
Abstract
Background
The overwhelming majority of dose-escalation clinical trials use methods that seek a maximum tolerable dose, including rule-based methods like the 3+3, and model-based methods like CRM and EWOC. These methods assume that the incidences of efficacy and toxicity always increase as dose is increased. This assumption is widely accepted with cytotoxic therapies. In recent decades, however, the search for novel cancer treatments has broadened, increasingly focusing on inhibitors and antibodies. The rationale that higher doses are always associated with superior efficacy is less clear for these types of therapies.
Methods
We extracted dose-level efficacy and toxicity outcomes from 115 manuscripts reporting dose-finding clinical trials in cancer between 2008 and 2014. We analysed the outcomes from each manuscript using flexible non-linear regression models to investigate the evidence supporting the monotonic efficacy and toxicity assumptions.
Results
We found that the monotonic toxicity assumption was well-supported across most treatment classes and disease areas. In contrast, we found very little evidence supporting the monotonic efficacy assumption.
Conclusions
Our conclusion is that dose-escalation trials routinely use methods whose assumptions are violated by the outcomes observed. As a consequence, dose-finding trials risk recommending unjustifiably high doses that may be harmful to patients. We recommend that trialists consider experimental designs that allow toxicity and efficacy outcomes to jointly determine the doses given to patients and recommended for further study.
Similar content being viewed by others


Background
A goal of dose-finding clinical trials is to evaluate outcomes under a set of investigational doses. A common general approach starts by giving a relatively low dose to a small cohort of patients. The outcomes of this cohort affect the dose given to the next. For instance, if no unacceptable toxic reactions are seen in this initial cohort, the next cohort is likely to be given a higher dose. However, if outcomes are adverse, the next cohort might be given the same dose or a lower dose, or the trial might be halted altogether. This sequential and adaptive approach continues until the experimental design identifies a suitable dose. This pattern of starting low and seeking to escalate dose justifies the descriptor dose-escalation trials.
The most common approach [1, 2] in dose-escalation trials is the family of rule-based A+B designs, the most famous example of which is the perennial 3+3 design [3]. It evaluates doses in cohorts of three patients, using a set of rules to escalate dose so long as an unacceptably high incidence of dose-limiting toxicity (DLT) is not seen.
The main alternative class of dose-escalation methodology comprises the so-called model-based designs. These use statistical models to estimate the dose-event curve. Whilst model-based methods are used far less frequently than rule-based methods, two designs that have seen relatively wide use in recent years [2] are the continual reassessment method (CRM) [4] and the escalation with overdose control (EWOC) method [5].
Biostatisticians have encouraged trialists to shift from rule-based to model-based methods, largely because they offer better performance [6–8]. The two approaches, however, share some fundamental assumptions. Firstly, they each assume that the probability of DLT increases as dose is increased. This reflects the toxicologists’ adage the dose makes the poison, a rule that is generally accepted without challenge. The 3+3, CRM and EWOC designs all select doses based only on binary DLT outcomes in pursuit of the maximum tolerable dose (MTD), the highest dose with toxicity probability less than some critical pre-specified value. So long as two doses are deemed tolerable, the higher dose is favoured. Efficacy outcomes like response or survival are not formally used in dose-selection decisions. There are variants of the CRM design [9, 10] and other statistical approaches [11–13] for dose-finding by co-primary toxicity and efficacy outcomes. However, these have experienced comparatively little use [2, 14].
In dose-finding trials, investigators seek to identify tolerable and efficacious doses. The reliance solely on a toxicity outcome in the majority of dose-finding trials dictates that an assumption is made about the efficacy outcome.
The assumption is that higher doses are always more efficacious.
We refer to this as the monotonic efficacy assumption. In MTD-seeking trials, investigators will escalate dose without formal reference to an efficacy outcome. When the monotonic efficacy assumption holds, the MTD maximises the expectation of efficacy for a specified risk of toxicity. However, a plausible way in which the monotonic efficacy assumption can be violated is when the probability of efficacy plateaus at some critical dose. Escalation beyond this point exposes patients to a greater risk of toxicity for no accompanying increase in the probability of efficacy. As such, the monotonic efficacy assumption is rather more controversial than the monotonic toxicity assumption.
Monotonic efficacy has a plausible rationale in the treatments that have formed the backbone of anti-cancer therapies for decades. Cytotoxic treatments like chemotherapy damage tumours and healthy tissue alike. The presence of toxicity is a sign that anti-tumour activity is probably happening as well. In this setting where toxicity and efficacy are broadly accepted to be coincident, the use of dose escalation designs became commonplace. In recent years, however, numerous targeted therapies, immunotherapies and cell therapies have been approved for use against cancer. In a recent systematic review of dose-finding methodologies used between 2008 and 2014, Chiuzan et al. [2] found that over half of the trials investigated a targeted therapy or immunotherapy and the overwhelming majority used an MTD-seeking method. With these novel treatment classes, the rationale for assuming that efficacy always increases in dose is less clear.
There are notable instances in the literature where a monotonic dose-efficacy relationship has not been observed. For instance, a major recent success in the development of novel anti-cancer drugs has been the PD-1 blockade antibody, pembrolizumab. A phase I trial investigated pembrolizumab doses of 1 mg/kg, 3 mg/kg and 10 mg/kg every 2 weeks, and 2 mg/kg and 10 mg/kg every 3 weeks in 30 patients with various malignancies [15]. Large expansion cohorts in non-small-cell lung cancer (NSCLC) further evaluated 495 patients at doses 2 mg/kg or 10 mg/kg every 3 weeks or 10 mg/kg every 2 weeks [16]. Subsequently, a phase III trial that contributed to a licensing application in NSCLC randomised 345 patients to pembrolizumab 2 mg/kg, 346 to pembrolizumab 10 mg/kg, and compared each of these experimental arms to a control arm comprising 343 patients randomised to docetaxel [17]. Despite the wide range of doses investigated in large sample sizes, the phase III trial observed very similar overall survival and RECIST response outcomes in the two pembrolizumab doses, with each producing materially better outcomes than the control arm. The drug was subsequently licensed at 200 mg (i.e. not adjusted for patient weight) every 3 weeks, reflecting the absence of extra efficacy at higher doses. Assuming an average adult weight of 70-80kg, the licensed dose is situated at the lower end of the doses investigated throughout these three clinical trials.
We naturally wonder about the suitability of the monotonic efficacy assumption in a wider sense. In this manuscript, we investigate two related questions. What evidence is there that the probabilities of a) toxicity, and b) efficacy increase in dose in modern cancer therapies?
Methods
We sought to identify a broad sample of manuscripts reporting recent dose-finding clinical trials in oncology.
Identifying manuscripts
Chiuzan et al. [2] conducted a systematic review of the methods used in cancer dose-finding trials. Their findings mirrored those of Rogatko et al. [1] from the previous decade that over 90% of dose-finding trials use a rule-based design like 3+3. The authors found 1,712 manuscripts published between 2008 and the first half of 2014. The authors published in their paper a large table summarising the trials that used model-based methods, like CRM or EWOC, of which there were 92 examples. Whilst extracting data from 1,712 papers would be infeasible, extracting data from 92 was possible. However, the subset of trials that use model-based methods may not be representative of the entire sample. For this reason, we supplemented the list of 92 model-based papers with 30 randomly-selected papers that used rule-based methods, stratified by year of publication. Combined, this produced a sample of 122 manuscripts.
Extracting data
Each of the 122 manuscripts [18–139] presented the results of at least one dose-finding experiment in humans. Some papers reported the results of more than one experiment. From each experiment, we extracted descriptive data pertaining to the patient population, the dose-varying treatment, and any concomitant treatments. Concerning outcomes, we extracted the dose-levels administered, the number of patients evaluated at each, and the number of DLT and objective response events recorded at each. These outcomes are explained and justified in the following sections.
Dose-level outcomes vs pooled outcomes
We only recorded outcomes broken down by dose-level because these would allow us to address the research question pertaining to the evidence for monotonically increasing toxicity and efficacy probabilities. We did not collect outcomes that were reported by pooling all dose-levels because this would not address our research question.
Toxicity outcomes
Dose-limiting toxicity (DLT) is the de-facto standard safety outcome in dose-finding trials. Manifestation of DLT involves the occurrence of pre-specified adverse events (AEs) that are serious enough that they would motivate the clinicians to rule out higher doses in the affected patient or consider the temporary suspension or complete cessation of current therapy. There is no standard definition of DLT across trials but the outcome would be defined in each trial protocol and remain consistent across the doses investigated within each trial. The definition of DLT in a given trial may reflect the clinical characteristics of the disease and the anticipated adverse events from the entire treatment ensemble (i.e. arising from the experimental therapy and concomitant therapies).
Data on DLT outcomes were sought in every manuscript. We analyse outcomes for DLT because it was the most widely-reported toxicity outcome measure.
Efficacy outcomes
The question motivating this research concerns drug efficacy and how this changes as dose is increased. Efficacy is only loosely defined in cancer. There is no single outcome that is unambiguously accepted as the variable best reflecting efficacy. Applications for drug licensing are generally supported by phase III trials that use survival outcomes like overall survival (OS) and progression-free survival (PFS). In contrast, early phase trials, when they evaluate efficacy, tend to use surrogate outcomes that can be evaluated over the short-term like disease response.
Assessing disease response generally involves comparing the extent of disease (e.g. tumour size or number of leukaemic cells) at baseline and after treatment administration to characterise the patient’s response to treatment using one of several categories. RECIST [140] is the most common response outcome categorisation used in solid tumour trials. RECIST categorises each disease assessment as one of: complete response (CR); partial response (PR); stable disease (SD); or progressive disease (PD).
Researchers have defined analogues to RECIST in other cancers, including blood cancers where diseased cells reside in the blood rather than a discrete measurable mass. An example of this is the Cheson criteria in acute myeloid leukaemia (AML) [141] and iwCLL criteria in chronic myeloid leukaemia [142]. These contain response categories that are similar to those in RECIST, with slight modifications to reflect the phenomena specific to the disease.
Under RECIST, an objective response (OR) is said to occur when a patient experiences CR or PR. Under the RECIST analogues, further response categories are included in OR. For instance, in AML, a patient with complete remission with incomplete blood count recovery would be considered to have experienced OR.
Data on OR outcomes were sought in every manuscript. We analyse outcomes for OR because it was the most widely-reported efficacy outcome measure.
Orderability of doses
Analysing how the probabilities of events change as dose increases requires that we are working with increasing doses. The general 3+3, CRM and EWOC methods require that the doses under investigation are fully orderable. That is, we need to be able to unambiguously say that di<dj or di>dj for each pair of doses in the set of doses under investigation.
When we encountered dose-levels that were not fully orderable, for the purposes of conducting statistical analysis we broke the doses up to form fully orderable subsets that we called analysis series.
There are many possible subsets of a set so the way we formed the analysis series was unavoidably subjective. To promote objectivity, we followed some simple rules. We sought to maximise the size of the largest fully orderable series. Furthermore, we avoided allocating a dose to several series unless repetition was the only way to avoid having an orphan dose (i.e. a series of size 1).
