
Abstract
Hypertrophic cardiomyopathy (HCM) is a cardiovascular disease with high heterogeneity. Limited knowledge concerning the genetic background of nearly 40% HCM cases indicates there is a clear need for further investigation to explore the genetic pathogenesis of the disease. In this study, we undertook a whole exome sequencing (WES) approach to identify novel candidate genes and mutations associated with HCM. The cohort consisted of 74 unrelated patients with sporadic HCM (sHCM) previously determined to be negative for mutations in eight sarcomere genes. The results showed that 7 of 74 patients (9.5%) had damaging mutations in 43 known HCM disease genes. Furthermore, after analysis combining the Transmission and De novo Association (TADA) program and the ToppGene program, 10 putative genes gained priority. A thorough review of public databases and related literature revealed that there is strong supporting evidence for most of the genes playing roles in various aspects of heart development. Findings from recent studies suggest that the putative and known disease genes converge on three functional pathways: sarcomere function, calcium signaling and metabolism pathway. This study illustrates the benefit of WES, in combination with rare variant analysis tools, in providing valuable insight into the genetic etiology of a heterogeneous sporadic disease.
Similar content being viewed by others

Introduction
Hypertrophic cardiomyopathy (HCM) is a relatively common genetic cardiac disorder, with a prevalence of 0.16% in Chinese population and 0.20% in European and North American populations1,2. Classically, it is defined by the presence of a hypertrophied, nondilated left ventricle (LV) in the absence of any cause capable of producing the magnitude of evident hypertrophy, such as pressure overload or storage/infiltrative diseases3,4. The clinical presentation of HCM varies considerably, from an asymptomatic or slightly symptomatic course to more serious symptoms, including dyspnea, palpitations, heart failure and even sudden cardiac death5. Although numerous patterns of LV wall thickening can be found, hypertrophy in the anterior septum occurs most frequently, creating marked asymmetry. Based upon the septal contour and the location and extent of hypertrophy, patients with HCM exhibit at least four major types of LV morphology: reverse curvature-, sigmoidal-, apical- and neutral septum-hypertrophy6.
Most cases of HCM are familial in nature and are transmitted in an autosomal dominant fashion. Previous studies have demonstrated that, in 50–60% of HCM cases, the disease is caused by mutations in at least eight genes coding for cardiac sarcomere proteins, including MYH7, MYBPC3, TNNT2, TNNI3, TPM1, ACTC1, MYL2 and MYL3, of which, the most commonly affected are the sarcomere genes MYH7 and MYBPC3. Mutations in genes encoding the Z-disc or calcium-handling proteins account for less than 1% of cases and a further 5% of patients have metabolic disorders, neuromuscular disease, chromosome abnormalities or genetic malformation syndromes7,8,9. All genes with strong evidence for their participation in the pathogenesis of HCM are shown in Supplement Table S1. Sporadic HCM (sHCM) refers to cases where clinical features of the disease are absent from both of the patient’s parents. It is possible that sHCM reflects an inaccurate family history, incomplete penetrance (absence of clinical expression despite the presence of a mutation) in family members, or a de novo mutation that can initiate new familial disease3,10. For this reason, the genetic background of this subtype needs to be investigated to see whether and to what extent it is different from that of other HCM cases.
With the advent of next-generation sequencing technologies, the ability to quickly and relatively inexpensively sequence an individual’s entire genome has placed them among the most effective tools for detecting the genetic causes of Mendelian diseases. This is especially true for whole exome sequencing (WES), which has shown remarkable efficiency in identifying genetic variants in all of an individual’s genes11. In this study, we sought to apply WES on sHCM patients to investigate possible novel causative genes and mutations. To maximize the specificity of the results, we selected sHCM patients without mutations in the eight sarcomere genes using the Sanger sequencing method. The clinical and genetic studies of patients with sHCM have been restricted to Chinese Han cohorts. Our study demonstrates the value of WES in uncovering genetic mechanisms involved in the development of HCM, but it also highlights the challenges and limitations of such studies and the need for further studies with larger patient and control sample sizes.
