitor VS4718 with anti-PD1 therapy in hepatocellular carcinoma resulted in decreased macrophage numbers and increased CD8+ T-cell numbers [141]. Additionally, when FAK inhibitors were combined with agents that induce T-cell costimulatory pathways in skin squamous cell carcinoma, the tumors became more sensitive to FAK inhibitors, and this effect was mediated by CD80. This suppressed tumor formation and even drove complete regression [118].
Lu et al. constituted a nanodrug PLGA-FAKi by encapsulated FAK inhibitor using poly(lactic-co-glycolic) acid (PLGA). PLGA-FAKi treatment increased ovalbumin-specific CTLs (OVA-CTLs) infiltration into B16-OVA tumors, leading to reduced immunosuppression and increased tumor microvessel permeability, and further inhibited tumor growth when combined with OVA-CTLs [70]. In mice with HGSOC, the combination of FAK inhibitor and anti-TIGIT therapy was able to prolong survival rates, increase the level of CXCL13, which is associated with tumor infiltrating lymphocytes (TLS) formation, and promote B and T-cell enrichment [142]. Mechanical stretching has also been shown to have a positive effect on melanoma cells: it enhances M1 polarization and antitumor effects. This effect is associated with the FAK/NF-kB signaling pathway [143]. It was found that ABCB1, CXCR4, and FAK were overexpressed in non-small cell lung cancer (NSCLC) patients and cell lines [144]. Therefore, targeting CXCR4 and FAK could be a way to overcome DOX resistance and enhance the anti-invasive effects of CXCR4 and FAK inhibitors in NSCLC cells.
Combination with targeted therapies
Research suggests that co-treatment using KRAS G12C inhibitors and IN10018 is likely to benefit cancer patients with mutated KRAS G12C and may also prevent resistance to KRAS G12C inhibition by targeting dysregulated FAK-YAP signaling and fibrogenesis [145]. Uveal melanoma (UM) patients with unresponsive liver metastases have a druggable downstream signaling hub from GNAQ mutations that activates YAP1 via FAK [146]. Co-targeting FAK and MEK using this approach could lead to novel precision therapy and inhibit tumor growth in UM cells and UM xenograft models. It is important to note that FAK is overexpressed in tumors and activated in iCCA lesions, which in turn contribute to cancer initiation and progression through the YAP proto-oncogene [52]. iCCA growth was dramatically inhibited by combination of FAK and CDK4/6 inhibitor. Overall, the study proved the role of FAK-YAP signaling and suggests its potential as a target for precision therapy in inhibiting cancer growth using various approaches.
Co-targeting this pathway using the FAK inhibitor PF562271 and the BRAF inhibitor vemurafenib could represent a promising therapeutic approach for BRAF-mutant colorectal cancer (CRC) patients, as they exhibit synergistic antitumor effects in vitro and in vivo [147]. Notably, the combination of small-molecule inhibitors of β-catenin or FAK along with vemurafenib not only inhibits the proliferation of BRAF V600E colon cancer cells in vitro but also prevents tumor formation in xenograft mice [148]. Overall, these findings emphasize the potential of combination therapy using several inhibitors in treating cancers with mutations in BRAF and highlights the potential of FAK inhibitors in several therapeutic approaches.
One study found that using TAE226 and sorafenib together effectively reduces hepatocellular carcinoma growth by changing gene expression and epigenetics through FAK nuclear interactome dysregulation [149]. Another study showed that combining VS-6063 and JQ1 to target integrins inhibits FAK signaling and PI3K/AKT, reducing survival in primary HGSOC tumors with co-amplification of FAK and c-Myc [150]. In squamous cell carcinoma cells with mutated FAK, HDAC and FAK inhibitors work synergistically to arrest cellular proliferation and tumor growth, emphasizing the importance of collaborations of multiple targets [151]. In addition, combining SFK/FAK inhibitors with osimertinib shows promise as a therapeutic approach to inhibit growth and resistance in EGFR-mutant lung cancer treatment [152]. Simultaneous targeting of the FAK and Janus kinase/STAT3 pathways produces a synergistic effect. This suggests that repressing STAT3 signals may overcome FAK inhibitor resistance in PDAC, as demonstrated in another study [153]. The studies described above provide evidence that FAK inhibitors, when combined with targeted therapy, present a new and promising avenue of tumor treatment.
Combination with chemotherapies
Chemotherapy is often ineffective against ovarian cancer; however, the hyaluronic acid-labeled two-in-one drug delivery system HA-PLGA-NPs, containing paclitaxel and FAK siRNA, has high binding efficiency to CD44-positive tumor cells, resulting in increased cytotoxicity and apoptosis in drug-resistant tumors, as demonstrated in experimental studies [154]. FAK inhibition has been identified to enhance chemotherapy sensitivity and promote anticancer effects, primarily through the activation of p53 transcriptional activity, making it a potential focal point for therapeutic strategies in gastric cancer management and a valuable prognostic indicator in clinical settings [155]. Inhibition of FAK, as observed in the study of phosphorylated kinases in PDAC, shows synergistic effects with nab-paclitaxel to reduce tumor growth and appears to be a promising potential treatment option [156]. Furthermore, endothelial cell focal adhesion kinase (EC-FAK) plays a significant role in the regulation of the chemotherapy response and the levels of endocrine factors, and the combination of FAK inhibitors with gemcitabine has the potential to serve as a promising strategy to control PDAC metastasis, as supported by studies that revealed reduced metastasis load and improved survival rates in gemcitabine-treated mice and patients with low levels of EC-FAK [157]. FAK regulates CSC activity in breast cancer, and inhibition of FAK suppresses self-renewal, leading to a reduced tumor size, thereby providing a promising strategy to improve survival by suppressing CSC activity; the approach is especially effective when combined with paclitaxel treatment [98]. In addition, inhibiting endothelial FAK enhances the response of B16 and CMT19T mouse tumors to adriamycin or radiotherapy by suppressing NF-κB activation and cytokine production, thereby improving the effectiveness of DNA damage therapy [158]. Furthermore, elevated EC-PY397-FAK expression levels are strongly correlated with advanced clinical parameters of breast cancer and poor treatment response and independently predict unfavorable five-year recurrence-free survival, which highlights the need to assess the role of FAK inhibitors in optimizing treatments and improve the response to various strategies [159].
Combination with radiotherapies
Combining FAK inhibitors with low-dose radiation in pancreatic cancer can regulate the TME through several mechanisms, including reducing hypoxia, boosting immune cell infiltration, and enhancing radiosensitivity [160]. FAK overexpression is known to be associated with treatment resistance and metastasis in pancreatic cancer. A database study identified VS-4718 as a potential inhibitor of FAK that can enhance radiosensitivity and inhibit ECM synthesis [161]. Combining FAK inhibition with radiotherapy may prove to be effective against this disease. Tests in a PDAC mouse model have shown that inhibiting FAK with IN10018 can enhance the anticancer effect of radiotherapy by decreasing suppressor granulocyte infiltration and increasing CD8+ T cell and macrophage [162]. This indicates that FAK inhibitors have the potential to enhance the radiosensitivity and immunomodulation of PDAC.
