
Abstract
The traditional oral pharmacological therapy for type 2 diabetes mellitus (T2DM) has been based on the prescription of metformin, a biguanide, as first line antihyperglycemic agent world over. It has been demonstrated that after 3 years of treatment, approximately 50 % of diabetic patients could achieve acceptable glucose levels with monotherapy; but by 9 years this had declined to only 25 %. Therefore, the implementation of a combined pharmacological therapy acting via different pathways becomes necessary, and its combination with a compound of the sulfonylurea group was along decades the most frequently employed prescription in routine clinical practice. Meglitinides, glitazones and alpha-glucosidase inhibitors were subsequently developed, but the five mentioned groups of oral antihyperglycemic agents are associated with variable degrees of undesirable or even severe cardiovascular events. The gliptins—also called dipeptidyl peptidase 4 (DPP4) inhibitors—are an additional group of antidiabetic compounds with increasing clinical use. We review the status of the gliptins with emphasis on their capabilities to positively or negatively affect the cardiovascular system, and their potential involvement in major adverse cardiovascular events (MACE). Alogliptin, anagliptin, linagliptin, saxagliptin, sitagliptin, teneligliptin and vildagliptin are the compounds currently in clinical use. Regardless differences in chemical structure and metabolic pathways, gliptins as a group exert favorable changes in experimental models. These changes, as an almost general rule, include improved endothelial function, reduction of inflammatory markers, oxidative stress ischemia/reperfusion injury and atherogenesis. In addition, increased adiponectin levels and modest decreases in lipidemia and blood pressure were reported. In clinical settings, several trials—notably the longer one, employing sitagliptin, with a mean follow-up period of 3 years—did not show an increased risk for ischemic events. Anyway, it should be emphasized that the encouraging results from basic science were not yet translated into clinical evidence, probably due the multiple and pleiotropic enzymatic effects of DPP4 inhibition. Moreover, when employing saxagliptin, while the drug was not associated with an augmented risk for ischemic events, it should be pinpointed that the rate of hospitalization for heart failure was significantly increased. Gliptins as a group constitute a widely accepted therapy for the management of T2DM, usually as a second-line medication. Nonetheless, for the time being, a definite relationship between gliptins treatment and improved cardiovascular outcomes remains uncertain and needs yet to be proven.
Similar content being viewed by others

Review
Background
The traditional oral pharmacological therapy for type 2 diabetes mellitus (T2DM) has been based on the prescription of metformin as first line antihyperglycemic agent world over. This biguanide derivate is the most widely prescribed drug to manage glucose metabolism in individuals with T2DM and is still recommended, in conjunction with lifestyle modification, as a first line medication in the joint guidelines of the American Diabetes Association and the European Association of the Study of Diabetes [1]. It has been demonstrated that after 3 years of treatment, approximately 50 % of diabetic patients could achieve acceptable glucose levels with monotherapy; but by 9 years this had declined to only 25 % [2]. Therefore, the implementation of a combined pharmacological therapy acting via different biochemical pathways becomes necessary, and its combination with a compound of the sulfonylurea group was along decades the most frequently employed prescription in routine clinical practice [3]. While it is generally considered a relatively safe drug, increased mortality associated with the use of metformin was reported during long [4] and even short-term follow-up [5]. Appearance of lactic acidosis has been reported as well, especially in the elderly and in patients with liver failure [6, 7].
Cardiovascular derangement has been widely described for most sulfonylureas, which exert their action by closing the ATP-dependent potassium channels; this feature is accountable for both the insulinotropic and the undesirable effects on the heart. During myocardial ischemia, sulfonylureas partially inhibit channels opening, avoiding so the required hyperpolarization that protects the cell by impeding calcium influx (the so-called “ischemic preconditioning”) [8, 9]. In a population of coronary patients, data from our laboratory indicated a huge increased all-cause crude mortality over a mean 7.7-year follow-up in diabetics on combined treatment with metformin and glibenclamide; figures on mortality in this group almost quadrupled those of nondiabetic coronary patients [10]. Similar findings have been reported in a general diabetic population [11], and the increased morbidity and mortality of this combined therapy was recently confirmed in a large nationwide Danish study [12]. Glinide compounds like repaglinide—which also act by closing the ATP-dependent potassium channels—appear to be associated with a similar risk of adverse cardiovascular sequelae than sulfonylureas [13].
Regarding glitazones, these compounds have been associated with a higher risk of developing stroke, heart failure and all-cause mortality [14–17]. These excess risks were largest in people aged 65 years or older, and especially in patients treated with rosiglitazone [15].
Concerning alpha-glucosidase inhibitors, voglibose was found to improve endothelial function in patients with type 2 diabetes [18] and miglitol enhanced postprandial endothelial function in patients with acute coronary syndrome and new-onset postprandial hyperglycemia [19]. Moreover, acarbose has been postulated as a potential agent for coronary disease secondary prevention [20, 21]. Despite these promising perspectives, no unequivocal benefits have been established since in a recent large intention-to-treat analyses, acarbose was associated with a higher risk of development of any cardiovascular event, heart failure, and ischemic stroke compared to metformin [22].
As stated above, the five mentioned groups of oral antihyperglycemic agents—biguanides, sulfonylureas, meglitinides, glitazones and alpha-glucosidase inhibitors—are associated with variable degrees of undesirable or even severe cardiovascular events. Moreover, the adequate treatment in case of comorbidities is not defined [23]. Taking these facts into consideration, the Food and Drug Administration (FDA) recommended that in order to establish the safety of a new antidiabetic therapy for T2DM, the manufacturers or sponsors should demonstrate that the new drug will not result in an unacceptable increase in cardiovascular risk and the trials population should also include high risk patients. Other criteria included trials duration of at least 2 years [24]. A sixth pharmacological group, the gliflozins, constitutes a new category, and its yet unknown long-term cardiovascular outcome is currently being investigated [25]. The gliptins—also called dipeptidyl peptidase-4 (DPP4) inhibitors—are an additional group of antidiabetic compounds with increasing worldwide clinical use since the first of them, sitagliptin, got the FDA approval in 2006 [26]. In this context, the purpose of the present article is to review the status of the gliptins currently in clinical use, with special emphasis on their capabilities to positively or negatively affect the cardiovascular system, and their potential involvement in major adverse cardiovascular events (MACE).
Biochemistry
Dipeptidyl peptidases are a family of several complex proteases with similar chemical structure; the biological roles and identity of their endogenous substrates remains poorly understood for the majority of them [27]. Therefore, a cautious evaluation of the selectivity and specificity of any pharmacological compound used to inhibit DPP4 activity is required [28]. For the time being, DPP4 is the best known member of the family and acts as a membrane-anchored cell surface peptidase transmitting intracellular signals through a short intracellular tail. In humans, the DPP4 gene is located on chromosome 2 locus 2q24.3 and composed of 26 exons that encode a protein of 766 amino acids [29]. DPP4 is widely expressed in several cell types, particularly in exocrine glands and absorptive epithelia. It is mainly found in the brush borders of epithelial cells of the proximal convoluted tubules in the kidney, in the small and large intestine, prostate tissue, hepatocytes, fibroblasts and in activated leukocytes [30–33], preferentially cleaving peptide hormones containing a position two alanine or proline [27].
The incretins are gut-derived hormones, constituents of the glucagon superfamily, released in response to nutrient ingestion, mainly sugars and fat. They put forth a wide range of effects, including stimulation of pancreatic insulin secretion in a glucose-dependent manner and play a central role in local gastrointestinal and whole-body physiology. The principal incretins are the glucose-dependent insulinotropic polypeptide (GIP) and the glucagon-like peptide-1 (GLP-1), representing the endogenous physiological substrates for DPP4 activity [34, 35]. GIP is secreted from the L-cells of the distal ileum and colon and GLP-1 from the K-cells in the duodenum and jejunum [36]. GLP-1 is stronger than GIP regarding insulinotropic activity, and their biological activity is cumulative. GIP has a half life of approximately 7 min, much longer than the 2 min documented for GLP-1 [27, 37, 38]. Therefore, taking into consideration the DPP4 mechanism of action, its pharmacological inhibition by gliptines will avoid interaction with its substrates, increasing plasmatic GIP and GLP-1 concentrations and stimulating insulin biosynthesis [39].
Gliptins comprise a diverse group of compounds, which from a pharmacokinetic point of view can be broadly divided into peptidomimetics and non-peptidomimetics. Both are competitive reversible inhibitors of the DPP4 substrate acting extracellularly. The former are nitrile-containing inhibitors that act on the DPP4 substrate involving production of a reversible covalent enzyme–inhibitor complex, which gradually and slowly dissociates from the substrate leading to a lasting inhibitory activity, even after the drug has been inactivated. This phenomenon explains why these compounds act for longer than the relatively short half-lives of most of them would suggest. On the contrary, non-peptidomimetic drugs yield to a non-covalent extracellular cross-talk with residues in the catalytic site of the DPP4 substrate, resulting in a strong and immediate inhibition [40–43]. It has been suggested that these differences between compounds may also be reflected in their cardiovascular effects [44]. The differences in the metabolic pathways are also expressed in the therapeutic doses, which may range from 5 mg for saxagliptin to 100 mg for sitagliptin, and in the dosing frequency. The currently most used compounds include alogliptin [45–48], anagliptin [49–52], linagliptin [44, 53–56], saxagliptin [57–59], sitagliptin [60], teneligliptin [61] and vildagliptin [62]. Data regarding approving authority, chemistry, half life, dosage and catabolic pathway are summarized in Table 1. The research regarding an additional drug, dutogliptin, was discontinued [63], while several another compounds, notably gemigliptin [64] (already approved in South Korea) are currently at diverse stages of development and evaluation. Trelagliptin, a compound that unlike other approved agents of its class—which are usually administered once daily—can be administered once weekly, has been recently approved in Japan [65], and studies on another long-acting drug, omarigliptin, are in progress [66].