Consider, for instance, the three dose scenario: dose 1 = 10mg of drug A + 20mg of drug B; dose 2 = 20mg A + 10mg B; dose 3 = 20mg A + 20mg B. This set of doses is not totally orderable because it is impossible to say whether dose 1 is higher or lower than dose 2. However, each of these doses is categorically less than dose 3. Thus, in this hypothetical scenario, we would have analysed outcomes of the two totally orderable subsets (dose 1, dose 3) and (dose 2, dose 3). In doing so, the outcomes at dose 3 would have featured in the analyses twice. The alternative would have been to arbitrarily throw away the outcomes at dose 1 or dose 2, an option we rejected because it is wasteful.
In summary, the data have been recorded in a way amenable to answering the research questions.
Database creation
Data were extracted from papers and recorded on prior-written standardised forms. The data were then recorded on sheets in an Excel file that was deposited in the University of Birmingham’s data repository [143].
Data were extracted by VH, GS, KB and CP. Data for 52 manuscripts were extracted by two different authors and differences were resolved by discussion. Data for 70 manuscripts were extracted by one author.
Analysis model
The DLT and OR outcomes we extracted were binary in nature. Outcomes were analysed within study using Emax models [144].
Emax is a flexible non-linear approach for fitting sigmoidal (i.e. S-shaped) dose-response curves to continuous or binary responses. In our analysis, the binary response variable was the patient-level presence of DLT or OR. The explanatory variable was the dose-level administered. Emax can capture relationships where the mean response increases in dose, decreases in dose, or is independent of dose. Separate models were fit to the collection of patient outcomes in each study. Outcomes from different studies were not pooled because of the disparate patient populations and definitions of DLT and OR. Each of these factors remained consistent within each analysis series. The fitted values from the Emax models represent the event probabilities at each dose, ranging from 0 to 1.
Binary outcomes might generally suggest analysis via logit linear regression models. We did not use logit models because they assume that the event probability invariably tends to 1 with large enough predictor values. This is inappropriate in our analysis that seeks to investigate how event probabilities vary with dose, including the possibility that event probabilities plateau at values less than 1. Whilst it may be acceptable to assume that a high enough dose will guarantee a toxic outcome, empirically it is inappropriate to assume that a high enough dose guarantees an efficacious response. The relative strength of the Emax model is that it allows the event probability to plateau at a value less than 1. It contains as a special case the scenario reflected by logit models where the event probability tends to 1 as dose is increased.
We used both maximum likelihood and Bayesian approaches to fit Emax models. Maximum likelihood models were fit because they do not require the specification of priors. However, the analysis series in this research were very small, with some data-sets including fewer than 10 patients. In several instances, maximum likelihood models failed to converge. In these circumstances, Bayesian models can be extremely valuable because the specification of very small amounts of information in prior distributions promotes model convergence. Bayesian models succeeded in estimating dose-event curves in all instances. Our chosen prior distributions are introduced briefly below and expanded in detail in the supplementary appendix.
The height of each dose-event curve was calculated as the model-fitted event probability at the highest dose under investigation minus that of the lowest dose. This concept is illustrated graphically in Figure 1 in the supplementary appendix.
Prior distributions
For the Bayesian analyses, we were required to specify prior distributions on the four parameters in the Emax model. We selected uniform priors on the parameters that reflect the minimum and maximum event probabilities, constrained to take values only in the region from 0 to 1. This meant that all event probabilities at the lowest and highest doses were equally likely, with no inclination towards a particular probability.
Prior distributions on the other two parameters, that control the location and steepness of the S-curve, were chosen to provide very modest information to constrain estimation to the region of feasible values. Full details on prior selection are given in the supplementary appendix, including in supplementary Figure 2 a plot of candidate event curves that are generated by the priors.
Appraising model fits
Model-generated dose-event curves were inspected visually alongside source data to verify the quality of model fits. Furthermore, we recorded metrics for all Bayesian models that can diagnose a potentially poor model fit. Further details are given in the supplementary appendix.
Software
Maximum-likelihood Emax models were fit using the DoseFinding package [145] and Bayesian models were fit using the brms package [146] in R [147]. Data processing was aided using tidyverse [148] packages, posterior samples were extracted using tidybayes [149], and plots were produced using ggplot2 [150].
Results
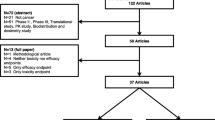
115 (94%) of the 122 examined manuscripts reported outcomes by dose-level. After extracting data and creating fully-orderable dose subsets, these yielded 155 analysis series for DLT, and 93 analysis series for OR (Fig. 1). Characteristic information is summarised in Table 1.

The data extraction process. The procedure for forming fully orderable subsets of doses is described in the text
Patient groups
Three-quarters of the data analysed came from non-haematological cancers. In approximately one third of cases, the patient group comprised different types of solid tumour. Most commonly, however, trials were conducted within specific cancer sites, with breast, gastrointestinal, and lung cancers featuring relatively frequently.
Approximately one quarter of the data came from trials in haematological malignancies. Once again, trials were sometimes conducted in fairly heterogeneous patient groups and sometimes in specific diseases like AML, CLL and lymphoma.
Experimental treatments
The treatment type most commonly undergoing dose-escalation was inhibitors, contributing 58 (37%) DLT series and 43 (46%) OR series. Chemotherapies were also common targets for dose-escalation, contributing 51 (33%) DLT series and 25 (27%) OR series. Monoclonal antibodies were fairly infrequent in this data set, contributing only 8 (5%) DLT series and zero OR series. Trials that escalated dose in two different treatment types were common, with chemotherapy plus inhibitor the most common pairing, yielding 11% of the DLT series and 13% of the OR series. The median of doses in an analysis series was 4 (IQR = 2, 5).
Monotonicity of DLT and OR in dose
Fitted curves for the dose-DLT series produced by the Bayesian Emax models are shown in Fig. 2. Each line reflects the best fit to all of the DLT outcomes observed in one analysis series. A separate panel is shown for each type of treatment that underwent dose-escalation. Information on patient group and concomitant therapies are not shown in this plot.

Fitted dose-DLT curves yielded by Bayesian Emax models. For presentation, doses are centralised at zero (i.e. the average dose-level for each series is subtracted) and up to the middle nine doses are shown, to allow the series to be visualised together
We see that the majority of the fitted DLT series show a positive relationship with dose, reflecting that DLT becomes more likely as dose is increased. This is seen in all types of therapy and matches our expectation that the dose makes the poison. The fitted series for inhibitors and chemotherapies appear to increase more steeply than for other therapies.
Fitted curves for the dose-OR series produced by the Bayesian Emax models are shown in Fig. 3. Contrasting to Fig. 2, we see that there are materially fewer OR curves than DLT curves. However, it is clear that the fitted OR curves are much less likely to show a strong positive association between dose and response. It is striking how few positive gradients there are. Even amongst chemotherapies, there is scant evidence of greater efficacy at higher doses. The single OR series for an antibody-drug conjugate and one of the series for an immunomodulatory drug show comparatively strong evidence of a positive dose-response effect.

Fitted dose-OR curves yielded by Bayesian Emax models. For presentation, doses are centralised at zero (i.e. the average dose-level for each series is subtracted) and up to the middle nine doses are shown, to allow the series to be visualised together
The heights of the fitted dose-DLT and dose-OR curves are shown in Fig. 4, with statistics for the two outcomes plotted side-by-side within treatment type. The dashed red line at zero reflects the null value where there is no relationship between dose and event. Positive curve heights indicate that the event became more likely as dose is increased, and vice-versa.

Heights of dose-DLT and dose-OR curves. The dashed red lines reflect a curve height of zero where there is no association between dose and event. Positive values reflect that event probabilities increase as dose is increased, and negative values reflect a decreasing probability as dose is increased
We see that the majority of DLT curves show a positive curve height in all classes of treatment. There are infrequent series that suggest no relationship, or a modestly negative relationship. In stark contrast, the same statistics for the OR series straddle the null line in the most frequent treatment categories, demonstrating a lack of evidence in support of the monotonic efficacy assumption.
Furthermore, the curve heights of the DLT series show much greater variability than those of the OR series. There are some instances where the probability of DLT increased quite rapidly as dose increased.
Curve heights for DLT and OR series are plotted by type of disease in Figure 3 in the supplementary appendix. We see that the phenomena we have described are broadly observed across most disease types.
Similarly, Figure 4 in the supplementary appendix shows the equivalent inferences split by class of dose-finding methodology (i.e. rule-based or model-based). The two OR series with the steepest positive relationship with dose both use a rule-based design. Generally, however, the heights of the DLT and OR curves from studies that used model-based designs were similar to those yielded by rule-based designs. In both groups, the observation remained that DLT curves commonly increased with dose whilst OR curves were mostly invariant in dose.
The inferences thus far have come from the Bayesian Emax models, where the statistical model fitting procedure succeeded in all instances. We also sought to analyse series using maximum likelihood Emax models. The maximum likelihood model fitting procedure failed in many instances. Examples of model-fitting failure were procedures that did not converge, or procedures that yielded extremely large estimates for the standard errors of model parameters. For completeness, we show the fitted DLT and OR series yielded by the maximum likelihood models in Figures 5 and 6 in the supplementary appendix. The fitted series produced by the maximum likelihood models are less smooth. Supplementary Figure 7 shows that the curve heights estimated by maximum likelihood models are generally more extreme than those produced by Bayesian models, with more values positive and negative values far from zero for DLT and OR series. In the most frequently investigated treatments, once again we see that the DLT series are overwhelmingly likely to have a positive curve height, whilst the heights of OR series cluster around zero.
Discussion
We collected dose-level toxicity and efficacy outcomes from 115 papers reporting early phase clinical trials of experimental anti-cancer therapies. These trials all used experimental methods that assume that higher doses are associated with greater probabilities of both toxicity and efficacy. We then analysed those outcomes using flexible non-linear regression methods. In summary, we found broad evidence that the probability of toxicity increased in dose in most treatment classes, in most types of cancer, in scenarios that use rule-based or model-based dose-escalation methodologies. In contrast, we found very little evidence that the probability of response increased as dose was increased.
On the face of it, the implication of our findings is that dose-escalation clinical trials commonly advocate doses that are unjustifiably high. For a treatment where toxicity incidence is positively associated with dose and response incidence is invariant to dose, lower doses should be preferred. However, by conducting dose-escalation experiments that recommend doses only by toxicity outcomes, explicitly assuming that higher doses are more efficacious and therefore preferable, many trials miss the opportunity to recommend a lower dose with less toxic effects and no commensurate cost to efficacy. This could help explain why dose-reduction occurs in phase II and III clinical trials. For example, a detailed account of post-phase I dose optimisation in an inhibitor drug is given by Lee et al. [151].
The statements above pertain to the ranges of doses investigated in phase I clinical trials. They do not apply to the ranges of all possible doses. Naturally, we acknowledge in all active therapies that there must be a dose so low that the attendant anti-tumour effect is negligible. That we found many series with non-trivial response rates at all doses is perhaps testament to the value of pre-clinical research and PK/PD modelling in locating dose ranges that are likely to be tolerable and active for phase I trials.
A logical remedy to the problems we have described would be to use so-called seamless phase I/II designs that recommend doses by toxicity and response outcomes, several examples of which have been published [9–13] and implemented in clinical trials [14, 152]. These designs bring their own challenges, the most notable of which are the extra statistical complexity and the requirement that the co-primary outcomes can be evaluated over a similar time horizon to allow dose-escalation decisions to be made in a timely manner.