The presence of CSCs in high-grade DCIS is associated with disease recurrence and resistance to radiotherapy via the FAK/Wnt pathway, and the use of FAK inhibitors can reduce cellular self-renewal while enhancing the effects of radiation in breast cancer [163]. Moreover, inhibiting FAK decreased tumor cell adhesion in a glioblastoma/breast cancer cell and endothelial cell coculture model after radiotherapy [164]. The evidence suggests that FAK inhibitors hold promise for enhancing radiosensitivity.
Similarly, a study demonstrated that the combination of FAK inhibition and carbon ion irradiation was effective in inhibiting metastasis in tongue squamous cell carcinoma. The treatment decreased colony formation, increased apoptosis, and reduced migration and invasion in CAL27 cells [165]. Furthermore, in HPV-negative HNSCC cells, FAK inhibition led to enhanced radiosensitivity by inducing G2/M arrest and DNA damage. The study also revealed that lower protein tyrosine kinase 2 (PTK2)/FAK mRNA expression was linked to better disease-free survival. Therefore, PTK2/FAK could be a potential biomarker for HNSCC patients who are susceptible to relapse after radiotherapy [5]. According to the available data, FAK inhibitors have shown promising results in sensitizing cancer cells to the effects of ionizing radiation, which helps reduce tumor burden and recurrence rates.
Conclusion
Cancer is a complex illness that arises due to diverse genetic and epigenetic alterations that cause abnormalities in multiple biological pathways. Although the development of molecularly targeted therapies has aided in their treatment, their frequent ineffectiveness and drug resistance pose significant challenges owing to recurrence and metastasis s; therefore, new therapeutic targets are urgently needed. Crucial cellular processes such as cell adhesion, migration, proliferation, and survival are regulated by FAK. Furthermore, FAK promotes cancer progression, including features such as tumor angiogenesis, EMT, cancer stemness, and immunomodulatory capacity [166, 167]. FAK is widely activated in multiple cancer types, such as colorectal, lung, ovarian, neck, bladder, breast, and esophageal cancers, and predicts a poor prognosis [98, 168, 169].
The FAK pathway has also been linked to the generation of CSCs [101, 103, 105, 114], which are responsible for tumor propagation, metastasis, and therapy resistance. FAK inhibition has been shown to decrease the number of CSCs, suggesting that FAK may represent a viable target for eliminating CSCs, thereby improving cancer therapy outcomes. In addition, the FAK signaling pathway plays a crucial role in regulating the complex TME [119, 125, 129, 170], which comprises cellular and noncellular components that promote tumor growth and metastasis. Gaining a comprehensive understanding of the involvement of FAK in tumor microenvironment (TME) remodeling is imperative in advancing cancer treatment outcomes to a higher level.
FAK has emerged as a promising target for cancer therapy owing to its key role in tumor cells and the TME. Various FAK inhibitors have demonstrated significant antitumor efficacy in diverse preclinical models and are currently being evaluated in clinical trials. Combining FAK inhibitors with standard cancer treatments has been shown to significantly enhance treatment efficacy and decrease chemotherapy resistance [12, 170], as FAK inhibition sensitizes cancer cells to chemotherapy, leading to better therapeutic outcomes. It is interesting that D-pinitol, a 3-methoxy analogue of d-chiro-inositol in soy foods and legumes, can reduce c-Src kinase activity and NF-kB activation through inhibiting FAK phosphorylation, resulting in decrease of prostate cancer metastasis [171, 172]. Recently, an effective FAK degradation agents have been developed that can selectively degrade FAK and showed outstanding inhibitory effects in triple-negative breast cancer and ovarian cancer cells [173, 174].
However, the clinical translation of FAK inhibitors has been hampered by several challenges. First, a uniform method for measuring the expression of FAK, whether phosphorylated FAK or total FAK, needs to be selected. Second, the selection of an appropriate FAK assay is necessary, and immunohistochemistry, western blotting, and RT‒PCR are the most commonly employed methodologies. Each method has advantages and limitations, and the selection must account for factors such as sensitivity, specificity, and reliability. Finally, there are challenges regarding combination therapies utilizing FAK, including reduced selectivity and specificity, drug resistance, and emerging molecular targets that impact efficacy. As a result, future research should aim to enhance the selectivity and specificity of FAK inhibitors and also develop novel combination therapies to overcome these therapeutic obstacles. By addressing these challenges, the clinical translational impact of FAK-targeted therapies in patients can be optimized, ultimately resulting in more effective and personalized cancer treatments.
Availability of data and materials
Not applicable.
Abbreviations
- ATE:
-
Atezolizumab
- CAFs:
-
Cancer-associated fibroblast
- CDK:
-
Cyclin-dependent kinase
- CIK:
-
Cytokine-induced killer
- CRC:
-
Colorectal cancer
- CSCs:
-
Cancer stem cells
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial–mesenchymal transformation
- EGFR:
-
Epidermal growth factor receptor
- FADD:
-
Fas-associated death domain
- FAK:
-
Focal adhesion kinase
- FAKi:
-
Focal adhesion kinase inhibitor
- FAT:
-
Focal adhesion-targeting
- FERM:
-
Four-point-one-ezrin-radixin-moesin
- HNSCC:
-
Neck squamous cell carcinoma
- iCCA:
-
Intrahepatic cholangiocarcinoma
- IGF-1:
-
Insulin-like growth factor-1
- IGF-1R:
-
Insulin-like growth factor-1 receptor system
- JNK:
-
Jun NH2-terminal kinase
- LncRNA:
-
Long non‑coding RNA
- MaCSC:
-
Mammary cancer stem cells
- Mdm2:
-
Murine double minute2
- MUCL1:
-
Mucin-like 1
- NES:
-
Nuclear export signal
- NES2:
-
Nuclear export signal 2
- NSCLC:
-
Non-small cell lung cancer
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDGF:
-
Platelet-derived growth factor
- PDGFR:
-
Platelet-derived growth factor receptor
- PTK2:
-
Protein tyrosine kinase 2
- PRRs:
-
Proline-rich regions
- PyK2:
-
Proline-rich tyrosine kinase 2
- RIP:
-
Receptor-interacting protein
- RTKs:
-
Receptor tyrosine kinases
- Runx1:
-
Runt-related transcription factor 1
- SH3:
-
Src homology 3
- TLS:
-
Tumor infiltrating lymphocytes
- TM4LFM5:
-
Tetraspanin transmembrane 4 L6 family member 5
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple-negative breast cancer
- UM:
-
Uveal melanoma
- VEGFR:
-
Vascular endothelial growth factor receptor
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
- YAP:
-
Yes-associated protein/yes-related protein
References
Guan JL, Shalloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature. 1992;358:690–2.