Pharmacological interactions
In daily clinical practice, gliptins are almost usually prescribed together with other antidiabetic, antihypertensive and antihyperlipidemic agents. Therefore, it is of utmost importance to establish whether potential undesirable interactions may be present. No adverse events were disclosed in the co-administration with metformin [67–69], glibenclamide [70], glitazones [71, 72], and simvastatin [73]. Moreover, in an experimental murine model, the combination with valsartan improved both pancreatic beta-cell function and insulin sensitivity, with a reduction of the inflammatory and cell stress milieu [74]. It should be mentioned that some gliptins, like linagliptin and saxagliptin may be metabolized via the CYP3A4 or CYP3A4/5 pathways, and this could result in a diminished clearance of other drugs using the same pathways. In addition, a reduction in the dose of sulfonylureas is usually recommended when a gliptin is added, since its pharmacodynamic interaction with the sulfonylurea may result in a higher risk of hypoglycemia. Anyway, this issue does not seem to be of clinical relevance [75]. As a general rule, there is no great propensity for the gliptins to be involved in any significant drug interaction with commonly prescribed antidiabetic medicines [42, 43].
Cardiovascular effects of currently used gliptins
Sitagliptin
The effects of this medication—a beta-amino acid—on cardiovascular outcomes in patients with T2DM have been recently reported by Green et al. in the Trial to Evaluate Cardiovascular Outcomes after Treatment with Sitagliptin (TECOS) [60]. This is a randomized double-blind open-label study including nearly 15,000 patients in which co-administration of either sitagliptin or placebo was implemented on top of their previous antidiabetic treatment. The included patients had T2DM and proven cardiovascular disease and were at least 50 years old, with glycated hemoglobin levels of 6.5–8.0 % when treated with stable doses of one or two oral antihyperglycemic agents (metformin, pioglitazone, or a sulfonylurea) or insulin (with or without metformin). Sitagliptin dose was of 100 mg daily, or 50 mg daily in cases of kidney dysfunction. A composite of cardiovascular death, nonfatal myocardial infarction (MI), nonfatal stroke, or hospitalization for unstable angina was established as primary cardiovascular outcome. The secondary composite cardiovascular outcome was defined as the first confirmed event of cardiovascular death, nonfatal MI, or nonfatal stroke. In addition, other secondary outcomes comprised occurrence of the individual components of the primary composite cardiovascular outcome, fatal and nonfatal MI, fatal and nonfatal stroke, death from any cause, and hospitalization for heart failure. The study showed that after a mean follow-up period of 3 years, the addition of sitagliptin to the conventional pharmacological treatment did not have a significant effect on rates of major adverse cardiovascular events or hospitalization for heart failure. This study confirmed the findings of an earlier pooled analysis of 25 randomised clinical trials totalizing—similarly to the TECOS trial—almost 15,000 patients, which did not indicate that treatment with sitagliptin increases cardiovascular risk in patients with T2DM [76].
Data from other studies reported several beneficial effects. Indices of glycemic control, such as hemoglobin A1c, glycated albumin, and 1.5-anhydro-d-glucitol were significantly improved after a 3-month treatment with sitagliptin, and serum adiponectin level was significantly increased without changes of body weight [77]. Endothelial function was improved as well [18]. It was also suggested that previous chronic treatment with sitagliptin may have cardioprotective effects in diabetic patients presenting with acute coronary syndrome [78]. Moreover, in patients who were at high risk of heart failure after acute coronary syndrome, sitagliptin exposure was not associated with an increased risk of de novo heart failure [79] or other adverse cardiovascular events [80].
Saxagliptin
Saxagliptin, a cyanopyrrolidine, was the second gliptin that obtained the FDA approval. A comprehensive study including about 16,500 patients followed for a median of 2.1 years and describing the cardiovascular outcomes of this medication was reported by Scirica et al. in the SAVOR-TIMI 53 trial [58]. Patients with T2DM with documented or at risk for cardiovascular events were randomly assigned to receive saxagliptin or placebo. The usual dose was 5 mg daily, and the primary end point a composite of cardiovascular death, MI or ischemic stroke, and the main secondary end point a composite of cardiovascular death, MI, stroke, hospitalization for unstable angina, coronary revascularization, or heart failure. The study demonstrated that saxagliptin did not increase or decrease the rate of ischemic events. Subsequently, an analysis of pooled data from 20 clinical trials comprising over 9000 patients with T2DM confirmed that the drug was not associated with an increased risk for ischemic events [57]. Anyway, it should be specially pinpointed that the rate of hospitalization for heart failure was increased [58, 81–83]—in contrast to the findings reported for sitagliptin [60, 79]. Moreover, the risk of heart failure hospitalization was augmented irrespective of age category [84], being highest among patients with elevated levels of natriuretic peptides, previous heart failure, or chronic kidney disease [85].
Linagliptin
Linagliptin is a potent gliptin with a xanthine-based molecular structure [86–88]. It inhibits DPP4 competitively and reversibly, showing a slow rate of dissociation from the active center of the DPP4 enzyme molecule, and regarding selectivity towards DPP4 in comparison to other enzymes of the DPP family, it is 40,000-fold higher towards DPP-4 than towards DPP-8 and >10,000-fold higher towards DPP-9 [89]. Linagliptin is rapidly absorbed after oral administration with a Tmax of 0.7–3 h that does not differ between healthy and T2DM subjects after single or multiple doses [90–92]. A pre-specified patient-level pooled analysis of all available double-blind, randomized, controlled trials evaluating the cardiovascular safety of the drug has been recently published [54]. The study encompassed 19 trials of ≥12 weeks’ duration, including nearly 9500 subjects receiving linagliptin versus placebo or another active treatment. Out of these patients, about 5850 received linagliptin; most of them 5 mg daily. The comparators were glimepiride, voglibose and placebo. The primary end point was time to the first occurrence of any components of the 4P-MACE composite, i.e. cardiovascular death (including fatal stroke and fatal MI), non-fatal MI (excluding silent MI), non-fatal stroke or hospitalization for unstable angina pectoris. The main limitation of this study is the mean duration of the included trials, considerably shorter than the recommended by the FDA [24], reducing so the extent of interpretations that can be made. Anyway, the incidence rate of 4P-MACE was 13.4 events per 1000 patient-years for linagliptin-treated patients compared with 18.9 in the active comparator group; therefore it should be pointed out that linagliptin was not associated with an increase in cardiovascular risk.
Two ongoing trials will provide a more definitive answer on the cardiovascular safety profile of linagliptin. The first is the CAROLINA trial [93, 94], started in 2010, that has randomized more than 6000 patients with early T2DM and predominantly medium cardiovascular risk, to treatment with either linagliptin or glimepiride. Therefore, it is the first head-to-head outcome trial of a gliptin versus an active comparator that is sufficiently powered to unveil potential differences in MACE occurrence between treatment groups [54]. The second one is the CARMELINA trial, initiated in 2013 and aimed to investigate the long term impact on cardiovascular morbidity, mortality and renal function in a selected population of 8300 patients with T2DM and renal compromise, comparing outcomes against placebo, on a background of standard of care [95]. The results of both trials are expected in 2018. For the time being, the available information regarding linagliptin and kidney function is encouraging [96–98]. In addition, it has been documented that linagliptin ameliorates cardiovascular injury in salt-sensitive hypertensive rats independently of blood glucose and blood pressure [44], and also attenuates neointima formation after vascular injury and vascular smooth muscle cells (VSMC) proliferation beyond its glucose-lowering effect [56].
Alogliptin
Alogliptin is a pyrimidinedione derivative highly selective gliptin, rapidly absorbed, with a mean time to maximum concentration (Cmax) of approximately 1–2 h [99–101]. Preliminary clinical studies reported favorable results [102, 103]. The EXAMINE trial was programmed in order to assess its cardiovascular safety in T2DM patients with acute coronary syndrome [104]. It was a multicentre, double-blind trial, into which 5380 subjects—enrolled from 49 countries—who underwent such an event event in the previous 15–90 days were randomly assigned to alogliptin 25 mg daily or placebo plus standard treatment for diabetes and cardiovascular disease prevention. The results have been recently published [105]. The pre-specified MACE endpoint was all-cause mortality, non-fatal MI, non-fatal stroke, urgent revascularization for unstable angina syndrome, and hospitalization for heart failure. Alogliptin neither increased cardiovascular morbidity or mortality, nor worsened pre-existing heart failure, including in those patients with a very recent acute coronary syndrome, after a median duration treatment of 18 months [106, 107]. In addition, assessment of N-terminal pro-BNP (NT-pro-BNP) concentration from baseline to 6 months did not reveal any significant changes.
