
Abstract
Breast cancer is the second leading cause of death for women worldwide. The heterogeneity of this disease presents a big challenge in its therapeutic management. However, recent advances in molecular biology and immunology enable to develop highly targeted therapies for many forms of breast cancer. The primary objective of targeted therapy is to inhibit a specific target/molecule that supports tumor progression. Ak strain transforming, cyclin-dependent kinases, poly (ADP-ribose) polymerase, and different growth factors have emerged as potential therapeutic targets for specific breast cancer subtypes. Many targeted drugs are currently undergoing clinical trials, and some have already received the FDA approval as monotherapy or in combination with other drugs for the treatment of different forms of breast cancer. However, the targeted drugs have yet to achieve therapeutic promise against triple-negative breast cancer (TNBC). In this aspect, immune therapy has come up as a promising therapeutic approach specifically for TNBC patients. Different immunotherapeutic modalities including immune-checkpoint blockade, vaccination, and adoptive cell transfer have been extensively studied in the clinical setting of breast cancer, especially in TNBC patients. The FDA has already approved some immune-checkpoint blockers in combination with chemotherapeutic drugs to treat TNBC and several trials are ongoing. This review provides an overview of clinical developments and recent advancements in targeted therapies and immunotherapies for breast cancer treatment. The successes, challenges, and prospects were critically discussed to portray their profound prospects.
Similar content being viewed by others


Introduction
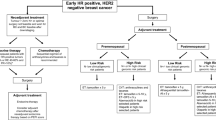
Despite ongoing medical advances, breast cancer remains the second most widespread and fatal malignancy in females [1]. During the past four decades, breast cancer incidences have risen alarmingly [2]. In 2020, there were approximately 2.3 million new cases of breast cancer worldwide, and there were also roughly 6,85,000 deaths from this illness, with notable geographic variations between various countries and regions [3]. Notably, high-economic nations represent a greater percentage of breast cancer fatalities. By 2040, it has been anticipated that there will be more than 3 million new instances of breast cancer each year with more than 1 million annual deaths [4]. There are several different forms of breast carcinoma. It has been found that steroid hormone receptors (HRs) are significant prognostic factors for breast cancer [5]. Breast cancer cells with HRs expressing estrogen (ER), progesterone (PR), or both are referred to as ER-positive (ER +), PR-positive (PR +), or ER/PR positive (ER + /PR +) breast cancers, respectively. Most breast carcinomas are ER + and of those, more than half are both ER + /PR + ; while, only about 2% are solely PR + . Triple-negative breast cancer (TNBC) refers to the cancerous cells lacking ER and PR as well as making too little or no expression of human epidermal growth receptor 2 (HER2). About 10–15% of all breast carcinomas are TNBC. In recent years immunogenic prospects of breast tumor have been revealed. The discovery of tumor-infiltrating lymphocytes (TILs) within breast cancers demonstrated the immunogenic character of breast cancer [6, 7]. Analysis of breast tumor samples revealed that individuals with HER2-positive (HER2 +) and TNBC had a greater number of TILs than those of HR-positive (HR +) subtypes [8, 9].
After decades of endocrine and other therapies, an improved understanding of immune evasion by cancerous cells and the creation of specific immune checkpoint antagonists have unveiled new therapeutic options [10,11,12,13,14]. Immunotherapy involves the enhancement of host immunity and admits cancer as a foreign antigen, which leads to the destruction of cancer cells. However, some adverse events have been advocated in the patients who received immunotherapies, which include fatigue, nausea, vomiting, dizziness, and pruritus [15]. Enterocolitis, endocrinopathies, liver anomalies, and uveitis are some additional immune-related side effects that can result from using immunotherapeutics like ipilimumab. Immune checkpoint blockers are being used in conjunction with chemotherapy, targeted therapy, and radiation therapy under intense examination due to the low efficacy of previous immunotherapeutic drugs in treating many kinds of breast carcinoma. On the other hand, breast cancer-targeted therapies use substances or drugs to inhibit disease progression by obstructing certain components essential for cancer cell survival, proliferation, and migration, as well as for angiogenesis [16]. Poly (ADP-ribose) polymerase (PARP), cyclin-dependent kinase (CDK) 4 and 6 (CDK4/6), Ak strain transforming (AKT), and fibroblast growth factor receptor (FGFR) are well-established therapeutic targets that have been the main focus of drug development for the treatment of breast cancer [17,18,19,20]. Patients under targeted therapy also deal with some common side effects such as nausea, vomiting, diarrhea, tiredness, and rashes. Thus, adequate attention must be given to address the toxicity concerns of both the targeted and immune therapies. This review will give a concise overview of the different targeted and immunotherapies used to treat breast cancer. The effectiveness and safety of immunotherapy in combination with chemotherapy and/or radiotherapy, HER2-targeted therapy, CDK4/6 inhibitors, angiogenesis inhibitors, PARP antagonists, and other treatments in the clinical setting were also covered in this review.
Crosstalk pathway in breast cancer
A growing body of evidence revealed that the activation of prolactin (PRL) receptor (PRLR) and erythroblastic leukemia viral oncogene homolog receptor (ErbBR) endorses oncogenesis in mammary glands, supports breast tumor growth, and induces chemoresistance [21, 22]. The amplification of the epidermal growth factor (EGF) receptor (EGFR), HER2, as well as PRLR in breast cancers, has been regarded to accelerate tumor growth by activating their downstream signaling pathways [23]. Secretion of PRL from breast cells leads to the activation of downstream PRLR signaling, involving the activation of Janus kinase (JAK)/signal transducer and activator of transcription (STAT), phosphoinositide 3-kinase (PI3K)/Ak strain transforming (AKT), and mitogen-activated protein kinase (MAPK) pathways, which have been implicated in mammary tumorigenesis [24, 25]. When EGF is released, the complete pathway is covered, which aids in the formation of a complex with specificity protein 1 (Sp1) and enhancer binding protein (EBP) resulting in activation of STAT5. Further overexpression of EGFR2 activates the RAS/focal adhesion kinase (FAK) signaling pathways, activating EGFR by cross-phosphorylation. Mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase 1/2 (ERK1/2) along with PI3K/AKT pathways are jointly induced by PRL and EGF through their corresponding receptors, promoting the proliferation, survival, and metastasis of breast cancer cells [26]. Thus, PRL factors as well as EGFR signaling pathways interact in breast cancer development and progression. In cell line-based assay involving breast cancer cells (MCF-7 and T47D), it has been found that the PRL/PRLR triggers EGFR2 phosphorylation (tyr1221 and 1222) via JAK2, activating the downstream PI3K/AKT signaling [26]. Crosstalk between PRLR and HER2 signaling allows the phosphorylation of ER, its attachment to the PRLR regulator, as well as a rise in PRLR dictation [21]. It has also been revealed that EGF/EGFR could phosphorylate ER (ser118 and 167) and initiate downstream MAPK signaling in breast cancer cells, as well as STAT5b and PRLR transcription [27]. Proto-oncogene tyrosine-protein kinase SRC (c-SRC) endorses EGFR phosphorylation (tyr845) and allows EGFR to indirectly bind to and activate STAT5b. This results in the activation of ER to the generic hPIII promoter and PRLR. It has been revealed that endogenous PRL is necessary for ER on PRLR activation [28]. EGFR can also endorse STAT5 activation directly via phosphorylation in an EGFR-dependent manner [21]. Activated STAT5 translocate to the nucleus, which endorses the transcription of certain genes important for proliferation, diversification, as well as survival. Additionally, it has been revealed that EGFR and HER2 activation are related to STAT3 activation, which also encourages tumor survival and development [21, 28]. Fig. 1 explains the crosstalk between PRLR and EGFR/HER2 signaling to accelerate the growth of breast tumor.

Crosstalk between PRLR and EGFR/HER2 signaling to promote breast cancer progression. PRL endorses PRLR activation, which recruits downstream pathways, such as JAK/STAT, MAPK/ERK, PI3K/AKT/mTOR, NEK3/VAV2/RhoA and TEC/VAV1/RAC1 involved in growth, survival, and migration of breast cancer. EGF/EGFR signaling somewhat overlaps with PRLR signaling to result in the activation of similar downstream events. PRLR/HER2 crosstalk endorses ER phosphorylation and promotes its attachment to the PRLR regulator and promotes PRLR transcription. EGF/EGFR can also trigger PRLR transcription in MAPK/PI3K-dependent manner. In addition, PRL/PRLR activation recruits HRE2 via JAK2 resulting an activation of FAC signaling that promotes cell adherence and induces metastasis. Arrows represent downstream events. AKT, Ak strain transforming; EGF, Epidermal growth factor; EGFR, Epidermal growth factor receptor; ER, Estrogen receptor; ERK, Extracellular signal-regulated kinase; GRB2, Growth factor receptor-bound protein 2; HER2, Human epidermal growth factor-2; JAK, Janus kinase, MAPK, Mitogen-activated protein kinase; mTOR, Mammalian target of rapamycin; NEK3, NIMA-related kinase 3; P, Phosphate; PI3K, Phosphoinositide 3-kinase; PRL, Prolactin; PRLR: Prolactin receptor; RhoA, Ras homolog family member A; STAT, Signal transducer and activator of transcription; TEK, TEK receptor tyrosine kinase; VAV, Vav guanine nucleotide exchange factor 1
Targeted therapy
Surgery followed by radiotherapy and chemotherapy is the standard treatment protocol for breast cancer management [29]. The aim of chemotherapy after surgery or radiotherapy is to reduce the likelihood of cancer recurrence. Targeted drug therapy aims at target proteins on breast cancer cells that support their growth, spread, and ability to proliferate [30]. The most effective therapies for breast cancer are those that specifically target the ER and HER2 receptors [31]. A growing body of evidence has proposed some promising novel therapies for several forms of breast cancer, including TNBC and HER2 + carcinomas [32]. Aromatase antagonists, endocrine treatment, selective ER modulators (SERMs), and selective estrogen down regulators (SERDs) are all well-known forms of personalized therapy for HER2 + breast cancer [33, 34]. The course of breast cancer is largely influenced by estrogen and ER [35, 36]. This is the rationale behind the long-standing practice of inhibiting the estrogen signaling pathway in breast cancer patients that are ER + . The first approved drug for treating ER + advanced breast cancer was tamoxifen, which reduces tumor recurrence by roughly 40–50% [37]. SERMs have indeed been utilized to limit cancer growth in estrogen-reliant forms of breast cancer [38]. Inhibitors of any of the targets, such as PARP, HER2, PI3K, AKT, mammalian target of rapamycin (mTOR), fibroblast growth factor (FGF) receptors (FGFRs), and vascular endothelial growth factor (VEGF), could potentially be used as therapeutic approaches to stop the progression of breast cancer due to their roles in various pathways of carcinogenesis, including the cell cycle, angiogenesis, metastasis, etc. [39, 40]. These inhibitors have already demonstrated clinical potential. An mTOR-inhibitor, zortress (42-O-(2-hydroxyethyl)rapamycin) has been authorized for advanced or metastatic aromatase antagonist-resistant ER + breast carcinoma. There is presently no approved targeted therapy for TNBC so far. Given the variety of breast cancer, it is unavoidable that the treatments will be more carefully individualized for each subtype, stage, and grade of breast cancer [41].
PARP inhibitors
PARP enzymes play crucial roles in many cellular events including the deoxyribonucleic acid (DNA) repairing process [42]. Two enzymes, PARP1 and PARP2, which are activated by DNA strand break, take a role in DNA repair. A growing body of evidence has highlighted PARP as a possible chemotherapeutic target [42]. Inhibition of PARP enzymes prevents DNA repair and causes DNA strands to break, posing a danger to cell survival [43]. Four PARP antagonists have been studied extensively in the clinical setting of breast cancers.
Olaparib, the first US Food and Drug Administration (FDA) approved PARP inhibitor, belongs to N-acyl piperazine class [44]. Olaparib inhibits the activity of DNA topoisomerase 2-binding protein 1 (TOPBP1) and WEE1 (nuclear kinase belonging to the ser/thr family). This results in the buildup of DNA damage that persists during mitosis and resulting in mitotic catastrophe and cell death.. It is one of the PARP inhibitors, which does not cause transaminitis [45]. Importantly, this drug is delivered through the oral route. In the phase III OlympiAD trial (NCT02000622), olaparib (300 mg, twice daily) monotherapy was found to be more effective than standard therapy in patients with metastatic HER2 − breast cancer and germline breast cancer gene (gBRCA) mutations in terms of prolonging progression-free survival (2.8 months) and reducing death (42%) [46]. However, olaparib has been mostly studied as an adjuvant or neoadjuvant in combination with other chemotherapeutic drugs. Olaparib has been used in combination with carboplatin, cyclophosphamide, dacarbazine, durvalumab, eribulin, gemcitabine, paclitaxel, and prexasertib in different clinical studies. In the phase I/II trial (NCT01445418), the olaparib/carboplatin combination is tolerable and exhibits moderate activity in women with sporadic TNBC with the maximum tolerated dose of olaparib was 400 mg, twice daily [47]. However, the majority of grade 3 and 4 adverse events in 36% of patients were neutropenia, followed by thrombocytopenia and anemia in 11% of patients. In another phase I/II trial (UMIN00009498), olaparib (300 mg twice daily)/eribulin combination exhibited antitumor activity against sporadic TNBC patients; though, caution has been recommended if febrile neutropenia is present [48]. 20.8% of patients in phase I and 33.3% of patients in phase II experienced febrile neutropenia. Importantly, PARP inhibition was assured even at the lowest dose of olaparib (25 mg, twice daily). In another phase I/II basket study (NCT02734004), a combination of olaparib (300 mg, twice daily) and durvalumab (1·5 g) exhibited promising anticancer efficacy and was well tolerated in gBRCA-mutated metastatic breast cancer patients [49]. Olaparib given to patients with high-risk, HER2 − early breast cancer and gGRCA-pathogenic or likely pathogenic breast cancer after completion of local treatment was associated with a significantly longer invasive disease-free endurance of patients, according to a phase III trial (NCT02032823) [50]. In a phase II trial (NCT02789332), it was found that individuals with primary HER2 − breast cancer and homologous recombination deficiency responded better to olaparib than carboplatinum as neoadjuvant with paclitaxel in terms of pathological complete response percentage, followed by epirubicin/cyclophosphamide chemotherapy [51]. However, if another drug is being taken that is subject to hepatic metabolism, the use of olaparib must be avoided.
Another potential orally effective PARP inhibitor is talazoparib which is efficacious against gBRCA-mutated, HER2 − breast cancer with an average tolerated dose of 0.60–1 mg/day [52]. In the phase I trial (NCT01286987) involving 18 gBRCA-mutated progressive breast cancer patients, talazoparib (1 mg once daily) monotherapy resulted in a significant therapeutic response rate of 50% and an excellent clinical benefit rate of 86% after 24 weeks of treatment with a good safety and tolerability profile [53]. Talazoparib (1 mg/day) treatment showed promising antitumor activity in a two-stage phase II study (NCT02034916), with manageable adverse effects for gBRCA-mutated progressive breast cancer subjects who had previously undergone platinum-based or non-platinum chemotherapy [54]. In an open-label, randomized, phase III trial (NCT01945775), talazoparib monotherapy significantly outperformed standard single agent-chemotherapy in terms of progression-free endurance among individuals with gBRCA-mutated advanced breast carcinoma [55]. Common side effects were mostly hematologic, rarely happened after medication withdrawal, and could be negotiated with dose adjustments or supportive treatment. In other reports, talazoparib has been revealed to be well tolerated and statistically more effective than other chemotherapeutic drugs in phase III clinical settings [56, 57].
Rucaparib is another orally effective well-tolerated PARP antagonist that ensured sustained PARP inhibition in the surrogate tissues even at the lower doses [58, 59]. In a phase II trial, a lack of objective response rate was seen after intravenous or oral rucaparib treatment within a 21-day cycle in the patients representing gBRCA-mutated locally advanced or metastatic breast tumor. In 39% of breast cancer patients, stable illness on/after 12 weeks was the best outcome [59]. Patsouris and colleagues reported that only a small population of breast cancer patients without gBRCA1/2 mutation represented a significant loss of heterozygosity scores and could be benefited from PARP inhibition by rucaparib [60]. In a phase I clinical trial (NCT01009190), it has been found that oral rucaparib (240 mg/day) can be safely combined with a clinically applicable dose of carboplatin [61]. The clinical observation did not indicate favorable results with cisplatin and low-dose rucaparib combination in BRCA-mutated TNBC [62, 63].
Niraparib is an orally effective PARP inhibitor with effective PARP-trapping activity and has exhibited potential anticancer effects against ovarian and prostate cancer in clinical settings [64, 65]. Niraparib has demonstrated a strong PARP inhibitory impact and improved clinical benefit rates and was well tolerated in patients with gBRAC-mutated locally advanced or metastatic breast cancer [59]. In a randomized, phase III trial (BRAVO), breast cancer patients representing gBRCA mutation were randomized 2:1 between niraparib and the physicians’ optimal chemotherapy [66]. Centrally measured progression-free survival served as the principal outcome. Global survival, progression-free survival by limited evaluation, and objective response rate were considered secondary goals in addition to safety assessment. Following the pre-planned interim investigation, enrollment was stopped due to the occurrence of a significant discrepancy between the local and central progression-free survival assessments in the control (physicians’ choice chemotherapy) arm, which caused informative withholding of the trial [67]. Some trials are ongoing to evaluate the clinical potential of niraparib in breast cancer management.
CDK4/6 inhibitors
CDK4/6 play important role in the proliferation of cancer cells (Fig. 2). Activation of CDK4/6 by D-type cyclins (cyclin D1/CDK4 and cyclin D3/CDK6) endorses phosphorylation of retinoblastoma-associated protein (Rb) [68]. By turning on E2F transcription, phospho-Rb enables the cell to advance through the cell cycle and divide. Since cyclin D upregulation and restoration phospho-Rb expression is common in HR + breast cancer cells, the G1/S checkpoint is a prime therapeutic target to arrest cancer cell proliferation [69]. In addition, the transcription factor Forkhead box M1 (FOXM1) is an important phosphorylation target of CDK4/6 (Fig. 2). CDK4 and CDK6 endorse the stabilization and activation of FOXM1 via phosphorylation leading to the activation of the G1/S phase gene expressions, the suppression of ROS, and the prevention of senescence in cancer cells [69]. The CDK4/6 inhibitors arrest the cell cycle through this checkpoint (Fig. 2). CDK 4/6 inhibitors are prescription drugs that are clinically effective along with hormonal therapies to treat HR + and HER2 − progressive or metastatic mammary cancer [69]. FDA-approved CDK4/6 antagonists for the treatment of various types of breast cancer include palbociclib, abemaciclib, and ribociclib.