Nader GPF, Ezratty EJ, Gundersen GG. FAK, talin and PIPKIγ regulate endocytosed integrin activation to polarize focal adhesion assembly. Nat Cell Biol. 2016;18:491–503.
Fan T, Chen J, Zhang L, Gao P, Hui Y, Xu P, et al. Bit1 knockdown contributes to growth suppression as well as the decreases of migration and invasion abilities in esophageal squamous cell carcinoma via suppressing FAK-paxillin pathway. Mol Cancer. 2016;15:23.
Balsas P, Palomero J, Eguileor Á, Rodríguez ML, Vegliante MC, Planas-Rigol E, et al. SOX11 promotes tumor protective microenvironment interactions through CXCR4 and FAK regulation in mantle cell lymphoma. Blood. 2017;130:501–13.
Skinner HD, Giri U, Yang L, Woo SH, Story MD, Pickering CR, et al. Proteomic profiling identifies PTK2/FAK as a driver of radioresistance in HPV-negative head and neck cancer. Clin Cancer Res. 2016;22:4643–50.
Yoon H, Dehart JP, Murphy JM, Lim S-TS. Understanding the roles of FAK in cancer: inhibitors, genetic models, and new insights. J Histochem Cytochem. 2015;63:114–28.
Chuang HH, Zhen YY, Tsai YC, Chuang CH, Hsiao M, Huang MS, et al. FAK in cancer: from mechanisms to therapeutic strategies. Int J Mol Sci. 2022;23:1726.
Rigiracciolo DC, Cirillo F, Talia M, Muglia L, Gutkind JS, Maggiolini M, et al. Focal adhesion kinase fine tunes multifaced signals toward breast cancer progression. Cancers. 2021;13:645.
Golubovskaya VM. Targeting FAK in human cancer: from finding to first clinical trials. Front Biosci. 2014;19:687–706.
Tremblay L, Hauck W, Aprikian AG, Begin LR, Chapdelaine A, Chevalier S. Focal adhesion kinase (pp125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int J Cancer. 1996;68:164–71.
Ossovskaya V, Lim ST, Ota N, Schlaepfer DD, Ilic D. FAK nuclear export signal sequences. FEBS Lett. 2008;582:2402–6.
Zhou J, Yi Q, Tang L. The roles of nuclear focal adhesion kinase (FAK) on cancer: a focused review. J Exp Clin Cancer Res. 2019;38:250.
Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22.
Constanzo JD, Tang K-J, Rindhe S, Melegari M, Liu H, Tang X, et al. PIAS1-FAK interaction promotes the survival and progression of non-small cell lung cancer. Neoplasia. 2016;18:282–93.
Tai H-C, Chang A-C, Yu H-J, Huang C-Y, Tsai Y-C, Lai Y-W, et al. Osteoblast-derived WNT-induced secreted protein 1 increases VCAM-1 expression and enhances prostate cancer metastasis by down-regulating miR-126. Oncotarget. 2014;5:7589–98.
Serrels B, McGivern N, Canel M, Byron A, Johnson SC, McSorley HJ, et al. IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks. Sci Signal. 2017;10: eaan8355.
Lim S-T, Miller NLG, Chen XL, Tancioni I, Walsh CT, Lawson C, et al. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J Cell Biol. 2012;197:907–19.
Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol. 2010;11:802–14.
Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35:347–67.
Sun X, Meng L, Qiao W, Yang R, Gao Q, Peng Y, et al. Vascular endothelial growth factor A/Vascular endothelial growth factor receptor 2 axis promotes human dental pulp stem cell migration via the FAK/PI3K/Akt and p38 MAPK signalling pathways. Int Endod J. 2019;52:1691–703.
Shen T, Guo Q. EGFR signaling pathway occupies an important position in cancer-related downstream signaling pathways of Pyk2. Cell Biol Int. 2019;44:2–13.
Rigiracciolo DC, Santolla MF, Lappano R, Vivacqua A, Cirillo F, Galli GR, et al. Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells. J Exp Clin Cancer Res. 2019;38:58.
Goñi GM, Epifano C, Boskovic J, Camacho-Artacho M, Zhou J, Bronowska A, et al. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc Natl Acad Sci USA. 2014;111:E3177–86.
Lim Y, Lim ST, Tomar A, Gardel M, Bernard-Trifilo JA, Chen XL, et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008;180:187–203.
Mousson A, Sick E, Carl P, Dujardin D, De Mey J, Rondé P. Targeting focal adhesion kinase using inhibitors of protein–protein interactions. Cancers. 2018;10:278.
Rashid M, Belmont J, Carpenter D, Turner CE, Olson EC. Neural-specific deletion of the focal adhesion adaptor protein paxillin slows migration speed and delays cortical layer formation. Development. 2017;144:4002–14.
Baumann K. Cell adhesion: FAK or talin: who goes first? Nat Rev Mol Cell Biol. 2012;13:138.
Brami-Cherrier K, Gervasi N, Arsenieva D, Walkiewicz K, Boutterin MC, Ortega A, et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. Embo J. 2014;33:356–70.
Liu Y, Loijens JC, Martin KH, Karginov AV, Parsons JT. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Mol Biol Cell. 2002;13:2147–56.
Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–8.
Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16.
Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–63.
Lim Y, Park H, Jeon J, Han I, Kim J, Jho EH, et al. Focal adhesion kinase is negatively regulated by phosphorylation at tyrosine 407. J Biol Chem. 2007;282:10398–404.
Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–91.
Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–84.
Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68.
Cai X, Lietha D, Ceccarelli DF, Karginov AV, Rajfur Z, Jacobson K, et al. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008;28:201–14.
Zhang N, Zhu H-P, Huang W, Wen X, Xie X, Jiang X, et al. Unraveling the structures, functions and mechanisms of epithelial membrane protein family in human cancers. Exp Hematol Oncol. 2022;11:69.
Jung O, Choi S, Jang SB, Lee SA, Lim ST, Choi YJ, et al. Tetraspan TM4SF5-dependent direct activation of FAK and metastatic potential of hepatocarcinoma cells. J Cell Sci. 2012;125:5960–73.
Lee E, Choi A, Jun Y, Kim N, Yook JI, Kim SY, et al. Glutathione peroxidase-1 regulates adhesion and metastasis of triple-negative breast cancer cells via FAK signaling. Redox Biol. 2020;29: 101391.
Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–56.
Chen TH, Chan PC, Chen CL, Chen HC. Phosphorylation of focal adhesion kinase on tyrosine 194 by Met leads to its activation through relief of autoinhibition. Oncogene. 2011;30:153–66.