The safety of the co-administration of alogliptin and pioglitazone has been documented in both clinical [48, 107] and experimental [108] settings. Moreover, additional beneficial effects have been described. The drug attenuates arterial inflammation and neointimal formation after injury in low-density lipoprotein receptor-deficient mice [109] and—besides the expected decrease in glucose and hemoglobin A1c—leads also to a decrease in serum lipids in humans [110]. A preliminary report indicates that in the postprandial state lipemia diminishes, and endothelial dysfunction improves in non-diabetic subjects [111]. An anti-arrhythmogenic effect was described in rabbits via augmentation of atrial angiogenesis [112].
Vildagliptin
Vildagliptin is a cyanopyrrolidine derivative [113–115]. A large retrospective meta-analysis of prospectively adjudicated cardiovascular events has been recently published, including data from 40 double-blind, randomised-controlled phase III and IV studies encompassing about 17,500 patients (mean age 57 years) receiving vildagliptin 50 mg once and twice daily, or comparators [116]. The primary endpoint was occurrence of any MACE. The duration of the trials was very variable, lasting from 12 up to 104 weeks, and the average length of treatment was about 1 year. The results indicated that the medication was not associated with an increased risk for adjudicated MACE relative to comparators. Moreover, the event rates for new onset of heart failure or hospitalization for its aggravation were relatively low (0.4 %) and similar both groups.
Another vildagliptin trial is the VISUAL study [117]. The study was aimed to compare the efficacy and safety of adding vildagliptin with sulfonylurea dose-increasing as an active comparator in patients with inadequately controlled T2DM using metformin plus sulfonylurea in a real clinical setting. The study demonstrated that the vildagliptin add-on group exhibited no clinically relevant weight gain and had a lower incidence of hypoglycemia compared with the sulfonylurea group, and the addition of vildagliptin to sulfonylurea could be considered as a treatment option prior to intensification with insulin [118].
In rodent models, vildagliptin enhanced blood flow recovery and capillary density in ischemic limbs with accompanying increases in endothelial nitric-oxide synthase [119] and prevented left ventricular hypertrophy caused by continuous beta-adrenergic stimulation by isoproterenol [120].
Anagliptin
Like saxagliptin and vildagliptin, anagliptin is a cyanopyrrolidine derivative possessing a prolylprolinenitrile scaffold [113, 121], available for clinical use in Japan since 2012. Since this compound is not generally used in countries other than Japan, there is a paucity of reports investigating its effects. The recommended dose of is 200 mg daily (100 mg bid), albeit increases in the dose up to 400 mg daily have been approved in cases in which the blood glucose-lowering effect is unsatisfactory [52].
Besides its hypoglycemic effects, a pooled analysis of data obtained from Phase III trials, the serum LDL-cholesterol, triglycerides, total cholesterol and non-high-density lipoprotein-cholesterol levels were significantly reduced after the administration of anagliptin at doses of 200 or 400 mg [122]. Moreover, it has been demonstrated in a model of apoE-deficient mice that anagliptin reduced the area of atherosclerotic lesions, suggesting thus a potential anti-atherogenic action via direct inhibition of smooth muscle cell proliferation and inflammatory reaction of monocytes [123].
Teneligliptin
Teneligliptin is a prolylthiazolidine [61] approved for the treatment of T2DM in Japan in 2012 and in South Korea in 2014 [124, 125]. A 4-week, randomized, double-blind, placebo-controlled trial showed a significantly improved 24-h blood glucose control [126], and a subsequent pooled post hoc analysis including more than 700 patients provides evidence of the safety and efficacy of a 52 weeks mean follow-up of use of teneligliptin 20 mg daily as monotherapy or in combination with sulfonylurea, glinide, biguanide or α-glucosidase inhibitor in Japanese patients with T2DM [127]. The dose can be increased up to 40 mg per day [128]. Teneligliptin was also used as an initial monotherapy for drug-naive newly diagnosed T2DM subjects. It activated beta-cell function and decreased insulin resistance, being rather effective in reducing both fasting blood glucose levels and HbA1c. However, a significant increase in uric acid concentration was observed in some patients [129].
Regarding the cardiovascular effects, it should be pinpointed that QT/QTc evaluations were performed for this compound [128]. No QT prolongations were detected with 40 mg daily of teneligliptin, which is the maximal dose in usual clinical practice. Anyway, a mild QTc transient prolongation was documented while using supraclinical dosages. Therefore, caution is needed if the drug is used for a long period or in co-administration with medications known to cause QT prolongation on their own [128]. On the other hand, teneligliptin treatment was associated with improvements in left ventricular function—particularly diastolic—and endothelial functions, as well as with an increase in serum adiponectin levels [130].
Mechanistic construal
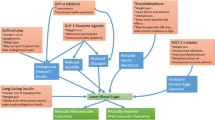
Which could be the molecular mechanisms for the favorable results of DPP4 inhibition in experimental settings? Several responses can be outlined. DPP4 is a transmembrane glycoprotein that cleaves N-terminal dipeptides from many substrates. It is ubiquitously expressed, including several growth factors, hormones, neuropeptides, and chemokines [30, 33, 131]. Using a proteomics approach, human adipocytes’ secretome has been unveiled, identifying DPP4 as a newfangled adipokine, produced particularly by fully differentiated adipocytes [132, 133]. Anyway, it should be emphasized that the main divergence with many other adipokines is that DPP4 is not directly secreted by adipocytes but released from the plasma membrane as soluble DPP4 following proteolytic cleavage [32, 134]. Soluble DPP4 is a striking activator of both mitogen-activated protein kinases (MAPK) and nuclear factor kappa B (NF-κB); these pro-inflammatory signaling pathways lead to proliferation and atherogenic transformation of VSMC [133]. A DPP-4 inhibitor exerts anti-inflammatory effects on macrophages and adipocytes, being able to suppress NF-κB activation [135]. In this context, soluble DPP4 also induces activation of extracellular-signal-regulated kinases (ERKs, a specific subset of the MAPK family) in VSMC, which was partially blocked by DPP4 inhibition [123]. The main effects of this inhibition are depicted in Fig. 1.

Schematic depiction of the main biochemical and clinical effects of DPP4 (dipeptidyl peptidase-4) inhibition. While both the increasing (green arrows) and decreasing (blue arrows) effects seem to be favorable, the overall influence on MACE (major adverse cardiovascular events) remains uncertain
DPP4 consistently impaired insulin signaling at the level of protein kinase B (the so-called Akt) in several primary cell types like adipocytes, skeletal muscle, and VSMC [132]. In addition, it has been shown that while a trend for higher DPP4 expression in visceral fat is present lean subjects, in obese subjects DPP4 is significantly higher in visceral adipose tissues than in its subcutaneous counterparts [132]. DPP4 expression in both adipose depots is significantly higher in obese compared with lean subjects, and its levels positively correlate with body mass index (BMI), subcutaneous and visceral adipocytes size, insulin, and leptin [132, 136], whereas a negative correlation with age and adiponectin was found [132, 137]. High DPP4 activity might induce subsequent accumulation of body fat and increased BMI [138], and in turn long-term obesity and lipid accumulation will lead to insulin resistance and hyperglycemia via abnormally obesity-linked down-regulation of adiponectin [139]. Moreover, an increased DPP4 production from adipocytes is observed in both type 1 and T2DM [140]. Together, all these biochemical facts represent a mechanistic construal reflecting a solid scaffold for the alleged beneficial cardiovascular outcomes obtained by inhibition of DPP4 activity.
Therefore, besides their glucose-lowering activity, gliptins have an intrinsic manifold cardiovascular impact. This impact is rooted in the fact that the presence of GLP-1 receptors in human cardiac myocytes has been demonstrated two decades ago [141] and subsequently confirmed in later years [142, 143], boosting the interest of researchers to properly establish the relevance of the gliptin-induced changes in clinical settings. As mentioned above, the changes are explained by reasonable molecular mechanisms and seem to be mainly beneficial. They include improvement of endothelial function, decrease of inflammatory markers, reduction of ischemia/reperfusion injury in experimental models, prevention of left ventricular remodeling, modest decreases in blood pressure and lipidemia, and a trend to lower MACE incidence.
Controversial issues
Unfortunately, the above mentioned favorable features are counterbalanced by other ones. For instance, in addition to inactivate the incretin hormones, DPP4 can also affect the orexigenic hormone neuropeptide Y, and its inhibition by gliptines enhances antilipolytic action in human adipose tissue leading to further accumulation of body fat [144, 145]. Additional important points represent a source of concern, such as the significantly higher rate of hospitalization for heart failure with saxagliptin reported in the SAVOR-TIMI 53 trial [58], the fact that the research regarding dutogliptin—initially considered a promising selective DPP4 inhibitor [146] was abruptly discontinued without giving any reason [63] and the finding of hyperuricemia in patients treated with teneligliptin [129]. A recent assessment of the results of the EXAMINE trial [105] points out that the rate of heart failure was increased in patients on alogliptin who had no previous history of this disorder [147]. Moreover, the co-administration of gliptins with ACE-inhibitors or angiotensin receptor blockers maybe associated with severe angioedema [148, 149], despite favorable metabolic effects in an experimental model [74].