The mechanistic insight of CDK4/6 inhibitors in the management of breast cancer. CDK4/6 activation leads to Rb activation via phosphorylation. Activated Rb enables cell cycle progression by turning on E2F transcription. In addition, CDK4 and CDK6 endorse the stabilization and activation of FOXM1 via phosphorylation, which in turn promotes the upregulation of the G1/S phase genes and the avoidance of cell senescence. CDK4/6 inhibitors, such as palbociclib, ribociclib, or abemaciclib arrest the cell cycle by suppressing CDK4/6 downstream signaling event and arrest. Grey arrows represent downstream events and red lines represent inhibition. CDK, Cyclin-dependent kinase; FOXM1, Forkhead box protein M1; Rb: Retinoblastoma-associated protein
Palbociclib, the first selective orally effective CDK4/6 inhibitor, has received approval for use in treating cancer [70]. It is effective against HR + and HER − breast cancer or metastatic breast carcinoma in pre- or postmenopausal women. It is mainly used in combination with hormonal therapeutics, such as aromatase inhibitor and estrogen receptor antagonist. In contrast, Mayer and colleagues reported that the inclusion of palbociclib did not improve the likelihood of disease-free survival of adjuvant endocrine therapy in phase III clinical settings (NCT02513394) [71]. In phase I trial (NCT00141297), palbociclib exhibited slow absorption (Tmax = 5.5 h) and elimination (t1/2 = 25.9 h) profile with a high distribution volume of 2,793 L. At a dose of 125 mg/day, neutropenia has been identified as the main toxicity [72]. In the phase II trial (UPCC03909; NCT01037790), endocrine-resistant, HR + and Rb + advanced breast cancer patients responded favorably to single-agent palbociclib (125 mg/day, 1–21 days of a 28-day cycle) [73]. Uncomplicated cytopenias have been identified as adverse effects, which are negotiable with dose reduction. In a phase II clinical study (NCT00721409), the inclusion of palbociclib (125 mg/day) to letrozole (2.5 mg/day) showed significant improvement in progression-free survival with progressive ER + and HER2 − breast cancer [74]. Pulmonary embolism, back pain, and diarrhea were recorded as the serious adverse effects of this combination therapy only in a limited number (8%) of patients. Palbociclib was given the FDA approval for the treatment of HR + metastatic breast cancer when used in conjunction with letrozole; studies also indicated that fulvestrant could improve clinical outcomes [75]. In a double-blind, placebo-controlled, randomized phase III trial (NCT01942135), the palbociclib-fulvestrant treatment led to a longer overall survival (though statistically insignificant) than treatment with placebo-fulvestrant among HR + and HER2 − advanced breast cancer patients who had shown sensitivity to prior endocrine therapy [76].
Ribociclib is a highly selective and orally effective CDK4/6 inhibitor used for the treatment of HR + and HER2 − breast cancers with manageable toxicity profiles. In general, ribociclib is administered along with a hormone-blocking agent in breast cancer management [77]. In a phase III trial (NCT01958021), ribociclib (600 mg/day; 1–21 days of a 28-day cycle) in combination with letrozole (2.5 mg/day, continuous treatment) significantly prolonged progression-free survival in postmenopausal women with HR + and HER2 − breast cancers compared with placebo plus letrozole (25.3 months vs. 16.0 months) treated patients [78]. According to a phase III clinical trial (NCT02278120),ribociclib + letrozole treatment improved progression-free survival for premenopausal women bearing HR + and HER2 − progressive breast carcinoma, while exhibited a tolerable safety profile [79].
Abemaciclib is the strongest of these three CDK4/6 inhibitors in terms of CDK4/6 enzyme inhibition potential in vitro. It also demonstrated inhibitory effects on other kinases, such as CDK9 and proto-oncogene serine/threonine-protein kinase (PIM1) [80]. Oral treatment, continuous dosing, effective target inhibition, and manageable toxicities make abemaciclib a new therapeutic option for the most prevalent forms of breast cancer [81]. The highest tolerated dose of abemaciclib was established to be 200 mg, twice daily accordance with a phase I trial (NCT01394016) [82]. Single-agent abemaciclib exhibited a higher clinical benefit rate in HR + breast cancer compared with HR − cases. Compared to monotherapy, combination with fulvestrant slightly improved the clinical benefit rate in HR + breast cancer cases. In an open-label phase II trial (NCT02102490), abemaciclib exhibited potential clinical efficacy and tolerability in HR + and HER2 − metastatic breast cancer management [83]. The FDA approved abemaciclib (200 mg, twice daily) monotherapy on September 28, 2017, for women with HR + progressive or metastatic breast cancer continuing on both hormone treatment and chemotherapy, despite substantial dose reduction or omission rates were seen in phase II trial at this dose range [81, 83]. Abemaciclib in combination with endocrine therapy with antiestrogenic drugs in breast cancer management has been found to be efficacious with manageable safety profile in clinical settings. In the phase III double-blind trial (NCT02107703), abemaciclib (150 mg, twice daily) in combination with fulvestrant was efficacious, dramatically enhancing progression-free survival and objective response rate and exhibiting a tolerable safety profile in women with HR + and HGR2 − progressive breast cancer who proceeded while receiving hormone therapy [84]. A similar observation has been recorded with abemaciclib (150 mg, twice daily) in combination with a non-steroidal aromatase inhibitor either anastrozole (1 mg/day) or letrozole (2.5 mg/day) in a phase III clinical study (NCT02246621) involving postmenopausal women with HR + and HGR2 − progressive breast cancer [85]. Abemaciclib plus fulvestrant has been revealed to improve therapeutic outcomes in clinical settings (NCT02107703) for premenopausal women with HR + and HGR2 − progressive breast carcinoma who are resistant to hormone therapy [86].
The toxicity profiles of all three CDK4/6 inhibitors are comparable [81]. The FDA has warned that certain patients with advanced breast cancer who are being treated with CDK4/6 inhibitor, palbociclib, ribociclib, or abemaciclib may experience a rare but serious lung inflammation.
AKT Inhibitors
AKT is a crucial transducer in PI3K/AKT/mTOR signaling pathway that promotes the growth, division, migration, survival, and senescence of cells as well as the progression of cancer (Fig. 3) [17, 87]. PI3K is the key component of AKT signaling, which is a heterodimer comprising two subunits, such as p85 (regulatory) and p110 (catalytic). PI3K is activated by different growth stimuli via phosphorylation of the p85 subunit. Upon activation, PI3K endorses PIP2 (phosphoinositol 4, 5-biphosphate) phosphorylation to form PIP3 (phosphoinositol 3, 4, 5-triphosphate) in the plasma membrane, which acts as a secondary messenger of AKT recruitment [88, 89]. Phosphatase and tensin homolog is a phosphatase (PTEN) that acts as a negative regulator of PI3K/AKT signaling. Activation of AKT via phosphorylation endorses phosphorylation of forkhead box O1 (FOXO1) resulting in transcriptional suppression of FOXO1 and inhibition of its proapoptotic role [89, 90]. In addition, activation of mTOR via TSC1/2 phosphorylation is endorsed by AKT resulting in suppression of cell death pathways and activation of cell proliferation. AKT activation is regarded to directly suppress the activation of pro-apoptotic factors like the Bcl-2-associated death promoter (Bad). In addition, mutation of the PI3K catalytic alpha (PIK3CA) gene is known to activate PI3K. PIK3CA gene mutation is abandoned in HR + and HER + breast cancer; while it is less frequent in TNBC. On the other hand, TNBC has been found to be associated with PTEN loss [91]. Eventually, breast cancer cells are associated with activated PI3K/AKT signaling associated with either PIK3CA point mutations or PTEN suppression depending upon the type of breast cancer. Thus, inhibition of PI3K/AKT signaling could be attributed to arresting breast cancer recurrence and can aid a dimension in breast cancer management.

AKT signaling pathway in breast cancer development and progression. It is a major intracellular pathway which leads to cell survival and cell proliferation. Activation of PI3K catalyzes the phosphorylation of PIP2 to PIP3, which further endorses PDK1 activation. Phosphorylation of FOXO1 by AKT inhibits its transcriptional activities resulting in cell growth, proliferation and survival. In addition, AKT inhibits TSC1/2 resulting in an activation of mTOR which simultaneously suppress autophagy and apoptosis and triggers proliferation. Gray arrows represent downstream events, gray lines represent inhibition, green up arrowheads represent activation/upregulation, and red down arrowheads represent suppression/downregulation. AKT, Ak strain transforming; Bad, Bcl-2-associated death promoter; FOXO, Forkhead box transcription factors; GPCR, G protein-coupled receptor; mTOR, Mammalian target of rapamycin; NO, Nitric oxide; PDK1, Phosphoinositide-dependent kinase-1; PI3K, Phosphoinositide 3-kinase; PIP2, Phosphoinositol 4, 5-biphosphate; PIP3, Phosphoinositol 3, 4, 5-triphosphate; PTEN, Phosphatase and tensin homolog is a phosphatase; TSC, Tuberous sclerosis complex
Different ATP-competitive and allosteric AKT inhibitors have been discovered so far and tested in clinical platforms [92]. Among allosteric inhibitors, only MK-2206 has been tested on breast cancer patients. In a phase I study (NCT00963547), a combination of MK-2206 and trastuzumab was well tolerated by patients with HER2 + tumors; however, MK-2206 treatment has been observed to support HER3 activation via feedback mechanisms, reducing anticancer efficacy [93]. In early-phase clinical trials, MK-2206 in combination with other drugs did not show a potential therapeutic response [94, 95]. In the I-SPY 2 trial (NCT01042379), MK-2206 (135 mg/week) and anthracycline- or taxane-based neoadjuvant chemotherapy improved complete pathologic response in HER2 + and/or HR − breast cancer patients [96].
ATP-competitive AKT inhibitors provide a superior therapeutic window than allosteric inhibitors. Two ATP-competitive AKT inhibitors, capivasertib and ipatasertib, were subjected to detailed phase I and II clinical trials as monotherapy or in combination with hormone therapeutics or chemotherapeutic drugs [97]. The first phase I open-label trial (NCT01226316) with capivasertib recommended the oral dose of 480 mg, twice daily (4/7 intermittent) for phase II trial [98]. In this trial, capivasertib showed a reduction in tumor size in PIK3CA-mutant breast cancer patients. In another phase I study (NCT01090960), capivasertib showed effective suppression of AKT signaling and disease control efficacy against solid tumors including breast tumors [99]. In these phase I trials, capivasertib has been well tolerated. In another phase I trial (NCT01226316), AKT1E17K-mutant and ER + metastatic breast cancer patients who had received extensive pretreatment with capivasertib alone or in combination with fulvestrant, including those whose disease had previously progressed on fulvestrant, exhibited clinically significant therapeutic efficacy [100]. Moreover, the activity and tolerability seemed to be enhanced by this combination.
Capivasertib and fulvestrant combination has also ensured enhanced progression-free survival of patients with ER + metastatic breast cancer in the phase II clinical setting (NCT01992952) [101]. In a randomized and double-blind phase II trial (NCT02423603), the inclusion of capivasertib with paclitaxel chemotherapy improved progression-free and overall survival of patients with metastatic TNBC [102]. In contrast, in a phase I/II trial (NCT01625286), a combination of capivasertib to paclitaxel failed to show improvement of the chemotherapeutic potential of paclitaxel among the patients with PIK3CA-mutated or overall ER +/HER2 − metastatic/progressive breast cancer [103]. In the phase II trial (NCT02077569), capivasertib (480 mg, twice daily) effectively suppressed primary end-point biomarkers of the AKT signaling pathway, such as phospho-GSK3β and phospho-PRAS40 as well as dropped Ki67, a proliferation marker after 4.5 days treatment in invasive breast cancer patients [104]. In addition, significant changes in the secondary biomarkers namely phospho-AKT, Forkhead box O3 and phospho-S6 were observed following capivasertib treatment. A few phase III trials (NCT03997123 and NCT04305496) to evaluate the clinical efficacy of capivasertib along with chemotherapeutic or anti-estrogenic drugs in different forms of breast cancer are underway. In addition, capivasertib could aid in radiotherapy as a radiosensitizer [105].
Ipatasertib is another orally effective ATP-competitive AKT inhibitor, which has demonstrated potential anticancer activity against solid tumors in both pre-clinical and clinical studies [106]. In the phase II LOTUS trial (NCT02162719), adding ipatasertib to paclitaxel improved the progression-free survival of patients with metastatic TNBC [107]. In another randomized phase II trial (NCT02301988), the inclusion of ipatasertib co-treatment with paclitaxel chemotherapy (12 weeks) did not cause a significant improvement in pathologic complete response rate in early TNBC patients; however, showed a good overall response rate and antitumor effect evidenced by magnetic resonance imaging and biomarker analyses [108]. In contrast, in a double-blind, randomized phase III trial (NCT03337724), the inclusion of ipatasertib to paclitaxel did not improvement of efficacy in terms of progression-free survival among HR + and HER2 − progressive breast cancer patients [109]. It is thought to be targeting ER along with AKT inhibition could offer a better therapeutic output in breast cancer management. Several phase III trials (NCT04650581, NCT04060862 and NCT04177108) are underway to evaluate the clinical efficacy of ipatasertib in combination with other drugs to treat different forms of breast cancer.
Angiogenesis inhibitors
Angiogenesis is the rapid increase in the formation of blood vessels to ensure sufficient oxygen supply to the tumor cells. The angiogenic switch in cancer cells is regulated by several mechanisms which encourage the growth of new blood vessels and raise the possibility of distant metastasis [110]. A variety of angiogenesis inhibitors can be therapeutically used to treat various types of advanced solid tumors [111]. These inhibitors include monoclonal antibodies or small-molecule tyrosine kinase inhibitors (TKIs), which primarily target the traditional VEGF and its receptors (VEGFRs). In TNBC treatment, angiogenesis inhibitors like VEGFR targeting agents have been the focus of most research [110]. It is important to mention that around 15–20% of all breast cancer patients represent HER2 + malignancy [112]. Increased angiogenesis and the expression of VEGF are strongly correlated with HER2 activation in cancer cells [113]. Thus, HER2 inhibition would serve as a potential treatment approach for the treatment of HER2 + tumors. Several anti-HER2 antibodies and HER2 TKIs have shown therapeutic promise in the management of HER2 + breast cancer.
Anti-angiogenesis monotherapy has a minimum effect on complex TNBC; however, it has been found to have a workable antitumor impact when added to standard chemotherapy [114]. Bevacizumab, an anti-VEGF monoclonal antibody has been studied in clinical settings to inhibit angiogenesis to treat metastatic and proliferative breast cancer. The clinical study (phase III) data demonstrated that adding bevacizumab to capecitabine significantly improved response rates but did not cause improvement in overall survival or progression-free survival of metastatic breast cancer patients [115]. In a randomized, open-label phase III E2100 trial (NCT00028990), the inclusion of bevacizumab along with paclitaxel chemotherapy improved progression-free survival but not overall survival of metastatic breast cancer patients compared to paclitaxel monotherapy [116]. Considering the outcome of this E2100 trial, the FDA approved bevacizumab for the treatment of breast cancer [117]. In the E2100 trial, Schneider and colleagues found a correlation between the VEGF genotype and overall survival as well as grade 3/4 hypertension in patients with metastatic breast cancer using bevacizumab. The result of the E2100 trail was further validated through several phase III clinical trials viz. AVADO (NCT00333775), RIBBON-1 (NCT00262067), and RIBBON-2 (NCT00281697). Overall survival was not improved in breast cancer patients in any of them, and progression-free survival was shown to be less than in the E2100 study. In the phase III AVEREL trial (NCT00391092), locally recurrent or metastatic HER2 + breast cancer patients treated with bevacizumab in conjunction with trastuzumab and docetaxel did not show significant improvement in progression-free survival [118]. Meta-analyses of randomized trials showed that the inclusion of bevacizumab with chemotherapeutic drugs improves the overall and progression-free survival of progressive and metastatic breast cancer patients [119,120,121]. Ramucirumab, an anti-VEGFR2 monoclonal antibody has been approved by the FDA in 2014 for the treatment of metastatic gastric cancer. A few phase II (NCT01234402 and NCT01427933) and III (NCT00703326) trials have been undertaken, where ramucirumab alone or in combination with other therapeutic drugs did not show any promising therapeutic outcome in breast cancer management [122]. Moreover, adding ramucirumab to docetaxel treatment has been found to increase the risk of toxicity in patients with metastatic breast cancer [123].
TKIs are a class of compounds that target the catalytic roles of the VEGFRs and other growth factor receptors associated with angiogenesis [124]. Sunitinib, sorafenib, axitinib, pazopanib, cediranib and vandetanib are the TKIs that have been studied in the clinical setting of breast cancer. Among them, cediranib (NCT00454805) and vandetanib (NCT00880334) exhibited poor therapeutic efficacy in phase II trials when combined with fulvestrant and docetaxel, respectively [125, 126]. Axitinib also did not show significant therapeutic output to metastatic breast cancer patients when combined with docetaxel in a double-blind, randomized phase II clinical trial (NCT00076024) [127]. Pazopanib, an orally effective multitarget TKI exhibited promising therapeutic efficacy in terms of disease stability in recurrent or metastatic invasive breast cancer patients with allowable untoward effects in the phase II clinical setting [128]. However, combining pazopanib with other anticancer drugs either did not demonstrate a potential therapeutic response or produced unacceptable toxicity [122]. Sorafenib, an orally effective multitarget kinase inhibitor has been approved for the treatment of different types of cancer. However, sorafenib demonstrated mixed observations in the clinical setting of breast cancer when combined with other drugs. In a double-blind, randomized phase II trial (NCT00493636), the combination of sorafenib with gemcitabine or capecitabin promoted progression-free survival moderately in HER2 − breast cancer patients with manageable toxicity [129]. In contrast, in some phase II (EudraCT ID 2007–000290-32) and phase III (NCT01234337) trials, the inclusion of sorafenib to capecitabin therapy did not improve survival advantage as well as imparted incidences of toxicities in HER2 − breast cancer patients [130, 131]. Similarly, other trials did not reveal any therapeutic benefit of sorafenib along with standard chemotherapy in the management of different forms of breast cancer [132]. Sunitinib is another multitarget kinase inhibitor that was approved by the FDA for the treatment of advanced renal cell carcinoma, pancreatic neuroendocrine, and gastrointestinal stromal cancers. Some clinical studies have been undertaken to see the effect of sunitinib in breast cancer management. However, in the clinical setting of breast cancer, sunitinib monotherapy or combination with other drugs failed to establish any promising outcome.
FGFR inhibitors
FGF/FGFR constitute an important signaling network involved in the regulation of growth, survival, differentiation, migration, and apoptosis of cancer cells (Fig. 4) [133]. When FGFs attach to FGFRs, the receptors become dimerized, which causes TGFR1 transphosphorylation through a kinase domain activation loop. Following this, FGFR substrate 2 (FRS2) and phospholipase C, two intracellular receptor substrates are activated via phosphorylation. Furthermore, active FRS2 encourages downstream signaling via the PI3K/AKT and/or RAS/MAPK pathways, which control cell proliferation, differentiation, as well as survival [134, 135]. Genetic shifts in FGFRs may lead to the development of tumors and unfavorable outcomes in women with breast carcinoma [19]. The FGFR system is frequently dysregulated in various cancers, however, most of the research focuses on FGFR1, 2, and 3. FGFR4 is also implicated in oncogenesis, tumor growth, and resistance to anti-tumor treatment in different types of cancer. A cohort of 391 tumor patients revealed that FGF/FGFR abnormalities are common in breast cancer (32.1%) and frequently co-exist with other abnormalities [19]. For instance, specific anomalies linked to changes in FGF/FGFR were found in 15 genes in a univariate analysis. Unusual activation FGF/FGFR axis has been revealed in various clinical subtypes of breast cancer, thus inhibition of FGFR would serve as an approach to cancer management.