Results
Study population
The study included 74 patients who had been previously diagnosed with HCM at ZhongDa Hospital or Nanjing General Hospital of Nanjing Military Command. All of them had been found to lack mutations in the eight sarcomere genes (MYH7, MYBPC3, TNNT2, TNNI3, TPM1, ACTC1, MYL2 and MYL3). The mean age at initial evaluation was 56.3 ± 14.0 years (range = 18–83 years); 50 (67.6%) were male. Mean maximal left ventricular wall thickness (MLVWT) was 19.1 ± 3.9 mm (range = 13.9–31.2 mm) and mean left ventricular ejection fraction (LVEF) was 66.8% ± 6.4% (range = 49%–78%). Analysis of echocardiograms of septal morphology revealed apical-HCM (44.6% of the cohort) and reversed curve-HCM (21.6% of the cohort) as the two most common anatomical subtypes of HCM in the cohort (Table 1).
WES on HCM cases
We obtained an average of 2,405 Mb initial reads per person and more than 99% of them were usable after removal of low-quality reads and adaptor or contaminant sequences. More than 98% exome regions were covered. About 94% of the targeted regions were covered at least four times with high coverage of the entire exon regions (Supplementary Table S2). Using the Genome Analysis Toolkit (GATK), the reads were aligned for single-nucleotide polymorphisms (SNPs) and insertion-deletions (InDels) calling and subsequent analysis12,13,14. A total of 3,286,164 SNPs and 220,355 InDels were identified. After removing all variants with minor allele frequency (MAF) > 0.1% in dbSNP138, 1000 Genome, ExAC and 2,000 in-house exome data (see Methods and Materials), the numbers were significantly reduced to 11018 for SNPs and 654 for InDels. For the functional filter, damaging exonic missense mutations, loss-of-function (LoF) mutations and splicing mutations (i.e., extreme mutations) were included in this candidate set, with 3265 SNPs and 477 InDels remaining (Fig. 1).

Flowchart of disease genes exploration of sporadic HCM patients.
Then we analyzed how many of our case samples harbored novel or rare variants in known HCM disease genes (Supplement Table S1). MYH7 damaging mutations, which should have been filtered in the selecting process, were identified in two patients. Among the other 72 out of 74 patients sequenced, we found rare or novel variants in 5 patients, suggesting that approximately 90.5% of the cases had no potential contributing genetic variants that could be identified from the known HCM disease genes using WES. Just as expected, the vast majority of the HCM cases could not be explained, consistent with results reported by other studies7,8,9.
Given that the genetic causes of most cases were unknown, we attempted to uncover novel HCM disease genes through a recently developed Transmission and De novo Association (TADA) program15 to compare case and control data. Interestingly, some novel genes did emerge and they are discussed in more detail below, together with other individual genes.
Known genes associated with HCM
Based on published reports, a total of 43 genes with strong evidence for their participation were considered known HCM disease genes (Supplement Table S1) and we tested whether we could uncover either known disease-causing mutations or novel mutations from the known HCM disease genes in our cohort. Genes that were listed in the Online Mendelian Inheritance in Man (OMIM) database (http://www.ncbi.nlm.nih.gov/omim) and Human Gene Mutation Database (HGMD) as causing or increasing susceptibility to HCM were referenced, as well as genes that were recently found to be tied to sHCM. Since the patients had been prescreened for mutations in the eight known sarcomere genes using the Sanger sequencing method, none of the patients was expected to carry damaging mutations in these common sarcomere genes. Still, two damaging mutations from sarcomere genes, p.Q1794K (rs397516247) and p.N1327K (rs141764279) in MYH7, were identified in two patients. Previously, rs141764279 had been reported in HCM patients16, while rs397516247 was first reported here in one HCM patient. In addition, we were also able to find several rare and novel mutations from other known disease genes in our HCM samples, as described below (Table 2).