Several explanations can be hypothesized regarding these conflicting results. In first place, gliptins are multi-target compounds, and therefore their activity is connected with the inhibition of various substrates, leading to undesirable effects [150]. Moreover, DPP4 inhibitors may exert differential effects on substrate activity in a diabetic versus a normoglycemic setting [151] and chronic treatment with gliptins exerts progressive changes in metabolic parameters beyond those detected in single-dose administration studies [152]. Finally, most of the trials were not long enough to comply with the FDA recommendations regarding cardiovascular outcomes [24].
Regardless the differences in their chemical structure and metabolic pathways, gliptins as a group constitute a widely accepted therapy for the management of T2DM, usually as a second-line medication [27, 42, 153–156]. It has neutral effects on weight [157, 158]; oral administration and low incidence of hypoglycemia are important advantages. On the other hand, while experimental and preliminary clinical studies on several surrogate end-points suggest that gliptins can exert cardioprotective actions, the results of placebo-controlled phase IV trials have been rather disappointing so far. In clinical settings, several trials—notably the longer one, with sitagliptin—did not show an increased risk for ischemic events [60], but the encouraging results from basic science were not yet translated into clinical evidence, probably due the pleiotropic enzymatic effects of DPP4 [159, 160]. The relatively rapid FDA approval of sitagliptin for clinical use has been criticized [161], post-marketing reports reveal safety aspects that need further investigation [162], and it has even been suggested to restrict the use of gliptins to Phase IV clinical trials until cardiovascular safety is clearly established [163].
Conclusion
For the time being, a definite relationship between gliptins treatment and improved cardiovascular outcomes remains uncertain and needs yet to be proven.
Abbreviations
- BMI:
-
body mass index
- DPP4:
-
dipeptidyl peptidase-4
- FDA:
-
Food and Drug Administration
- GIP:
-
glucose-dependent insulinotropic polypeptide
- GLP-1:
-
glucagon-like peptide-1
- MACE:
-
major adverse cardiovascular events
- MI:
-
myocardial infarction
- NF-κB:
-
nuclear factor kappa B
- T2DM:
-
type 2 diabetes mellitus
- VSMC:
-
vascular smooth muscle cells
References
Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, Zinman B. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2009;32:193–203.
Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin or insulin in patients with type 2 diabetes: progressive requirement for multiple therapies (UKPDS 49). UK Diabetes Prospective Study (UKPDS) Group. JAMA. 1999;281:2005–12.
Fisman EZ, Motro M, Tenenbaum A. Non-insulin antidiabetic therapy in cardiac patients: current problems and future prospects. Adv Cardiol. 2008;45:154–70.
Fisman EZ, Tenenbaum A, Benderly M, Goldbourt U, Behar S, Motro M. Antihyperglycemic treatment in diabetics with coronary disease: increased metformin-associated mortality over a 5-year follow-up. Cardiology. 1999;91:195–202.
Innerfield RJ. Metformin-associated mortality in U.S. studies. N Engl J Med. 1996;334:1611–3.
Misbin RI, Green L, Stadel BV, Gueriguian JL, Gubbi A, Fleming GA. Lactic acidosis in patients with diabetes treated with metformin. N Engl J Med. 1998;338:265–6.
Lalau JD. Lactic acidosis induced by metformin: incidence, management and prevention. Drug Saf. 2010;33:727–40.
Brady PA, Terzic A. The sulfonylurea controversy: more questions from the heart. J Am Coll Cardiol. 1998;31:950–6.
Kottenberg E, Thielmann M, Kleinbongard P, Frey UH, Heine T, Jakob H, Heusch G, Peters J. Myocardial protection by remote ischaemic pre-conditioning is abolished in sulphonylurea-treated diabetics undergoing coronary revascularisation. Acta Anaesthesiol Scand. 2014;58:453–62.
Fisman EZ, Tenenbaum A, Boyko V, Benderly M, Adler Y, Friedensohn A, Kohanovski M, Rotzak R, Schneider H, Behar S, Motro M. Oral antidiabetic treatment in patients with coronary disease: time-related increased mortality on combined glyburide/metformin therapy over a 7.7-year follow-up. Clin Cardiol. 2001;24:151–8.
Olsson J, Lindberg G, Gottsater M, Lindwall K, Sjostrand A, Tisell A, Melander A. Increased mortality in Type II diabetic patients using sulphonylurea and metformin in combination: a population-based observational study. Diabetologia. 2000;43:558–60.
Mogensen UM, Andersson C, Fosbøl EL, Schramm TK, Vaag A, Scheller NM, Torp-Pedersen C, Gislason G, Køber L. Metformin in combination with various insulin secretagogues in type 2 diabetes and associated risk of cardiovascular morbidity and mortality—a retrospective nationwide study. Diabetes Res Clin Pract. 2015;107:104–12.
Huang Y, Abdelmoneim AS, Light P, Qiu W, Simpson SH. Comparative cardiovascular safety of insulin secretagogues following hospitalization for ischemic heart disease among type 2 diabetes patients: a cohort study. J Diabetes Complicat. 2015;29:196–202.
Lu CJ, Sun Y, Muo CH, Chen RC, Chen PC, Hsu CY. Risk of stroke with thiazolidinediones: a 10-year nationwide population-based cohort study. Cerebrovasc Dis. 2013;36:145–51.
Gallagher AM, Smeeth L, Seabroke S, Leufkens HG, van Staa TP. Risk of death and cardiovascular outcomes with thiazolidinediones: a study with the general practice research database and secondary care data. PLoS One. 2011;6(12):e28157.
Filion KB, Joseph L, Boivin JF, Suissa S, Brophy JM. Thiazolidinediones and the risk of incident congestive heart failure among patients with type 2 diabetes mellitus. Pharmacoepidemiol Drug Saf. 2011;20:785–96.
Berthet S, Olivier P, Montastruc JL, Lapeyre-Mestre M. Drug safety of rosiglitazone and pioglitazone in France: a study using the French PharmacoVigilance database. BMC Clin Pharmacol. 2011;11:5.
Nakamura K, Oe H, Kihara H, Shimada K, Fukuda S, Watanabe K, Takagi T, Yunoki K, Miyoshi T, Hirata K, Yoshikawa J, Ito H. DPP-4 inhibitor and alpha-glucosidase inhibitor equally improve endothelial function in patients with type 2 diabetes: EDGE study. Cardiovasc Diabetol. 2014;13:110.
Kitano D, Chiku M, Li Y, Okumura Y, Fukamachi D, Takayama T, Hiro T, Saito S, Hirayama A. Miglitol improves postprandial endothelial dysfunction in patients with acute coronary syndrome and new-onset postprandial hyperglycemia. Cardiovasc Diabetol. 2013;12:92.
Holman RR, Bethel MA, Chan JC, Chiasson JL, Doran Z, Ge J, Gerstein H, Huo Y, McMurray JJ, Ryden L, Liyanage W, Schröder S, Tendera M, Theodorakis MJ, Tuomilehto J, Yang W, Hu D, Pan C, ACE Study Group. Rationale for and design of the Acarbose Cardiovascular Evaluation (ACE) trial. Am Heart J. 2014;168(23–9):e2.
Standl E, Theodorakis MJ, Erbach M, Schnell O, Tuomilehto J. On the potential of acarbose to reduce cardiovascular disease. Cardiovasc Diabetol. 2014;13:81.
Chang CH, Chang YC, Lin JW, Chen ST, Chuang LM, Lai MS. Cardiovascular risk associated with acarbose versus metformin as the first-line treatment in patients with type 2 diabetes: a nationwide cohort study. J Clin Endocrinol Metab. 2015;100:1121–9.
Tschöpe D, Hanefeld M, Meier JJ, Gitt AK, Halle M, Bramlage P, Schumm-Draeger PM. The role of co-morbidity in the selection of antidiabetic pharmacotherapy in type-2 diabetes. Cardiovasc Diabetol. 2013;12:62.
FDA. Guidance for industry: diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. 2008. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071627.pdf. Accessed 19 July 2015.
Zinman B, Inzucchi SE, Lachin JM, Wanner C, Ferrari R, Fitchett D, Bluhmki E, Hantel S, Kempthorne-Rawson J, Newman J, Johansen OE, Woerle HJ, Broedl UC. Rationale, design, and baseline characteristics of a randomized, placebo-controlled cardiovascular outcome trial of empagliflozin (EMPA-REG OUTCOME™). Cardiovasc Diabetol. 2014;13:102.
FDA. FDA approves new treatment for diabetes. First in a new class of diabetes drug. 2015. http://www.fda.gov//NewsEventsNewsroom/PressAnnouncements/2006/ucm108770.htm. Accessed 19 July 2015.
Drucker DJ. Dipeptidyl peptidase-4 inhibition and the treatment of type 2 diabetes preclinical biology and mechanisms of action. Diabetes Care. 2007;30:1335–43.