FGFR signaling pathway in breast cancer as a potential therapeutic target. Attachment of FGFs to FGFRs causes their dimerization, which encourages TGFR1 activation through kinase domain activation loop with activation of FRS2, PLCγ and downstream transduction pathways, such as PI3K/AKT/mTOR, PKC, RAS/MAPK pathways, which potentiate proliferation, differentiation, migration, angiogenesis, and survival process. Arrows represent downstream events and the line represents inhibition. AKT, Ak strain transforming; ERK, Extracellular signal-regulated kinase; FGF, Fibroblast growth factor; FGFR, Fibroblast growth factor receptor; FRS2, Fibroblast growth factor receptor substrate 2; GRB2, Growth factor receptor-bound protein 2; JAK, Janus kinase; MEK, Mitogen-activated protein kinase kinase; mTOR, Mammalian target of rapamycin; PI3K, Phosphoinositide 3-kinase; PKC, Protein kinase C; RAF, Rapidly accelerated fibrosarcoma; RAS, Rat sarcoma; SOS, Son of sevenless; STAT, signal transducer and activator of transcription
Among FGFR inhibitors, TKIs are important. The therapeutic roles of some important non-selective TKIs on breast cancer patients have been discussed in the earlier section. Dovitinib, lucitanib, and lenvatinib are some additional members of non-selective TKIs, which have been subjected to clinical trials in breast cancer setting. Dovitinib in association with fulvestrant showed promising clinical efficacy to ER + or HER2 + breast cancer in postmenopausal women in a phase II clinical setting (NCT01528345); however, the trial was discontinued in response to the poor enrollment of FGF-expressing patients [136]. Lucitanib is another orally effective and multitarget TKI that has been subjected to a phase II clinical study (NCT02202746) involving patients with/without FGFR1-amplified metastatic breast cancer. Lenvatinib is now being tested in a phase II clinical trial (NCT03168074) for the treatment of patients with early-stage ER + breast cancer.
To understand the on-target FGFR inhibition in patients with FGF/FGFR anomalies, selective FGFR inhibitors have been developed. Among selective FGFR inhibitors, infigratinib, erdafitinib, AZD4547, Debio-1347, and TAS-120 have been subjected to clinical trials in breast cancer settings. In the phase I clinical trial (NCT01928459), the combination of infigratinib (125 mg/day), a pan-FGFR inhibitor, with alpelisib, a PIK3CA inhibitor failed to reveal any conclusive evidence of synergistic effect in the patients with PIK3CA-mutant progressive solid tumors (including metastatic breast cancer), with/without FGFR/2/3 amplifications [137]. In the phase II trial (NCT01795768), AZD4547, an FGFR1/2/3 inhibitor exhibited a potential therapeutic effect on FGFR1-amplified breast tumor patients [138]. A phase II clinical trial (NCT01791985) involving patients with ER + breast cancer has been carried out to assess the therapeutic potential of AZD4547 when combined with letrozole or anastrozole, however, the results are not yet available. The trials with Debio-1347 (NCT03344536), TAS-120 (NCT04024436) and erdafitinib (NCT03238196) on patients bearing different subtypes of breast cancer are ongoing.
Although FGFR antagonists has received a lot of attention, evaluating the efficacy of monoclonal antibodies or ligand-trapping agents has been simultaneously taken into consideration to achieve anti-FGFR effect. GP369, FPA144, and MFGR1877S are a few monoclonal antibodies that have been developed to block FGFR2-IIIb, FGFR2, and FGFR, respectively. FP-1039 (GSK3052230), a ligand trap has been developed to sequester FGFs and thus inhibits FGFR signaling. In a phase I trial (NCT00687505), FP-1039 exhibited good tolerability and acceptable safety profile on solid tumor (including breast tumor) patients. However, more clinical research is required to develop them as drug candidates for breast cancer management. Table 1 shows some important FDA-approved small molecules for targeted therapy of breast cancer.
Immunotherapy in breast cancer management
The typical management plan for breast cancer involves surgery, radiation, and chemotherapy [29]. However, no optimal chemotherapy has yet been discovered to treat all subtypes of breast cancer. Condensed chemotherapy looks to be more effective than standard therapies; nonetheless, these chemotherapies demand the use of growth factors, which significantly increases the cost of treatment [148, 149]. In addition, the adverse effect of chemotherapeutic drugs is a big challenge in cancer management [150, 151]. Although hormone therapy is a popular non-targeted treatment for breast cancer, it is also associated with serious adverse effects [152]. Targeted therapy, which focuses on specific pathophysiological targets, offers new hope for a more effective and novel therapeutic approach to combat various breast cancer subtypes [152]. However, the efficacy and tolerance of target agents needed to be extensively evaluated in clinical settings. Similarly, immunotherapy has recently been regarded as a potential therapeutic tool to target a particular protein expressed in cancer cells offering enormous hopes for breast cancer treatment.
Despite the fact that breast cancer is not typically a highly immunogenic disease, strategies to modulate the immune system are being tested in clinical settings [153, 154]. Microarray-based investigations of immune-related tumor gene expression showed that the immune signatures influence the clinical outcomes, particularly with HER2 + breast tumors and TNBC [155]. However, immune responses varied significantly according to the subtypes of breast cancer, and it is anticipated that not all breast cancer patients will benefit from the same immunotherapeutic strategy. Thus, beyond the known breast cancer subtypes, it is an important aspect to discover prognostic biomarkers to customize immunotherapies. Programmed death-ligand 1 (PD-L1) is the most recurrently used biomarker even for TNBC. The tumor mutational burden (TMB) is a predictive marker for immunogenicity and foreignness [156]. Tumor-infiltrating lymphocytes (TILs), interferon γ (IFN-γ), programmed cell death ligand-1 (PD-L1), and human leukocyte antigen-I (HLA-I) have also been regarded as predictive markers of immunotherapy.
The presence of negligible effector tumor-infiltrating lymphocytes in the tumor microenvironment forms an obstacle to T-cell-based therapy for certain subtypes of breast cancer. Thus, the development of strategies to improve the number of immune cell infiltration to tumor microenvironment may serve as a potential tool for breast cancer immunotherapy. Breast cancer patients respond well toward immunotherapy in the presence of breast calcifications that are mainly associated with immune dysregulation and Erb-B2 receptor tyrosine kinase 2 (ERBB2) hyperactivation. Induction of hyperthermia in the breast tumor milieu has been regarded as another potential immunotherapeutic approach to directly kill tumor cells [157, 158]. Hyperthermia sensitizes cancer cells toward natural killer (NK) cells and CD8 + cells in a human leukocyte antigen-I (HLA-I) polypeptide-dependent manner. Since estrogen suppresses HLA-I, anti-estrogenic agents could potentiate the action of immunotherapeutic drugs. Thus, HLA-I expression is a key consideration in breast cancer immunotherapy. This section deals with the current developments in immunotherapy for breast cancer, counting immune checkpoint blockades, adoptive T-cell immunotherapy, anti-cancer vaccines, etc.
Immune checkpoint blockers
Immune checkpoints involve both T-cell activation and tolerance. Under normal physiological circumstances, they are essential for preserving immunological homeostasis and self-tolerance. Immune inhibitory signals from tumors may result in an immune escape of tumor antigens. PD-1 (programmed cell death protein-1)/PD-L1 (programmed cell death ligand-1) axis and cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) are inhibitory signals suppressing the immune response of T-cells [159]. CTLA-4 signaling plays a greater role in preventing the commencement of T-cell response; while PD-1 plays a more significant role later on and serves to abstract T-cell activity in the immunological setting of tumor microenvironment. CTLA-4 binds to CD80 (B7-1) and CD86 (B7-2) expressed on dendritic cells, thus attenuating T-cell-provoked immune reaction. On the other hand, PD-1 activation by PD-L1 persuades inhibition of T-cell immune activity, activation of T-cell death, suppression of pro-inflammatory cytokine production, and induction of antigen tolerance. Inhibiting immune checkpoints by impeding the CTLA-4 or PD-1/PD-L1 axis could therefore reduce the immune escaping of tumor cells and represent a potential immunotherapeutic approach (Fig. 5) [159, 160]. AntiPD-1, anti-PD-L1, or anti-CTLA-4 monoclonal antibodies are among the immune checkpoint blockers being used in clinical practice and some others are still under development. Anti-PD-1 antibodies, such as cemiplimab, pembrolizumab, and nivolumab; anti-PD-L1 antibodies, such as avelumab, atezolizumab, and durvalumab; and anti-CTLA-4, such as tremelimumab and ipilimumab have demonstrated therapeutic potential and were approved for the treatment of various malignancies, including solid tumors [155]. The therapeutic roles of these immune checkpoint blockers based on monoclonal antibodies in breast cancer management have been described in the section below.

Immune escape mechanism of breast cancer cells and therapeutic role of immune checkpoint blockers in breast cancer treatment. When cytotoxic T-cells in the tumor microenvironment cannot be activated by immunological checkpoints or by the suppressive effect of Tregs, cancer cells are able to withstand the immune assault, survive, and proliferate. CTLA-4 is able to endorse Treg activity leading to an immunosuppressive effect. CTLA-4 binds to B7 (CD80 and CD86) expressed on APCs, such as DCs and inhibits T-cell-mediated immune response. In addition, the binding of CD28 with B7 on APCs suppresses T-cell activity. PD-1/PD-L1 system plays an important role later on and serves to abstract T-cell activity. When PD-1 binds to PD-L1, cytotoxic T-cells become anergic, which further encourages inhibitory signals. APC, Antigen presenting cell; CTLA-4, Cytotoxic T-lymphocytic antigen-4; DC, Dendritic cell; MHC, Major histocompatibility complex; PD-1, Programmed cell death-1; PD-L1. Programmed cell death-1 ligand; TCR: T-cell receptor; Treg, Regulatory T-cell
PD-1/PD-L1 blockers as immunotherapeutic agents
Breast cancer cells especially TNBC cells are abundantly expressing PD-1 and the degree of PD-1 expression is connected to the extent of malignancy [161]. The activation of PD-1 is directly associated with PD-L1. Conventional cancer therapies often endorse PD-L1 activation. A growing body of evidence revealed that blockades of the PD-1/PD-L1 axis stand as a potential therapeutic strategy mainly to treat TNBC. Cemiplimab, pembrolizumab, and nivolumab are some monoclonal antibodies that target PD-1; while avelumab, atezolizumab, and durvalumab target PD-L1.
Cemiplimab is an immunoglobulin G4 (IgG4) monoclonal antibody that targets PD-1 and has been approved by the FDA for the treatment of skin cancer. It has also been found to be efficacious and safe in the clinical setting (NCT03836105) of metastatic cutaneous squamous cell carcinoma [162]. A phase II clinical trial (NCT04243616) is ongoing to check therapeutic responses upon neoadjuvant chemotherapy and cemiplimab treatment in patients with high-risk or progressive HR + and HER2 − breast cancer or TNBC. According to the phase II I-SPY2 trial (NCT01042379), the combination of cemiplimab and a lymphocyte activation gene-3 (LAG-3) inhibitor, fianlimab along with paclitaxel exhibited a significant pathologic complete response than paclitaxel alone in HR + and HER2 − breast cancer or TNBC patients [163].
Pembrolizumab, a humanized monoclonal IgG4 antibody binds to the PD-1 receptor and prevents its communication with PD-L1/2. Pembrolizumab monotherapy (10 mg/kg every two weeks, i.v.) showed a tolerable safety profile in patients with advanced TNBC in the phase Ib KEYNOTE-012 (NCT01848834) study [164]. In the phase II KEYNOTE-086 trial (NCT02447003), pembrolizumab monotherapy also demonstrated potential antitumor efficacy with a manageable safety profile against PD-L1-positive (PD-L1 +) metastatic TNBC [165]. However, the therapeutic response to pembrolizumab is largely dependent on the degree of PD-L1 expression. In the phase III KEYNOTE-119 trial (NCT02555657), even the patients with a PD-L1 combined positive score of ≥ 1 or ≥ 10, pembrolizumab monotherapy for previously treated metastatic TNBC did not increase overall survival significantly in comparison to chemotherapy [166]. Only patients with significantly higher PD-L1 expression responded well to pembrolizumab treatment. Interestingly, some findings proposed that chemotherapeutic drugs can endorse the anti-cancer effects of immune checkpoint blockers through their immunomodulatory responses. In agreement, a phase III KEYNOTE-355 trial (NCT02819518) demonstrated that a combination of pembrolizumab with standard chemotherapy could improve progression-free survival in patients with metastatic TNBC compared to the patients who received only chemotherapy [167]. In the phase Ib/II trial (NCT02513472), combination of eribulin with pembrolizumab was mostly well-tolerated and displayed promising antitumor efficacy in metastatic TNBC patients; however, the effect was found to be largely associated with the presentation of PD-L1 expression [168]. In another phase II study (NCT03222856), the addition of pembrolizumab to eribulin confirmed promising therapeutic efficacy in patients with highly pre-treated, HR + and HER2 − metastatic or locally recurrent breast cancer [169]. In contrast, in an earlier phase II clinical trial (NCT03051659), pembrolizumab plus eribulin did not show any promising clinical outcome in HR + or ERBB2 − metastatic breast cancer patients of either intention-to-treat or PD-L1 + groups compared to only eribulin-treated patients [170]. Furthermore, pembrolizumab plus radiotherapy was found to be safe and efficacious in patients with metastatic TNBC in a phase II clinical setting (NCT02730130) [171]. An ongoing cTRACK-TN trial (NCT03145961) would enlighten the advantages of a watchful increase of pembrolizumab treatment for TNBC patients with measurable circulating tumor DNA.
Nivolumab, a humanized IgG4 monoclonal antibody, is an inhibitor of the PD-1 receptor located at the T-cell surface and prevents its interaction with PD-L1. Nivolumab received the FDA approval for the treatment of melanoma, lung cancer, and renal cell carcinoma [172]. In a phase II TONIC trial (NCT02499367), a significant portion of patients who received the induction treatment with temporary chemotherapy or radiation followed by nivolumab experienced a therapeutic effect that was higher than anticipated in metastatic TNBC patients [173]. In this trial, cisplatin and doxorubicin priming before nivolumab treatment showed a superior therapeutic response, which has been shown to be associated with the immunoregulatory features of these chemotherapeutic drugs to develop favorable tumor microenvironment for PD-1 blockers. Like other immune checkpoint blockers, the therapeutic effects seemed to be linked to PD-1 expression in TNBC despite only a small number of patients representing PD-1 + tumors. However, this finding motivates additional clinical research using nivolumab as a monotherapy or in conjunction with other drugs on breast cancer patients. In contrast, in a phase II study (NCT02834013) comprising 17 patients, a combination of nivolumab with ipilimumab (an anti-CTLA-4 antibody) was found to be associated with extraordinary outcomes in a small subset of patients with chemotherapy-refractory rare metaplastic breast cancer versus no activity [174]. However, in another phase II trial (NCT03316586), cabozantinib plus nivolumab did not demonstrate any promising clinical outcomes in metastatic TNBC patients [175].
Avelumab is a humanized monoclonal IgG1 antibody that targets PD-L1 and prevents PD-1/PD-L1 communication. It endorses NK cell-mediated cytotoxicity and cytokine production to kill tumor cells [176]. In the phase I (NCT01772004) trial, avelumab demonstrated acceptable tolerability and modest therapeutic efficacy in a comprehensively pretreated group of metastatic breast cancer patients [177]. However, a higher possibility of clinical response to avelumab in could be achieved with a higher count of tumor-related immune cells with higher PD-L1 expression specifically in TNBC. In randomized phase III A-BRAVE trial (NCT02926196) is ongoing, which would allow us to interpret the clinical benefits of avelumab among serious TNBC patients. Another phase II trial (NCT03971409) is underway to check the clinical efficacy of avelumab plus liposomal doxorubicin with/without binimetinib or avelumab plus sacituzumab govitecan on patients with stage 4 or inoperable, recurrent TNBC.
Atezolizumab is another ant-PD-L1 monoclonal IgG1 antibody. Atezolizumab monotherapy exhibited acceptable safety with durable therapeutic benefit in metastatic TNBC patients in phase I study (NCT01375842) [178]. Among combination therapy, atezolizumab with nab-paclitaxel exhibited prolongation of recurrence-free survival of metastatic TNBC patients compared to only nab-paclitaxel-treated subgroup in a phase III IMpassion130 clinical setting (NCT02425891) [179]. In the IMpassion130 trial, overall survival for the intention-to-treat population was statistically insignificant, but the PD-L1 + subgroup demonstrated considerable therapeutic effects [180]. Based on IMpassion130 observation, FDA-approved atezolizumab plus paclitaxel therapy for adult patients with locally progressed or metastatic TNBC with tumor-infiltrating immune cells expressing PD-L1. Results from an ongoing trial (IMpassion131, NCT03125902) involving patients with inoperable, previously untreated, locally progressive, or metastatic TNBC may give confirmation of the therapeutic roles of the combination of atezolizumab and paclitaxel. However, the primary observation of the IMpassion131 trial has been disappointing in terms of overall or progression-free survival [181]
Durvalumab, an IgG1 monoclonal antibody, exhibits a strong affinity for PD-L1 binding. In an open-label phase I/II trial (NCT02734004) comprising metastatic breast cancer patients carrying gBRCA mutation, durvalumab (1.5 g every 4 weeks, i.v. infusion) in combination with a PARP inhibitor, olaparib (300 mg, twice daily for 4 weeks) exhibited a favorable antitumor effect with an acceptable safety profile [49]. Durvalumab also exhibited clinical benefits in breast cancer patients when combined with chemotherapeutic drugs. In the phase II GeparNuevo trial (NCT02685059), the inclusion of durvalumab with anthracycline- or taxane-based neoadjuvant chemotherapy to early TNBC patients slightly improved pathological complete response compared to the placebo group; however, the effect was greater for the patients who received durvalumab monotherapy two weeks before the combination therapy [182]. The addition of durvalumab and olaparib to paclitaxel neoadjuvant chemotherapy exhibited better clinical efficacy in a phase II I-SPY2 trial (NCT01042379) over paclitaxel neoadjuvant chemotherapy in HER2 − breast cancer, mainly in serious HR + and HER2 − patients [183]. For patients with stages 1–3 TNBC, durvalumab combined with nab-paclitaxel and dose-dense cyclophosphamide/doxorubicin neoadjuvant chemotherapy led to a 44% pathologic complete response rate in a phase II trial (NCT02489448) [184]. However, the pathologic complete response rate remained higher in PD-L1 + patients. In the SAFIR02-BREAST IMMUNO (NCT02299999) substudy, durvalumab maintenance in metastatic breast cancer patients whose disease remained stable after 6–8 rounds of chemotherapy did not increase progression-free or overall survival in comparison to maintenance chemotherapy. However, maintenance therapy with durvalumab as a single agent improved overall survival in both PD-L1 + and PD-L1 − TNBC patients. These findings support testing durvalumab monotherapy in TNBC patients as maintenance treatment following chemotherapy. The observation anticipated CD274 activation might identify a subset of individuals who would respond well to this anti-PD-L1 antibody; however, further studies are required to reveal the exact reason.