A simpler solution would require early phase trialists to address the assumptions made by their phase I designs and discuss the appropriateness of their final dose recommendations in light of toxicity and efficacy outcomes. By reporting suitable efficacy and toxicity outcomes by dose, researchers would allow the community to assess the suitability of the methodological assumptions and identify doses most appropriate for further study. Arguably, this already occurs in practice when trialists use dose-escalation methods like 3+3, CRM or EWOC cognisant of the possibility that higher doses might not bring greater efficacy. The drawback of this putative approach is that it risks allocating patients in the dose-finding trial to inappropriate doses. Trialists that regard this as unethical would be encouraged to use seamless phase I/II methodologies, described above.
It is possible that monotonic efficacy effects in dose are observed in long-term clinical outcomes like OS and PFS, even if they are not seen in surrogate outcomes like response. We cannot provide any evidence to support or refute this hypothesis since survival outcomes were reported by dose extremely infrequently. If this hypothesis is true, however, it calls into question the validity of objective response as a surrogate outcome for clinical outcomes.
The sample size of the trials included in this research is small. Researchers might legitimately ponder the feasibility of detecting strong dose-event relationships with such small sample sizes. It was advantageous, then, that we included and analysed DLT outcomes because they have shown that evidence of stark relationships can be garnered from small trials, particularly when analysed together. Within study, OR outcomes routinely failed to show the strength of relationship with dose shown by DLT outcomes.
It is perhaps self-fulfilling that we have observed stark monotonic effects in toxicity because dose-finding trials escalate dose in the absence of unacceptable toxicity and de-escalate dose or halt the trial when toxicity manifests. For this reason, it is plausible that the toxicity relationships we have presented here may be biased upwards. Nevertheless, this does not explain the lack of evidence for a relationship between dose and efficacy.
Recently, Hazim et al. [153] investigated the relationship between dose and efficacy in single-agent phase I trials in oncology. They did so by calculating aggregate response rates at dose categories defined by the ratio of the given dose to the recommended phase II dose (RP2D). They found evidence that response rates increased modestly as doses approached the RP2D. Compared to their study, this research benefits from including treatment combinations, including both efficacy and toxicity outcomes, and using statistical modelling.
How could dose-finding be conducted in modern cancer therapeutics?
We conclude this article with some discussion on the design, conduct and reporting of dose-finding clinical trials.
It is our opinion that early phase clinical trials of modern cancer therapies should seek to optimise the delivery of treatments, balancing the risk of harmful toxicity with the expectation of improving meaningful clinical outcomes. This same sentiment was recently advocated by early phase trial methodologists at MD Anderson [154]. Naturally, this requires that toxicity and efficacy outcomes are evaluated, and used to adapt the doses given to future patients.
Investigators should be clear on what constitutes a meaningful clinical outcome. OS and quality of life measures are widely seen as gold standard outcomes for demonstrating benefit to patients and, in many settings, PFS is also considered a valid clinical outcome. Overwhelmingly, however, we have seen that efficacy is measured in dose-finding trials only by categorical response outcomes.
Where response has been validated as a reliable surrogate for a clinical outcome in a patient group, it should continue to be used to measure efficacy in dose-finding trials. In this scenario, we recommend using co-primary toxicity and response outcomes in a phase I/II dose-finding design. These designs are unified by the belief that the MTD may not be the best dose. Many examples of such designs have been published [9–13] and more will surely be added. More recently, methods have been developed for modeling toxicity and time to event outcomes like PFS [155, 156].
Comparing to traditional phase I designs that only assess toxicity, seamless phase I/II designs will likely take longer to run and require more patients. However, as phase I/II designs also answer the question typically investigated in a traditional phase II trial, the more appropriate comparison is the total time and sample size required to run a phase I followed by a phase II trial. By this comparison, when considering that a seamless design requires only one protocol and no break between trials, a seamless design may not require more resource than separate trials.
Where response has not been validated as a reliable surrogate for a clinical outcome in a patient group, the situation is much more challenging. If investigators seek to avoid using a surrogate of unknown utility, the entire clinical trial program will amount to contrasting clinically meaningful outcomes at several different doses, perhaps compared to a shared control, incorporating pre-specified decisions to narrow the experimental doses, culminating in a final test of superiority. These goals can be achieved using the multi-arm multi-stage design [157] if we treat the doses as experimental arms. Other methodologists have also recently addressed the topic of broad seamless dose-finding trials [158]. These proposals are obviously ambitious, covering the combined goals of trials at phases I, II and III. However, they illustrate how dose-finding could be embedded into an entire drug development program to maximise the chances of identifying the best dose for patients.
Justifiable operating characteristics and statistical error rates will naturally vary with the incidence of the disease. Logically, overall probabilities of failing to stop in genuinely toxic or ineffective scenarios should be similar to type I error rates typically used in trials, and the probabilities of correctly identifying one of the most superior doses should be comparable to conventional power.
Without doubt, this would require larger dose-finding trials. However, our proposals could be viewed as reallocating some patients from late phase trials to early phase stages of seamless trials, so that total sample sizes are not necessarily increased. Relating to this theme, [154] extolled that a dose-finding trial should not be seen as a challenge to be overcome as quickly as possible, but as an opportunity to optimise the way that an experimental treatments is given and maximise benefit for patients.
Regarding trial reporting, we advise that investigators identify the assumptions implicit in their design and justify them with reference to the nature of the treatment. We recommend that outcomes are always reported broken down by the doses investigated. A goal of dose-finding trials is to evaluate outcomes under a set of investigational doses and pooling outcomes across doses obfuscates that goal. Furthermore, investigators should discuss how the data concur or refute the assumptions, and the implications of violated assumptions on conclusions and recommendations.
Conclusion
We conclude that dose-escalation clinical trials routinely use methods whose assumptions are violated by the outcomes observed. Specifically, methods that implicitly assume that efficacy monotonically increases in dose are very commonly used. However, we have demonstrated that the probabilities of disease response frequently do not increase with dose. We have shown this across a range of treatment types, including monotherapies and combinations. Consequently, dose-finding trials risk recommending unjustifiably high doses that may be harmful to patients. We recommend that trialists consider experimental designs that allow toxicity and efficacy outcomes to jointly determine the doses given to patients and recommended for further study.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the UBIRA eData repository, https://doi.org/10.25500/edata.bham.00000337
R code to reproduce all statistics, tables and figures is freely available at
Abbreviations
- A+B designs:
-
A family of rule-based phase I clinical trial designs
- AML:
-
Acute myeloid leukaemia
- BOIN:
-
Bayesian Optimal Interval design, a phase I clinical trial design
- CLL:
-
Chronic lymphocytic leukaemia CR: Complete response
- CRM:
-
Continual Reassessment Method, a phase I clinical trial design
- DLT:
-
Dose limiting toxicity
- IQR:
-
Inter-quartile range
- iwCLL:
-
International Workshop in Chronic Lymphocytic Leukaemia
- NSCLC:
-
Non-small-cell lung cancer
- MTD:
-
Maximum tolerable dose
- OR:
-
Objective response
- OS:
-
Overall survival
- PD:
-
Progressive disease
- PD-1:
-
Programmed cell death protein 1
- PFS:
-
Progression-free survival
- PK/PD:
-
Pharmacokinetics / pharmacodynamics
- PR:
-
Partial response
- RECIST:
-
Response Evaluation Criteria in Solid Tumours
- RP2D:
-
Recommended phase 2 dose
- SD:
-
Stable disease
References
Rogatko A, Schoeneck D, Jonas W, Tighiouart M, Khuri FR, Porter A. Translation of innovative designs into phase I trials. J Clin Oncol. 2007; 25(31):4982–6. https://doi.org/10.1200/JCO.2007.12.1012.
Chiuzan C, Shtaynberger J, Manji GA, Duong JK, Schwartz GK, Ivanova A, Lee SM. Dose-finding designs for trials of molecularly targeted agents and immunotherapies. J Biopharm Stat. 2017; 27(3):477–94. https://doi.org/10.1080/10543406.2017.1289952. Publisher: Taylor & Francis.
Le Tourneau C, Lee JJ, Siu LL. Dose escalation methods in phase i cancer clinical trials. J Natl Cancer Inst. 2009; 101(10):708–20. https://doi.org/10.1093/jnci/djp079.
O’Quigley J, Pepe M, Fisher L. Continual reassessment method: A practical design for phase 1 clinical trials in cancer. Biometrics. 1990; 46(1):33–48. https://doi.org/10.2307/2531628.
Tighiouart M, Rogatko A. Dose Finding with Escalation with Overdose Control (EWOC) in Cancer Clinical Trials. Stat Sci. 2010; 25(2):217–26. https://doi.org/10.1214/10-STS333.
O’Quigley J, Zohar S. Experimental designs for phase i and phase i/ii dose-finding studies. Br J Cancer. 2006; 94(5):609–13.
Iasonos A, Wilton AS, Riedel ER, Seshan VE, Spriggs DR. A comprehensive comparison of the continual reassessment method to the standard 3+ 3 dose escalation scheme in phase i dose-finding studies. Clin Trials. 2008; 5(5):465–77.
Wheeler GM, Mander AP, Bedding A, Brock K, Cornelius V, Grieve AP, Jaki T, Love SB, Weir CJ, Yap C, et al. How to design a dose-finding study using the continual reassessment method. BMC Med Res Methodol. 2019; 19(1):1–15.
Braun TM. The bivariate continual reassessment method: Extending the CRM to phase I trials of two competing outcomes. Control Clin Trials. 2002; 23(3):240–56. https://doi.org/10.1016/S0197-2456(01)00205-7.
Zhang W, Sargent DJ, Mandrekar S. An adaptive dose-finding design incorporating both toxicity and efficacy. Stat Med. 2006; 25(14):2365–83. https://doi.org/10.1002/sim.2325.
Ivanova A. A new dose-finding design for bivariate outcomes. Biometrics. 2003; 59(4):1001–7.
Thall P, Cook J. Dose-Finding Based on Efficacy-Toxicity Trade-Offs. Biometrics. 2004; 60(3):684–93. https://doi.org/10.1111/j.0006-341X.2004.00218.x.
Wages NA, Tait C. Seamless phase i/ii adaptive design for oncology trials of molecularly targeted agents. J Biopharm Stat. 2015; 25(5):903–20.
Brock K, Billingham L, Copland M, Siddique S, Sirovica M, Yap C. Implementing the efftox dose-finding design in the matchpoint trial. BMC Med Res Methodol. 2017; 17(1):112.
Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, Drengler R, Chen C, Smith L, Espino G, et al. Phase i study of pembrolizumab (mk-3475; anti–pd-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res. 2015; 21(19):4286–93.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. Pembrolizumab for the treatment of non–small-cell lung cancer. New Engl J Med. 2015; 372(21):2018–28.
Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y, Molina J, Kim J-H, Arvis CD, Ahn M-J, et al. Pembrolizumab versus docetaxel for previously treated, pd-l1-positive, advanced non-small-cell lung cancer (keynote-010): a randomised controlled trial. Lancet. 2016; 387(10027):1540–50.