Lankas GR, Leiting B, Roy RS, Eiermann GJ, Beconi MG, Biftu T, Chan CC, Edmondson S, Feeney WP, He H, Ippolito DE, Kim D, Lyons KA, Ok HO, Patel RA, Petrov AN, Pryor KA, Qian X, Reigle L, Woods A, Wu JK, Zaller D, Zhang X, Zhu L, Weber AE, Thornberry NA. Dipeptidyl peptidase IV inhibition for the treatment of type 2 diabetes: potential importance of selectivity over dipeptidyl peptidases 8 and 9. Diabetes. 2005;54:2988–94.
Abbott CA, Baker E, Sutherland GR, Mc-Caughan GW. Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics. 1994;40:331–8.
Darmoul D, Lacasa M, Baricault L, Marguet D, Sapin C, Trotot P, Barbat A, Trugnan G. Dipeptidyl peptidase IV (CD 26) gene expression in enterocyte-like colon cancer cell lines HT-29 and Caco-2. Cloning of the complete human coding sequence and changes of dipeptidyl peptidase IV mRNA levels during cell differentiation. J Biol Chem. 1992;267:4824–33.
Ginés S, Mariño M, Mallol J, Canela EI, Morimoto C, Callebaut C, Hovanessian A, Casadó V, Lluis C, Franco R. Regulation of epithelial and lymphocyte cell adhesion by adenosine deaminase-CD26 interaction. Biochem J. 2002;361(Pt 2):203–9.
Simeoni L, Rufini A, Moretti T, Forte P, Aiuti A, Fantoni A. Human CD26 expression in transgenic mice affects murine T-cell populations and modifies their subset distribution. Hum Immunol. 2002;63:719–30.
Girardi AC, Fukuda LE, Rossoni LV, Malnic G, Rebouças NA. Dipeptidyl peptidase IV inhibition downregulates Na+-H+ exchanger NHE3 in rat renal proximal tubule. Am J Physiol Renal Physiol. 2008;294:F414–22.
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–57.
Cernea S, Raz I. Therapy in the early stage: incretins. Diabetes Care. 2011;34(Suppl 2):S264–71.
Mortensen K, Christensen LL, Holst JJ, Orskov C. GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regul Pept. 2003;114:189–96.
Meier JJ, Nauck MA. Incretins and the development of type 2 diabetes. Curr Diabetes Rep. 2006;6:194–201.
Holst JJ. The physiology and pharmacology of incretins in type 2 diabetes mellitus. Diabetes Obes Metab. 2008;10(Suppl 3):14–21.
Gerick J. DPP-4 inhibitors. What may be the clinical differentiators? Diabetes Res Cin Pract. 2010;90:131–40.
Vincent SH, Reed JR, Bergman AJ, Elmore CS, Zhu B, Xu S, Ebel D, Larson P, Zeng W, Chen L, Dilzer S, Lasseter K, Gottesdiener K, Wagner JA, Herman GA. Metabolism and excretion of the dipeptidyl peptidase 4 inhibitor [14C] sitagliptin in humans. Drug Metab Dispos. 2007;35:533–8.
Gupta V, Kalra S. Choosing a gliptin. Indian J Endocrinol Metab. 2011;15:298–308.
Deacon CF. Dipeptidyl peptidase-4 inhibitors in the treatment of type 2 diabetes: a comparative review. Diabetes Obes Metab. 2011;13:7–18.
Blech S, Ludwig-Schwellinger E, Gräfe-Mody EU, Withopf B, Wagner K. The metabolism and disposition of the oral dipeptidyl peptidase-4 inhibitor, linagliptin, in humans. Drug Metab Dispos. 2010;38:667–78.
Koibuchi N, Hasegawa Y, Katayama T, Toyama K, Uekawa K, Sueta D, Kusaka H, Ma M, Nakagawa T, Lin B, Kim-Mitsuyama S. DPP-4 inhibitor linagliptin ameliorates cardiovascular injury in salt-sensitive hypertensive rats independently of blood glucose and blood pressure. Cardiovasc Diabetol. 2014;13:157.
Saisho Y. Alogliptin benzoate for management of type 2 diabetes. Vasc Health Risk Manag. 2015;11:229–43.
Chen XW, He ZX, Zhou ZW, Yang T, Zhang X, Yang YX, Duan W, Zhou SF. An update on the clinical pharmacology of the dipeptidyl peptidase 4 inhibitor alogliptin used for the treatment of type 2 diabetes mellitus. Clin Exp Pharmacol Physiol. 2015. doi:10.1111/1440-1681.12469.
Kusunoki M, Sato D, Nakamura T, Oshida Y, Tsutsui H, Natsume Y, Tsutsumi K, Miyata T. The beneficial effects of the DPP-4 inhibitor Alogliptin on hemoglobin A1c and Serum lipids in Japanese patients with type 2 diabetes. Drug Res (Stuttg). 2015. doi:10.1055/s-0035-1547254.
Scheen AJ. Pharmacokinetics and clinical evaluation of the alogliptin plus pioglitazone combination for type 2 diabetes. Expert Opin Drug Metab Toxicol. 2015;11:1005–20.
Furuta S, Smart C, Hackett A, Benning R, Warrington S. Pharmacokinetics and metabolism of [14C] anagliptin, a novel dipeptidyl peptidase-4 inhibitor, in humans. Xenobiotica. 2013;43:432–42.
Watanabe YS, Yasuda Y, Kojima Y, Okada S, Motoyama T, Takahashi R, Oka M. Anagliptin, a potent dipeptidyl peptidase IV inhibitor: its single-crystal structure and enzyme interactions. J Enzyme Inhib Med Chem. 2015;1–8. doi:10.3109/14756366.2014.1002402.
Aoki K, Ijima T, Kamiyama H, Kamiko K, Terauchi Y. Anagliptin decreases serum lathosterol level in patients with type 2 diabetes: a pilot study. Expert Opin Pharmacother. 2015;16:1749–54.
Nishio S, Abe M, Ito H. Anagliptin in the treatment of type 2 diabetes: safety, efficacy, and patient acceptability. Diabetes Metab Syndr Obes. 2015;8:163–71.
Inzucchi SE, Nauck MA, Hehnke U, Woerle HJ, von Eynatten M, Henry RR. Improved glucose control with reduced hypoglycaemic risk when linagliptin is added to basal insulin in elderly patients with type 2 diabetes. Diabetes Obes Metab. 2015. doi:10.1111/dom.12490 [Epub ahead of print].
Rosenstock J, Marx N, Neubacher D, Seck T, Patel S, Woerle HJ, Johansen OE. Cardiovascular safety of linagliptin in type 2 diabetes: a comprehensive patient-level pooled analysis of prospectively adjudicated cardiovascular events. Cardiovasc Diabetol. 2015;14:57.
Pichereau S, Zhao X, Cui Y, Zhao S, Hohl K, Meinicke T, Friedrich C. Relative bioavailability study of linagliptin/metformin tablets in healthy Chinese subjects. Int J Clin Pharmacol Ther. 2015;53:582–93.
Terawaki Y, Nomiyama T, Kawanami T, Hamaguchi Y, Takahashi H, Tanaka T, Murase K, Nagaishi R, Tanabe M, Yanase T. Dipeptidyl peptidase-4 inhibitor linagliptin attenuates neointima formation after vascular injury. Cardiovasc Diabetol. 2014;13:154.
Iqbal N, Parker A, Frederich R, Donovan M, Hirshberg B. Assessment of the cardiovascular safety of saxagliptin in patients with type 2 diabetes mellitus: pooled analysis of 20 clinical trials. Cardiovasc Diabetol. 2014;13:33.
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I, SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–26.
Saine ME, Carbonari DM, Newcomb CW, Nezamzadeh MS, Haynes K, Roy JA, Cardillo S, Hennessy S, Holick CN, Esposito DB, Gallagher AM, Bhullar H, Strom BL, Lo Re V. Determinants of saxagliptin use among patients with type 2 diabetes mellitus treated with oral anti-diabetic drugs. BMC. Pharmacol Toxicol. 2015;16:8.
Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, Josse R, Kaufman KD, Koglin J, Korn S, Lachin JM, McGuire DK, Pencina MJ, Standl E, Stein PP, Suryawanshi S, Van de Werf F, Peterson ED, Holman RR, TECOS Study Group. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med. 2015;373:232–42.
Kadowaki T, Marubayashi F, Yokota S, Katoh M, Iijima H. Safety and efficacy of teneligliptin in Japanese patients with type 2 diabetes mellitus: a pooled analysis of two Phase III clinical studies. Expert Opin Pharmacother. 2015;16:971–81.
Odawara M, Yoshiki M, Sano M, Hamada I, Lukashevich V, Kothny W. Efficacy and safety of a single-pill combination of vildagliptin and metformin in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled trial. Diabetes Ther. 2015;6:17–27.
UK Medicines Information. New Drugs Online Report for dutogliptin. 2015. http://www.ukmi.nhs.uk/applications/ndo/record_view_open.asp?newDrugID=4904. Accessed 5 September 2015.
Jung E, Kim J, Kim SH, Kim S, Cho MH. Gemigliptin, a novel dipeptidyl peptidase-4 inhibitor, exhibits potent anti-glycation properties in vitro and in vivo. Eur J Pharmacol. 2014;744:98–102.
Trelagliptin McKeage K. First global approval. Drugs. 2015;75:1161–4.