CTLA-4 blockers as immunotherapeutic agents
A key suppressive ligand, CTLA-4 is present on effector T-cells [185]. T-cell activation and growth are usually encouraged by CD28 binding, which results in CTLA-4 translocation to and expression on the T-cell surface. Increased surface expression of CTLA-4 following T-cell activation suppresses the effector function of T-cells by inhibiting signaling through the CD28 receptor. The inhibitory signals of CTLA-4 are propagated through the binding of B7-1 (CD80) and B7-2 (CD86) on antigen-presenting cells (APCs). It is worth mentioning that CTLA-4 exhibits higher binding affinity with B7s than CD28. Apart from blocking co-stimulation, CTLA-4 binding blocks IL-2 transcription, which is essential for T-cell growth and NK-based cytotoxic effects. Thus, blocking CTLA-4 by monoclonal antibodies could increase the effector functions of T-cells to kill cancer cells.
Tremelimumab is a CTLA-4-targeted humanized IgG2 monoclonal antibody, which inhibits tumor growth by preventing the interaction between CTLA-4 and B7s and thereby allowing T-cell activation [186]. So far, its clinical efficacy and safety have been assessed in different types of cancer. The FDA approved tremelimumab plus durvalumab combination therapy for inoperable hepatocellular carcinoma. Recently, it received approval from the FDA in combination with durvalumab and platinum-based chemotherapeutic drugs for the treatment of non-small cell lung cancer (metastatic) without EGFR mutation [187]. Although clinical evidence is yet to be available about the effectiveness and safety of pembrolizumab monotherapy in the setting of breast cancer, these issues have been addressed in various combinations comprising pembrolizumab as a component of treatment for breast cancer. In a phase I trial comprising 26 HR + advanced breast cancer patients, pembrolizumab in combination with an aromatase inhibitor, exemestane was well-tolerated with an overall response of ≥ 12 weeks in 42% of patients [188]. Interestingly, CTLA-4 blocking caused immune activation by enhancing inducible co-stimulator activation on CD8 + and CD4 + T-cells. In a pilot study, the combination of tremelimumab with an anti-PD-L1 drug, durvalumab, exhibited poor outcomes in overall metastatic breast cancer patients; however, TNBC patients achieved better therapeutic responses [189]. Surprisingly, TNBC patients who received anti-PD-L1 (durvalumab) monotherapy had a superior response. Pembrolizumab along with radiation therapy showed an acceptable safety profile in a phase I trial even after an extended period [190].
Ipilimumab is a CTLA-4 targeting humanized IgG1 monoclonal antibody, which demonstrates significant anti-tumor activity against a variety of cancers. Preclinical data revealed that ipilimumab promotes the release of IL-2 by TNBC cells to the tumor microenvironment, which endorses the immune response [191]. The FDA approved ipilimumab for the treatment of renal cell cancer, colorectal cancer, malignant pleural mesothelioma, hepatocellular carcinoma, melanoma, and colorectal cancer [172]. In a Phase I trial (NCT01502592), preoperative cryoablation or ipilimumab monotherapy, or their combination in initial stage/operable breast cancer patients exhibited an acceptable safety profile and no delay in surgery was experienced [192]. The outcomes anticipated the possibility of this approach to synergize the antitumor effect through activating anti-tumor immunity. In an open-label and two-stage phase II trial (NCT02834013), nivolumab plus ipilumumab exhibited remarkable responses in 3 out of 17 patients with chemotherapy-nonresponsive, metaplastic breast cancer [174]. This dichotomization with this combination might be associated with specific biomarker/s presented by 18% of patients who received excellent therapeutic benefits and this must be taken into account before executing further trials. Ipilimumab plus nivolumab with neoadjuvant paclitaxel demonstrated prospective overall and pathological complete responses in early-stage TNBC patients in the phase II clinical settings (ACTRN12617000651381) and the observed clinical response was not found to be linked with PD-L1 status [193].
Other immune checkpoint inhibitors
Some additional molecules exist that target immunological checkpoints to stop T-cell suppression, such as LAG3, and T-cell immunoglobulin and ITIM domain (TIGIT), or to activate T-cells and boost their cytotoxic function, such as OX-40 (CD134) and 4-1BB [160]. LAG-3 is a novel inhibitory receptor that is exceedingly expressed in regulatory and disabled T-cells. Unlike PD-1/PD-L1 and CTLA-1 blockers, LAG3 blockers can additionally inhibit regulatory T-cells (Tregs) along with endorsing effector T-cell activity. In a phase I trial (NCT00349934), combining the standard chemotherapeutic drug, paclitaxel with the recombinant LAG-3Ig fusion protein, IMP321 showed excellent improvement in objective response rate in patients with metastatic breast cancer in terms of boosting APC, NK cell, and CD8 T-cell counts with excellent safety [194]. A phase IIb AIPAC study (NCT02614833) has been conducted to check the clinical efficacy and safety outcomes of immunotherapy with IMP321 in combination with adjunctive paclitaxel chemotherapy in patients with stage 4 breast adenocarcinoma. The overall outcomes are yet to be published. Several other immune checkpoint targets have been developed, such as B-cell maturation antigen (BCMA), CD19, CD20, CD47, colony-stimulating factor 1 receptor (CSF1R), indoleamine 2,3-dioxygenase (IDO), transmembrane glycoprotein mucin 1 (MUC1), New York esophageal squamous cell carcinoma 1 (NY-ESO1), stimulator of interferon genes (STING), Wilms' tumor gene 1 (WT1), human papillomavirus (HPV), T-cell immunoglobulin domain and mucin domain 3 (TIM3), etc.; however, no specific target is yet to be recognized with promising therapeutic effect in treating breast cancer [195]. Table 2 represents some selected ongoing trials using different immune checkpoint blockers in the context of breast cancer.
Anticancer vaccines
Anticancer vaccines aim at endorsing antigen-specific T-cell-based activation of the immune system to target and eliminate cancer cells [196]. Among different types of vaccines, peptide, protein-based, recombinant DNA, carbohydrate antigen, granulocyte macrophage colony-stimulating factor (GM-CSF)-secreting tumor cell, dendritic cell-based, and dendritic cell-tumor cell fusion vaccines are important in the context of breast cancer [197]. Based on the target, breast cancer vaccines are broadly divided into two categories, such as the vaccines that target HER2 or HER2-associated antigens and the vaccines that target non-HER2-related antigens. The efficacy and safety issues of different HER2 and non-HER2 targeting vaccines in the context of breast cancer under different phases of clinical trials have been discussed in the subsequent section.
HER2 targeting breast cancer vaccines
Protein/peptide vaccines that precisely target HER2 are the main focus of developing breast cancer vaccines. E75 (nelipepimut-S, HER2 369–377, or NeuVax), an immunogenic peptide (human leukocyte antigen-A2/A3-constricted) generated from the HER2 protein, can induce a potent anti-HER2 immune response when paired with the immunoadjuvant granulocyte–macrophage colony-stimulating factor [198]. The E75 vaccine has been regarded as a safe and effective immunotherapeutic tool to elicit a peptide-based immune response. In a phase I trial (NCT00841399), it appeared to dramatically lower the risk of cancer recurrence in patients with lymph node-negative (LN–) and HER2 + advanced breast cancer [199]. United States Military Cancer Institute-based trials demonstrated that lymph node-positive (LN +) or LN- breast cancer patients who received an optimal dose of the E75 vaccine experienced similar safety issues to those vaccinated with suboptimal doses; however, exhibited superior HER-2/neu-endorsed immunity to lessen the chance of breast tumor recurrence [200]. However, the study requires a substantially long follow-up to attain conclusive evidence. Final report of phase I/II clinical trials (NCT00841399 and NCT00584789) of the E75 vaccine along with booster immunizations significantly mitigated cancer recurrence in 95.2% of participants with HER2-expressing high-risk breast cancer [201]. Interestingly, the E75 vaccine appeared to be a safe and well-tolerated immunotherapy. Only low-grade local and rare systemic (mild) toxicities were recorded. In a short-term study of a double-blind and randomized phase III trial (NCT01479244) involving patients with LN + and low HER2 + breast tumors, an interim study did not reveal any difference in disease-free survival between E75-vaccinated and placebo-vaccinated patients [202]. Therefore, the trial was suspended at an early phase. A meta-analysis of 24 clinical studies, added to the confusion by revealing that the E75 vaccine significantly reduced disease recurrence but had no meaningful impact on overall survival [203]. Additionally, those who received the E75 vaccine showed moderate variability in their disease-free survival rate.
GP2 (HER2 654–662) is another human leukocyte antigen-A2/3-restricted immunogenic peptide, which is derived from HER2 transmembrane domain. Despite GP2 exhibiting lower affinity toward HLA-A2 compared to E75, however, it could be more immunogenic than E75 [197]. In the United States Military Cancer Institute-based phase I trial, the GP2 vaccine appeared to be well-tolerated and safe, as well as capable of triggering GP2-induced T-cell responses and delayed-type hypersensitivity responses when administered with GM-CSF in the patients with high-risk, LN– breast cancer [204]. This finding encourages additional investigation of the GP2 vaccine to reveal its therapeutic potential to prevent breast cancer recurrence. It has been reported that completion of primary immunization series with GP2 plus GM-CSF entirely stopped cancer recurrence throughout a 5-year follow-up period in HER2/3 + patients, who received trastuzumab treatment following surgery. A phase IIb trial confirmed the outcomes of phase I regarding the safety profile of GP2 plus GM-CSF immunization in women with HER2 + operable breast cancer [205]. This trial demonstrated that the inclusion of GM-CSF did not impart any additional adverse event.
Another peptide vaccine linked to HER2 called AE37 (HER2 776–790) is used as an adjuvant immunotherapy for breast cancer. It is obtained from HER2’s intracellular domain. As a major histocompatibility complex (MHC) class-II epitope, AE37, in contrast to E75 and GP2, primarily endorses CD4 + T cell activation [197]. In the phase I clinical settings, AE37 plus GM-CSF immunotherapy appeared to be well-tolerated and safe, as well as capable of eliciting adjuvant-independent HER-2/neu-mediated immune reaction in disease-free, LN– breast cancer patients [206]. A single-blinded phase II trial (NCT00524277) has been undertaken to compare the efficacy of two HER2 vaccines, viz, GP2 and AE37 in breast cancer patients representing HER2 expression [207]. Both vaccines were found to be safe but effective against only certain subtypes of breast cancer. AE37 vaccination only achieved therapeutic benefit in patients representing low HER2 expressing and TNBC patients; while GP2 vaccination imparted recurrence-free survival only to HER2 overexpressing subgroups. Thus, the extent of HER2 expression is an important marker in selecting peptide-based immunization.
Vaccination with HER2 protein (25, 150, and 900 µg) intracellular domain (amino acids 676–1255) with GM-CSF has been found to elicit a sustained (9–12 months) and dose-dependent HER2-specific T-cell-based and antibody-based immunity in a phase I study involving 29 patients bearing stages 2–4 breast and ovarian cancers with high expression of HER2 [208]. Recombinant-HER2 protein vaccine (20, 100, and 500 µg) in conjugation with an immunostimulant (AS15) also exhibited a satisfactory safety profile in HER2 + breast cancer patients in a phase I trial (NCT00058526). In addition, this vaccine can generate sustained HER2-specific humoral immune responses and prolong overall and disease-free survival [209].
HER2-plasmid DNA vaccination in combination with IL-2, GM-CSF, and trastuzumab treatment exhibited a good safety profile among patients with metastatic HER2 + breast cancer. Although there were no T-cell responses against HER2 immediately following immunization, a significant rise in MHC class-II-mediated T-cell responses against HER2 was found after a specific interval [210]. Vaccination with V930, a DNA vaccine containing plasmid DNA encoding trans-membrane and extracellular domains of human HER2, alone (NCT00250419) or in combination (NCT00647114) with a viral vector vaccine encoding HER2, V932 demonstrated acceptable safety in patients bearing solid tumors; however, cell-based immune response to carcinoembryonic antigen or HER2 was not detected [211]. A couple of trials (NCT00436254 and NCT00393783) with DNA vaccines encoding different forms of HER2-derived proteins are ongoing to measure efficacy and safety issues with them in breast cancer patients.
It has been found that almost one-half of the patients with metastatic TNBC and HER + breast cancers develop brain metastases. Loss of anti-HER2/3 immunity is thought to be associated with this disease progression. A HER2/3 targeting dendritic cell vaccine, alpha-DC1, has been subjected to a phase II trial (NCT04348747) on TNBC and HER2 + breast cancer patients who have brain metastases [212]. After surgery and adjuvant therapy, treatment with autologous dendritic cells pulsed with a HER2 intracellular domain peptide significantly reduced the risk of cancer recurrence in all seven patients in a small clinical setting (NCT00005956) with high HER2 expressing breast cancer (stage II-IV) and six patients showed anti-HER2 antibody [213]. In addition, GM-CSF-secreting HER2 + tumor cell vaccines have been evaluated in clinical trials (NCT00399529 and NCT00093834). These tumor cell vaccines along with chemotherapeutic drugs are safe and capable of imparting anti-HER2 immunity [214, 215].
Non-HER2 targeting breast cancer vaccines
Certain subtypes of breast cancer, such as TNBC which represents a lack of ER, PR, and HER2 expressions, provide clinical challenges with HER2-targeting breast cancer vaccines [197]. TNBC expresses a number of non-HER2 tumor-associated antigens, which could be successfully aimed at developing cancer vaccines for clinical applications specifically for TNBC. The cancer-testis antigens (CTAs) may be the most regularly targeted non-HER2 tumor-associated antigens for cancer vaccination [155]. Some notable CTAs, such as New York esophageal squamous cell carcinoma 1 (NY-ESO-1), Wilms' tumor protein (WT1), folate receptor alpha (FRα), melanoma antigen gene protein-12 (MAGE-12), brachyury protein, and p53 could be targeted for developing TNBC vaccines. However, the management of TNBC using the aforementioned CTAs as the targets have not yet shown the anticipated success, and considerable clinical research is required to develop clinically effective CTA-targeting vaccines for TNBC.
Mucin 1 (MUC1) is a protein that is immunologically inaccessible and hyperglycosylated in several epithelial cells; however, an immunologically accessible hypoglycosylated form of this protein is expressed in different malignancies including TNBC. It is thought to be an intriguing tumor-associated antigen for developing TNBC vaccines [216]. Synthetic MUC1 peptide vaccine carrying a toll-like receptor (TLR) 7 agonist has shown preclinical success and raised the prospect for further clinical translation [217]. PANVAC, a recombinant viral vaccine comprises a recombinant fowl pox vector, encoding tumor-associated antigens like carcinoembryonic antigen and MUC1 and another viral vector vaccine containing cancer-associated antigen transgenes as well as different co-stimulatory agent, TRICOM. In a phase II trial (NCT00179309), immunization with PANVAC along with docetaxel treatment to the patients with metastatic breast cancer improved progression-free survival to more than 2 folds over control [218]. Another breast cancer-associated antigen is mammaglobin-A (MAM-A) which is abundantly expressed in 40–80% of breast cancer [197]. In the phase I clinical trial (NCT00807781), the MAM-A DNA vaccine showed satisfactory safety and early signs of improved progression-free survival in metastatic breast cancer patients [219]. Moreover, MAM-A vaccination in breast cancer patients demonstrated activation of inducible co-stimulatory molecules on CD4 + T cells with a substantial reduction of Treg occurrence [220].
Human telomerase reverse transcriptase (hTERT) which plays a key role in oncogenesis, is almost always overexpressed in human malignancies, including breast cancer [221]. Cytotoxic CD8 + T cells can recognize them and endorse the lysis of tumor cells. Thus, hTERT can serve as a cellular immune surveillance target. In a study involving 19 breast cancer patients, the hTERT peptide vaccine was able to elicit CD8 + T cell-based immune response in about 47% of patients [222]. Some phase I trials (NCT01660529, NCT01660529, and NCT02960594) with hTERT are underway.
Tumor-associated carbohydrate antigens are generally regarded as poorly immunogenic, which can be aimed to employ mimicking peptides (mimotopes) to induce anti-human tumor-associated carbohydrate antibodies [223]. Such a mimotope vaccine called P10s-PADRE was subjected to phase II clinical involving metastatic breast cancer patients; however, lack of funding compelled to suspend the patient recruitment. In a phase Ib trial (NCT02229084), 3 weekly immunizations that preceded the first dose of chemotherapy were found to be safe and immunologically promising. However, more trial is required with this combination to address therapeutic benefits [224]. Sialyl-TN (STn) is a carbohydrate epitope, which has been coupled to the keyhole limpet hemocyanin (KLH) carrier protein to develop a vaccine. In a randomized phase II trial, low-dose cyclophosphamide followed for 3 days followed by STn-KLH (100 μg) plus an adjuvant, Enhanzyn™ (DETOX-B) vaccination (on day 0, 2, 5, and 9) to metastatic breast cancer patients exhibited a significantly higher antibody level compared to only vaccine treated group [225]. Even though STn-KLH was well tolerated by patients with metastatic breast cancer, the overall improvement in survival or time to progression has been disappointing in phase III clinical settings. However, in a phase III trial (NCT00003638), STn-KLH in combination with endocrine therapy showed some therapeutic prospects in metastatic breast cancer patients [226]. Additionally, a vaccine that targets the Globo H glycosphingolipid antigen has been clinically studied in patients with different subtypes of breast cancer. In a phase II trial (NCT01516307), treatment with a synthetic Globo H vaccine conjugated with KHL, adagloxad simolenin with adjuvant OBI-821 was well-tolerated and demonstrated anti-Globo H humoral immune responses in patients with Globo H-positive (Globo H +) metastatic overexpressing breast cancer patients. Adagloxad simolenin plus OBI-821 has entered the phase III clinical trial (NCT03562637) to find the efficacy, safety, and tolerability of the vaccine in Globo H + TNBC patients [227].
Even though a number of vaccines have been developed and trialed to improve immunogenicity to different breast cancer subtypes, clinical success has not yet been attained. Thus, substantial research is required to identify the precise target to develop a specific vaccine for the exact subtypes of breast cancer patients.