Sharma S, de Vries EG, Infante JR, Oldenhuis CN, Gietema JA, Yang L, Bilic S, Parker K, Goldbrunner M, Scott JW, Burris HA. Safety, pharmacokinetics, and pharmacodynamics of the DR5 antibody LBY135 alone and in combination with capecitabine in patients with advanced solid tumors. Investig New Drugs. 2013; 32(1):135–44. https://doi.org/10.1007/s10637-013-9952-9.
Popovtzer A, Normolle D, Worden FP, Prince ME, Chepeha DB, Wolf GT, Bradford CR, Lawrence TS, Eisbruch A. Phase i trial of radiotherapy concurrent with twice-weekly gemcitabine for head and neck cancer: Translation from preclinical investigations aiming to improve the therapeutic ratio. Transl Oncol. 2014; 7(4):479–83. https://doi.org/10.1016/j.tranon.2014.04.016.
Kobayashi S, Nagano H, Sakai D, Eguchi H, Hatano E, Kanai M, Seo S, Taura K, Fujiwara Y, Ajiki T, Takemura S, Kubo S, Yanagimoto H, Toyokawa H, Tsuji A, Terajima H, Morita S, Ioka T. Phase i study of adjuvant gemcitabine or s-1 in patients with biliary tract cancers undergoing major hepatectomy: KHBO1003 study. Cancer Chemother Pharmacol. 2014; 74(4):699–709. https://doi.org/10.1007/s00280-014-2543-4.
Khouri IF, Wei W, Korbling M, Turturro F, Ahmed S, Alousi A, Anderlini P, Ciurea S, Jabbour E, Oran B, Popat UR, Rondon G, Bassett RL, Gulbis A. BFR (bendamustine, fludarabine, and rituximab) allogeneic conditioning for chronic lymphocytic leukemia/lymphoma: reduced myelosuppression and GVHD. Blood. 2014; 124(14):2306–12. https://doi.org/10.1182/blood-2014-07-587519.
Ando Y, Inada-Inoue M, Mitsuma A, Yoshino T, Ohtsu A, Suenaga N, Sato M, Kakizume T, Robson M, Quadt C, Doi T. Phase i dose-escalation study of buparlisib (BKM120), an oral pan-class i PI3k inhibitor, in japanese patients with advanced solid tumors. Cancer Sci. 2014; 105(3):347–53. https://doi.org/10.1111/cas.12350.
Shaw AT, Kim D-W, Mehra R, Tan DSW, Felip E, Chow LQM, Camidge DR, Vansteenkiste J, Sharma S, Pas TD, Riely GJ, Solomon BJ, Wolf J, Thomas M, Schuler M, Liu G, Santoro A, Lau YY, Goldwasser M, Boral AL, Engelman JA. Ceritinib in ALK-rearranged non–small-cell lung cancer. New Engl J Med. 2014; 370(13):1189–97. https://doi.org/10.1056/nejmoa1311107.
Saura C, Bendell J, Jerusalem G, Su S, Ru Q, Buck SD, Mills D, Ruquet S, Bosch A, Urruticoechea A, Beck JT, Tomaso ED, Sternberg DW, Massacesi C, Hirawat S, Dirix L, Baselga J. Phase ib study of buparlisib plus trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on trastuzumab-based therapy. Clin Cancer Res. 2014; 20(7):1935–45. https://doi.org/10.1158/1078-0432.ccr-13-1070.
Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J, Sarantopoulos J, Mahalingam D, Shou Y, Moles MA, Yang L, Granvil C, Hurh E, Rose KL, Amakye DD, Dummer R, Mita AC. A phase i, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res. 2014; 20(7):1900–9. https://doi.org/10.1158/1078-0432.ccr-13-1710.
Faderl S, Balakrishnan K, Thomas DA, Ravandi F, Borthakur G, Burger J, Ferrajoli A, Cortes J, O’Brien S, Kadia T, Feliu J, Plunkett W, Gandhi V, Kantarjian HM. Phase i and extension study of clofarabine plus cyclophosphamide in patients with relapsed/refractory acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk. 2014; 14(3):231–8. https://doi.org/10.1016/j.clml.2013.12.001.
Boulin M, Hillon P, Cercueil JP, Bonnetain F, Dabakuyo S, Minello A, Jouve JL, Lepage C, Bardou M, Wendremaire M, Guerard P, Denys A, Grandvuillemin A, Chauffert B, Bedenne L, Guiu B. Idarubicin-loaded beads for chemoembolisation of hepatocellular carcinoma: results of the IDASPHERE phase i trial. Aliment Pharmacol Ther. 2014; 39(11):1301–13. https://doi.org/10.1111/apt.12746.
Rodon J, Braña I, Siu LL, Jonge MJD, Homji N, Mills D, Tomaso ED, Sarr C, Trandafir L, Massacesi C, Eskens F, Bendell JC. Phase i dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-class i PI3k inhibitor, in patients with advanced solid tumors. Investig New Drugs. 2014; 32(4):670–81. https://doi.org/10.1007/s10637-014-0082-9.
Doi T, Onozawa Y, Fuse N, Yoshino T, Yamazaki K, Watanabe J, Akimov M, Robson M, Boku N, Ohtsu A. Phase i dose-escalation study of the HSP90 inhibitor AUY922 in japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014; 74(3):629–36. https://doi.org/10.1007/s00280-014-2521-x.
Infante JR, Dees EC, Olszanski AJ, Dhuria SV, Sen S, Cameron S, Cohen RB. Phase i dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2014; 32(28):3103–10. https://doi.org/10.1200/jco.2013.52.3993.
Tsimberidou AM, Keating MJ, Jabbour EJ, Ravandi-Kashani F, O’Brien S, Estey E, Bekele N, Plunkett WK, Kantarjian H, Borthakur G. A phase i study of fludarabine, cytarabine, and oxaliplatin therapy in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leukemia. 2014; 14(5):395–4001. https://doi.org/10.1016/j.clml.2014.01.009.
Iyer SP, Beck JT, Stewart AK, Shah J, Kelly KR, Isaacs R, Bilic S, Sen S, Munshi NC. A phase IB multicentre dose-determination study of BHQ880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br J Haematol. 2014; 167(3):366–75. https://doi.org/10.1111/bjh.13056.
Brana I, Berger R, Golan T, Haluska P, Edenfield J, Fiorica J, Stephenson J, Martin LP, Westin S, Hanjani P, Jones MB, Almhanna K, Wenham RM, Sullivan DM, Dalton WS, Gunchenko A, Cheng JD, Siu LL, Gray JE. A parallel-arm phase i trial of the humanised anti-IGF-1r antibody dalotuzumab in combination with the AKT inhibitor MK-2206, the mTOR inhibitor ridaforolimus, or the NOTCH inhibitor MK-0752, in patients with advanced solid tumours. Br J Cancer. 2014; 111(10):1932–44. https://doi.org/10.1038/bjc.2014.497.
Isakoff SJ, Wang D, Campone M, Calles A, Leip E, Turnbull K, Bardy-Bouxin N, Duvillié L, Calvo E. Bosutinib plus capecitabine for selected advanced solid tumours: results of a phase 1 dose-escalation study. Br J Cancer. 2014; 111(11):2058–66. https://doi.org/10.1038/bjc.2014.508.
Brennan RC, Furman W, Mao S, Wu J, Turner DC, Stewart CF, Santana V, McGregor LM. Phase i dose escalation and pharmacokinetic study of oral gefitinib and irinotecan in children with refractory solid tumors. Cancer Chemother Pharmacol. 2014; 74(6):1191–8. https://doi.org/10.1007/s00280-014-2593-7.
Gandhi L, Bahleda R, Tolaney SM, Kwak EL, Cleary JM, Pandya SS, Hollebecque A, Abbas R, Ananthakrishnan R, Berkenblit A, Krygowski M, Liang Y, Turnbull KW, Shapiro GI, Soria J-C. Phase i study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2–dependent and other solid tumors. J Clin Oncol. 2014; 32(2):68–75. https://doi.org/10.1200/jco.2012.47.2787.
Fanale M, Assouline S, Kuruvilla J, Solal-Céligny P, Heo DS, Verhoef G, Corradini P, Abramson JS, Offner F, Engert A, Dyer MJS, Carreon D, Ewald B, Baeck J, Younes A, Freedman AS. Phase IA/II, multicentre, open-label study of the CD40 antagonistic monoclonal antibody lucatumumab in adult patients with advanced non-hodgkin or hodgkin lymphoma. Br J Haematol. 2013; 164(2):258–65. https://doi.org/10.1111/bjh.12630.
Das P, Eng C, Rodriguez-Bigas MA, Chang GJ, Skibber JM, You YN, Maru DM, Munsell MF, Clemons MV, Kopetz SE, Garrett CR, Shureiqi I, Delclos ME, Krishnan S, Crane CH. Preoperative radiation therapy with concurrent capecitabine, bevacizumab, and erlotinib for rectal cancer: A phase 1 trial. Int J Radiat Oncol Biol Phys. 2014; 88(2):301–5. https://doi.org/10.1016/j.ijrobp.2013.10.034.
Besse B, Heist RS, Papadmitrakopoulou VA, Camidge DR, Beck JT, Schmid P, Mulatero C, Miller N, Dimitrijevic S, Urva S, Pylvaenaeinen I, Petrovic K, Johnson BE. A phase ib dose-escalation study of everolimus combined with cisplatin and etoposide as first-line therapy in patients with extensive-stage small-cell lung cancer. Ann Oncol. 2014; 25(2):505–11. https://doi.org/10.1093/annonc/mdt535.
Cruz CRY, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S, Shpall EJ, Krance RA, Kamble RT, Carrum G, Hosing CM, Gee AP, Mei Z, Grilley BJ, Heslop HE, Rooney CM, Brenner MK, Bollard CM, Dotti G. Infusion of donor-derived CD19-redirected virus-specific t cells for b-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013; 122(17):2965–73. https://doi.org/10.1182/blood-2013-06-506741.
Thornton KA, Chen AR, Trucco MM, Shah P, Wilky BA, Gul N, Carrera-Haro MA, Ferreira MF, Shafique U, Powell JD, Meyer CF, Loeb DM. A dose-finding study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcoma. Int J Cancer. 2013; 133(4):997–1005. https://doi.org/10.1002/ijc.28083.
Sharma S, Beck J, Mita M, Paul S, Woo MM, Squier M, Gadbaw B, Prince HM. A phase i dose-escalation study of intravenous panobinostat in patients with lymphoma and solid tumors. Investig New Drugs. 2013; 31(4):974–85. https://doi.org/10.1007/s10637-013-9930-2.
Sessa C, Shapiro GI, Bhalla KN, Britten C, Jacks KS, Mita M, Papadimitrakopoulou V, Pluard T, Samuel TA, Akimov M, Quadt C, Fernandez-Ibarra C, Lu H, Bailey S, Chica S, Banerji U. First-in-human phase i dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res. 2013; 19(13):3671–80. https://doi.org/10.1158/1078-0432.ccr-12-3404.
Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, Paskett LA, Wong H, Dobrolecki LE, Lewis MT, Froehlich AM, Paranilam J, Hayes DF, Wicha MS, Chang JC. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res. 2013; 19(6):1512–24. https://doi.org/10.1158/1078-0432.ccr-11-3326.