Biftu T, Sinha-Roy R, Chen P, Qian X, Feng D, Kuethe JT, Scapin G, Gao YD, Yan Y, Krueger D, Bak A, Eiermann G, He J, Cox J, Hicks J, Lyons K, He H, Salituro G, Tong S, Patel S, Doss G, Petrov A, Wu J, Xu SS, Sewall C, Zhang X, Zhang B, Thornberry NA, Weber AE. Omarigliptin (MK-3102): a novel long-acting DPP-4 inhibitor for once-weekly treatment of type 2 diabetes. J Med Chem. 2014;57:3205–12.
Herman GA, Bergman A, Yi B, Kipnes M. Tolerability and pharmacokinetics of metformin and the dipeptidyl peptidase-4 inhibitor sitagliptin when co-administered in patients with type 2 diabetes. Curr Med Res Opin. 2006;22:1939–47.
He YL, Sabo R, Picard F, et al. Study of the pharmacokinetic interaction of vildagliptin and metformin in patients with type 2 diabetes. Curr Med Res Opin. 2009;25:1265–72.
Berndt-Zipfel C, Michelson G, Dworak M, Mitry M, Löffler A, Pfützner A, Forst T. Vildagliptin in addition to metformin improves retinal blood flow and erythrocyte deformability in patients with type 2 diabetes mellitus—results from an exploratory study. Cardiovasc Diabetol. 2013;12:59.
Mistry GC, Bergman AJ, Zheng W, Hreniuk D, Zinny MA, Gottesdiener KM, Wagner JA, Herman GA, Ruddy M. Sitagliptin, an dipeptidyl peptidase-4 inhibitor, does not alter the pharmacokinetics of the sulphonylurea, glyburide, in healthy subjects. Br J Clin Pharmacol. 2008;66:36–42.
Karim A, Laurent A, Munsaka M, Wann E, Fleck P, Mekki Q. Coadministration of pioglitazone or glyburide and alogliptin: pharmacokinetic drug interaction assessment in healthy participants. J Clin Pharmacol. 2009;49:1210–9.
Mistry GC, Bergman AJ, Luo WL, Cilissen C, Haazen W, Davies MJ, Gottesdiener KM, Wagner JA, Herman GA. Multiple-dose administration of sitagliptin, a dipeptidyl peptidase-4 inhibitor, does not alter the single dose pharmacokinetics of rosiglitazone in healthy subjects. J Clin Pharmacol. 2007;47:159–64.
Graefe-Mody U, Huettner S, Stähle H, Ring A, Dugi KA. Effect of linagliptin (BI 1356) on the steady-state pharmacokinetics of simvastatin. Int J Clin Pharmacol Ther. 2010;48:367–74.
Miyagawa K, Kondo T, Goto R, Matsuyama R, Ono K, Kitano S, Kawasaki S, Igata M, Kawashima J, Matsumura T, Motoshima H, Araki E. Effects of combination therapy with vildagliptin and valsartan in a mouse model of type 2 diabetes. Cardiovasc Diabetol. 2013;12:160.
Scheen AJ. Dipeptidylpeptidase-4 inhibitors (gliptins): focus on drug-drug interactions. Clin Pharmacokinet. 2010;49:573–88.
Engel SS, Golm GT, Shapiro D, Davies MJ, Kaufman KD, Goldstein BJ. Cardiovascular safety of sitagliptin in patients with type 2 diabetes mellitus: a pooled analysis. Cardiovasc Diabetol. 2013;12:3.
Hibuse T, Maeda N, Kishida K, Kimura T, Minami T, Takeshita E, Hirata A, Nakagawa Y, Kashine S, Oka A, Hayashi M, Nishizawa H, Funahashi T, Shimomura I. A pilot 3-month sitagliptin treatment increases serum adiponectin level in Japanese patients with type 2 diabetes mellitus—a randomized controlled trial START-J study. Cardiovasc Diabetol. 2014;13:96.
Leibovitz E, Gottlieb S, Goldenberg I, Gevrielov-Yusim N, Matetzky S, Gavish D. Sitagliptin pretreatment in diabetes patients presenting with acute coronary syndrome: results from the Acute Coronary Syndrome Israeli Survey (ACSIS). Cardiovasc Diabetol. 2013;12:53.
Eurich DT, Weir DL, Simpson SH, Senthilselvan A, McAlister FA. Risk of new-onset heart failure in patients using sitagliptin: a population-based cohort study. Diabet Med. 2015. doi:10.1111/dme.12867 [Epub ahead of print].
Wang SH, Chen DY, Lin YS, Mao CT, Tsai ML, Hsieh MJ, Chou CC, Wen MS, Wang CC, Hsieh IC, Hung KC, Chen TH. Cardiovascular outcomes of sitagliptin in type 2 diabetic patients with acute myocardial infarction, a population-based cohort study in Taiwan. PLoS One. 2015;10(6):e0131122. doi:10.1371/journal.pone.0131122 eCollection 2015.
Udell JA, Bhatt DL, Braunwald E, Cavender MA, Mosenzon O, Steg PG, Davidson JA, Nicolau JC, Corbalan R, Hirshberg B, Frederich R, Im K, Umez-Eronini AA, He P, McGuire DK, Leiter LA, Raz I, Scirica BM, SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes and moderate or severe renal impairment: observations from the SAVOR-TIMI 53 Trial. Diabetes Care. 2015;38:696–705.
Wang KL, Liu CJ, Chao TF, Huang CM, Wu CH, Chen SJ, Yeh CM, Chen TJ, Lin SJ, Chiang CE. Sitagliptin and the risk of hospitalization for heart failure: a population-based study. Int J Cardiol. 2014;177:86–90.
Gilbert RE, Krum H. Heart failure in diabetes: effects of anti-hyperglycaemic drug therapy. Lancet. 2015;385:2107–17.
Leiter LA, Teoh H, Braunwald E, Mosenzon O, Cahn A, Kumar KM, Smahelova A, Hirshberg B, Stahre C, Frederich R, Bonnici F, Scirica BM, Bhatt DL, Raz I, SAVOR-TIMI 53 Steering Committee and Investigators. Efficacy and safety of saxagliptin in older participants in the SAVOR-TIMI 53 trial. Diabetes Care. 2015;38:1145–53.
Scirica BM, Braunwald E, Raz I, Cavender MA, Morrow DA, Jarolim P, Udell JA, Mosenzon O, Im K, Umez-Eronini AA, Pollack PS, Hirshberg B, Frederich R, Lewis BS, McGuire DK, Davidson J, Steg PG, Bhatt DL, SAVOR-TIMI 53 Steering Committee and Investigators. Heart failure, saxagliptin, and diabetes mellitus: observations from the SAVOR-TIMI 53 randomized trial. Circulation. 2014;130:1579–88.
Gallwitz B. Emerging DPP-4 inhibitors: focus on linagliptin for type 2 diabetes. Diabetes Metab Syndr Obes Targets Ther. 2013;6:1–9.
Guedes EP, Hohl A, de Melo TG, Lauand F. Linagliptin: farmacology, efficacy and safety in type 2 diabetes treatment. Diabetol Metab Syndr. 2013;5:25.
Retlich S, Duval V, Graefe-Mody U, Friedrich C, Patel S, Jaehde U, Staab A. Population pharmacokinetics and pharmacodynamics of Linagliptin in patients with type 2 diabetes mellitus. Clin Pharmacokinet. 2015;54:737–50.
Thomas L, Eckhardt M, Langkopf E, Tadayyon M, Himmelsbach F, Mark M. (R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methylquinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J Pharmacol Exp Ther. 2008;325:175–82.
He YL, Sadler BM, Sabo R. The absolute oral bioavailability and population-based pharmacokinetic modelling of a novel dipeptidylpeptidase-IV inhibitor, vildagliptin, in healthy volunteers. Clin Pharmacokinet. 2007;46:787–802.
Herman GA, Stein PP, Thornberry NA, Wagner JA. Dipeptidyl peptidase-4 inhibitors for the treatment of type 2 diabetes: focus on sitagliptin. Clin Pharmacol Ther. 2007;81:761–7.
Deacon CF, Holst JJ. Linagliptin, a xanthine-based dipeptidyl peptidase-4 inhibitor with an unusual profile for the treatment of type 2 diabetes. Expert Opin Investig Drugs. 2010;19:133–40.
Rosenstock J, Marx N, Kahn SE, Zinman B, Kastelein JJ, Lachin JM, Bluhmki E, Patel S, Johansen OE, Woerle HJ. Cardiovascular outcome trials in type 2 diabetes and the sulphonylurea controversy: Rationale for the active-comparator CAROLINA trial. Diab Vasc Dis Res. 2013;10:289–301.
Marx N, Rosenstock J, Kahn SE, Zinman B, Kastelein JJ, Lachin JM, Espeland MA, Bluhmki E, Mattheus M, Ryckaert B, Patel S, Johansen OE, Woerle HJ. Design and baseline characteristics of the CARdiovascular outcome trial of LINAgliptin versus glimepiride in type 2 diabetes: CAROLINA®. Diab Vasc Dis Res. 2015;12:164–74.
FDA doctor. Clinical Trial CARMELINA Study with Linagliptin in Patients with Type II Diabetes. 2015. http://fdadoctor.com/clinical-trial-carmelina-study-with-linagliptin-in-patients-with-type-ii-diabetes/. Accessed 27 August 2015.