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906.
Choi CH, Webb BA, Chimenti MS, Jacobson MP, Barber DL. pH sensing by FAK-His58 regulates focal adhesion remodeling. J Cell Biol. 2013;202:849–59.
Xing P, Wang Y, Zhang L, Ma C, Lu J. Knockdown of lncRNA MIR44352HG and ST8SIA1 expression inhibits the proliferation, invasion and migration of prostate cancer cells in vitro and in vivo by blocking the activation of the FAK/AKT/betacatenin signaling pathway. Int J Mol Med. 2021;47(6):93.
Gu W, Zhang L, Zhang X, Wang B, Shi X, Hu K, et al. MiR-15p-5p mediates the coordination of ICAM-1 and FAK to promote endothelial cell proliferation and migration. Inflammation. 2022;45:1402–17.
Nunez RE, Del Valle MM, Ortiz K, Almodovar L, Kucheryavykh L. Microglial cytokines induce invasiveness and proliferation of human glioblastoma through Pyk2 and FAK activation. Cancers. 2021;13(24):6160.
Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273–91.
Luo X, Cao M, Gao F, He X. YTHDF1 promotes hepatocellular carcinoma progression via activating PI3K/AKT/mTOR signaling pathway and inducing epithelial-mesenchymal transition. Exp Hematol Oncol. 2021;10:35.
Paul R, Luo M, Mo X, Lu J, Yeo SK, Guan J-L. FAK activates AKT-mTOR signaling to promote the growth and progression of MMTV-Wnt1-driven basal-like mammary tumors. Breast Cancer Res. 2020;22:59.
Rigiracciolo DC, Nohata N, Lappano R, Cirillo F, Talia M, Scordamaglia D, et al. IGF-1/IGF-1R/FAK/YAP transduction signaling prompts growth effects in triple-negative breast cancer (TNBC) cells. Cells. 2020;9(4):1010.
Song X, Xu H, Wang P, Wang J, Affo S, Wang H, et al. Focal adhesion kinase (FAK) promotes cholangiocarcinoma development and progression via YAP activation. J Hepatol. 2021;75:888–99.
Zhao JH, Reiske H, Guan JL. Regulation of the cell cycle by focal adhesion kinase. J Cell Biol. 1998;143:1997–2008.
Conley SJ, Bosco EE, Tice DA, Hollingsworth RE, Herbst R, Xiao Z. HER2 drives Mucin-like 1 to control proliferation in breast cancer cells. Oncogene. 2016;35:4225–34.
Zhao J, Pestell R, Guan JL. Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol Biol Cell. 2001;12:4066–77.
Alza L, Nàger M, Visa A, Cantí C, Herreros J. FAK inhibition induces glioblastoma cell senescence-like state through p62 and p27. Cancers. 2020;12:1086.
Jeong K, Murphy JM, Ahn EE, Lim SS. FAK in the nucleus prevents VSMC proliferation by promoting p27 and p21 expression via Skp2 degradation. Cardiovasc Res. 2022;118:1150–63.
Canel M, Byron A, Sims AH, Cartier J, Patel H, Frame MC, et al. Nuclear FAK and Runx1 cooperate to regulate IGFBP3, cell-cycle progression, and tumor growth. Cancer Res. 2017;77:5301–12.
Sharma BK, Mureb D, Murab S, Rosenfeldt L, Francisco B, Cantrell R, et al. Fibrinogen activates focal adhesion kinase (FAK) promoting colorectal adenocarcinoma growth. J Thromb Haemost. 2021;19:2480–94.
Kim EY, Cha YJ, Jeong S, Chang YS. Overexpression of CEACAM6 activates Src-FAK signaling and inhibits anoikis, through homophilic interactions in lung adenocarcinomas. Transl Oncol. 2022;20:101402.
Kurenova E, Xu LH, Yang X, Baldwin AS, Craven RJ, Hanks SK, et al. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol Cell Biol. 2004;24:4361–71.
Huang C, Wang Y, Huang J-H, Liu W. Sema3A drastically suppresses tumor growth in oral cancer Xenograft model of mice. BMC Pharmacol Toxicol. 2017;18:55.
Liao M, Qin R, Huang W, Zhu H-P, Peng F, Han B, et al. Targeting regulated cell death (RCD) with small-molecule compounds in triple-negative breast cancer: a revisited perspective from molecular mechanisms to targeted therapies. J Hematol Oncol. 2022;15:44.
Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–9.
da Silva SD, Xu B, Maschietto M, Marchi FA, Alkailani MI, Bijian K, et al. TRAF2 Cooperates with Focal Adhesion Signaling to Regulate Cancer Cell Susceptibility to Anoikis. Mol Cancer Ther. 2019;18:139–46.
Alanko J, Mai A, Jacquemet G, Schauer K, Kaukonen R, Saari M, et al. Integrin endosomal signalling suppresses anoikis. Nat Cell Biol. 2015;17:1412–21.
Alanko J, Ivaska J. Endosomes: Emerging Platforms for Integrin-Mediated FAK Signalling. Trends Cell Biol. 2016;26:391–8.
Zhao M, Finlay D, Kwong E, Liddington R, Viollet B, Sasaoka N, et al. Cell adhesion suppresses autophagy via Src/FAK-mediated phosphorylation and inhibition of AMPK. Cell Signal. 2022;89:110170.
Lu D, Wang Y, Zhang T, Wang F, Li K, Zhou S, et al. Metabolic radiolabeling and in vivo PET imaging of cytotoxic T lymphocytes to guide combination adoptive cell transfer cancer therapy. J Nanobiotechnol. 2021;19:175.
Xu LH, Yang X, Bradham CA, Brenner DA, Baldwin AS, Craven RJ, et al. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J Biol Chem. 2000;275:30597–604.
Sonoda Y, Matsumoto Y, Funakoshi M, Yamamoto D, Hanks SK, Kasahara T. Anti-apoptotic role of focal adhesion kinase (FAK). Induction of inhibitor-of-apoptosis proteins and apoptosis suppression by the overexpression of FAK in a human leukemic cell line, HL-60. J Biol Chem. 2000;275:16309–15.
Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280:25008–21.
Shi Q, Boettiger D. A novel mode for integrin-mediated signaling: tethering is required for phosphorylation of FAK Y397. Mol Biol Cell. 2003;14:4306–15.
Vicente-Manzanares M, Webb DJ, Horwitz AR. Cell migration at a glance. J Cell Sci. 2005;118:4917–9.
Cary LA, Han DC, Polte TR, Hanks SK, Guan JL. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J Cell Biol. 1998;140:211–21.
Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6:154–61.
Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. JNK phosphorylates paxillin and regulates cell migration. Nature. 2003;424:219–23.