Mangiacavalli S, Pochintesta L, Pascutto C, Cocito F, Cazzola M, Corso A. Good clinical activity and favorable toxicity profile of once weekly bortezomib, fotemustine, and dexamethasone (b-MuD) for the treatment of relapsed multiple myeloma. Am J Hematol. 2012; 88(2):102–6. https://doi.org/10.1002/ajh.23358.
Larocca A, Montefusco V, Bringhen S, Rossi D, Crippa C, Mina R, Galli M, Marcatti M, Verde GL, Giuliani N, Magarotto V, Guglielmelli T, Rota-Scalabrini D, Omedé P, Santagostino A, Baldi I, Carella AM, Boccadoro M, Corradini P, Palumbo A. Pomalidomide, cyclophosphamide, and prednisone for relapsed/refractory multiple myeloma: a multicenter phase 1/2 open-label study. Blood. 2013; 122(16):2799–806. https://doi.org/10.1182/blood-2013-03-488676.
Harvey RD, Owonikoko TK, Lewis CM, Akintayo A, Chen Z, Tighiouart M, Ramalingam SS, Fanucchi MP, Nadella P, Rogatko A, Shin DM, El-Rayes B, Khuri FR, Kauh JS. A phase 1 bayesian dose selection study of bortezomib and sunitinib in patients with refractory solid tumor malignancies. Br J Cancer. 2013; 108(4):762–5. https://doi.org/10.1038/bjc.2012.604.
Finn RS, Poon RTP, Yau T, Klümpen H-J, Chen L-T, Kang Y-K, Kim T-Y, Gomez-Martin C, Rodriguez-Lope C, Kunz T, Paquet T, Brandt U, Sellami D, Bruix J. Phase i study investigating everolimus combined with sorafenib in patients with advanced hepatocellular carcinoma. J Hepatol. 2013; 59(6):1271–7. https://doi.org/10.1016/j.jhep.2013.07.029.
DeAngelo DJ, Spencer A, Bhalla KN, Prince HM, Fischer T, Kindler T, Giles FJ, Scott JW, Parker K, Liu A, Woo M, Atadja P, Mishra KK, Ottmann OG. Phase ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia. 2013; 27(8):1628–36. https://doi.org/10.1038/leu.2013.38.
Chiappella A, Tucci A, Castellino A, Pavone V, Baldi I, Carella AM, Orsucci L, Zanni M, Salvi F, Liberati AM, Gaidano G, Bottelli C, Rossini B, Perticone S, Masi PD, Ladetto M, Ciccone G, Palumbo A, Rossi G, and UV. Lenalidomide plus cyclophosphamide, doxorubicin, vincristine, prednisone and rituximab is safe and effective in untreated, elderly patients with diffuse large b-cell lymphoma: a phase i study by the fondazione italiana linfomi. Haematologica. 2013; 98(11):1732–8. https://doi.org/10.3324/haematol.2013.085134.
Cannon DM, Mehta MP, Adkison JB, Khuntia D, Traynor AM, Tomé WA, Chappell RJ, Tolakanahalli R, Mohindra P, Bentzen SM, Cannon GM. Dose-limiting toxicity after hypofractionated dose-escalated radiotherapy in non–small-cell lung cancer. J Clin Oncol. 2013; 31(34):4343–8. https://doi.org/10.1200/jco.2013.51.5353.
Angevin E, Lopez-Martin JA, Lin C-C, Gschwend JE, Harzstark A, Castellano D, Soria J-C, Sen P, Chang J, Shi M, Kay A, Escudier B. Phase i study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin Cancer Res. 2013; 19(5):1257–68. https://doi.org/10.1158/1078-0432.ccr-12-2885.
Foster MC, Amin C, Voorhees PM, van Deventer HW, Richards KL, Ivanova A, Whitman J, Chiu WM, Barr ND, Shea T. A phase i dose-escalation study of clofarabine in combination with fractionated gemtuzumab ozogamicin in patients with refractory or relapsed acute myeloid leukemia. Leuk Lymphoma. 2012; 53(7):1331–7. https://doi.org/10.3109/10428194.2011.647313.
Tsien CI, Brown D, Normolle D, Schipper M, Piert M, Junck L, Heth J, Gomez-Hassan D, Haken RKT, Chenevert T, Cao Y, Lawrence T. Concurrent temozolomide and dose-escalated intensity-modulated radiation therapy in newly diagnosed glioblastoma. Clin Cancer Res. 2011; 18(1):273–9. https://doi.org/10.1158/1078-0432.ccr-11-2073.
Tevaarwerk A, Wilding G, Eickhoff J, Chappell R, Sidor C, Arnott J, Bailey H, Schelman W, Liu G. Phase i study of continuous MKC-1 in patients with advanced or metastatic solid malignancies using the modified time-to-event continual reassessment method (TITE-CRM) dose escalation design. Investig New Drugs. 2011; 30(3):1039–45. https://doi.org/10.1007/s10637-010-9629-6.
Sinha R, Kaufman JL, Khoury HJ, King N, Shenoy PJ, Lewis C, Bumpers K, Hutchison-Rzepka A, Tighiouart M, Heffner LT, Lechowicz MJ, Lonial S, Flowers CR. A phase 1 dose escalation study of bortezomib combined with rituximab, cyclophosphamide, doxorubicin, modified vincristine, and prednisone for untreated follicular lymphoma and other low-grade b-cell lymphomas. Cancer. 2012; 118(14):3538–48. https://doi.org/10.1002/cncr.26660.
Schneider BJ, Kalemkerian GP, Bradley D, Smith DC, Egorin MJ, Daignault S, Dunn R, Hussain M. Phase i study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Investig New Drugs. 2010; 30(1):249–57. https://doi.org/10.1007/s10637-010-9503-6.
Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DCS, Xiong H, Cui Y, Busman TA, McKeegan EM, Krivoshik AP, Enschede SH, Humerickhouse R. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase i study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012; 30(5):488–96. https://doi.org/10.1200/jco.2011.34.7898.
Reardon DA, Vredenburgh JJ, Desjardins A, Peters KB, Sathornsumetee S, Threatt S, Sampson JH, Herndon JE, Coan A, McSherry F, Rich JN, McLendon RE, Zhang S, Friedman HS. Phase 1 trial of dasatinib plus erlotinib in adults with recurrent malignant glioma. J Neuro-Oncol. 2012; 108(3):499–506. https://doi.org/10.1007/s11060-012-0848-x.
Moulder S, Gladish G, Ensor J, Gonzalez-Angulo AM, Cristofanilli M, Murray JL, Booser D, Giordano SH, Brewster A, Moore J, Rivera E, Hortobagyi GN, Tran HT. A phase 1 study of weekly everolimus (RAD001) in combination with docetaxel in patients with metastatic breast cancer. Cancer. 2011; 118(9):2378–84. https://doi.org/10.1002/cncr.26571.
Mazard T, Ychou M, Thezenas S, Poujol S, Pinguet F, Thirion A, Bleuse JP, Portales F, Samalin E, Assenat E. Feasibility of biweekly combination chemotherapy with capecitabine, irinotecan, and oxaliplatin in patients with metastatic solid tumors: results of a two-step phase i trial: XELIRI and XELIRINOX. Cancer Chemother Pharmacol. 2011; 69(3):807–14. https://doi.org/10.1007/s00280-011-1764-z.
Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC, Mita M, Cuero MM, Stutvoet S, Birle D, Anak O, Hackl W, Baselga J. Phase i safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol. 2012; 23(9):2399–408. https://doi.org/10.1093/annonc/mds011.
Lu C, Stewart DJ, Lee JJ, Ji L, Ramesh R, Jayachandran G, Nunez MI, Wistuba II, Erasmus JJ, Hicks ME, Grimm EA, Reuben JM, Baladandayuthapani V, Templeton NS, McMannis JD, Roth JA. Phase i clinical trial of systemically administered TUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PLoS ONE. 2012; 7(4):34833. https://doi.org/10.1371/journal.pone.0034833.
Kawahara M, Kubo A, Komuta K, Fujita Y, Sasaki Y, Fukushima M, Daimon T, Furuse K, Mishima M, Mio T. A phase i study of amrubicin and fixed dose of irinotecan (CPT-11) in relapsed small cell lung cancer: Japan multinational trial organization LC0303. J Thorac Oncol. 2012; 7(12):1845–9. https://doi.org/10.1097/jto.0b013e3181e47a62.
Jones DR, Moskaluk CA, Gillenwater HH, Petroni GR, Burks SG, Philips J, Rehm PK, Olazagasti J, Kozower BD, Bao Y. Phase i trial of induction histone deacetylase and proteasome inhibition followed by surgery in non–small-cell lung cancer. J Thor Oncol. 2012; 7(11):1683–90. https://doi.org/10.1097/jto.0b013e318267928d.
Jakubowiak AJ, Dytfeld D, Griffith KA, Lebovic D, Vesole DH, Jagannath S, Al-Zoubi A, Anderson T, Nordgren B, Detweiler-Short K, Stockerl-Goldstein K, Ahmed A, Jobkar T, Durecki DE, McDonnell K, Mietzel M, Couriel D, Kaminski M, Vij R. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood. 2012; 120(9):1801–9. https://doi.org/10.1182/blood-2012-04-422683.
Geletneky K, Huesing J, Rommelaere J, Schlehofer JR, Leuchs B, Dahm M, Krebs O, von Knebel Doeberitz M, Huber B, Hajda J. Phase i/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of parvovirus h-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer. 2012; 12(1). https://doi.org/10.1186/1471-2407-12-99.
Feng M, Smith DE, Normolle DP, Knol JA, Pan CC, Ben-Josef E, Lu Z, Feng MR, Chen J, Ensminger W, Lawrence TS. A phase i clinical and pharmacology study using amifostine as a radioprotector in dose-escalated whole liver radiation therapy. Int J Radiat Oncol Biol Phys. 2012; 83(5):1441–7. https://doi.org/10.1016/j.ijrobp.2011.10.020.
Farid M, Chowbay B, Chen X, Tan SH, Ramasamy S, Koo WH, Toh HC, Choo SP, Ong SYK. Phase i pharmacokinetic study of chronomodulated dose-intensified combination of capecitabine and oxaliplatin (XELOX) in metastatic colorectal cancer. Cancer Chemother Pharmacol. 2012; 70(1):141–50. https://doi.org/10.1007/s00280-012-1895-x.
Crew KD, Brown P, Greenlee H, Bevers TB, Arun B, Hudis C, McArthur HL, Chang J, Rimawi M, Vornik L, Cornelison TL, Wang A, Hibshoosh H, Ahmed A, Terry MB, Santella RM, Lippman SM, Hershman DL. Phase IB randomized, double-blinded, placebo-controlled, dose escalation study of polyphenon e in women with hormone receptor-negative breast cancer. Cancer Prev Res. 2012; 5(9):1144–54. https://doi.org/10.1158/1940-6207.capr-12-0117.
Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, Buck SSD, Ru QC, Peters M, Goldbrunner M, Baselga J. Phase i, dose-escalation study of BKM120, an oral pan-class i PI3k inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012; 30(3):282–90. https://doi.org/10.1200/jco.2011.36.1360.