Groop P-H, Cooper ME, Perkovic V, Emser A, Woerle H-J, von Eynatten M. Linagliptin lowers albuminuria on top of recommended standard treatment in patients with type 2 diabetes and renal dysfunction. Diabetes Care. 2013;36:3460–8.
Von Eynatten M, Gong Y, Emser A, Woerle H-J. Efficacy and safety of linagliptin in type 2 diabetes subjects at high risk for renal and cardiovascular disease: a pooled analysis of six phase III clinical trials. Cardiovasc Diabetol. 2013;12:60.
McGill JB, Sloan L, Newman J, et al. Long-term efficacy and safety of linagliptin in patients with type 2 diabetes and severe renal impairment: a 1-year, randomized, double-blind, placebo-controlled study. Diabetes Care. 2013;36:237–44.
Covington P, Christopher R, Davenport M, Fleck P, Karim A. Pharmacokinetic, pharmacodynamic, and tolerability profiles of the dipeptidyl peptidase-4 inhibitor alogliptin: a randomized, double-mind, placebo-controlled, multiple-dose study in adult patients with type 2 diabetes. Clin Ther. 2008;30:499–512.
Jarvis CI, Cabrera A, Charron D. Alogliptin: a new dipeptidyl peptidase-4 inhibitor for type 2 diabetes mellitus. Ann Pharmacother. 2013;47:1532–9.
Ndefo UA, Okoli O, Erowele G. Alogliptin: a new dipeptidyl peptidase-4 inhibitor for the management of type 2 diabetes mellitus. Am J Health Syst Pharm. 2014;71:103–9.
Pratley RE. Alogliptin: a new, highly selective dipeptidyl peptidase-4 inhibitor for the treatment of type 2 diabetes. Expert Opin Pharmacother. 2009;10:503–12.
Scott LJ. Alogliptin: a review of its use in the management of type 2 diabetes mellitus. Drugs. 2010;70(2051–72):58.
White WB, Bakris GL, Bergenstal RM, Cannon CP, Cushman WC, Fleck P, Heller S, Mehta C, Nissen SE, Perez A, Wilson C, Zannad F. EXamination of cArdiovascular outcoMes with alogliptIN versus standard of carE in patients with type 2 diabetes mellitus and acute coronary syndrome (EXAMINE): a cardiovascular safety study of the dipeptidyl peptidase 4 inhibitor alogliptin in patients with type 2 diabetes with acute coronary syndrome. Am Heart J. 2011;162:620–6.
Zannad F, Cannon CP, Cushman WC, Bakris GL, Menon V, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Lam H, White WB, EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067–76.
Keating GM. Alogliptin: a review of its use in patients with type 2 diabetes mellitus. Drugs. 2015;75:777–96.
Kaku K, Katou M, Igeta M, Ohira T, Sano H. Efficacy and safety of pioglitazone added to alogliptin in Japanese patients with type 2 diabetes mellitus: a multicenter, randomized, double-blind, parallel-group, comparative study. Diabetes Obes Metab. 2015. doi:10.1111/dom.12555 [Epub ahead of print].
Cai Y, Lydic TA, Turkette T, Reid GE, Olson LK. Impact of alogliptin and pioglitazone on lipid metabolism in islets of prediabetic and diabetic Zucker Diabetic Fatty rats. Biochem Pharmacol. 2015;95:46–57.
Akita K, Isoda K, Shimada K, Daida H. Dipeptidyl-peptidase-4 inhibitor, alogliptin, attenuates arterial inflammation and neointimal formation after injury in low-density lipoprotein (LDL) receptor-deficient mice. J Am Heart Assoc. 2015;4(3):e001469. doi:10.1161/JAHA.114.001469.
Kusunoki M, Sato D, Nakamura T, Oshida Y, Tsutsui H, Natsume Y, Tsutsumi K, Miyata T. The Beneficial Effects of the DPP-4 Inhibitor Alogliptin on Hemoglobin A1c and Serum Lipids in Japanese Patients with Type 2 Diabetes. Drug Res (Stuttg). 2015. [Epub ahead of print].
Noda Y, Miyoshi T, Oe H, Ohno Y, Nakamura K, Toh N, Kohno K, Morita H, Kusano K, Ito H. Alogliptin ameliorates postprandial lipemia and postprandial endothelial dysfunction in non-diabetic subjects: a preliminary report. Cardiovasc Diabetol. 2013;12:8.
Yamamoto T, Shimano M, Inden Y, Takefuji M, Yanagisawa S, Yoshida N, Tsuji Y, Hirai M, Murohara T. Alogliptin, a dipeptidyl peptidase-4 inhibitor, regulates the atrial arrhythmogenic substrate in rabbits. Heart Rhythm. 2015;12:1362–9.
Peters JU. 11 years of cyanopyrrolidines as DPP-IV inhibitors. Curr Top Med Chem. 2007;7:579–95.
Li C, Lu W, Lu C, Xiao W, Shen X, Huang J, Liu G, Tang Y. Identification of diverse dipeptidyl peptidase IV inhibitors via structure-based virtual screening. J Mol Model. 2012;18:4033–42.
Fukushima H, Hiratate A, Takahashi M, Saito-Hori M, Munetomo E, Kitano K, Saito H, Takaoka Y, Yamamoto K. Synthesis and structure-activity relationships of potent 1-(2-substituted-aminoacetyl)-4-fluoro-2-cyanopyrrolidine dipeptidyl peptidase IV inhibitors. Chem Pharm Bull (Tokyo). 2008;56:1110–7.
McInnes G, Evans M, Del Prato S, Stumvoll M, Schweizer A, Lukashevich V, Shao Q, Kothny W. Cardiovascular and heart failure safety profile of vildagliptin: a meta-analysis of 17,000 patients. Diabetes Obes Metab. 2015. doi:10.1111/dom.12548 [Epub ahead of print].
Hong AR, Lee J, Ku EJ, Hwangbo Y, Kim KM, Moon JH, Choi SH, Jang HC, Lim S. Comparison of vildagliptin as an add-on therapy and sulfonylurea dose-increasing therapy in patients with inadequately controlled type 2 diabetes using metformin and sulfonylurea (VISUAL study): a randomized trial. Diabetes Res Clin Pract. 2015;109:141–8.
Forst T, Koch C, Dworak M. Vildagliptin versus insulin in patients with type 2 diabetes mellitus inadequately controlled with sulfonylurea: results from a randomized, 24 week study. Curr Med Res Opin. 2015;31:1079–84.
Ishii M, Shibata R, Kondo K, Kambara T, Shimizu Y, Tanigawa T, Bando YK, Nishimura M, Ouchi N, Murohara T. Vildagliptin stimulates endothelial cell network formation and ischemia-induced revascularization via an endothelial nitric-oxide synthase-dependent mechanism. J Biol Chem. 2014;289:27235–45.
Miyoshi T, Nakamura K, Yoshida M, Miura D, Oe H, Akagi S, Sugiyama H, Akazawa K, Yonezawa T, Wada J, Ito H. Effect of vildagliptin, a dipeptidyl peptidase 4 inhibitor, on cardiac hypertrophy induced by chronic beta-adrenergic stimulation in rats. Cardiovasc Diabetol. 2014;13:43.
Kondo T, Sugimoto I, Nekado T, Ochi K, Ohtani T, Tajima Y, Yamamoto S, Kawabata K, Nakai H, Toda M. Design and synthesis of long-acting inhibitors of dipeptidyl peptidase IV. Bioorg Med Chem. 2007;15:2715–35.
Kakuda H, Kobayashi J, Kakuda M, Yamakawa J, Takekoshi N. The effect of anagliptin treatment on glucose metabolism and lipid metabolism, and oxidative stress in fasting and postprandial states using a test meal in Japanese men with type 2 diabetes. Endocrine. 2015;48:1005–9.
Ervinna N, Mita T, Yasunari E, Azuma K, Tanaka R, Fujimura S, Sukmawati D, Nomiyama T, Kanazawa A, Kawamori R, Fujitani Y, Watada H. Anagliptin, a DPP-4 inhibitor, suppresses proliferation of vascular smooth muscles and monocyte inflammatory reaction and attenuates atherosclerosis in male apo E-deficient mice. Endocrinology. 2013;154:1260–70.
Morishita R, Nakagami H. Teneligliptin: expectations for its pleiotropic action. Expert Opin Pharmacother. 2015;16:417–26.
Goda M, Kadowaki T. Teneligliptin for the treatment of type 2 diabetes. Drugs Today (Barc). 2013;49:615–29.
Eto T, Inoue S, Kadowaki T. Effects of once-daily teneligliptin on 24-h blood glucose control and safety in Japanese patients with type 2 diabetes mellitus: a 4-week, randomized, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2012;14:1040–6.
Kadowaki T, Marubayashi F, Yokota S, Katoh M, Iijima H. Safety and efficacy of teneligliptin in Japanese patients with type 2 diabetes mellitus: a pooled analysis of two Phase III clinical studies. Expert Opin Pharmacother. 2015;16(7):971–81. doi:10.1517/14656566.2015.1032249 Epub 2015 Apr 10.