Adoptive T-cell therapy
The objective of adoptive T-cell therapy is to endorse cell-based anti-tumor immunity in cancer patients by transferring lymphocytes. Adoptive T-cell therapy includes TIL-based, engineered T-cell receptor (TCR)-based, chimeric antigen receptor (CAR)-based, dendritic cell-based, NK cell-based therapies [228, 229]. It has been regarded as an emerging anticancer immunotherapy achieved by boosting host immunity, particularly in patients representing low immunity. Rosenberg and associates initially introduced the adoptive infusion of autologous TILs as a strategic therapy in 1987. Since then, adoptive TIL therapy has been used in the clinical setting of different types of cancers. However, the therapeutic responses have been heterogeneous based on the cancer types. It has been regarded that recognition of TIL-responsive tumor antigens can improve TIL therapy by endorsing tumor recognition and killing capacity. In the breast cancer setting, it has been revealed that TILs isolated from a specific subtype of tumor can recognize the immunogenic mutations in this tumor milieu and could serve as an immunotherapeutic agent for this specific tumor subtype [230]. In a case report, treatment with TILs reactive against mutant versions of KIAA0368, SLC3A2, CTSB, and CADPS2 proteins in combination with a PD1 blocker and IL-2 demonstrated a durable and complete cancer regression in a patient with chemoresistant, HR + and HER2– metastatic breast cancer [231]. The safety and efficacy of TILs as a monotherapy or in combination with other therapies in the setting of breast cancer may be clarified by the findings of phase I (NCT04111510 and NCT00301730) and phase II (NCT01174121) clinical trials.
CAR T cell therapy is one of the most recent and effective adoptive T-cell therapy in cancers, which utilizes the specificity of an antibody to target a specific tumor antigen to regulate the cytotoxic capacity of T-cells [229, 232]. Several antigen targets for CAR-T therapy of breast cancer have been discovered through preclinical studies, which include FRα, EGFR, AXL receptor tyrosine kinase (AXL), NKG2D, integrin αvβ3, c-Met, HER2, MUC1, mesothelin, receptor tyrosine kinase-like orphan receptor 1 (ROR1), tumor endothelial marker 8 (TEM8), trophoblast cell-surface antigen 2 (TROP2), etc. [233]. Though TNBC lacks expressions of three major antigens associated with breast cancer, CAR-T therapy still exhibits promising outcomes against the disease. Antigens like MUC-1, NKG2D, AXL, ROR1, c-Met, FRα, mesothelin, etc. have been successfully targeted in various in vitro and in vivo models of TNBC by engineered CAR-T cells [234]. In this context, an encouraging result in the phase I clinical trial (NCT01837602) has already been achieved with c-Met-CAR-T cells. Intratumoral injection of c-Met-CAR-T cells demonstrated an acceptable safety profile in TNBC patients and was found to elicit intratumoral inflammatory response [235]. However, CAR-T cell therapy has also experienced its ups and downs with regard to toxicities and efficacy shortcomings. MUC1-CAR-T cells were mostly studied in early-phase clinical trials (NCT04020575, NCT02587689, and NCT04025216) in TNBC patients. Among them, MUC1-CAR-T cells were mostly studied in early-phase clinical trials (NCT04020575, NCT02587689, and NCT04025216) in TNBC patients. Among them, the NCT04025216 trial has been suspended and the recruitment status of the NCT02587689 trial is unknown. Only the phase I/II trial (NCT04020575) is still recruiting patients to evaluate the safety of MUC1-CAR-T cell therapy. A couple of phase I trials (NCT02580747 and NCT02792114) have been initiated to check the safety of mesothelin-CAR-T cell therapy; however, the results have not been published yet. The phase I trial with NKG2D-CAR-T (NCT04107142) was initiated but the status is unknown, while the phase I trial (NCT02706392) with ROR1-CAR-T (NCT02706392) was terminated. In addition, a few clinical trials had to be abandoned due to unacceptable damage to healthy tissues [236, 237]. In such cases, the target antigen has been observed to be expressed simultaneously by both tumor cells and healthy cells, thus attracting CAR-T cells to both types of tissues consequently resulting in toxicities. A very interesting development to prevent such possibilities has come up in the form of targeting tumor-specific glycoforms of tumor-associated antigens. CAR-T cells have been designed to target aberrantly glycosylated glycoforms of tumor-associated antigens, in addition to the tumor antigens (also expressed by some healthy cells). Certain antigens e.g. Tn, T, and sialyl-Tn are involved in the progression and metastasis of different types of cancers including breast cancer. Underglycosylation of these antigens is a tumor-specific feature owing to the dysregulation of chaperone, Cosmc, and glycosyltransferases [238]. Hence, it is not surprising that targeting these tumor-associated, low-sugar antigens alongside the tumor-specific target antigens by CAR-T cells has expressed futuristic promise to overcome non-selective adverse events. Thus, overcoming the present shortcomings of CAR-T cell therapy might potentially establish it as one of the prime modes of cancer treatment [239].
Additionally, newer prospects for adoptive cell therapy are developing with the discovery of different neoantigens and the application of different immune cells, including NK cells and dendritic cells in cancer management. The clinical results of dendritic cell-based vaccination therapy have been discussed in the earlier section. NK cell-based treatment has not been studied comprehensively in the clinical setting of breast cancer. In only one phase II trial, the adoptive transfer of allogeneic NK cells following lymphodepleting chemotherapy did not demonstrate clinical significance in recurrent breast cancer patients [240]. However, preclinical studies are continuing to develop a successful NK cell-based therapy for breast cancer management. Table 3 represents some selected ongoing trials using adoptive cell therapy in the context of breast cancer.
Immune-associated adverse effects with aforementioned immune therapies are mostly unpredictable. Severe endocrine irregularities and albeit rare are the most regular and consistently mentioned adverse reactions with immune checkpoint inhibitors. Despite the toxicities associated with vaccines being acceptable and manageable; they are yet to achieve therapeutic success in breast cancer management. Adoptive cell-based immunotherapy also demonstrated a diverse portrait of adverse events including multiorgan failure, neurotoxicity, cardiac distress, bone marrow challenges and respiratory failure mainly associated with lymphodepletion, immune-mediated side effects including cytokine storm in the clinical setting of breast cancer. Thus, achieving therapeutic success largely depends on careful monitoring pathophysiological status of patients before the execution of immunotherapy or during the course of immune-directed treatment.
Perspectives
Breast cancer is a heterogeneous pool of diseases that can be divided into various subtypes based on the origin, course, and molecular markers. Protein-gene products that unswervingly influence the biological and clinical traits of cancer cells are prospective targets for the development of novel treatments. Gene signatures have been thought to be the predictors of therapeutic response. Thus, to ascertain appropriate therapy, it is critical to consider the aforementioned factors. In response to chemoresistance and recurrence, scientists have developed targeted drugs for the management of different forms of breast cancer. PARP inhibitors, PI3K/AKT/mTOR inhibitors, CDK4/6 inhibitors and HER2 TKIs emerged as potential targeted therapies of gBRCA-mutated, PIK3CA-mutated, ER + , and HER2 + subtypes of breast cancer, respectively. Some promising clinical outcomes with some of these targeted drugs as monotherapy or in combination with other drugs have been already accredited by regulatory bodies through approval for the treatment of specific subtypes of breast cancer. However, a target-specific therapy could not ensure complete clinical success for a specific subtype of breast cancer. Drug resistance represents a key challenge with current targeted agents. Inclusion of a co-drug that antagonizes drug resistance mechanism and minimizes escape pathway would serve as a potential solution. In this aspect, it is important to ensure that the disease characteristics concerning specific clinical/genetic markers to achieve expected therapeutic success. Thus, it is imperative to ascertain the molecular, genetic, and immune signatures of tumor cells as well as tumor microenvironments before the execution of targeted therapy. Many small molecules have shown promising results in preclinical assays, which may be subjected to further clinical translation. The analyses of cutting-edge approaches, specifically those that prioritize gene/molecular-level analyses of breast cancer subtypes in clinical settings in response to treatment, must be the main focus of upcoming clinical research to reveal a more accurate conclusion. In addition, the adverse reaction to targeted therapeutics must be critically scrutinized, specifically the serious toxicities associated with the treatment require thorough critical interpretation. On the other hand, so far, chemotherapy is the only form of existing treatment for TNBC patients. Extensive clinical development in the field of immunotherapy would expectedly raise new hope in breast cancer management, specifically in TNBC. The introduction of immune-checkpoint blockers and other immunotherapeutic agents to conventional breast cancer therapies enables a higher therapeutic response in terms of progression-free and overall survival including for patients with TNBC. The regulatory body approval of immune-checkpoint blockers in combination with other therapies in TNBC would reveal the prospect of immunotherapy. Moreover, the increasing number of clinical trials demonstrates that the inclusion of immune-checkpoint blockers with other therapeutic drugs would come up as an efficacious therapeutic strategy in breast cancer management especially for TNBC patients. Although the advancement of immune checkpoint blockers is a significant achievement in TNBC treatment, the scope is limited for PD-L1 overexpressing tumors. Thus, more research is required to address this issue. A thorough understanding of tumor subtypes, as well as tumor microenvironments with respect to molecular, genetic, and immune standpoints, would enhance the scope of developing specific immunotherapy directed toward specific breast tumor subtypes to achieve better therapeutic efficacy.
Conclusion
The recent advancements in targeted therapy have presented a more specific and effective therapeutic option in breast cancer management. Inhibition of specific molecules that promote tumor growth and survival remains the main objective of targeted therapy. Some targeted drugs alone or in combination with other drugs have already got FDA approval for the treatment of different breast cancer subtypes and many of them are under clinical trials. However, drug resistance is a big challenge associated with these drugs. Moreover, the majority of targeted therapy is yet to find clinical success in TNBC patients. On the other hand, immune therapies have emerged as promising targeted therapies specifically for TNBC patients. Some immune-checkpoint blockers in combination with other drugs have already received FDA approval for TNBC treatment. An increasing number of ongoing trials also demonstrates the interest in this field. However, considering the breast cancer heterogeneity, a complete understanding of the molecular, genetic, and immunological landscape of tumor cells as well as tumor microenvironment is the most important aspect to achieving desired therapeutic success with targeted or immune therapy. Thus, it is urgently required to discover/develop specific markers for a clear understanding of breast cancer subtypes to decide on an accurate therapeutic regime.
Availability of data and materials
Not applicable.
Abbreviations
- AKT:
-
Ak strain transforming
- BRCA:
-
Breast cancer gene
- CD28:
-
Cluster of differentiation 28
- CDK:
-
Cyclin-dependent kinase
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated antigen 4
- DC:
-
Dendritic cell; EGFR, Epidermal Growth Factor Receptor
- ER:
-
Estrogen Receptors
- ERK:
-
Extracellular signal-related kinases
- FDA:
-
United States-food and drug administration
- FGFR:
-
Fibroblast growth factor receptor
- HER2:
-
Human epidermal growth factor receptor-2
- JAK2:
-
Janus kinase 2
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
Mitogen-activated protein kinase kinase
- ERK:
-
Extracellular signal-regulated kinase
- mTOR:
-
Mammalian target of rapamycin
- NK:
-
Natural killer
- PARP:
-
Poly (ADP-ribose) polymerase
- PD-1:
-
Programmed death-1
- PD-L1:
-
Programmed cell death ligand-1
- PI3K:
-
Phosphoinositide 3-kinase
- PR:
-
Progesterone receptors
- PTEN:
-
Phosphatase and tensin homolog
- STAT:
-
Signal transducer and activator of transcription
- TILs:
-
Tumor infiltrating lymphocytes
- TNBC:
-
Triple-negative breast cancer
References
Bodai BI, Tuso P. Breast cancer survivorship: a comprehensive review of long-term medical issues and lifestyle recommendations. Perm J. 2015;19(2):48. https://doi.org/10.7812/TPP/14-241.
Giaquinto AN, Sung H, Miller KD, Kramer JL, Newman LA, Minihan A, et al. Breast Cancer Statistics 2022. CA Cancer J Clin. 2022;72(6):524–41. https://doi.org/10.3322/caac.21754.
Li Y, Zheng J, Deng Y, Deng X, Lou W, Wei B, et al. Global Burden of Female Breast Cancer: Age-Period-Cohort Analysis of Incidence Trends From 1990 to 2019 and Forecasts for 2035. Front. Oncol. 2022;12:891824. https://doi.org/10.3389/fonc.2022.891824.
Arnold M, Morgan E, Rumgay H, Mafra A, Singh D, Laversanne M, et al. Current and future burden of breast cancer: Global statistics for 2020 and 2040. Breast J. 2022;66:15–23. https://doi.org/10.1016/j.breast.2022.08.010.
Bae SY, Kim S, Lee JH, Lee HC, Lee SK, Kil WH, et al. Poor prognosis of single hormone receptor- positive breast cancer: similar outcome as triple-negative breast cancer. BMC Cancer. 2015;15:138. https://doi.org/10.1186/s12885-015-1121-4.
Ayoub NM, Al-Shami KM, Yaghan RJ. Immunotherapy for HER2-positive breast cancer: recent advances and combination therapeutic approaches. Breast Cancer Targets Ther. 2019;11:53–69. https://doi.org/10.2147/BCTT.S175360.
Luque M, Sanz-Álvarez M, Morales-Gallego M, Madoz-Gúrpide J, Zazo S, Domínguez C, et al. Tumor-Infiltrating Lymphocytes and Immune Response in HER2-Positive Breast Cancer. Cancers. 2022;14(24):6034. https://doi.org/10.3390/cancers14246034.
Čelešnik H, Potočnik U. Peripheral blood transcriptome in breast cancer patients as a source of less invasive immune biomarkers for personalized medicine, and implications for triple negative breast cancer. Cancers. 2022;14(3):591. https://doi.org/10.3390/cancers14030591.
Mercogliano MF, Bruni S, Elizalde PV, Schillaci R. Tumor necrosis factor α blockade: an opportunity to tackle breast cancer. Front Oncol. 2020;10:584. https://doi.org/10.3389/fonc.2020.00584.
Mehanna J, Haddad FG, Eid R, Lambertini M, Kourie HR. Triple-negative breast cancer: current perspective on the evolving therapeutic landscape. Int J Womens Health. 2019;11:431–7. https://doi.org/10.2147/IJWH.S178349.
Boyero L, Sánchez-Gastaldo A, Alonso M, Noguera-Uclés JF, Molina-Pinelo S, Bernabé-Caro R. Primary and acquired resistance to immunotherapy in lung cancer: unveiling the mechanisms underlying of immune checkpoint blockade therapy. Cancers. 2020;12(12):3729. https://doi.org/10.3390/cancers12123729.
Mukherjee AG, Wanjari UR, Prabakaran D, Ganesan R, Renu K, Dey A, et al. The Cellular and Molecular Immunotherapy in Prostate Cancer. Vaccines. 2022;10(8):1370. https://doi.org/10.3390/vaccines10081370.
Rahman MM, Behl T, Islam MR, Alam MN, Islam MM, Albarrati A, et al. Emerging management approach for the adverse events of immunotherapy of cancer. Molecules. 2022;27(12):3798. https://doi.org/10.3390/molecules27123798.
Ghosh S, Javia A, Shetty S, Bardoliwala D, Maiti K, Banerjee S, et al. Triple negative breast cancer and non-small cell lung cancer: Clinical challenges and nano-formulation approaches. J Control Release. 2021;337:27–58. https://doi.org/10.1016/j.jconrel.2021.07.014.
Choi J, Lee SY. Clinical Characteristics and Treatment of Immune-Related Adverse Events of Immune Checkpoint Inhibitors. Immune Netw. 2020;20(1):e9. https://doi.org/10.4110/in.2020.20.e9.
Tufail M, Cui J, Wu C. Breast cancer: molecular mechanisms of underlying resistance and therapeutic approaches. Am J Cancer Res. 2022;12(7):2920–49.
Ortega MA, Fraile-Martínez O, Asúnsolo Á, Buján J, García-Honduvilla N, Coca S. Signal transduction pathways in breast cancer: the important role of PI3K/Akt/mTOR. J Oncol. 2020;2020:9258396. https://doi.org/10.1155/2020/9258396.
Presti D, Quaquarini E. The PI3K/AKT/mTOR and CDK4/6 pathways in endocrine resistant HR+/HER2− metastatic breast cancer: biological mechanisms and new treatments. Cancers. 2019;11(9):1242. https://doi.org/10.3390/cancers11091242.
Santolla MF, Maggiolini M. The FGF/FGFR system in breast cancer: oncogenic features and therapeutic perspectives. Cancers. 2020;12(10):3029. https://doi.org/10.3390/cancers12103029.
El-Tanani M, Al Khatib AO, Al-Najjar BO, Shakya AK, El-Tanani Y, Lee Y-F, et al. Cellular and molecular basis of therapeutic approaches to breast cancer. Cell signal. 2022;101:110492. https://doi.org/10.1016/j.cellsig.2022.110492.
Kavarthapu R, Anbazhagan R, Dufau ML. Crosstalk between PRLR and EGFR/HER2 signaling pathways in breast cancer. Cancers. 2021;13(18):4685. https://doi.org/10.3390/cancers13184685.
Mei L, Borg JP. ERBB2 oncogenicity: ERBIN helps to perform the job. Mol Cell Oncol. 2015; 2(3):e995033. https://doi.org/10.4161/23723556.2014.995033.
Rusidzé M, Adlanmérini M, Chantalat E, Raymond Letron I, Cayre S, Arnal JF, et al. Estrogen receptor-α signaling in post-natal mammary development and breast cancers. Cell Mol Life Sci. 2021;78(15):5681–705. https://doi.org/10.1007/s00018-021-03860-4.
Menendez JA, Peirce SK, Papadimitropoulou A, Cuyàs E, Vander Steen T, Verdura S, et al. Progesterone receptor isoform-dependent cross-talk between prolactin and fatty acid synthase in breast cancer. Aging. 2020;12(24):24671–24692. https://doi.org/10.18632/aging.202289.
Kavarthapu R, Dufau ML. Prolactin receptor gene transcriptional control, regulatory modalities relevant to breast cancer resistance and invasiveness. Front Endocrinol (Lausanne). 2022;13:949396. https://doi.org/10.3389/fendo.2022.949396.
He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal transduct target Ther. 2021;6(1):425. https://doi.org/10.1038/s41392-021-00828-5.
Carvajal A, Espinoza N, Kato S, Pinto M, Sadarangani A, Monso C, et al. Progesterone pre-treatment potentiates EGF pathway signaling in the breast cancer cell line ZR-75. Breast Cancer Res Treat. 2005;94(2):171–83. https://doi.org/10.1007/s10549-005-7726-6.
Kavarthapu R, Dufau ML. Essential role of endogenous prolactin and CDK7 in estrogen-induced upregulation of the prolactin receptor in breast cancer cells. Oncotarget. 2017;8(16):27353–27363. https://doi.org/10.18632/oncotarget.16040.
Moo TA, Sanford R, Dang C, Morrow M. Overview of Breast Cancer Therapy. PET Clin. 2018;13(3):339–54. https://doi.org/10.1016/j.cpet.2018.02.006.
Baudino TA. Targeted cancer therapy: the next generation of cancer treatment. Curr Drug Discov Technol. 2015;12(1):3–20. https://doi.org/10.2174/1570163812666150602144310.