Chu P-Y, Huang L-Y, Hsu C-H, Liang C-C, Guan J-L, Hung T-H, et al. Tyrosine phosphorylation of growth factor receptor-bound protein-7 by focal adhesion kinase in the regulation of cell migration, proliferation, and tumorigenesis. J Biol Chem. 2009;284:20215–26.
Shen T-L, Han DC, Guan J-L. Association of Grb7 with phosphoinositides and its role in the regulation of cell migration. J Biol Chem. 2002;277:29069–77.
Carragher NO, Westhoff MA, Fincham VJ, Schaller MD, Frame MC. A novel role for FAK as a protease-targeting adaptor protein: regulation by p42 ERK and Src. Curr Biol. 2003;13:1442–50.
Kerstein PC, Patel KM, Gomez TM. Calpain-mediated proteolysis of Talin and FAK regulates adhesion dynamics necessary for axon guidance. J Neurosci. 2017;37:1568–80.
Chan KT, Bennin DA, Huttenlocher A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J Biol Chem. 2010;285:11418–26.
McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer—a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–15.
Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33.
Wang T, Rao D, Yu C, Sheng J, Luo Y, Xia L, et al. RHO GTPase family in hepatocellular carcinoma. Exp Hematol Oncol. 2022;11:91.
Krugmann S, Jordens I, Gevaert K, Driessens M, Vandekerckhove J, Hall A. Cdc42 induces filopodia by promoting the formation of an IRSp53: Mena complex. Curr Biol. 2001;11:1645–55.
Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–9.
Huang Y, Hong W, Wei X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J Hematol Oncol. 2022;15:129.
Dong B, Li S, Zhu S, Yi M, Luo S, Wu K. MiRNA-mediated EMT and CSCs in cancer chemoresistance. Exp Hematol Oncol. 2021;10:12.
Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. 2013;126:393–401.
Avizienyte E, Wyke AW, Jones RJ, McLean GW, Westhoff MA, Brunton VG, et al. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat Cell Biol. 2002;4:632–8.
Canel M, Serrels A, Miller D, Timpson P, Serrels B, Frame MC, et al. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: effects on E-cadherin dynamics. Cancer Res. 2010;70:9413–22.
Serrels A, Canel M, Brunton VG, Frame MC. Src/FAK-mediated regulation of E-cadherin as a mechanism for controlling collective cell movement: insights from in vivo imaging. Cell Adh Migr. 2011;5:360–5.
Gayrard C, Bernaudin C, Déjardin T, Seiler C, Borghi N. Src- and confinement-dependent FAK activation causes E-cadherin relaxation and β-catenin activity. J Cell Biol. 2018;217:1063–77.
Hauck CR, Hsia DA, Schlaepfer DD. The focal adhesion kinase—a regulator of cell migration and invasion. IUBMB Life. 2002;53:115–9.
Ma R-R, Zhang H, Chen H-F, Zhang G-H, Tian Y-R, Gao P. MiR-19a/miR-96-mediated low expression of KIF26A suppresses metastasis by regulating FAK pathway in gastric cancer. Oncogene. 2021;40:2524–38.
Saito D, Kyakumoto S, Chosa N, Ibi M, Takahashi N, Okubo N, et al. Transforming growth factor-β1 induces epithelial-mesenchymal transition and integrin α3β1-mediated cell migration of HSC-4 human squamous cell carcinoma cells through Slug. J Biochem. 2013;153:303–15.
Timbrell S, Aglan H, Cramer A, Foden P, Weaver D, Pachter J, et al. FAK inhibition alone or in combination with adjuvant therapies reduces cancer stem cell activity. NPJ Breast Cancer. 2021;7:65.
Tancioni I, Miller NLG, Uryu S, Lawson C, Jean C, Chen XL, et al. FAK activity protects nucleostemin in facilitating breast cancer spheroid and tumor growth. Breast Cancer Res. 2015;17:47.
Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S, et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009;69:466–74.
Thiagarajan PS, Sinyuk M, Turaga SM, Mulkearns-Hubert EE, Hale JS, Rao V, et al. Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nat Commun. 2018;9:578.
Fan H, Zhao X, Sun S, Luo M, Guan J-L. Function of focal adhesion kinase scaffolding to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J Biol Chem. 2013;288:3322–33.
Kolev VN, Tam WF, Wright QG, McDermott SP, Vidal CM, Shapiro IM, et al. Inhibition of FAK kinase activity preferentially targets cancer stem cells. Oncotarget. 2017;8:51733–47.
Moon JH, Rho YS, Lee SH, Koo BS, Lee HJ, Do SI, et al. Role of integrin β1 as a biomarker of stemness in head and neck squamous cell carcinoma. Oral Oncol. 2019;96:34–41.
Ming X-Y, Fu L, Zhang L-Y, Qin Y-R, Cao T-T, Chan KW, et al. Integrin α7 is a functional cancer stem cell surface marker in oesophageal squamous cell carcinoma. Nat Commun. 2016;7:13568.
Wang L, He T, Liu J, Tai J, Wang B, Chen Z, et al. Pan-cancer analysis reveals tumor-associated macrophage communication in the tumor microenvironment. Exp Hematol Oncol. 2021;10:31.
Zhu S, Yi M, Wu Y, Dong B, Wu K. Roles of tumor-associated macrophages in tumor progression: implications on therapeutic strategies. Exp Hematol Oncol. 2021;10:60.
Yan Y, Huang L, Liu Y, Yi M, Chu Q, Jiao D, et al. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J Hematol Oncol. 2022;15:104.
Lei X, Lei Y, Li J-K, Du W-X, Li R-G, Yang J, et al. Immune cells within the tumor microenvironment: biological functions and roles in cancer immunotherapy. Cancer Lett. 2020;470:126–33.
Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009;11:R68.
Walsh C, Tanjoni I, Uryu S, Tomar A, Nam J-O, Luo H, et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Ther. 2010;9:778–90.
Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10:2135–45.
Huehn J, Hamann A. Homing to suppress: address codes for Treg migration. Trends Immunol. 2005;26:632–6.
Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell. 2015;163:160–73.
Griffith BGC, Upstill-Goddard R, Brunton H, Grimes GR, Biankin AV, Serrels B, et al. FAK regulates IL-33 expression by controlling chromatin accessibility at c-Jun motifs. Sci Rep. 2021;11:229.
Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–8.
Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635–47.
Canel M, Taggart D, Sims AH, Lonergan DW, Waizenegger IC, Serrels A. T-cell co-stimulation in combination with targeting FAK drives enhanced anti-tumor immunity. Elife. 2020;9: e48092.
Llewellyn RA, Gutknecht MF, Thomas KS, Conaway MR, Bouton AH. Focal adhesion kinase (FAK) deficiency in mononuclear phagocytes alters murine breast tumor progression. Am J Cancer Res. 2018;8:675–87.
Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18:99–115.
Wang X, Zhou Q, Yu Z, Wu X, Chen X, Li J, et al. Cancer-associated fibroblast-derived Lumican promotes gastric cancer progression via the integrin β1-FAK signaling pathway. Int J Cancer. 2017;141:998–1010.
Begum A, McMillan RH, Chang Y-T, Penchev VR, Rajeshkumar NV, Maitra A, et al. Direct interactions with cancer-associated fibroblasts lead to enhanced pancreatic cancer stem cell function. Pancreas. 2019;48:329–34.
Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–60.
Begum A, Ewachiw T, Jung C, Huang A, Norberg KJ, Marchionni L, et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS ONE. 2017;12: e0180181.
Wu H-J, Hao M, Yeo SK, Guan J-L. FAK signaling in cancer-associated fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs-mediated intercellular communication. Oncogene. 2020;39:2539–49.
Demircioglu F, Wang J, Candido J, Costa ASH, Casado P, de Luxan DB, et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat Commun. 2020;11:1290.
Rizzo MT. Focal adhesion kinase and angiogenesis. Where do we go from here? Cardiovasc Res. 2004;64:377–8.
Peng X, Ueda H, Zhou H, Stokol T, Shen T-L, Alcaraz A, et al. Overexpression of focal adhesion kinase in vascular endothelial cells promotes angiogenesis in transgenic mice. Cardiovasc Res. 2004;64:421–30.
Tavora B, Batista S, Reynolds LE, Jadeja S, Robinson S, Kostourou V, et al. Endothelial FAK is required for tumour angiogenesis. EMBO Mol Med. 2016;8:1229.
Kurio N, Shimo T, Fukazawa T, Okui T, Hassan NMM, Honami T, et al. Anti-tumor effect of a novel FAK inhibitor TAE226 against human oral squamous cell carcinoma. Oral Oncol. 2012;48:1159–70.
Rothhut B, Ghoneim C, Antonicelli F, Soula-Rothhut M. Epidermal growth factor stimulates matrix metalloproteinase-9 expression and invasion in human follicular thyroid carcinoma cells through Focal adhesion kinase. Biochimie. 2007;89:613–24.
Wu Q, Qian W, Sun X, Jiang S. Small-molecule inhibitors, immune checkpoint inhibitors, and more: FDA-approved novel therapeutic drugs for solid tumors from 1991 to 2021. J Hematol Oncol. 2022;15:143.
Marin-Acevedo JA, Kimbrough EO, Lou Y. Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol. 2021;14:45.
Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y, et al. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J Hematol Oncol. 2022;15:24.
Wu H, Gong Y, Ji P, Xie Y, Jiang Y-Z, Liu G. Targeting nucleotide metabolism: a promising approach to enhance cancer immunotherapy. J Hematol Oncol. 2022;15:45.
Dai M, Liu M, Yang H, Küçük C, You H. New insights into epigenetic regulation of resistance to PD-1/PD-L1 blockade cancer immunotherapy: mechanisms and therapeutic opportunities. Exp Hematol Oncol. 2022;11:101.
Zhu S, Wu Y, Song B, Yi M, Yan Y, Mei Q, et al. Recent advances in targeted strategies for triple-negative breast cancer. J Hematol Oncol. 2023;16:100.
Mohan N, Hosain S, Zhao J, Shen Y, Luo X, Jiang J, et al. Atezolizumab potentiates Tcell-mediated cytotoxicity and coordinates with FAK to suppress cell invasion and motility in PD-L1+ triple negative breast cancer cells. Oncoimmunology. 2019;8: e1624128.
Gao X, Mi Y, Guo N, Xu H, Xu L, Gou X, et al. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front Immunol. 2017;8:774.
Pan M-R, Wu C-C, Kan J-Y, Li Q-L, Chang S-J, Wu C-C, et al. Impact of FAK expression on the cytotoxic effects of CIK therapy in triple-negative breast cancer. Cancers. 2019;12:94.
Wei Y, Wang Y, Liu N, Qi R, Xu Y, Li K, et al. A FAK inhibitor boosts anti-PD1 immunotherapy in a hepatocellular carcinoma mouse model. Front Pharmacol. 2021;12: 820446.
Ozmadenci D, Shankara Narayanan JS, Andrew J, Ojalill M, Barrie AM, Jiang S, et al. Tumor FAK orchestrates immunosuppression in ovarian cancer via the CD155/TIGIT axis. Proc Natl Acad Sci USA. 2022;119: e2117065119.
Shan S, Fang B, Zhang Y, Wang C, Zhou J, Niu C, et al. Mechanical stretch promotes tumoricidal M1 polarization via the FAK/NF-κB signaling pathway. FASEB J. 2019;33:13254–66.
Dragoj M, Milosevic Z, Bankovic J, Tanic N, Pesic M, Stankovic T. Targeting CXCR4 and FAK reverses doxorubicin resistance and suppresses invasion in non-small cell lung carcinoma. Cell Oncol. 2017;40:47–62.
Zhang B, Zhang Y, Zhang J, Liu P, Jiao B, Wang Z, et al. Focal adhesion kinase (FAK) inhibition synergizes with KRAS G12C inhibitors in treating cancer through the regulation of the FAK-YAP signaling. Adv Sci. 2021;8: e2100250.
Paradis JS, Acosta M, Saddawi-Konefka R, Kishore A, Gomes F, Arang N, et al. Synthetic lethal screens reveal cotargeting FAK and MEK as a multimodal precision therapy for GNAQ-driven uveal melanoma. Clin Cancer Res. 2021;27:3190–200.
Chen G, Gao C, Gao X, Zhang DH, Kuan S-F, Burns TF, et al. Wnt/β-catenin pathway activation mediates adaptive resistance to BRAF inhibition in colorectal cancer. Mol Cancer Ther. 2018;17:806–13.
Taylor KN, Schlaepfer DD. Adaptive resistance to chemotherapy, a multi-FAK-torial linkage. Mol Cancer Ther. 2018;17:719–23.
Romito I, Porru M, Braghini MR, Pompili L, Panera N, Crudele A, et al. Focal adhesion kinase inhibitor TAE226 combined with Sorafenib slows down hepatocellular carcinoma by multiple epigenetic effects. J Exp Clin Cancer Res. 2021;40:364.
Xu B, Lefringhouse J, Liu Z, West D, Baldwin LA, Ou C, et al. Inhibition of the integrin/FAK signaling axis and c-Myc synergistically disrupts ovarian cancer malignancy. Oncogenesis. 2017;6: e295.
Dawson JC, Serrels B, Byron A, Muir MT, Makda A, García-Muñoz A, et al. A synergistic anticancer FAK and HDAC inhibitor combination discovered by a novel chemical-genetic high-content phenotypic screen. Mol Cancer Ther. 2020;19:637–49.