Ben-Josef E, Schipper M, Francis IR, Hadley S, Ten-Haken R, Lawrence T, Normolle D, Simeone DM, Sonnenday C, Abrams R, Leslie W, Khan G, Zalupski MM. A phase i/II trial of intensity modulated radiation (IMRT) dose escalation with concurrent fixed-dose rate gemcitabine (FDR-g) in patients with unresectable pancreatic cancer. Int J Radiat Oncol Biol Phys. 2012; 84(5):1166–71. https://doi.org/10.1016/j.ijrobp.2012.02.051.
Stasi AD, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK. Inducible apoptosis as a safety switch for adoptive cell therapy. New Engl J Med. 2011; 365(18):1673–83. https://doi.org/10.1056/nejmoa1106152.
Warren KE, Goldman S, Pollack IF, Fangusaro J, Schaiquevich P, Stewart CF, Wallace D, Blaney SM, Packer R, MacDonald T, Jakacki R, Boyett JM, Kun LE. Phase i trial of lenalidomide in pediatric patients with recurrent, refractory, or progressive primary CNS tumors: Pediatric brain tumor consortium study PBTC-018. J Clin Oncol. 2011; 29(3):324–9. https://doi.org/10.1200/jco.2010.31.3601.
Vansteenkiste J, Solomon B, Boyer M, Wolf J, Miller N, Scala LD, Pylvaenaeinen I, Petrovic K, Dimitrijevic S, Anrys B, Laack E. Everolimus in combination with pemetrexed in patients with advanced non-small cell lung cancer previously treated with chemotherapy: A phase i study using a novel, adaptive bayesian dose-escalation model. J Thor Oncol. 2011; 6(12):2120–9. https://doi.org/10.1097/jto.0b013e3182307ede.
Terakura S, Atsuta Y, Sawa M, Ohashi H, Kato T, Nishiwaki S, Imahashi N, Yasuda T, Murata M, Miyamura K, Suzuki R, Naoe T, Ito T, Morishita Y. A prospective dose-finding trial using a modified continual reassessment method for optimization of fludarabine plus melphalan conditioning for marrow transplantation from unrelated donors in patients with hematopoietic malignancies. Annals Oncol. 2011; 22(8):1865–71. https://doi.org/10.1093/annonc/mdq673.
Smith BD, Jones RJ, Cho E, Kowalski J, Karp JE, Gore SD, Vala M, Meade B, Baker SD, Zhao M, Piantadosi S, Zhang Z, Blumenthal G, Warlick ED, Brodsky RA, Murgo A, Rudek MA, Matsui WH. Differentiation therapy in poor risk myeloid malignancies: Results of a dose finding study of the combination bryostatin-1 and GM-CSF. Leuk Res. 2011; 35(1):87–94. https://doi.org/10.1016/j.leukres.2010.06.001.
Satoh T, Ura T, Yamada Y, Yamazaki K, Tsujinaka T, Munakata M, Nishina T, Okamura S, Esaki T, Sasaki Y, Koizumi W, Kakeji Y, Ishizuka N, Hyodo I, Sakata Y. Genotype-directed, dose-finding study of irinotecan in cancer patients with UGT1a1*28 and/or UGT1a1*6 polymorphisms. Cancer Sci. 2011; 102(10):1868–73. https://doi.org/10.1111/j.1349-7006.2011.02030.x.
Mehnert JM, Tan AR, Moss R, Poplin E, Stein MN, Sovak M, Levinson K, Lin H, Kane M, Gounder M, Lin Y, Shih WJ, White E, Rubin EH, Karantza V. Rationally designed treatment for solid tumors with MAPK pathway activation: A phase i study of paclitaxel and bortezomib using an adaptive dose-finding approach. Mol Cancer Ther. 2011; 10(8):1509–19. https://doi.org/10.1158/1535-7163.mct-10-0944.
Magenau J, Tobai H, Pawarode A, Braun T, Peres E, Reddy P, Kitko C, Choi S, Yanik G, Frame D, Harris A, Erba H, Kujawski L, Elenitoba-Johnson K, Sanks J, Jones D, Paczesny S, Ferrara J, Levine J, Mineishi S. Clofarabine and busulfan conditioning facilitates engraftment and provides significant antitumor activity in nonremission hematologic malignancies. Blood. 2011; 118(15):4258–64. https://doi.org/10.1182/blood-2011-06-358010.
Koolen SLW, Witteveen PO, Jansen RS, Langenberg MHG, Kronemeijer RH, Nol A, Garcia-Ribas I, Callies S, Benhadji KA, Slapak CA, Beijnen JH, Voest EE, Schellens JHM. Phase i study of oral gemcitabine prodrug (LY2334737) alone and in combination with erlotinib in patients with advanced solid tumors. Clin Cancer Res. 2011; 17(18):6071–82. https://doi.org/10.1158/1078-0432.ccr-11-0353.
Kim KB, Chesney J, Robinson D, Gardner H, Shi MM, Kirkwood JM. Phase i/II and pharmacodynamic study of dovitinib (TKI258), an inhibitor of fibroblast growth factor receptors and VEGF receptors, in patients with advanced melanoma. Clin Cancer Res. 2011; 17(23):7451–61. https://doi.org/10.1158/1078-0432.ccr-11-1747.
Jerusalem G, Fasolo A, Dieras V, Cardoso F, Bergh J, Vittori L, Zhang Y, Massacesi C, Sahmoud T, Gianni L. Phase i trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat. 2010; 125(2):447–55. https://doi.org/10.1007/s10549-010-1260-x.
Jakubowiak AJ, Griffith KA, Reece DE, Hofmeister CC, Lonial S, Zimmerman TM, Campagnaro EL, Schlossman RL, Laubach JP, Raje NS, Anderson T, Mietzel MA, Harvey CK, Wear SM, Barrickman JC, Tendler CL, Esseltine D-L, Kelley SL, Kaminski MS, Anderson KC, Richardson PG. Lenalidomide, bortezomib, pegylated liposomal doxorubicin, and dexamethasone in newly diagnosed multiple myeloma: a phase 1/2 multiple myeloma research consortium trial. Blood. 2011; 118(3):535–43. https://doi.org/10.1182/blood-2011-02-334755.
Geoerger B, Hargrave D, Thomas F, Ndiaye A, Frappaz D, Andreiuolo F, Varlet P, Aerts I, Riccardi R, Jaspan T, Chatelut E, Deley M-CL, Paoletti X, Saint-Rose C, Leblond P, Morland B, Gentet J-C, Meresse V, and GV. Innovative therapies for children with cancer pediatric phase i study of erlotinib in brainstem glioma and relapsing/refractory brain tumors. Neuro-Oncology. 2010; 13(1):109–18. https://doi.org/10.1093/neuonc/noq141.
Gandhi L, Camidge DR, de Oliveira MR, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM, Litvinovich E, Hemken PM, Dive C, Enschede SH, Nolan C, Chiu Y-L, Busman T, Xiong H, Krivoshik AP, Humerickhouse R, Shapiro GI, Rudin CM. Phase i study of navitoclax (ABT-263), a novel bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011; 29(7):909–16. https://doi.org/10.1200/jco.2010.31.6208.
Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, Packer RJ, Goldman S, Gururangan S, Gajjar A, Demuth T, Kun LE, Boyett JM, Gilbertson RJ. Phase i trial of MK-0752 in children with refractory CNS malignancies: A pediatric brain tumor consortium study. J Clin Oncol. 2011; 29(26):3529–34. https://doi.org/10.1200/jco.2011.35.7806.
Rathkopf D, Wong BY, Ross RW, Anand A, Tanaka E, Woo MM, Hu J, Dzik-Jurasz A, Yang W, Scher HI. A phase i study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2010; 66(1):181–9. https://doi.org/10.1007/s00280-010-1289-x.
Peereboom DM, Supko JG, Carson KA, Batchelor T, Phuphanich S, Lesser G, Mikkelson T, Fisher J, Desideri S, He X, Grossman SA. A phase i/II trial and pharmacokinetic study of ixabepilone in adult patients with recurrent high-grade gliomas. J Neuro-Oncol. 2010; 100(2):261–8. https://doi.org/10.1007/s11060-010-0190-0.
O’Donnell PH, Artz AS, Undevia SD, Pai RK, Cerro PD, Horowitz S, Godley LA, Hart J, Innocenti F, Larson RA, Odenike OM, Stock W, Besien KV. Phase i study of dose-escalated busulfan with fludarabine and alemtuzumab as conditioning for allogeneic hematopoietic stem cell transplant: reduced clearance at high doses and occurrence of late sinusoidal obstruction syndrome/veno-occlusive disease. Leuk Lymphoma. 2010; 51(12):2240–9. https://doi.org/10.3109/10428194.2010.520773.
Furman RR, Martin P, Ruan J, Cheung Y-KK, Vose JM, LaCasce AS, Elstrom R, Coleman M, Leonard JP. Phase 1 trial of bortezomib plus r-CHOP in previously untreated patients with aggressive non-hodgkin lymphoma. Cancer. 2010; 116(23):5432–9. https://doi.org/10.1002/cncr.25509.
Fouladi M, Stewart CF, Blaney SM, Onar-Thomas A, Schaiquevich P, Packer RJ, Gajjar A, Kun LE, Boyett JM, Gilbertson RJ. Phase i trial of lapatinib in children with refractory CNS malignancies: A pediatric brain tumor consortium study. J Clin Oncol. 2010; 28(27):4221–7. https://doi.org/10.1200/jco.2010.28.4687.
Andre F, Campone M, O’Regan R, Manlius C, Massacesi C, Tarek S, Mukhopadhyay P, Soria J-C, Naughton M, Hurvitz SA. Phase i study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol. 2010; 28(34):5110–5. https://doi.org/10.1200/jco.2009.27.8549.
Loeb DM, Garrett-Mayer E, Hobbs RF, Prideaux AR, Sgouros G, Shokek O, Wharam MD, Scott T, Schwartz CL. Dose-finding study of153sm-EDTMP in patients with poor-prognosis osteosarcoma. Cancer. 2009; 115(11):2514–22. https://doi.org/10.1002/cncr.24286.
Li B. -s., Zhou T, Wang Z. -t., Li H. -s., Sun H. -f., Zhang Z. -c., Lin H. -q., Wei Y. -m., Gong H. -y., Huang W, Yi Y, Wang L. -y.Phase i study of concurrent selective lymph node late course accelerated hyper-fractionated radiotherapy and capecitabine, cisplatin for locally advanced esophageal squamous cell carcinoma. Radiother Oncol. 2009; 93(3):458–61. https://doi.org/10.1016/j.radonc.2009.08.002.
Demetri GD, Russo PL, MacPherson IRJ, Wang D, Morgan JA, Brunton VG, Paliwal P, Agrawal S, Voi M, Evans TRJ. Phase i dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res. 2009; 15(19):6232–40. https://doi.org/10.1158/1078-0432.ccr-09-0224.
Borghaei H, Alpaugh K, Hedlund G, Forsberg G, Langer C, Rogatko A, Hawkins R, Dueland S, Lassen U, Cohen RB. Phase i dose escalation, pharmacokinetic and pharmacodynamic study of naptumomab estafenatox alone in patients with advanced cancer and with docetaxel in patients with advanced non–small-cell lung cancer. J Clin Oncol. 2009; 27(25):4116–23. https://doi.org/10.1200/jco.2008.20.2515.