Stravodimou A, Voutsadakis IA. The Future of ER+/HER2− metastatic breast cancer therapy: beyond PI3K Inhibitors. Anticancer Res. 2020;40(9):4829–4841. https://doi.org/10.21873/anticanres.14486.
Yin L, Duan JJ, Bian XW, Yu Sc. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Canc Res. 2020;22(1):61. https://doi.org/10.1186/s13058-020-01296-5.
Fan W, Chang J, Fu P. Endocrine therapy resistance in breast cancer: current status, possible mechanisms and overcoming strategies. Future Med Chem. 2015;7(12):1511–9. https://doi.org/10.4155/fmc.15.93.
Rani A, Stebbing J, Giamas G, Murphy J. Endocrine resistance in hormone receptor positive breast cancer–from mechanism to therapy. Front Endocrinol. 2019;10:245. https://doi.org/10.3389/fendo.2019.00245.
Saha T, Makar S, Swetha R, Gutti G, Singh SK. Estrogen signaling: An emanating therapeutic target for breast cancer treatment. Eur J Med Chem. 2019;177:116–43. https://doi.org/10.1016/j.ejmech.2019.05.023.
Shuai C, Yang X, Pan H, Han W. Estrogen receptor downregulates expression of PD-1/PD-L1 and infiltration of CD8+ T cells by inhibiting IL-17 signaling transduction in breast cancer. Front Oncol. 2020;10:582863. https://doi.org/10.3389/fonc.2020.582863.
Howell A, Howell SJ. Tamoxifen evolution. Br J Cancer. 2023;128(3):421–5. https://doi.org/10.1038/s41416-023-02158-5.
Patel HK, Bihani T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol Ther. 2018;186:1–12. https://doi.org/10.1016/j.pharmthera.2017.12.012.
Tung N, Garber JE. PARP inhibition in breast cancer: Progress made and future hopes. NPJ Breast Cancer. 2022;8(1):47. https://doi.org/10.1038/s41523-022-00411-3.
Zhu K, Wu Y, He P, Fan Y, Zhong X, Zheng H, Luo T. PI3K/AKT/mTOR-targeted therapy for breast cancer. Cells. 2022;11(16):2508. https://doi.org/10.3390/cells11162508.
Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thürlimann B, Senn HJ. Strategies for subtypes—dealing with the diversity of breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann oncol. 2011;22(8):1736–47. https://doi.org/10.1093/annonc/mdr304.
Sunkara T, Bandaru SS, Boyilla R, Kunadharaju R, Kukkadapu P, Chennamadhavuni A, et al. Poly adenosine diphosphate-ribose polymerase (PARP) inhibitors in pancreatic cancer. Cureus. 2022;14(2):e22575. https://doi.org/10.7759/cureus.22575.
Groelly FJ, Fawkes M, Dagg RA, Blackford AN, Tarsounas M. Targeting DNA damage response pathways in cancer. Nat Rev Cancer. 2023;23(2):78–94. https://doi.org/10.1038/s41568-022-00535-5.
Bornstein E, Jimeno A. Olaparib for the treatment of ovarian cancer. Drugs Today (Barc). 2016;52(1):17–28. https://doi.org/10.1358/dot.2016.52.1.2440714.
LaFargue CJ, Dal Molin GZ, Sood AK, Coleman RL. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019;20(1):e15–28. https://doi.org/10.1016/S1470-2045(18)30786-1.
Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017;377(6):523–33. https://doi.org/10.1056/NEJMoa1706450.
Lee JM, Hays JL, Chiou VL, Annunziata CM, Swisher EM, Harrell MI, et al. Phase I/Ib study of olaparib and carboplatin in women with triple negative breast cancer. Oncotarget. 2017;8(45):79175–79187. https://doi.org/10.18632/oncotarget.16577.
Yonemori K, Shimomura A, Yasojima H, Masuda N, Aogi K, Takahashi M, et al. A phase I/II trial of olaparib tablet in combination with eribulin in Japanese patients with advanced or metastatic triple-negative breast cancer previously treated with anthracyclines and taxanes. Eur J Cancer. 2021;109:84–91. https://doi.org/10.1016/j.ejca.2018.11.014.
Domchek SM, Postel-Vinay S, Im S-A, Park YH, Delord J-P, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21(9):1155–64. https://doi.org/10.1016/S1470-2045(20)30324-7.
Tutt AN, Garber JE, Kaufman B, Viale G, Fumagalli D, Rastogi P, et al. Adjuvant olaparib for patients with BRCA1-or BRCA2-mutated breast cancer. N Engl J Med. 2021;384(25):2394–405. https://doi.org/10.1056/NEJMoa2105215.
Fasching P, Link T, Hauke J, Seither F, Jackisch C, Klare P, Schmatloch S, Hanusch C, Huober J, Stefek A, and Seiler S. Neoadjuvant paclitaxel/olaparib in comparison to paclitaxel/carboplatinum in patients with HER2-negative breast cancer and homologous recombination deficiency (GeparOLA study). Ann Oncol. 2021;32(1),:49–57. https://doi.org/10.1016/j.annonc.2020.10.471.
Hoy SM. Talazoparib: first global approval. Drugs. 2018;78(18):1939–46. https://doi.org/10.1007/s40265-018-1026-z.
de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017;7(6):620–9. https://doi.org/10.1158/2159-8290.CD-16-1250.
Turner NC, Telli ML, Rugo HS, Mailliez A, Ettl J, Grischke EM, et al. A Phase II Study of Talazoparib after Platinum or Cytotoxic Nonplatinum Regimens in Patients with Advanced Breast Cancer and Germline BRCA1/2 Mutations (ABRAZO) Talazoparib in gBRCA1/2-Mutated Advanced Breast Cancer. Clin Cancer Res. 2019;25(9):2717–24. https://doi.org/10.1158/1078-0432.CCR-18-1891.
Hurvitz SA, Gonçalves A, Rugo HS, Lee KH, Fehrenbacher L, Mina LA, et al. Talazoparib in Patients with a Germline BRCA-Mutated Advanced Breast Cancer: Detailed Safety Analyses from the Phase III EMBRACA Trial. Oncologist. 2020;25(3):439–50. https://doi.org/10.1634/theoncologist.2019-0493.
Litton JK, Hurvitz SA, Mina LA, Rugo HS, Lee KH, Gonçalves A, et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: final overall survival results from the EMBRACA trial. Ann Oncol. 2020;31(11):1526–35. https://doi.org/10.1016/j.annonc.2020.08.2098.
Ettl J, Quek RGW, Lee KH, Rugo HS, Hurvitz S, Gonçalves A, et al. Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: patient-reported outcomes from the EMBRACA phase III trial. Ann Oncol. 2018;29(9):1939–47. https://doi.org/10.1093/annonc/mdy257.
Jenner ZB, Sood AK, Coleman RL. Evaluation of rucaparib and companion diagnostics in the PARP inhibitor landscape for recurrent ovarian cancer therapy. Future Oncol. 2016;12(12):1439–56. https://doi.org/10.2217/fon-2016-0002.
Drew Y, Ledermann J, Hall G, Rea D, Glasspool R, Highley M, et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer. 2016;114(7):723–30. https://doi.org/10.1038/bjc.2016.133.
Patsouris A, Diop K, Tredan O, Nenciu D, Gonçalves A, Arnedos M, et al. Rucaparib in patients presenting a metastatic breast cancer with homologous recombination deficiency, without germline BRCA1/2 mutation. Eur J Cancer. 2021;159:283–95. https://doi.org/10.1016/j.ejca.2021.09.028.
Wilson RH, Evans T, Middleton MR, Molife L, Spicer J, Dieras V, et al. A phase I study of intravenous and oral rucaparib in combination with chemotherapy in patients with advanced solid tumours. Br J Cancer. 2017;116(7):884–92. https://doi.org/10.1038/bjc.2017.36.
Miller K, Tong Y, Jones DR, Walsh T, Danso MA, Ma CX, et al. Cisplatin with or without rucaparib after preoperative chemotherapy in patients with triple negative breast cancer: Final efficacy results of Hoosier Oncology Group BRE09–146. Am J Clin Oncol. 2015;33(15_suppl):1082. https://doi.org/10.1200/jco.2015.33.15_suppl.1082.
Kalra M, Tong Y, Jones DR, Walsh T, Danso MA, Ma CX, et al. Cisplatin +/- rucaparib after preoperative chemotherapy in patients with triple-negative or BRCA mutated breast cancer. NPJ Breast Cancer. 2021;7(1):29. https://doi.org/10.1038/s41523-021-00240-w.
Wang M, Chen S, Ao D. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. MedComm. 2021;2(4):654–91. https://doi.org/10.1002/mco2.103.
Cortesi L, Rugo HS, Jackisch C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target Oncol. 2021;16(3):255–82. https://doi.org/10.1007/s11523-021-00796-4.
Balmana J, Tryfonidis K, Audeh W, Goulioti T, Slaets L, Agarwal S, et al. A phase III, randomized, open label, multicenter, controlled trial of niraparib versus physician's choice in previously-treated, HER2 negative, germline BRCA mutation-positive breast cancer patients. An EORTC-BIG intergroup study (BRAVO study). Cancer Res., 2016;76(4_Supplement):OT1–03–05. https://doi.org/10.1158/1538-7445.SABCS15-OT1-03-05.
Turner NC, Balmaña J, Poncet C, Goulioti T, Tryfonidis K, Honkoop AH, et al. Niraparib for Advanced Breast Cancer with Germline BRCA1 and BRCA2 Mutations: the EORTC 1307-BCG/BIG5–13/TESARO PR-30–50–10-C BRAVO Study. Clin Cancer Res. 2021;27(20):5482–91. https://doi.org/10.1158/1078-0432.CCR-21-0310.
Hu Y, Gao J, Wang M, Li M. Potential prospect of CDK4/6 inhibitors in triple-negative breast cancer. Cancer Manag Res. 2021;13:5223–37. https://doi.org/10.2147/CMAR.S310649.
Shah M, Nunes MR, Stearns V. CDK4/6 Inhibitors: Game Changers in the Management of Hormone Receptor-Positive Advanced Breast Cancer? Oncology (Williston Park). 2018;32(5):216–22.
McCain J. First-in-Class CDK4/6 Inhibitor Palbociclib Could Usher in a New Wave of Combination Therapies for HR+, HER2- Breast Cancer. P T. 2015;40(8):511–20.
Mayer EL, Dueck AC, Martin M, Rubovszky G, Burstein HJ, Bellet-Ezquerra M, et al. Palbociclib with adjuvant endocrine therapy in early breast cancer (PALLAS): interim analysis of a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2021;22(2):212–22. https://doi.org/10.1016/S1470-2045(20)30642-2.
Flaherty KT, LoRusso PM, DeMichele A, Abramson VG, Courtney R, Randolph SS, et al. Dose-Escalation Trial of the Oral Cyclin-Dependent Kinase 4/6 Inhibitor PD 0332991, Administered Using a 21-Day Schedule in Patients with Advanced CancerPhase I, Dose-Escalation Trial of PD 0332991, 4-Week Cycle. Clin Cancer Res. 2012;8(2):568–76. https://doi.org/10.1158/1078-0432.CCR-11-0509.
DeMichele A, Clark AS, Tan KS, Heitjan DF, Gramlich K, Gallagher M, et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res. 2015;21(5):995–1001. https://doi.org/10.1158/1078-0432.CCR-14-2258.
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16(1):25–35. https://doi.org/10.1016/S1470-2045(14)71159-3.
Vidula N, Rugo HS. Cyclin-dependent kinase 4/6 inhibitors for the treatment of breast cancer: a review of preclinical and clinical data. Clin Breast Cancer. 2016;16(1):8–17. https://doi.org/10.1016/j.clbc.2015.07.005.
Cristofanilli M, Rugo HS, Im SA, Slamon DJ, Harbeck N, Bondarenko I, et al. Overall Survival with Palbociclib and Fulvestrant in Women with HR+/HER2- ABC: Updated Exploratory Analyses of PALOMA-3, a Double-blind, Phase III Randomized Study. Clin Cancer Res. 2022;28(16):3433–42. https://doi.org/10.1158/1078-0432.CCR-22-0305.
Rascon K, Flajc G, De Angelis C, Liu X, Trivedi MV, Ekinci E. Ribociclib in HR+/HER2- Advanced or Metastatic Breast Cancer Patients. Ann Pharmacother. 2019;53(5):501–9. https://doi.org/10.1177/1060028018817904.
Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–48. https://doi.org/10.1056/NEJMoa1609709.
Tripathy D, Im SA, Colleoni M, Franke F, Bardia A, Harbeck N, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. 2018;19(7):904–15. https://doi.org/10.1016/S1470-2045(18)30292-4.
Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014;32(5):825–37. https://doi.org/10.1007/s10637-014-0120-7.
Martin JM, Goldstein LJ. Profile of abemaciclib and its potential in the treatment of breast cancer. Onco Targets Ther. 2018;11:5253–9. https://doi.org/10.2147/OTT.S149245.
Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016;6(7):740–53. https://doi.org/10.1158/2159-8290.CD-16-0095.
Dickler MN, Tolaney SM, Rugo HS, Cortés J, Diéras V, Patt D, et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR+/HER2- Metastatic Breast Cancer. Clin Cancer Res. 2017;23(17):5218–24. https://doi.org/10.1158/1078-0432.CCR-17-0754.
Sledge GW Jr, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: Abemaciclib in Combination with Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J Clin Oncol. 2017;35(25):2875–84. https://doi.org/10.1200/JCO.2017.73.7585.
Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, et al. MONARCH 3: Abemaciclib as Initial Therapy for Advanced Breast Cancer. J Clin Oncol. 2017;35(32):3638–46. https://doi.org/10.1200/JCO.2017.75.6155.
Neven P, Rugo HS, Tolaney SM, Iwata H, Toi M, Goetz MP, et al. Abemaciclib plus fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer in premenopausal women: subgroup analysis from the MONARCH 2 trial. Breast Cancer Res. 2021;23(1):87. https://doi.org/10.1186/s13058-021-01463-2.
Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin Cancer Biol. 2019;59:125–32. https://doi.org/10.1016/j.semcancer.2019.07.009.
Miricescu D, Totan A, Stanescu-Spinu I, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR signaling pathway in breast cancer: from molecular landscape to clinical aspects. Int J Mol Sci. 2020;22(1):173. https://doi.org/10.3390/ijms22010173.
Nitulescu GM, Van De Venter M, Nitulescu G, Ungurianu A, Juzenas P, Peng Q, et al. The Akt pathway in oncology therapy and beyond (Review). Int J Oncol. 2018;53(6):2319–31. https://doi.org/10.3892/ijo.2018.4597.
Bhattacharjee N, Barma S, Konwar N, Dewanjee S, Manna P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: An update. Eur J Pharmacol. 2016;791:8–24. https://doi.org/10.1016/j.ejphar.2016.08.022.
Nitulescu GM, Margina D, Juzenas P, Peng Q, Olaru OT, Saloustros E, et al. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use. Int J Oncol. 2016;48(3):869–85. https://doi.org/10.3892/ijo.2015.3306.
Doi T, Tamura K, Tanabe Y, Yonemori K, Yoshino T, Fuse N, et al. Phase 1 pharmacokinetic study of the oral pan-AKT inhibitor MK-2206 in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2015;76(2):409–416. https://doi.org/10.1007/s00280-015-2810-z.
Hudis C, Swanton C, Janjigian YY, Lee R, Sutherland S, Lehman R, et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013;15(6):R110. https://doi.org/10.1186/bcr3577.
Wisinski KB, Tevaarwerk AJ, Burkard ME, Rampurwala M, Eickhoff J, Bell MC, et al. Phase I Study of an AKT Inhibitor (MK-2206) Combined with Lapatinib in Adult Solid Tumors Followed by Dose Expansion in Advanced HER2+ Breast Cancer. Clin Cancer Res. 2016;22(11):2659–67. https://doi.org/10.1158/1078-0432.CCR-15-2365.
Xing Y, Lin NU, Maurer MA, Chen H, Mahvash A, Sahin A, et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019;21(1):78. https://doi.org/10.1186/s13058-019-1154-8.
Chien AJ, Tripathy D, Albain KS, Symmans WF, Rugo HS, Melisko ME, et al. I-SPY 2 Consortium. MK-2206 and Standard Neoadjuvant Chemotherapy Improves Response in Patients With Human Epidermal Growth Factor Receptor 2-Positive and/or Hormone Receptor-Negative Breast Cancers in the I-SPY 2 Trial. J Clin Oncol. 2020;38(10):1059–1069. https://doi.org/10.1200/JCO.19.01027.
Martorana F, Motta G, Pavone G, Motta L, Stella S, Vitale SR, et al. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front Pharmacol. 2021;12:662232. https://doi.org/10.3389/fphar.2021.662232.
Banerji U, Dean EJ, Pérez-Fidalgo JA, Batist G, Bedard PL, You B, et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and in PIK3CA-Mutated Breast and Gynecologic Cancers. Clin Cancer Res. 2018;24(9):2050–9. https://doi.org/10.1158/1078-0432.CCR-17-2260.
Saura C, Roda D, Roselló S, Oliveira M, Macarulla T, Pérez-Fidalgo JA, et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2017;7(1):102–13. https://doi.org/10.1158/2159-8290.CD-16-0512.
Smyth LM, Tamura K, Oliveira M, Ciruelos EM, Mayer IA, Sablin MP, et al. Capivasertib, an AKT Kinase Inhibitor, as Monotherapy or in Combination with Fulvestrant in Patients with AKT1E17K-Mutant, ER-Positive Metastatic Breast Cancer. Clin Cancer Res. 2020;26(15):3947–57. https://doi.org/10.1158/1078-0432.CCR-19-3953.
Jones RH, Casbard A, Carucci M, Cox C, Butler R, Alchami F, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020;21(3):345–57. https://doi.org/10.1016/S1470-2045(19)30817-4.
Schmid P, Abraham J, Chan S, Wheatley D, Brunt AM, Nemsadze G, et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel as First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J Clin Oncol. 2020;38(5):423–33. https://doi.org/10.1200/JCO.19.00368.
Turner NC, Alarcón E, Armstrong AC, Philco M, López Chuken YA, Sablin MP, et al. BEECH: a dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA mutant sub-population. Ann Oncol. 2019;30(5):774–80. https://doi.org/10.1093/annonc/mdz086.
Robertson JFR, Coleman RE, Cheung KL, Evans A, Holcombe C, Skene A, et al. Proliferation and AKT Activity Biomarker Analyses after Capivasertib (AZD5363) Treatment of Patients with ER+ Invasive Breast Cancer (STAKT). Clin Cancer Res. 2020;26(7):1574–85. https://doi.org/10.1158/1078-0432.CCR-19-3053.
Lang L, Lam T, Chen A, Jensen C, Duncan L, Kong FC, et al. Circumventing AKT-Associated Radioresistance in Oral Cancer by Novel Nanoparticle-Encapsulated Capivasertib. Cells. 2020;9(3):533. https://doi.org/10.3390/cells9030533.