Ichihara E, Westover D, Meador CB, Yan Y, Bauer JA, Lu P, et al. SFK/FAK signaling attenuates osimertinib efficacy in both drug-sensitive and drug-resistant models of EGFR-mutant lung cancer. Cancer Res. 2017;77:2990–3000.
Jiang H, Liu X, Knolhoff BL, Hegde S, Lee KB, Jiang H, et al. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut. 2020;69:122–32.
Byeon Y, Lee J-W, Choi WS, Won JE, Kim GH, Kim MG, et al. CD44-targeting PLGA nanoparticles incorporating paclitaxel and FAK siRNA overcome chemoresistance in epithelial ovarian cancer. Cancer Res. 2018;78:6247–56.
Hou J, Tan Y, Su C, Wang T, Gao Z, Song D, et al. Inhibition of protein FAK enhances 5-FU chemosensitivity to gastric carcinoma via p53 signaling pathways. Comput Struct Biotechnol J. 2020;18:125–36.
Le Large TYS, Bijlsma MF, El Hassouni B, Mantini G, Lagerweij T, Henneman AA, et al. Focal adhesion kinase inhibition synergizes with nab-paclitaxel to target pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2021;40:91.
Roy-Luzarraga M, Reynolds LE, de Luxán-Delgado B, Maiques O, Wisniewski L, Newport E, et al. Suppression of endothelial Cell FAK expression reduces pancreatic ductal adenocarcinoma metastasis after gemcitabine treatment. Cancer Res. 2022;82:1909–25.
Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, et al. Endothelial–cell FAK targeting sensitizes tumours to DNA–damaging therapy. Nature. 2014;514:112–6.
Roy-Luzarraga M, Abdel-Fatah T, Reynolds LE, Clear A, Taylor JG, Gribben JG, et al. Association of low tumor endothelial cell pY397-focal adhesion kinase expression with survival in patients with neoadjuvant-treated locally advanced breast cancer. JAMA Netw Open. 2020;3: e2019304.
Chen H, Tu W, Lu Y, Zhang Y, Xu Y, Chen X, et al. Low-dose X-ray irradiation combined with FAK inhibitors improves the immune microenvironment and confers sensitivity to radiotherapy in pancreatic cancer. Biomed Pharmacother. 2022;151: 113114.
Mohamed AA, Thomsen A, Follo M, Zamboglou C, Bronsert P, Mostafa H, et al. FAK inhibition radiosensitizes pancreatic ductal adenocarcinoma cells in vitro. Strahlenther Onkol. 2021;197:27–38.
Osipov A, Blair AB, Liberto J, Wang J, Li K, Herbst B, et al. Inhibition of focal adhesion kinase enhances antitumor response of radiation therapy in pancreatic cancer through CD8+ T cells. Cancer Biol Med. 2021;18:206–14.
Williams KE, Bundred NJ, Landberg G, Clarke RB, Farnie G. Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem Cells. 2015;33:327–41.
NguemgoKouam P, Bühler H, Hero T, Adamietz IA. The increased adhesion of tumor cells to endothelial cells after irradiation can be reduced by FAK-inhibition. Radiat Oncol. 2019;14:25.
Si Q, Ye Q, Bing Z, Fan R, Hu X, Liu B, et al. Carbon ion irradiation enhances the anti-tumor efficiency in tongue squamous cell carcinoma via modulating the FAK signaling. Front Public Health. 2021;9: 631118.
Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610.
Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–23.
Diaz Osterman CJ, Ozmadenci D, Kleinschmidt EG, Taylor KN, Barrie AM, Jiang S, et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife. 2019;8: e47327.
Qin Z, Zhou C. HOXA13 promotes gastric cancer progression partially via the FN1-mediated FAK/Src axis. Exp Hematol Oncol. 2022;11:7.
Dawson JC, Serrels A, Stupack DG, Schlaepfer DD, Frame MC. Targeting FAK in anticancer combination therapies. Nat Rev Cancer. 2021;21:313–24.
Xiong J, Yan L, Zou C, Wang K, Chen M, Xu B, et al. Integrins regulate stemness in solid tumor: an emerging therapeutic target. J Hematol Oncol. 2021;14:177.
Lin T-H, Tan T-W, Tsai T-H, Chen C-C, Hsieh T-F, Lee S-S, et al. d-Pinitol inhibits prostate cancer metastasis through inhibition of αVβ3 integrin by modulating FAK, c-Src and NF-κB pathways. Int J Mol Sci. 2013;14:9790–802.
Cromm PM, Samarasinghe KTG, Hines J, Crews CM. Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J Am Chem Soc. 2018;140:17019–26.
Li H, Dong J, Cai M, Xu Z, Cheng X-D, Qin J-J. Protein degradation technology: a strategic paradigm shift in drug discovery. J Hematol Oncol. 2021;14:138.
Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Can Res. 2008;68:1935–44.
Sun H, Pisle S, Gardner ER, Figg WD. Bioluminescent imaging study: FAK inhibitor, PF-562,271, preclinical study in PC3M-luc-C6 local implant and metastasis xenograft models. Cancer Biol Ther. 2010;10:38–43.
Slack-Davis JK, Hershey ED, Theodorescu D, Frierson HF, Parsons JT. Differential requirement for focal adhesion kinase signaling in cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol Cancer Ther. 2009;8:2470–7.
Serrels A, McLeod K, Canel M, Kinnaird A, Graham K, Frame MC, et al. The role of focal adhesion kinase catalytic activity on the proliferation and migration of squamous cell carcinoma cells. Int J Cancer. 2012;131:287–97.
Bagi CM, Christensen J, Cohen DP, Roberts WG, Wilkie D, Swanson T, et al. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biol Ther. 2009;8:856–65.
Kessler BE, Sharma V, Zhou Q, Jing X, Pike LA, Kerege AA, et al. FAK expression, not kinase activity, is a key mediator of thyroid tumorigenesis and protumorigenic processes. Mol Cancer Res. 2016;14:869–82.
Newport E, Pedrosa AR, Lees D, Dukinfield M, Carter E, Gomez-Escudero J, et al. Elucidating the role of the kinase activity of endothelial cell focal adhesion kinase in angiocrine signalling and tumour growth. J Pathol. 2022;256:235–47.
Stone RL, Baggerly KA, Armaiz-Pena GN, Kang Y, Sanguino AM, Thanapprapasr D, et al. Focal adhesion kinase: an alternative focus for anti-angiogenesis therapy in ovarian cancer. Cancer Biol Ther. 2014;15:919–29.
Wang S, Hwang EE, Guha R, O’Neill AF, Melong N, Veinotte CJ, et al. High-throughput chemical screening identifies focal adhesion kinase and aurora kinase B inhibition as a synergistic treatment combination in Ewing sarcoma. Clin Cancer Res. 2019;25:4552–66.