Bailey DB, Rassnick KM, Dykes NL, Pendyala L. Phase i evaluation of carboplatin by use of a dosing strategy based on a targeted area under the platinum concentration-versus-time curve and individual glomerular filtration rate in cats with tumors. Am J Vet Res. 2009; 70(6):770–6. https://doi.org/10.2460/ajvr.70.6.770.
Trümper L, Zwick C, Ziepert M, Hohloch K, Schmits R, Mohren M, Liersch R, Bentz M, Graeven U, Wruck U, Hoffmann M, Metzner B, Hasenclever D, Loeffler M, Pfreundschuh M. Dose-escalated CHOEP for the treatment of young patients with aggressive non-hodgkin’s lymphoma: I. a randomized dose escalation and feasibility study with bi- and tri-weekly regimens. Ann Oncol. 2008; 19(3):538–44. https://doi.org/10.1093/annonc/mdm497.
Saji S, Toi M, Morita S, Iwata H, Ito Y, Ohno S, Kobayashi T, Hozumi Y, Sakamoto J. Dose-finding phase i and pharmacokinetic study of capecitabine (xeloda) in combination with epirubicin and cyclophosphamide (CEX) in patients with inoperable or metastatic breast cancer. Oncology. 2007; 72(5-6):330–7. https://doi.org/10.1159/000113062.
Rao S, Starling N, Cunningham D, Benson M, Wotherspoon A, Lüpfert C, Kurek R, Oates J, Baselga J, Hill A. Phase i study of epirubicin, cisplatin and capecitabine plus matuzumab in previously untreated patients with advanced oesophagogastric cancer. Br J Cancer. 2008; 99(6):868–74. https://doi.org/10.1038/sj.bjc.6604622.
Neuenschwander B, Branson M, Gsponer T. Critical aspects of the bayesian approach to phase i cancer trials. Stat Med. 2008; 27(13):2420–39. https://doi.org/10.1002/sim.3230.
MacDonald TJ, Stewart CF, Kocak M, Goldman S, Ellenbogen RG, Phillips P, Lafond D, Poussaint TY, Kieran MW, Boyett JM, Kun LE. Phase i clinical trial of cilengitide in children with refractory brain tumors: Pediatric brain tumor consortium study PBTC-012. J Clin Oncol. 2008; 26(6):919–24. https://doi.org/10.1200/jco.2007.14.1812.
Jimeno A, Rudek MA, Kulesza P, Ma WW, Wheelhouse J, Howard A, Khan Y, Zhao M, Jacene H, Messersmith WA, Laheru D, Donehower RC, Garrett-Mayer E, Baker SD, Hidalgo M. Pharmacodynamic-guided modified continuous reassessment method–based, dose-finding study of rapamycin in adult patients with solid tumors. J Clin Oncol. 2008; 26(25):4172–9. https://doi.org/10.1200/jco.2008.16.2347.
Gururangan S, Turner CD, Stewart CF, O’Shaughnessy M, Kocak M, Poussaint TY, Phillips PC, Goldman S, Packer R, Pollack IF, Blaney SM, Karsten V, Gerson SL, Boyett JM, Friedman HS, Kun LE. Phase i trial of VNP40101m (cloretazine) in children with recurrent brain tumors: A pediatric brain tumor consortium study. Clin Cancer Res. 2008; 14(4):1124–30. https://doi.org/10.1158/1078-0432.ccr-07-4242.
Guillot B, Khamari A, Cupissol D, Delaunay M, Bedane C, Dreno B, Picot MC, Dereure O. Temozolomide associated with PEG-interferon in patients with metastatic melanoma: a multicenter prospective phase i/II study. Melanoma Res. 2008; 18(2):141–6. https://doi.org/10.1097/cmr.0b013e3282f6309c.
Grossman SA, Carson KA, Phuphanich S, Batchelor T, Peereboom D, Nabors LB, Lesser G, Hausheer F, Supko JG. Phase i and pharmacokinetic study of karenitecin in patients with recurrent malignant gliomas. Neuro-Oncology. 2008; 10(4):608–16. https://doi.org/10.1215/15228517-2008-030.
de Bono JS, Kristeleit R, Tolcher A, Fong P, Pacey S, Karavasilis V, Mita M, Shaw H, Workman P, Kaye S, Rowinsky EK, Aherne W, Atadja P, Scott JW, Patnaik A. Phase i pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin Cancer Res. 2008; 14(20):6663–73. https://doi.org/10.1158/1078-0432.ccr-08-0376.
Adkison JB, Khuntia D, Bentzen SM, Cannon GM, Tome WA, Jaradat H, Walker W, Traynor AM, Weigel T, Mehta MP. Dose escalated, hypofractionated radiotherapy using helical tomotherapy for inoperable non-small cell lung cancer: Preliminary results of a risk-stratified phase i dose escalation study. Technol Cancer Res Treat. 2008; 7(6):441–7. https://doi.org/10.1177/153303460800700605.
Atzori F, Tabernero J, Cervantes A, Prudkin L, Andreu J, Rodriguez-Braun E, Domingo A, Guijarro J, Gamez C, Rodon J, Cosimo SD, Brown H, Clark J, Hardwick JS, Beckman RA, Hanley WD, Hsu K, Calvo E, Rosello S, Langdon RB, Baselga J. A phase i pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2011; 17(19):6304–12. https://doi.org/10.1158/1078-0432.ccr-10-3336.
Berenson JR, Yellin O, Patel R, Duvivier H, Nassir Y, Mapes R, Abaya CD, Swift RA. A phase i study of samarium lexidronam/bortezomib combination therapy for the treatment of relapsed or refractory multiple myeloma. Clin Cancer Res. 2009; 15(3):1069–75. https://doi.org/10.1158/1078-0432.ccr-08-1261.
Conroy T, Viret F, François E, Seitz JF, Boige V, Ducreux M, Ychou M, Metges JP, Giovannini M, Yataghene Y, Peiffert D. Phase i trial of oxaliplatin with fluorouracil, folinic acid and concurrent radiotherapy for oesophageal cancer. Br J Cancer. 2008; 99(9):1395–401. https://doi.org/10.1038/sj.bjc.6604708.
Day FL, Leong T, Ngan S, Thomas R, Jefford M, Zalcberg JR, Rischin D, McKendick J, Milner AD, Iulio JD, Matera A, Michael M. Phase i trial of docetaxel, cisplatin and concurrent radical radiotherapy in locally advanced oesophageal cancer. Br J Cancer. 2010; 104(2):265–71. https://doi.org/10.1038/sj.bjc.6606051.
Dong M, Ning Z-Q, Xing P-Y, Xu J-L, Cao H-X, Dou G-F, Meng Z-Y, Shi Y-K, Lu X-P, Feng F-Y. Phase i study of chidamide (CS055/HBI-8000), a new histone deacetylase inhibitor, in patients with advanced solid tumors and lymphomas. Cancer Chemother Pharmacol. 2012; 69(6):1413–22. https://doi.org/10.1007/s00280-012-1847-5.
Forsyth P, Roldán G, George D, Wallace C, Palmer CA, Morris D, Cairncross G, Matthews MV, Markert J, Gillespie Y, Coffey M, Thompson B, Hamilton M. A phase i trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008; 16(3):627–32. https://doi.org/10.1038/sj.mt.6300403.
Frost A, Mross K, Steinbild S, Hedbom S, Unger C, Kaiser R, Trommeshauser D, Munzert G. Phase i study of the plk1 inhibitor BI 2536 administered intravenously on three consecutive days in advanced solid tumours. Curr Oncol. 2012; 19(1). https://doi.org/10.3747/co.19.866.
Gojo I, Tan M, Fang H-B, Sadowska M, Lapidus R, Baer MR, Carrier F, Beumer JH, Anyang BN, Srivastava RK, et al. Translational phase i trial of vorinostat (suberoylanilide hydroxamic acid) combined with cytarabine and etoposide in patients with relapsed, refractory, or high-risk acute myeloid leukemia. Clin Cancer Res. 2013; 19(7):1838–51.
Kantarjian HM, Padmanabhan S, Stock W, Tallman MS, Curt GA, Li J, Osmukhina A, Wu K, Huszar D, Borthukar G, Faderl S, Garcia-Manero G, Kadia T, Sankhala K, Odenike O, Altman JK, Minden M. Phase i/II multicenter study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of AZD4877 in patients with refractory acute myeloid leukemia. Investig New Drugs. 2011; 30(3):1107–15. https://doi.org/10.1007/s10637-011-9660-2.
Kim KB, Hwu W-J, Papadopoulos NE, Bedikian AY, Camacho LH, Ng C, Hernandez IM, Frost AM, Jack MA, Hwu P. Phase i study of the combination of docetaxel, temozolomide and cisplatin in patients with metastatic melanoma. Cancer Chemother Pharmacol. 2008; 64(1):161–7. https://doi.org/10.1007/s00280-008-0861-0.
Kimball KJ, Preuss MA, Barnes MN, Wang M, Siegal GP, Wan W, Kuo H, Saddekni S, Stockard CR, Grizzle WE, Harris RD, Aurigemma R, Curiel DT, Alvarez RD. A phase i study of a tropism-modified conditionally replicative adenovirus for recurrent malignant gynecologic diseases. Clin Cancer Res. 2010; 16(21):5277–87. https://doi.org/10.1158/1078-0432.ccr-10-0791.
Kunz PL, He AR, Colevas AD, Pishvaian MJ, Hwang JJ, Clemens PL, Messina M, Kaleta R, Abrahao F, Sikic BI, Marshall JL. Phase i trial of ixabepilone administered as three oral doses each separated by 6 hours every 3 weeks in patients with advanced solid tumors. Investig New Drugs. 2012; 30(6):2364–70. https://doi.org/10.1007/s10637-012-9800-3.
Lee K-W, Park SR, Oh D-Y, Park Y-I, Khosravan R, Lin X, Lee S-Y, Roh E-J, Valota O, Lechuga MJ, Bang Y-J. Phase i study of sunitinib plus capecitabine/cisplatin or capecitabine/oxaliplatin in advanced gastric cancer. Investig New Drugs. 2013; 31(6):1547–58. https://doi.org/10.1007/s10637-013-0032-y.
LoRusso P, Venkatakrishnan K, Chiorean EG, Noe D, Wu J-T, Sankoh S, Corvez M, Sausville EA. Phase 1 dose-escalation, pharmacokinetic, and cerebrospinal fluid distribution study of TAK-285, an investigational inhibitor of EGFR and HER2. Investig New Drugs. 2013; 32(1):160–70. https://doi.org/10.1007/s10637-013-9988-x.
Luke JJ, D’Adamo DR, Dickson MA, Keohan ML, Carvajal RD, Maki RG, de Stanchina E, Musi E, Singer S, Schwartz GK. The cyclin-dependent kinase inhibitor flavopiridol potentiates doxorubicin efficacy in advanced sarcomas: Preclinical investigations and results of a phase i dose-escalation clinical trial. Clin Cancer Res. 2012; 18(9):2638–47. https://doi.org/10.1158/1078-0432.ccr-11-3203.