Buckingham L, Hao T, O’Donnell J, Zhao Z, Zhang X, Fan Y, et al. Ipatasertib, an oral AKT inhibitor, inhibits cell proliferation and migration, and induces apoptosis in serous endometrial cancer. Am J Cancer Res. 2022;12(6):2850–62.
Kim SB, Dent R, Im SA, Espié M, Blau S, Tan AR, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360–72. https://doi.org/10.1016/S1470-2045(17)30450-3.
Oliveira M, Saura C, Nuciforo P, Calvo I, Andersen J, Passos-Coelho JL, et al. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann Oncol. 2019;30(8):1289–97. https://doi.org/10.1093/annonc/mdz177.
Turner N, Dent RA, O’Shaughnessy J, Kim SB, Isakoff SJ, Barrios C, et al. Ipatasertib plus paclitaxel for PIK3CA/AKT1/PTEN-altered hormone receptor-positive HER2-negative advanced breast cancer: primary results from cohort B of the IPATunity130 randomized phase 3 trial. Breast Cancer Res Treat. 2022;191(3):565–76. https://doi.org/10.1007/s10549-021-06450-x.
Shah AA, Kamal MA, Akhtar S. Tumor angiogenesis and VEGFR-2: mechanism, pathways and current biological therapeutic interventions. Curr Drug Metab. 2021;22(1):50–9. https://doi.org/10.2174/1389200221666201019143252.
Madu CO, Wang S, Madu CO, Lu Y. Angiogenesis in breast cancer progression, diagnosis, and treatment. J Cancer. 2020;11(15):4474–94. https://doi.org/10.7150/jca.44313.
Ahn S, Woo JW, Lee K, Park SY. HER2 status in breast cancer: changes in guidelines and complicating factors for interpretation. J Pathol Transl Med. 2020;54(1):34–44. https://doi.org/10.4132/jptm.2019.11.03.
Kumar R, Yarmand-Bagheri R. The role of HER2 in angiogenesis. Semin Oncol. 2001;28(5 Suppl 16):27–32. https://doi.org/10.1016/s0093-7754(01)90279-9.
Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. 2014;17(3):471–94. https://doi.org/10.1007/s10456-014-9420-y.
Miller KD, Chap LI, Holmes FA, Cobleigh MA, Marcom PK, Fehrenbacher L, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23(4):792–9. https://doi.org/10.1200/JCO.2005.05.098.
Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–76. https://doi.org/10.1056/NEJMoa072113.
Schneider BP, Wang M, Radovich M, Sledge GW, Badve S, Thor A, et al. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J Clin Oncol. 2008;26(28):4672–8. https://doi.org/10.1200/JCO.2008.16.1612.
Gianni L, Romieu GH, Lichinitser M, Serrano SV, Mansutti M, Pivot X, et al. AVEREL: a randomized phase III Trial evaluating bevacizumab in combination with docetaxel and trastuzumab as first-line therapy for HER2-positive locally recurrent/metastatic breast cancer. J Clin Oncol. 2013;31(14):1719–25. https://doi.org/10.1200/JCO.2012.44.7912.
Cuppone F, Bria E, Vaccaro V, Puglisi F, Fabi A, Sperduti I, et al. Magnitude of risks and benefits of the addition of bevacizumab to chemotherapy for advanced breast cancer patients: Meta-regression analysis of randomized trials. J Exp Clin Cancer Res. 2011;30(1):54. https://doi.org/10.1186/1756-9966-30-54.
Lee JB, Woo OH, Park KH, Woo SU, Yang DS, Kim AR, et al. Bevacizumab for salvage treatment of metastatic breast cancer: a systemic review and meta-analysis of randomized controlled trials. Invest New Drugs. 2011;29(1):182–8. https://doi.org/10.1007/s10637-009-9310-0.
Miles DW, Diéras V, Cortés J, Duenne AA, Yi J, O’Shaughnessy J. First-line bevacizumab in combination with chemotherapy for HER2-negative metastatic breast cancer: pooled and subgroup analyses of data from 2447 patients. Ann Oncol. 2013;24(11):2773–80. https://doi.org/10.1093/annonc/mdt276.
Ayoub NM, Jaradat SK, Al-Shami KM, Alkhalifa AE. Targeting Angiogenesis in Breast Cancer: Current Evidence and Future Perspectives of Novel Anti-Angiogenic Approaches. Front Pharmacol. 2022;13:838133. https://doi.org/10.3389/fphar.2022.838133.
Mackey JR, Ramos-Vazquez M, Lipatov O, McCarthy N, Krasnozhon D, Semiglazov V, et al. Primary results of ROSE/TRIO-12, a randomized placebo-controlled phase III trial evaluating the addition of ramucirumab to first-line docetaxel chemotherapy in metastatic breast cancer. J Clin Oncol. 2015;33(2):141–8. https://doi.org/10.1200/JCO.2014.57.1513.
Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13(1):1–14. https://doi.org/10.1007/s10456-009-9160-6.
Boér K, Láng I, Llombart-Cussac A, Andreasson I, Vivanco GL, Sanders N, et al. Vandetanib with docetaxel as second-line treatment for advanced breast cancer: a double-blind, placebo-controlled, randomized Phase II study. Invest New Drugs. 2012;30(2):681–7. https://doi.org/10.1007/s10637-010-9538-8.
Hyams DM, Chan A, de Oliveira C, Snyder R, Vinholes J, Audeh MW, et al. Cediranib in combination with fulvestrant in hormone-sensitive metastatic breast cancer: a randomized Phase II study. Invest New Drugs. 2013;31(5):1345–54. https://doi.org/10.1007/s10637-013-9991-2.
Rugo HS, Stopeck AT, Joy AA, Chan S, Verma S, Lluch A, et al. Randomized, placebo-controlled, double-blind, phase II study of axitinib plus docetaxel versus docetaxel plus placebo in patients with metastatic breast cancer. J Clin Oncol. 2011;29(18):2459–65. https://doi.org/10.1200/JCO.2010.31.2975.
Taylor SK, Chia S, Dent S, Clemons M, Agulnik M, Grenci P, et al. A phase II study of pazopanib in patients with recurrent or metastatic invasive breast carcinoma: a trial of the Princess Margaret Hospital phase II consortium. Oncologist. 2010;15(8):810–8. https://doi.org/10.1634/theoncologist.2010-0081.
Schwartzberg LS, Tauer KW, Hermann RC, Makari-Judson G, Isaacs C, Beck JT, et al. Sorafenib or placebo with either gemcitabine or capecitabine in patients with HER-2-negative advanced breast cancer that progressed during or after bevacizumab. Clin Cancer Res. 2013;19(10):2745–54. https://doi.org/10.1634/theoncologist.2010-0081.
Baselga J, Zamagni C, Gómez P, Bermejo B, Nagai SE, Melichar B, et al. RESILIENCE: Phase III Randomized, Double-Blind Trial Comparing Sorafenib with Capecitabine Versus Placebo with Capecitabine in Locally Advanced or Metastatic HER2-Negative Breast Cancer. Clin Breast Cancer. 2017;17(8):585-594.e4. https://doi.org/10.1016/j.clbc.2017.05.006.
Baselga J, Segalla JG, Roché H, Del Giglio A, Pinczowski H, Ciruelos EM, et al. Sorafenib in combination with capecitabine: an oral regimen for patients with HER2-negative locally advanced or metastatic breast cancer. J Clin Oncol. 2012;30(13):1484–91. https://doi.org/10.1200/JCO.2011.36.7771.
Fakhrejahani E, Toi M. Antiangiogenesis therapy for breast cancer: an update and perspectives from clinical trials. Jpn J Clin Oncol. 2014;44(3):197–207. https://doi.org/10.1093/jjco/hyt201.
Farooq M, Khan AW, Kim MS, Choi S. The role of fibroblast growth factor (FGF) signaling in tissue repair and regeneration. Cells. 2021;10(11):3242. https://doi.org/10.3390/cells10113242.
Francavilla C, O'Brien CS. Fibroblast growth factor receptor singaling dysregulation and targeting in breast cancer. Open Biol. 2022;12(2):210373. https://doi.org/10.1098/rsob.210373.
Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16(2):105–22. https://doi.org/10.1038/s41571-018-0115-y.
Musolino A, Campone M, Neven P, Denduluri N, Barrios CH, Cortes J, et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR+, HER2- breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res. 2017;19(1):18. https://doi.org/10.1186/s13058-017-0807-8.
Hyman DM, Tran B, Paz-Ares L, Machiels JP, Schellens JH, Bedard PL, et al. Combined PIK3CA and FGFR Inhibition With Alpelisib and Infigratinib in Patients With PIK3CA-Mutant Solid Tumors, With or Without FGFR Alterations. JCO Precis Oncol. 2019. https://doi.org/10.1200/P0.19.00221.
Smyth EC, Turner NC, Peckitt C, Pearson A, Brown G, Chua S, et al. Phase II multicenter proof of concept study of AZD4547 in FGFR amplified tumours. J Clin Oncol. 2015;33(15_suppl):2508. https://doi.org/10.1200/jco.2015.33.15_suppl.2508.
Verret B, Cortes J, Bachelot T, Andre F, Arnedos M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann Oncol. 2019;30(Suppl_10):x12-x20. https://doi.org/10.1093/annonc/mdz381.
Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–9. https://doi.org/10.1056/NEJMoa1109653.
Johnston SRD, Toi M, O’Shaughnessy J, Rastogi P, Campone M, Neven P, et al. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol. 2023;24:77–90. https://doi.org/10.1016/S1470-2045(22)00694-5.
Wedam S, Fashoyin-Aje L, Bloomquist E, Tang S, Sridhara R, Goldberg KB, et al. FDA Approval Summary: Palbociclib for Male Patients with Metastatic Breast Cancer. Clin Cancer Res. 2020;26(6):1208–12. https://doi.org/10.1158/1078-0432.CCR-19-2580.
Shah A, Bloomquist E, Tang S, Fu W, Bi Y, Liu Q, et al. FDA Approval: Ribociclib for the Treatment of Postmenopausal Women with Hormone Receptor-Positive, HER2-Negative Advanced or Metastatic Breast Cancer. Clin Cancer Res. 2018;24(13):2999–3004. https://doi.org/10.1158/1078-0432.CCR-17-2369.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J Clin Oncol. 2018;36(24):2465–72. https://doi.org/10.1200/JCO.2018.78.9909.
Arora S, Narayan P, Osgood CL, Wedam S, Prowell TM, Gao JJ, et al. U.S. FDA Drug Approvals for Breast Cancer: A Decade in Review. Clin Cancer Res. 2022;28(6):1072–1086. https://doi.org/10.1158/1078-0432.CCR-21-2600.
Lee A. Tucatinib: First Approval. Drugs. 2020;80(10):1033–8. https://doi.org/10.1007/s40265-020-01340-w.
Dai MS, Feng YH, Chen SW, Masuda N, Yau T, Chen ST, et al. Analysis of the pan-Asian subgroup of patients in the NALA Trial: a randomized phase III NALA Trial comparing neratinib+capecitabine (N+C) vs lapatinib+capecitabine (L+C) in patients with HER2+metastatic breast cancer (mBC) previously treated with two or more HER2-directed regimens. Breast Cancer Res Treat. 2021;189(3):665–76. https://doi.org/10.1007/s10549-021-06313-5.
Nounou MI, ElAmrawy F, Ahmed N, Abdelraouf K, Goda S, Syed-Sha-Qhattal H. Breast Cancer: Conventional Diagnosis and Treatment Modalities and Recent Patents and Technologies. Breast Cancer (Auckl). 2015;9(Suppl 2):17–34. https://doi.org/10.4137/BCBCR.S29420.
Pucci C, Martinelli C, Ciofani G. Innovative approaches for cancer treatment: current perspectives and new challenges. Ecancermedicalscience. 2019;13:961. https://doi.org/10.3332/ecancer.2019.961.
Manna P, Dewanjee S, Joardar S, Chakraborty P, Bhattacharya H, Bhanja S, et al. Carnosic acid attenuates doxorubicin-induced cardiotoxicity by decreasing oxidative stress and its concomitant pathological consequences. Food Chem Toxicol. 2022;166:113205. https://doi.org/10.1016/j.fct.2022.113205.
Nurgali K, Jagoe RT, Abalo R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front Pharmacol. 2018;9:245. https://doi.org/10.3389/fphar.2018.00245.
Masoud V, Pagès G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J Clin Oncol. 2017;8(2):120–34. https://doi.org/10.5306/wjco.v8.i2.120.
Baxevanis CN, Fortis SP, Perez SA. The balance between breast cancer and the immune system: Challenges for prognosis and clinical benefit from immunotherapies. Semin Cancer Biol. 2021;72:76–89. https://doi.org/10.1016/j.semcancer.2019.12.018.
Denkert C, Liedtke C, Tutt A, von Minckwitz G. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389(10087):2430–42. https://doi.org/10.1016/S0140-6736(16)32454-0.
Debien V, De Caluwé A, Wang X, Piccart-Gebhart M, TuohyVK, Romano E, and Buisseret L. Immunotherapy in breast cancer: an overview of current strategies and perspectives. npj Breast Cancer. 2023; 9(1):7. https://doi.org/10.1038/s41523-023-00508-3.
Esteva FJ, Hubbard-Lucey VM, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019;20(3):175–86. https://doi.org/10.1016/S1470-2045(19)30026-9.
Toraya-Brown S, Fiering S. Local tumour hyperthermia as immunotherapy for metastatic cancer. Int J Hyperthermia. 2014;30(8):531–9. https://doi.org/10.3109/02656736.2014.968640.
Simonian M, Haji Ghaffari M, Negahdari B. Immunotherapy for Breast Cancer Treatment. Iran Biomed J. 2021;25(3):140–156. https://doi.org/10.29252/ibj.25.3.140.
Schütz F, Stefanovic S, Mayer L, von Au A, Domschke C, Sohn C. PD-1/PD-L1 Pathway in Breast Cancer. Oncol Res Treat. 2017;40(5):294–7. https://doi.org/10.1159/000464353.
Li Y, Miao W, He D, Wang S, Lou J, Jiang Y, and Wang S. Recent Progress on Immunotherapy for Breast Cancer: Tumor Microenvironment, Nanotechnology and More. Front Bioeng Biotechnol. 2021;9:680315. https://doi.org/10.3389/fbioe.2021.680315.
Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2(4):361–70. https://doi.org/10.1158/2326-6066.CIR-13-0127.
Migden MR, Chandra S, Rabinowits G, Chen CI, Desai J, Seluzhytsky A, Sasane M, Campanelli B, Chen Z, Freeman ML, Ibrahim SF, Khushalani NI, Andria M, Ruiz E. CASE (CemiplimAb-rwlc Survivorship and Epidemiology) study in advanced cutaneous squamous cell carcinoma. Future Oncol. 2020;16(4):11–9.
Isaacs C, Nanda R, Chien J, Trivedi MS, Stringer-Reasor E, Vaklavas C, et al. Evaluation of anti-PD-1 Cemiplimab plus anti-LAG-3 REGN3767 in early-stage, high-risk HER2-negative breast cancer: Results from the neoadjuvant I-SPY 2 TRIAL. Cancer Res 2023;83(5 Suppl):Abstract nr GS5–03. https://doi.org/10.1158/1538-7445.SABCS22-GS5-03.
Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 study. J Clin Oncol. 2016;34(21):2460–7. https://doi.org/10.1200/JCO.2015.64.8931.
Adams S, Schmid P, Rugo HS, Winer EP, Loirat D, Awada A, et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort A of the phase II KEYNOTE-086 study. Ann Oncol. 2019;30(3):397–404. https://doi.org/10.1093/annonc/mdy517.
Winer EP, Lipatov O, Im SA, Goncalves A, Muñoz-Couselo E, Lee KS, et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(4):499–511. https://doi.org/10.1016/S1470-2045(20)30754-3.
Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396(10265):1817–28. https://doi.org/10.1016/S0140-6736(20)32531-9.
Tolaney SM, Kalinsky K, Kaklamani VG, D’Adamo DR, Aktan G, Tsai ML, et al. Eribulin Plus Pembrolizumab in Patients with Metastatic Triple-Negative Breast Cancer (ENHANCE 1): A Phase Ib/II Study. Clin Cancer Res. 2021;27(11):3061–8. https://doi.org/10.1158/1078-0432.CCR-20-4726.
Pérez-García JM, Llombart-Cussac A, Cortés MG, Curigliano G, López-Miranda E, Alonso JL, et al. Pembrolizumab plus eribulin in hormone-receptor-positive, HER2-negative, locally recurrent or metastatic breast cancer (KELLY): An open-label, multicentre, single-arm, phase II trial. Eur J Cancer. 2021;148:382–94. https://doi.org/10.1016/j.ejca.2021.02.028.
Tolaney SM, Barroso-Sousa R, Keenan T, Li T, Trippa L, Vaz-Luis I, et al. Effect of Eribulin With or Without Pembrolizumab on Progression-Free Survival for Patients With Hormone Receptor-Positive, ERBB2-Negative Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol. 2020;6(10):1598–605. https://doi.org/10.1001/jamaoncol.2020.3524.
Ho AY, Barker CA, Arnold BB, Powell SN, Hu ZI, Gucalp A, et al. A phase 2 clinical trial assessing the efficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer. 2020;126(4):850–60. https://doi.org/10.1002/cncr.32599.
Wesolowski J, Tankiewicz-Kwedlo A, Pawlak D. Modern Immunotherapy in the Treatment of Triple-Negative Breast Cancer. Cancers (Basel). 2022;14(16):3860. https://doi.org/10.3390/cancers14163860.
Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25(6):920–8. https://doi.org/10.1038/s41591-019-0432-4.
Adams S, Othus M, Patel SP, Miller KD, Chugh R, Schuetze SM, et al. A Multicenter Phase II Trial of Ipilimumab and Nivolumab in Unresectable or Metastatic Metaplastic Breast Cancer: Cohort 36 of Dual Anti-CTLA-4 and Anti-PD-1 Blockade in Rare Tumors (DART, SWOG S1609). Clin Cancer Res. 2022;28(2):271–8. https://doi.org/10.1158/1078-0432.CCR-21-2182.
Barroso-Sousa R, Keenan TE, Li T, Tayob N, Trippa L, Pastorello RG, et al. Nivolumab in combination with cabozantinib for metastatic triple-negative breast cancer: a phase II and biomarker study. NPJ Breast Cancer. 2021. https://doi.org/10.1038/s41523-021-00287-9.
Juliá EP, Amante A, Pampena MB, Mordoh J, Levy EM. Avelumab, an IgG1 anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front Immunol. 2018;9:2140. https://doi.org/10.3389/fimmu.2018.02140.
Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau H-T, Forero-Torres A, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast cancer Res Treat. 2018;167(3):671–86. https://doi.org/10.1007/s10549-017-4537-5.
Emens LA, Cruz C, Eder JP, Braiteh F, Chung C, Tolaney SM, et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019;5(1):74–82. https://doi.org/10.1001/jamaoncol.2018.4224.