Lander VE, Belle JI, Kingstonl NL, Herndon JM, Hogg GD, Liu X, et al. Stromal reprogramming by FAK inhibition overcomes radiation resistance to allow for immune priming and response to checkpoint blockade. Cancer Discov. 2022;12:2774–99.
Fu Y, Zhang Y, Lei Z, Liu T, Cai T, Wang A, et al. Abnormally activated OPN/integrin αVβ3/FAK signalling is responsible for EGFR-TKI resistance in EGFR mutant non-small-cell lung cancer. J Hematol Oncol. 2020;13:169.
Zhang Y, Cheng K, Xu B, Shi J, Qiang J, Shi S, et al. Epigenetic input dictates the threshold of targeting of the integrin-dependent pathway in non-small cell lung cancer. Front Cell Dev Biol. 2020;8:652.
Tong X, Tanino R, Sun R, Tsubata Y, Okimoto T, Takechi M, et al. Protein tyrosine kinase 2: a novel therapeutic target to overcome acquired EGFR-TKI resistance in non-small cell lung cancer. Respir Res. 2019;20:270.
Lin H-M, Lee BY, Castillo L, Spielman C, Grogan J, Yeung NK, et al. Effect of FAK inhibitor VS-6063 (defactinib) on docetaxel efficacy in prostate cancer. Prostate. 2018;78:308–17.
Kang Y, Hu W, Ivan C, Dalton HJ, Miyake T, Pecot CV, et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J Natl Cancer Inst. 2013;105:1485–95.
Huo X, Zhang W, Zhao G, Chen Z, Dong P, Watari H, et al. FAK PROTAC inhibits ovarian tumor growth and metastasis by disrupting kinase dependent and independent pathways. Front Oncol. 2022;12: 851065.
Zhang L, Zhao D, Wang Y, Zhang W, Zhang J, Fan J, et al. Focal adhesion kinase (FAK) inhibitor-defactinib suppresses the malignant progression of human esophageal squamous cell carcinoma (ESCC) cells via effective blockade of PI3K/AKT axis and downstream molecular network. Mol Carcinog. 2021;60:113–24.
Lopez-Mejia IC, Pijuan J, Navaridas R, Santacana M, Gatius S, Velasco A, et al. Oxidative stress-induced FAK activation contributes to uterine serous carcinoma aggressiveness. Mol Oncol. 2023;17:98–118.
Tiede S, Meyer-Schaller N, Kalathur RKR, Ivanek R, Fagiani E, Schmassmann P, et al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis. 2018;7:73.
Li H, Gao Y, Ren C. Focal adhesion kinase inhibitor BI 853520 inhibits cell proliferation, migration and EMT process through PI3K/AKT/mTOR signaling pathway in ovarian cancer. Discov Oncol. 2021;12:29.
Hirt UA, Waizenegger IC, Schweifer N, Haslinger C, Gerlach D, Braunger J, et al. Efficacy of the highly selective focal adhesion kinase inhibitor BI 853520 in adenocarcinoma xenograft models is linked to a mesenchymal tumor phenotype. Oncogenesis. 2018;7:21.
Kanteti R, Mirzapoiazova T, Riehm JJ, Dhanasingh I, Mambetsariev B, Wang J, et al. Focal adhesion kinase a potential therapeutic target for pancreatic cancer and malignant pleural mesothelioma. Cancer Biol Ther. 2018;19:316–27.
Chuang H-H, Wang P-H, Niu S-W, Zhen Y-Y, Huang M-S, Hsiao M, et al. Inhibition of FAK signaling elicits lamin A/C-associated nuclear deformity and cellular senescence. Front Oncol. 2019;9:22.
Golubovskaya VM, Virnig C, Cance WG. TAE226-Induced apoptosis in breast cancer cells with overexpressed Src or EGFR. Mol Carcinog. 2008;47:222–34.
Kurio N, Shimo T, Fukazawa T, Takaoka M, Okui T, Hassan NMM, et al. Anti-tumor effect in human breast cancer by TAE226, a dual inhibitor for FAK and IGF-IR in vitro and in vivo. Exp Cell Res. 2011;317:1134–46.
Liu T-J, LaFortune T, Honda T, Ohmori O, Hatakeyama S, Meyer T, et al. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol Cancer Ther. 2007;6:1357–67.
Storch K, Sagerer A, Cordes N. Cytotoxic and radiosensitizing effects of FAK targeting in human glioblastoma cells in vitro. Oncol Rep. 2015;33:2009–16.
Wang ZG, Fukazawa T, Nishikawa T, Watanabe N, Sakurama K, Motoki T, et al. TAE226, a dual inhibitor for FAK and IGF-IR, has inhibitory effects on mTOR signaling in esophageal cancer cells. Oncol Rep. 2008;20:1473–7.
Watanabe N, Takaoka M, Sakurama K, Tomono Y, Hatakeyama S, Ohmori O, et al. Dual tyrosine kinase inhibitor for focal adhesion kinase and insulin-like growth factor-I receptor exhibits anticancer effect in esophageal adenocarcinoma in vitro and in vivo. Clin Cancer Res. 2008;14:4631–9.
Golubovskaya VM, Figel S, Ho BT, Johnson CP, Yemma M, Huang G, et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo [3.3.1.1(3,7)]decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis. 2012;33:1004–13.
Haemmerle M, Bottsford-Miller J, Pradeep S, Taylor ML, Choi H-J, Hansen JM, et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J Clin Invest. 2016;126:1885–96.
Tanjoni I, Walsh C, Uryu S, Tomar A, Nam J-O, Mielgo A, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther. 2010;9:764–77.
Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, et al. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J Med Chem. 2008;51:7405–16.
Zhang H, Shao H, Golubovskaya VM, Chen H, Cance W, Adjei AA, et al. Efficacy of focal adhesion kinase inhibition in non-small cell lung cancer with oncogenically activated MAPK pathways. Br J Cancer. 2016;115:203–11.
Kurenova EV, Hunt DL, He D, Magis AT, Ostrov DA, Cance WG. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J Med Chem. 2009;52:4716–24.
Golubovskaya VM, Ho B, Zheng M, Magis A, Ostrov D, Morrison C, et al. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC Cancer. 2013;13:342.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 82272794, 82073370).
Author information
Authors and Affiliations
Contributions
XT drafted the manuscript and prepared the figures and tables. YY, BS, and SZ collected the related references and participated in the discussion. KW and QM designed this review and revised the manuscript. All authors contributed to this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tan, X., Yan, Y., Song, B. et al. Focal adhesion kinase: from biological functions to therapeutic strategies. Exp Hematol Oncol 12, 83 (2023). https://doi.org/10.1186/s40164-023-00446-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-023-00446-7