Michaelson MD, Zhu AX, Ryan DP, McDermott DF, Shapiro GI, Tye L, Chen I, Stephenson P, Patyna S, Ruiz-Garcia A, Schwarzberg AB. Sunitinib in combination with gemcitabine for advanced solid tumours: a phase i dose-finding study. Br J Cancer. 2013; 108(7):1393–401. https://doi.org/10.1038/bjc.2013.96.
Mita M, Kelly KR, Mita A, Ricart AD, Romero O, Tolcher A, Hook L, Okereke C, Krivelevich I, Rossignol DP, Giles FJ, Rowinsky EK, Takimoto C. Phase i study of e7820, an oral inhibitor of integrin -2 expression with antiangiogenic properties, in patients with advanced malignancies. Clin Cancer Res. 2011; 17(1):193–200. https://doi.org/10.1158/1078-0432.ccr-10-0010.
Oki Y, Buglio D, Fanale M, Fayad L, Copeland A, Romaguera J, Kwak LW, Pro B, de Castro Faria S, Neelapu S, Fowler N, Hagemeister F, Zhang J, Zhou S, Feng L, Younes A. Phase i study of panobinostat plus everolimus in patients with relapsed or refractory lymphoma. Clin Cancer Res. 2013; 19(24):6882–90. https://doi.org/10.1158/1078-0432.ccr-13-1906.
Pollyea DA, Kohrt HE, Gallegos L, Figueroa ME, Abdel-Wahab O, Zhang B, Bhattacharya S, Zehnder J, Liedtke M, Gotlib JR, Coutre S, Berube C, Melnick A, Levine R, Mitchell BS, Medeiros BC. Safety, efficacy and biological predictors of response to sequential azacitidine and lenalidomide for elderly patients with acute myeloid leukemia. Leukemia. 2011; 26(5):893–901. https://doi.org/10.1038/leu.2011.294.
Rao AV, Rizzieri DA, DeCastro CM, Diehl LF, Lagoo AS, Moore JO, Gockerman JP. Phase i study of dose dense induction and consolidation with gemtuzumab ozogamicin and high dose cytarabine in older adults with AML. J Geriatr Oncol. 2012; 3(3):220–7. https://doi.org/10.1016/j.jgo.2012.02.002.
Sanborn SL, Cooney MM, Dowlati A, Brell JM, Krishnamurthi S, Gibbons J, Bokar JA, Nock C, Ness A, Remick SC. Phase i trial of docetaxel and thalidomide: a regimen based on metronomic therapeutic principles. Investig New Drugs. 2008; 26(4):355–62. https://doi.org/10.1007/s10637-008-9137-0.
Shapiro GI, McCallum S, Adams LM, Sherman L, Weller S, Swann S, Keer H, Miles D, Müller T, LoRusso P. A phase 1 dose-escalation study of the safety and pharmacokinetics of once-daily oral foretinib, a multi-kinase inhibitor, in patients with solid tumors. Investig New Drugs. 2012; 31(3):742–50. https://doi.org/10.1007/s10637-012-9881-z.
Srivastava S, Jones D, Wood LL, Schwartz JE, Nelson RP, Abonour R, Secrest A, Cox E, Baute J, Sullivan C, Kane K, Robertson MJ, Farag SS. A phase i trial of high-dose clofarabine, etoposide, and cyclophosphamide and autologous peripheral blood stem cell transplantation in patients with primary refractory and relapsed and refractory non-hodgkin lymphoma. Biol Blood Marrow Transpl. 2011; 17(7):987–94. https://doi.org/10.1016/j.bbmt.2010.10.016.
Tsimberidou AM, Camacho LH, Verstovsek S, Ng C, Hong DS, Uehara CK, Gutierrez C, Daring S, Stevens J, Komarnitsky PB, Schwartz B, Kurzrock R. A phase i clinical trial of darinaparsin in patients with refractory solid tumors. Clin Cancer Res. 2009; 15(14):4769–76. https://doi.org/10.1158/1078-0432.ccr-08-2984.
Yamamoto K, Kokawa K, Umesaki N, Nishimura R, Hasegawa K, Konishi I, Saji F, Nishida M, Noguchi H, Takizawa K. Phase i study of combination chemotherapy with irinotecan hydrochloride and nedaplatin for cervical squamous cell carcinoma: Japanese gynecologic oncology group study. Oncol Rep. 2009; 21(4):1005–9.
Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A. Brentuximab vedotin (sgn-35) for relapsed cd30-positive lymphomas. New Engl J Med. 2010; 363(19):1812–21.
Garcia JA, Mekhail T, Elson P, Wood L, Bukowski RM, Dreicer R, Rini BI. Phase i/II trial of subcutaneous interleukin-2, granulocyte-macrophage colony-stimulating factor and interferon-alpha in patients with metastatic renal cell carcinoma. BJU Int. 2011; 109(1):63–69. https://doi.org/10.1111/j.1464-410x.2010.10011.x.
Saraiya B, Gounder M, Dutta J, Saleem A, Collazo C, Zimmerman L, Nazar A, Gharibo M, Schaar D, Lin Y, Shih W, Aisner J, Strair RK, Rubin EH. Sequential topoisomerase targeting and analysis of mechanisms of resistance to topotecan in patients with acute myelogenous leukemia. Anti-Cancer Drugs. 2008; 19(4):411–20. https://doi.org/10.1097/cad.0b013e3282f5218b.
Spunt SL, Grupp SA, Vik TA, Santana VM, Greenblatt DJ, Clancy J, Berkenblit A, Krygowski M, Ananthakrishnan R, Boni JP, Gilbertson RJ. Phase i study of temsirolimus in pediatric patients with recurrent/refractory solid tumors. J Clin Oncol. 2011; 29(21):2933–40. https://doi.org/10.1200/jco.2010.33.4649.
Yu KH, Yu SCH, Hui EP, Kam MKM, Vlantis AC, Yuen E, Chan ATC. Accelerated fractionation radiotherapy and late intensification with 2 intra-arterial cisplatin infusions for locally advanced head and neck squamous cell carcinoma. Head Neck. 2009. https://doi.org/10.1002/hed.21279.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45(2):228–47. https://doi.org/10.1016/j.ejca.2008.10.026. Publisher: Elsevier Ltd ISBN: 1879-0852 (Electronic).
Cheson BD, Bennett JM, Kopecky KJ, Büchner T, Willman CL, Estey EH, Schiffer CA, Doehner H, Tallman MS, Lister TA, Lo-Coco F, Willemze R, Biondi A, Hiddemann W, Larson RA, Löwenberg B, Sanz MA, Head DR, Ohno R, Bloomfield CD, LoCocco F, International Working Group for Diagnosis StandardizationofResponseCriteria Treatment Outcomes. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol Off J Am Soc Clin Oncol. 2003; 21(24):4642–9. https://doi.org/10.1200/JCO.2003.04.036.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating M, Montserrat E, Chiorazzi N, Stilgenbauer S, Rai KR, Byrd JC, Eichhorst B, O’Brien S, Robak T, Seymour JF, Kipps TJ. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018; 131(25):2745–60. https://doi.org/10.1182/blood-2017-09-806398.
Brock K, Homer V, Soul G, Potter C, Chiuzan C, Lee S. Dose-level toxicity and efficacy outcomes from dose-finding clinical trials in oncology. 2019. https://doi.org/10.25500/edata.bham.00000337.
Macdougall J. Analysis of Dose–Response Studies–Emax Model. In: Dose Finding in Drug Development. New York: Springer: 2006.
Bornkamp B. DoseFinding: Planning and Analyzing Dose Finding Experiments. 2019. R package version 0.9-17. https://CRAN.R-project.org/package=DoseFinding. Accessed 14 Dec 2020.
Bürkner P-C. Advanced Bayesian multilevel modeling with the R package brms. The R Journal. 2018; 10(1):395–411. https://doi.org/10.32614/RJ-2018-017.
R Core Team R. A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2020. R Foundation for Statistical Computing. https://www.R-project.org/. Accessed 14 Dec 2020.
Wickham H, Averick M, Bryan J, Chang W, McGowan LD, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen TL, Miller E, Bache SM, Mus̈ller K, Ooms J, Robinson D, Seidel DP, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H. Welcome to the tidyverse. J Open Source Softw. 2019; 4(43):1686. https://doi.org/10.21105/joss.01686.
Kay M. tidybayes: Tidy Data and Geoms for Bayesian Models. 2020. https://doi.org/10.5281/zenodo.1308151. R package version 2.0.3. http://mjskay.github.io/tidybayes/. Accessed 14 Dec 2020.
Wickham H. Ggplot2: Elegant Graphics for Data Analysis: Springer; 2016. https://ggplot2.tidyverse.org. Accessed 14 Dec 2020.
Lee SM, Backenroth D, Cheung YKK, Hershman DL, Vulih D, Anderson B, Ivy P, Minasian L. Case example of dose optimization using data from bortezomib dose-finding clinical trials. J Clin Oncol. 2016; 34(12):1395.
Zohar S, Baldi I, Forni G, Merletti F, Masucci G, Gregori D. Planning a bayesian early-phase phase i/ii study for human vaccines in her2 carcinomas. Pharm Stat. 2011; 10(3):218–26.
Hazim A, Mills G, Prasad V, Haslam A, Chen EY. Relationship Between Response and Dose in Published, Contemporary Phase I Oncology Trials. J Natl Compr Cancer Netw JNCCN. 2020; 18(4):428–33. https://doi.org/10.6004/jnccn.2019.7375.
Yuan Y, Nguyen HQ, Thall PF. Bayesian Designs for Phase I-II Clinical Trials. Boca Raton: CRC Press; 2017.
Yuan Y, Yin G. Journal of the Royal Statistical Society: Series C (Applied Statistics). 2009; 58(5):719–736.
Zhang Y, Cao S, Zhang C, Jin IH, Zang Y. A bayesian adaptive phase i/ii clinical trial design with late-onset competing risk outcomes. Biometrics. 2020. https://doi.org/10.1111/biom.13347.
Sydes MR, Parmar MK, James ND, Clarke NW, Dearnaley DP, Mason MD, Morgan RC, Sanders K, Royston P. Issues in applying multi-arm multi-stage methodology to a clinical trial in prostate cancer: the mrc stampede trial. Trials. 2009; 10(1):1–16.
Chapple AG, Thall PF. A hybrid phase i-ii/iii clinical trial design allowing dose re-optimization in phase iii. Biometrics. 2019; 75(2):371–81.
Acknowledgements
The authors gratefully acknowledge the participation of the patients in the constituent clinical trials meta-analysed in this research.
Funding
KB and VH were funded by the University of Birmingham. GS was funded by a Cancer Research UK Centre Scholarship in undertaking the MSc Clinical Oncology at the University of Birmingham. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
KB wrote the manuscript. KB, VH and GS analysed the data. KB, VH, GS, CP, CC, and SL contributed data. KB, VH, GS, CP, CC, and SL contributed to the design of study and interpretation of results. KB, VH, GS, CP, CC, and SL read the manuscript and agreed to its publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
Supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Brock, K., Homer, V., Soul, G. et al. Is more better? An analysis of toxicity and response outcomes from dose-finding clinical trials in cancer. BMC Cancer 21, 777 (2021). https://doi.org/10.1186/s12885-021-08440-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08440-0