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2018;379(22):2108–21. https://doi.org/10.1056/NEJMoa1809615.
Emens LA, Adams S, Barrios CH, Diéras V, Iwata H, Loi S, et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann Oncol. 2021;32(8):983–93. https://doi.org/10.1016/j.annonc.2021.05.355.
Miles D, Gligorov J, André F, Cameron D, Schneeweiss A, Barrios C, et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann Oncol. 2021;32(8):994–1004. https://doi.org/10.1016/j.annonc.2021.05.801.
Loibl S, Untch M, Burchardi N, Huober J, Sinn BV, Blohmer JU, et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: clinical results and biomarker analysis of GeparNuevo study. Ann Oncol. 2019;30(8):1279–88. https://doi.org/10.1093/annonc/mdz158.
Pusztai L, Yau C, Wolf DM, Han HS, Du L, Wallace AM, et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell. 2021;39(7):989-998.e5. https://doi.org/10.1016/j.ccell.2021.05.009.
Foldi J, Silber A, Reisenbichler E, Singh K, Fischbach N, Persico J, et al. Neoadjuvant durvalumab plus weekly nab-paclitaxel and dose-dense doxorubicin/cyclophosphamide in triple-negative breast cancer. NPJ Breast Cancer. 2021;7(1):9. https://doi.org/10.1038/s41523-021-00219-7.
Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Investig. 2015;125(9):3377–83. https://doi.org/10.1172/JCI80012.
Comin-Anduix B, Escuin-Ordinas H, Ibarrondo FJ. Tremelimumab: research and clinical development. Onco Targets Ther. 2016;9:1767–76. https://doi.org/10.2147/OTT.S65802.
Keam SJ. Tremelimumab: First Approval. Drugs. 2023;83(1):93–102. https://doi.org/10.1007/s40265-022-01827-8.
Vonderheide RH, LoRusso PM, Khalil M, Gartner EM, Khaira D, Soulieres D, et al. Tremelimumab in combination with exemestane in patients with advanced breast cancer and treatment-associated modulation of inducible costimulator expression on patient T cells. Clin Cancer Res. 2010;16(13):3485–94. https://doi.org/10.1158/1078-0432.CCR-10-0505.
Santa-Maria CA, Kato T, Park JH, Kiyotani K, Rademaker A, Shah AN, et al. A pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Oncotarget. 2018;9(27):18985–18996. https://doi.org/10.18632/oncotarget.24867.
Jiang DM, Fyles A, Nguyen LT, Neel BG, Sacher A, Rottapel R, et al. Phase I study of local radiation and tremelimumab in patients with inoperable locally recurrent or metastatic breast cancer. Oncotarget. 2019;10(31):2947–2958. https://doi.org/10.18632/oncotarget.26893.
Navarrete-Bernal MGC, Cervantes-Badillo MG, Martínez-Herrera JF, Lara-Torres CO, Gerson-Cwilich R, Zentella-Dehesa A, et al. Biological Landscape of Triple Negative Breast Cancers Expressing CTLA-4. Front Oncol. 2020;10:1206. https://doi.org/10.3389/fonc.2020.01206.
McArthur HL, Diab A, Page DB, Yuan J, Solomon SB, Sacchini V, et al. A Pilot Study of Preoperative Single-Dose Ipilimumab and/or Cryoablation in Women with Early-Stage Breast Cancer with Comprehensive Immune Profiling. Clin Cancer Res. 2016;22(23):5729–37. https://doi.org/10.1158/1078-0432.CCR-16-0190.
Loi S, Francis PA, Zdenkowski N, Gebski V, Fox SB, White M, et al. Neoadjuvant ipilimumab and nivolumab in combination with paclitaxel following anthracycline-based chemotherapy in patients with treatment resistant early-stage triple-negative breast cancer (TNBC): A single-arm phase 2 trial. J Clin Oncol 2022;40(16_suppl):602. https://doi.org/10.1200/JCO.2022.40.16_suppl.602.
Brignone C, Gutierrez M, Mefti F, Brain E, Jarcau R, Cvitkovic F, et al. First-line chemoimmunotherapy in metastatic breast carcinoma: combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J Transl Med. 2010;8:71. https://doi.org/10.1186/1479-5876-8-71.
Chaudhuri S, Thomas S, Munster P. Immunotherapy in breast cancer: A clinician’s perspective. Journal of the National Cancer Center. 2021;1(2):47–57. https://doi.org/10.1016/j.jncc.2021.01.001.
Smith PL, Piadel K, Dalgleish AG. Directing T-Cell Immune Responses for Cancer Vaccination and Immunotherapy. Vaccines (Basel). 2021;9(12):1392. https://doi.org/10.3390/vaccines9121392.
Zhu SY, Yu KD. Breast Cancer Vaccines: Disappointing or Promising? Front Immunol. 2022;13:828386. https://doi.org/10.3389/fimmu.2022.828386.
Clifton GT, Peoples GE, Mittendorf EA. The development and use of the E75 (HER2 369–377) peptide vaccine. Future Oncol. 2016;12(11):1321–9. https://doi.org/10.2217/fon-2015-0054.
Peoples GE, Gurney JM, Hueman MT, Woll MM, Ryan GB, Storrer CE, et al. Clinical trial results of a HER2/neu (E75) vaccine to prevent recurrence in high-risk breast cancer patients. J Clin Oncol. 2005;23(30):7536–45. https://doi.org/10.1200/JCO.2005.03.047.
Holmes JP, Gates JD, Benavides LC, Hueman MT, Carmichael MG, et al. Optimal dose and schedule of an HER-2/neu (E75) peptide vaccine to prevent breast cancer recurrence: from US Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Cancer. 2008;113(7):1666–75. https://doi.org/10.1002/cncr.23772.
Mittendorf EA, Clifton GT, Holmes JP, Schneble E, van Echo D, Ponniah S, et al. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann Oncol. 2014;25(9):1735–42. https://doi.org/10.1093/annonc/mdu211.
Mittendorf EA, Lu B, Melisko M, Price Hiller J, Bondarenko I, Brunt AM, et al. Efficacy and Safety Analysis of Nelipepimut-S Vaccine to Prevent Breast Cancer Recurrence: A Randomized, Multicenter, Phase III Clinical Trial. Clin Cancer Res. 2019;25(14):4248–54. https://doi.org/10.1158/1078-0432.CCR-18-2867.
You Z, Zhou W, Weng J, Feng H, Liang P, Li Y, et al. Application of HER2 peptide vaccines in patients with breast cancer: a systematic review and meta-analysis. Cancer Cell Int. 2021;21(1):489. https://doi.org/10.1186/s12935-021-02187-1.
Carmichael MG, Benavides LC, Holmes JP, Gates JD, Mittendorf EA, Ponniah S, et al. Results of the first phase 1 clinical trial of the HER-2/neu peptide (GP2) vaccine in disease-free breast cancer patients: United States Military Cancer Institute Clinical Trials Group Study I-04. Cancer. 2010;116(2):292–301. https://doi.org/10.1002/cncr.24756.
Patel S, McWilliams D, Fischette CT, Thompson J, Patel M, Daugherty FJ. Final five-year median follow-up safety data from a prospective, randomized, placebo-controlled, single-blinded, multicenter, phase IIb study evaluating the use of HER2/neu peptide GP2 + GM-CSF vs. GM-CSF alone after adjuvant trastuzumab in HER2-positive women with operable breast cancer. J Clin Oncol. 2021;39(15_suppl):542. https://doi.org/10.1200/JCO.2021.39.15_suppl.542.
Holmes JP, Benavides LC, Gates JD, Carmichael MG, Hueman MT, Mittendorf EA, et al. Results of the first phase I clinical trial of the novel II-key hybrid preventive HER-2/neu peptide (AE37) vaccine. J Clin Oncol. 2008;26(20):3426–33. https://doi.org/10.1200/JCO.2007.15.7842.
Brown TA 2nd, Mittendorf EA, Hale DF, Myers JW 3rd, Peace KM, Jackson DO, et al. Prospective, randomized, single-blinded, multi-center phase II trial of two HER2 peptide vaccines, GP2 and AE37, in breast cancer patients to prevent recurrence. Breast Cancer Res Treat. 2020;181(2):391–401. https://doi.org/10.1007/s10549-020-05638-x.
Disis ML, Schiffman K, Guthrie K, Salazar LG, Knutson KL, Goodell V, et al. Effect of dose on immune response in patients vaccinated with an her-2/neu intracellular domain protein-based vaccine. J Clin Oncol. 2004;22(10):1916–25. https://doi.org/10.1200/JCO.2004.09.005.
Limentani SA, Campone M, Dorval T, Curigliano G, de Boer R, Vogel C, White S, et al. A non-randomized dose-escalation Phase I trial of a protein-based immunotherapeutic for the treatment of breast cancer patients with HER2-overexpressing tumors. Breast Cancer Res Treat. 2016;156(2):319–30. https://doi.org/10.1007/s10549-016-3751-x.
Norell H, Poschke I, Charo J, Wei WZ, Erskine C, Piechocki MP, et al. Vaccination with a plasmid DNA encoding HER-2/neu together with low doses of GM-CSF and IL-2 in patients with metastatic breast carcinoma: a pilot clinical trial. J Transl Med. 2010;8:53. https://doi.org/10.1186/1479-5876-8-53.
Diaz CM, Chiappori A, Aurisicchio L, Bagchi A, Clark J, Dubey S, et al. Phase 1 studies of the safety and immunogenicity of electroporated HER2/CEA DNA vaccine followed by adenoviral boost immunization in patients with solid tumors. J Transl Med. 2013;11:62. https://doi.org/10.1186/1479-5876-11-62.
Gandhi S, Forsyth P, Opyrchal M, Ahmed K, Khong H, Attwood K, et al. Phase IIa study of alpha-DC1 vaccine against HER2/HER3, chemokine modulation regimen and pembrolizumab in patients with asymptomatic brain metastasis from triple negative or HER2+ breast cancer. Journal for ImmunoTherapy of Cancer 2020;8(supple 3):A196-A197. https://doi.org/10.1136/jitc-2020-SITC2020.0320.
Morse MA, Hobeika A, Osada T, Niedzwiecki D, Marcom PK, Blackwell KL, et al. Long term disease-free survival and T cell and antibody responses in women with high-risk Her2+ breast cancer following vaccination against Her2. J Transl Med. 2007;5:42. https://doi.org/10.1186/1479-5876-5-42.
Chen G, Gupta R, Petrik S, Laiko M, Leatherman JM, Asquith JM, et al. A feasibility study of cyclophosphamide, trastuzumab, and an allogeneic GM-CSF-secreting breast tumor vaccine for HER2+ metastatic breast cancer. Cancer Immunol Res. 2014;2(10):949–61. https://doi.org/10.1158/2326-6066.CIR-14-0058.
Emens LA, Asquith JM, Leatherman JM, Kobrin BJ, Petrik S, Laiko M, et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: a chemotherapy dose-ranging factorial study of safety and immune activation. J Clin Oncol. 2009;27(35):5911–8. https://doi.org/10.1200/JCO.2009.23.3494.
Kimura T, Finn OJ. MUC1 immunotherapy is here to stay. Expert Opin Biol Ther. 2013;13(1):35–49. https://doi.org/10.1517/14712598.2012.725719.
Liu Y, Tang L, Gao N, Diao Y, Zhong J, Deng Y, et al. Synthetic MUC1 breast cancer vaccine containing a Toll-like receptor 7 agonist exerts antitumor effects. Oncol Lett. 2020;20(3):2369–77. https://doi.org/10.3892/ol.2020.11762.
Heery CR, Ibrahim NK, Arlen PM, Mohebtash M, Murray JL, Koenig K, et al. Docetaxel Alone or in Combination With a Therapeutic Cancer Vaccine (PANVAC) in Patients With Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol. 2015;1(8):1087–95. https://doi.org/10.1001/jamaoncol.2015.2736.
Tiriveedhi V, Tucker N, Herndon J, Li L, Sturmoski M, Ellis M, et al. Safety and preliminary evidence of biologic efficacy of a mammaglobin-a DNA vaccine in patients with stable metastatic breast cancer. Clin Cancer Res. 2014;20(23):5964–75. https://doi.org/10.1158/1078-0432.CCR-14-0059.
Tiriveedhi V, Fleming TP, Goedegebuure PS, Naughton M, Ma C, Lockhart C, et al. Mammaglobin-A cDNA vaccination of breast cancer patients induces antigen-specific cytotoxic CD4+ICOShi T cells. Breast Cancer Res Treat. 2013;138(1):109–18. https://doi.org/10.1007/s10549-012-2110-9.
Zhang Y, Toh L, Lau P, Wang X. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/β-catenin pathway in human cancer. J Biol Chem. 2012;287(39):32494–324511. https://doi.org/10.1074/jbc.M112.368282.
Domchek SM, Recio A, Mick R, Clark CE, Carpenter EL, Fox KR, et al. Telomerase-specific T-cell immunity in breast cancer: effect of vaccination on tumor immunosurveillance. Cancer Res. 2007;67(21):10546–55. https://doi.org/10.1158/0008-5472.CAN-07-2765.
Kieber-Emmons T, Saha S, Pashov A, Monzavi-Karbassi B, Murali R. Carbohydrate-mimetic peptides for pan anti-tumor responses. Front Immunol. 2014;5:308. https://doi.org/10.3389/fimmu.2014.00308.
Makhoul I, Ibrahim SM, Abu-Rmaileh M, Jousheghany F, Siegel ER, Rogers LJ, et al. P10s-PADRE vaccine combined with neoadjuvant chemotherapy in ER-positive breast cancer patients induces humoral and cellular immune responses. Oncotarget. 2021;12(22):2252–2265. https://doi.org/10.18632/oncotarget.28083.
Miles D, Roché H, Martin M, Perren TJ, Cameron DA, Glaspy J, et al. Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist. 2011;16(8):1092–100. https://doi.org/10.1634/theoncologist.2010-0307.
Ibrahim NK, Murray JL, Zhou D, Mittendorf EA, Sample D, Tautchin M, et al. Survival Advantage in Patients with Metastatic Breast Cancer Receiving Endocrine Therapy plus Sialyl Tn-KLH Vaccine: Post Hoc Analysis of a Large Randomized Trial. J Cancer. 2013;4(7):577–84. https://doi.org/10.7150/jca.7028.
Rugo HS, Cortes J, Barrios CH, Cabrera P, Xu B, Huang CS, et al. GLORIA: phase III, open-label study of adagloxad simolenin/OBI-821 in patients with high-risk triple-negative breast cancer. Future Oncol. 2022. https://doi.org/10.2217/fon-2022-0812.
Fuentes-Antrás J, Guevara-Hoyer K, Baliu-Piqué M, García-Sáenz JÁ, Pérez-Segura P, Pandiella A, et al. Adoptive Cell Therapy in Breast Cancer: A Current Perspective of Next-Generation Medicine. Front Oncol. 2020;10:605633. https://doi.org/10.3389/fonc.2020.605633.
Huang Z, Dewanjee S, Chakraborty P, Jha NK, Dey A, Gangopadhyay M, et al. CAR T Cells: Engineering Immune Cells to Treat Brain Cancers and Beyond. Mol Cancer. 2023;22(1):22. https://doi.org/10.1186/s12943-022-01712-8.
Assadipour Y, Zacharakis N, Crystal JS, Prickett TD, Gartner JJ, Somerville RPT, et al. Characterization of an Immunogenic Mutation in a Patient with Metastatic Triple-Negative Breast Cancer. Clin Cancer Res. 2017;23(15):4347–53. https://doi.org/10.1158/1078-0432.CCR-16-1423.
Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 2018;24(6):724–30. https://doi.org/10.1038/s41591-018-0040-8.
Safarzadeh Kozani P, Naseri A, Mirarefin SMJ, Salem F, Nikbakht M, Evazi Bakhshi S, et al. Nanobody-based CAR-T cells for cancer immunotherapy. Biomark Res. 2022;10(1):24. https://doi.org/10.1186/s40364-022-00371-7.
Dees S, Ganesan R, Singh S, Grewal IS. Emerging CAR-T Cell Therapy for the Treatment of Triple-Negative Breast Cancer. Mol Cancer Ther. 2020;19(12):2409–21. https://doi.org/10.1158/1535-7163.MCT-20-0385.
Nasiri F, Kazemi M, Mirarefin SMJ, Mahboubi Kancha M, Ahmadi Najafabadi M, Salem F, et al. CAR-T cell therapy in triple-negative breast cancer: Hunting the invisible devil. Front Immunol. 2022;13:1018786. https://doi.org/10.3389/fimmu.2022.1018786.
Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol Res. 2017;5(12):1152–61. https://doi.org/10.1158/2326-6066.CIR-17-0189.
Garber K. Driving T-cell immunotherapy to solid tumors. Nat Biotechnol. 2018;36(3):215–9. https://doi.org/10.1038/nbt.4090.
Safarzadeh Kozani P, Safarzadeh Kozani P, Ahmadi Najafabadi M, Yousefi F, Mirarefin SMJ, Rahbarizadeh F. Recent Advances in Solid Tumor CAR-T Cell Therapy: Driving Tumor Cells From Hero to Zero? Front Immunol. 2022;13:795164. https://doi.org/10.3389/fimmu.2022.795164.
Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. CAR T cells redirected against tumor-specific antigen glycoforms: can low-sugar antigens guarantee a sweet success? Front Med. 2022;16(3):322–38. https://doi.org/10.1007/s11684-021-0901-2.
Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F, Khoshtinat Nikkhoi S. Strategies for Dodging the Obstacles in CAR T Cell Therapy. Front Oncol. 2021;11:627549. https://doi.org/10.3389/fonc.2021.627549.
Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. 2011;13(1):98–107. https://doi.org/10.3109/14653249.2010.515582.
Funding
This work was supported by funds from the National Natural Science Foundation of China (82203130, Feng Ye), the Science and Technology Program of Guangzhou (2023A04J1785, Feng Ye), the Key Research Project of Hunan Provincial Education Department (21A0270, Yuehua Li), and China Postdoctoral Science Foundation (2022M711541, Yuehua Li).
Author information
Authors and Affiliations
Contributions
Conceptualization, Hailin Tang, Tapan Behl, Yuehua Li and Saurabh Kumar Jha; data accumulation, writing, and original draft preparation: Saikat Dewanjee, Ankush Kumar, Vishakha, Zhe-Sheng Chen, Saurabh Kumar Jha and Tapan Behl; editing: Saikat Dewanjee, Zhe-Sheng Chen, Saurabh Kumar Jha and Tapan Behl; figure preparation: Niraj Kumar Jha and Saikat Dewanjee; revision: Saikat Dewanjee; supervision: Hailin Tang, Tapan Behl, Yuehua Li, Saikat Dewanjee and Saurabh Kumar Jha. All authors have read and approved the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This is a review article; data is not related to ethics approval.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ye, F., Dewanjee, S., Li, Y. et al. Advancements in clinical aspects of targeted therapy and immunotherapy in breast cancer. Mol Cancer 22, 105 (2023). https://doi.org/10.1186/s12943-023-01805-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01805-y