Kishimoto M. Teneligliptin: a DPP-4 inhibitor for the treatment of type 2 diabetes. Diabetes Metab Syndr Obes. 2013;6:187–95.
Kutoh E, Hirate M, Ikeno Y. Teneligliptin as an initial therapy for newly diagnosed, drug naive subjects with type 2 diabetes. J Clin Med Res. 2014;6:287–94.
Hashikata T, Yamaoka-Tojo M, Kakizaki R, Nemoto T, Fujiyoshi K, Namba S, Kitasato L, Hashimoto T, Kameda R, Maekawa E, Shimohama T, Tojo T, Ako J. Teneligliptin improves left ventricular diastolic function and endothelial function in patients with diabetes. Heart Vessels. 2015. doi:10.1007/s00380-015-0724-7.
Yazbeck R, Howarth GS, Abbott CA. Dipeptidyl peptidase inhibitors, an emerging drug class for inflammatory disease? Trends Pharmacol Sci. 2009;30:600–7.
Lamers D, Famulla S, Wronkowitz N, Hartwig S, Lehr S, Ouwens DM, Eckardt K, Kaufman JM, Ryden M, Müller S, Hanisch FG, Ruige J, Arner P, Sell H, Eckel J. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60:1917–25.
Wronkowitz N, Görgens SW, Romacho T, Villalobos LA, Sánchez-Ferrer CF, Peiró C, Sell H, Eckel J. Soluble DPP4 induces inflammation and proliferation of human smooth muscle cells via protease-activated receptor 2. Biochim Biophys Acta. 2014;1842:1613–21.
Ikushima H, Munakata Y, Iwata S, Ohnuma K, Kobayashi S, Dang NH, Morimoto C. Soluble CD26/dipeptidyl peptidase IV enhances transendothelial migration via its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cell Immunol. 2002;215:106–10.
Shinjo T, Nakatsu Y, Iwashita M, Sano T, Sakoda H, Ishihara H, Kushiyama A, Fujishiro M, Fukushima T, Tsuchiya Y, Kamata H, Nishimura F, Asano T. DPP-IV inhibitor anagliptin exerts anti-inflammatory effects on macrophages, adipocytes, and mouse livers by suppressing NF-κB activation. Am J Physiol Endocrinol Metab. 2015;309(3):E214–23.
Rüter J, Hoffmann T, Demuth HU, Moschansky P, Klapp BF, Hildebrandt M. Evidence for an interaction between leptin, T cell costimulatory antigens CD28, CTLA-4 and CD26 (dipeptidyl peptidase IV) in BCG-induced immune responses of leptin- and leptin receptor-deficient mice. Biol Chem. 2004;385:537–41.
Aso Y, Terasawa T, Kato K, Jojima T, Suzuki K, Iijima T, Kawagoe Y, Mikami S, Kubota Y, Inukai T, Kasai K. The serum level of soluble CD26/dipeptidyl peptidase 4 increases in response to acute hyperglycemia after an oral glucose load in healthy subjects: association with high-molecular weight adiponectin and hepatic enzymes. Transl Res. 2013;162:309–16.
Kirino Y, Sei M, Kawazoe K, Minakuchi K, Sato Y. Plasma dipeptidyl peptidase 4 activity correlates with body mass index and the plasma adiponectin concentration in healthy young people. Endocr J. 2012;59:949–53.
Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–51.
Das SS, Hayashi H, Sato T, Yamada R, Hiratsuka M, Hirasawa N. Regulation of dipeptidyl peptidase 4 production in adipocytes by glucose. Diabetes Metab Syndr Obes. 2014;7:185–94.
Bullock BP, Heller RS, Habener JF. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology. 1996;137:2968–78.
Luque MA, González N, Márquez L, Acitores A, Redondo A, Morales M, Valverde I, Villanueva-Peñacarrillo ML. Glucagon-like peptide-1 (GLP-1) and glucose metabolism in human myocytes. J Endocrinol. 2002;173:465–73.
Pyke C, Heller RS, Kirk RK, Ørskov C, Reedtz-Runge S, Kaastrup P, Hvelplund A, Bardram L, Calatayud D, Knudsen LB. GLP-1 receptor localization in monkey and human tissue: novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. 2014;155:1280–90.
Kos K, Harte AL, James S, Snead DR, O’Hare JP, McTernan PG, Kumar S. Secretion of neuropeptide Y in human adipose tissue and its role in maintenance of adipose tissue mass. Am J Physiol Endocrinol Metab. 2007;293:E1335–40.
Kos K, Baker AR, Jernas M, Harte AL, Clapham JC, O’Hare JP, Carlsson L, Kumar S, McTernan PG. DPP-IV inhibition enhances the antilipolytic action of NPY in human adipose tissue. Diabetes Obes Metab. 2009;11:285–92.
Pattzi HM, Pitale S, Alpizar M, Bennett C, O’Farrell AM, Li J, Cherrington JM. Guler HP; PHX1149-PROT202 Study Group. Dutogliptin, a selective DPP4 inhibitor, improves glycaemic control in patients with type 2 diabetes: a 12-week, double-blind, randomized, placebo-controlled, multicentre trial. Diabetes Obes Metab. 2010;12:348–55.
Standl E, Schnell O. DPP-4 inhibitors and risk of heart failure EXAMINEd. Lancet. 2015;385:2022–4.
Brown NJ, Byiers S, Carr D, Maldonado M, Warner BA. Dipeptidyl peptidase-IV inhibitor use associated with increased risk of ACE inhibitor-associated angioedema. Hypertension. 2009;54:516–23.
Gosmanov AR, Fontenot EC. Sitagliptin-associated angioedema. Diabetes Care. 2012;35:e60.
Costante R, Stefanucci A, Carradori S, Novellino E, Mollica A. DPP-4 inhibitors: a patent review (2012–2014). Expert Opin Ther Pat. 2015;25:209–36.
Hansotia T, Baggio LL, Delmeire D, Hinke SA, Yamada Y, Tsukiyama K, Seino Y, Holst JJ, Schuit F, Drucker DJ. Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes. 2004;53:1326–35.
Pospisilik JA, Stafford SG, Demuth HU, Brownsey R, Parkhouse W, Finegood DT, McIntosh CH, Pederson RA. Long-term treatment with the dipeptidyl peptidase IV inhibitor P32/98 causes sustained improvements in glucose tolerance, insulin sensitivity, hyperinsulinemia, and β-cell glucose responsiveness in VDF (fa/fa) Zucker rats. Diabetes. 2002;51:943–50.
Pathak R, Bridgeman MB. Dipeptidyl peptidase-4 (DPP-4) inhibitors in the management of diabetes. Pharm Ther. 2010;35:509–13.
Crepaldi G, Carruba M, Comaschi M, Del Prato S, Frajese G, Paolisso G. Dipeptidyl peptidase 4 (DPP-4) inhibitors and their role in Type 2 diabetes management. J Endocrinol Invest. 2007;30:610–4.
Godinho R, Mega C, Teixeira-de-Lemos E, Carvalho E, Teixeira F, Fernandes R, Reis F. The Place of dipeptidyl peptidase-4 inhibitors in type 2 diabetes therapeutics: a “me too” or “the special one” antidiabetic class? J Diabetes Res. 2015;2015:806979. doi:10.1155/2015/806979 Epub 2015 May 17.
Craddy P, Palin HJ, Johnson KI. Comparative effectiveness of dipeptidylpeptidase-4 inhibitors in type 2 diabetes: a systematic review and mixed treatment comparison. Diabetes Ther. 2014;5:1–41.
Amori RE, Lau J, Pittas AG. Efficacy and safety of incretin therapy in type 2 diabetes: systematic review and meta-analysis. JAMA. 2007;298:194–200.
Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE, Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care. 2007;30:1979–87.
Röhrborn D, Wronkowitz N, Eckel J. DPP4 in diabetes. Front Immunol. 2015;6:386. doi:10.3389/fimmu.2015.00386.
Fadini GP, Albiero M, Avogaro A. Direct effects of DPP-4 inhibition on the vasculature. Reconciling basic evidence with lack of clinical evidence. Vasc Pharmacol. 2015. doi:10.1016/j.vph.2015.08.004 [Epub ahead of print].
Nathan DM. Finding new treatments for diabetes—how many, how fast… how good? N Engl J Med. 2007;356:437–40.
Filippatos TD, Athyros VG. Elisaf MS The pharmacokinetic considerations and adverse effects of DPP-4 inhibitors. Expert Opin Drug Metab Toxicol. 2014;10:787–812.
Doggrell SA, Dimmitt SB. Gliptins—do they increase cardiovascular risk or benefit? Expert Opin Drug Saf. 2014;13:675–80.
Authors’ contributions
EZF and AT have equally contributed in the conception and drafting of the manuscript. Both authors read and approved the final manuscript.
Acknowledgements
This work was supported in part by the Cardiovascular Diabetology Research Foundation (RA 58-040-684-1), Holon, Israel.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fisman, E.Z., Tenenbaum, A. Antidiabetic treatment with gliptins: focus on cardiovascular effects and outcomes. Cardiovasc Diabetol 14, 129 (2015). https://doi.org/10.1186/s12933-015-0294-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-015-0294-0