ACTN2 and VCL
Mutations in two Z-disk genes, ACTN2 and VCL, have previously been linked to HCM and dilated cardiomyopathy (DCM)17,18,19. We found a novel mutation in ACTN2, p.D857H (NM_001103: c.G2569C), which is present in the calcium-insensitive C-terminal EF-hand domain of the protein. An interaction between the negatively charged asp 857 in ACTN2 and the positively charged arg 671 of the seventh Z-repeat of titin was considered from the structure in solution of the Act-EF34–Zr7 complex (Protein Data Bank entry code 1h8b). We also found a novel mutation in VCL, p.I321fs (NM_014000: c.962delT), which is supposed to lead to a complete loss of the actin binding site in the C-terminal tail domain of metavinculin, a muscle-specific isoform of vinculin mainly expressed in smooth and cardiac muscle tissue20. In a young patient with severely obstructive HCM, the VCL mutation was identified to be responsible for a serious phenotype, just as predicted. The patient showed decreased LVEF (49%) with an early onset age of 18 years. Previous studies suggest that sarcomere protein mutations are much more frequently seen in HCM patients with the reverse septal curvature type of hypertrophy, while Z-disc–associated HCM tends to develop the sigmoidal type21,22,23. Nevertheless, diverse types of LV hypertrophic morphology have also been identified in patients with mutations in ACTN217,19. Our study identified an ACTN2 mutation p.D857H and a VCL mutation p.I321fs in two patients with the neutral septum hypertrophy and the apical hypertrophy, respectively.
JPH2 and PLN
Recently, calcium handling proteins encoded by JPH2 and PLN have been found to be associated with HCM in several studies24,25. In our study, a splicing mutation, c.2011-1G>T in JPH2 (NM_020433), was identified in one patient with apical HCM. The mutation was predicted to affect the transmembrane domain of junctophilin-2. As a key regulator of cardiac diastolic function, phospholamban encoded by PLN, has been reported to modulate calcium re-uptake during muscle relaxation and play an important role in calcium homeostasis in the heart muscle26. We also found a missense mutation p.V49M in PLN (NM_002667) localized in a functional transmembrane domain of phospholamban.
MYH6
MYH6 encodes myosin-6, a component of the thick filament and has been previously reported to be a rare gene causing HCM27. Here we identified a heterozygous 4727G-A transversion of the MYH6 gene (NM_002471), resulting in an arg1576-to-gln (p.R1576Q) substitution at a highly conserved residue of the rod domain. The novel mutation was found in an 83-year-old woman with late-onset HCM. It is possible that the mild heterozygous point mutation may be causative for adult onset sHCM.
Putative genes associated with HCM
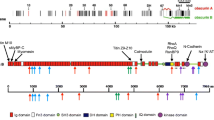
TADA was applied to evaluate the case-control difference15. The data we used were all extreme mutations from 74 HCM cases and 2000 controls (see Methods and Materials). We identified a total of 92 candidate genes with PTADA ≤ 0.001 (Fig. 1, Supplementary Table S3), most of which harbor recurrent extreme mutations. The ToppGene program28 was then used and ten genes with high priority were selected for further analysis (Supplementary Table S4). The numbers of pathogenic mutations in the top 10 putative HCM associated genes (TTN, RYR2, NEB, OBSCN, CMYA5, PLAC4, NES, CAP1, CFLAR and MYH15) are shown in Fig. 2a.

(a) Number of pathogenic mutations in top 10 putative HCM disease genes. (b) Schematic diagram of the conserved domains of four proteins. Newly found LoF mutations in our study were marked in orange. RYR2 protein: showing the MIR, RyR (ryanodine receptor), SPRY (SPIa/ryanodine receptor) and TM (transmembrane region). CMYA5 protein: showing the TRIM-like region consists of: B-box, BBC (B-box coiled coil), FN3 repeats (fibronectin 3 repeat) and SPRY. MYH15 protein: MYSc (myosin motor) and IQ (isoleucine-glutamine calmodulin-binding motif). OBSCN protein: showing the Ig (immunoglobulin), FN3 repeats, IQ, SH3 (src-homology 3) and DH (dbl homology), PH (pleckstrin homology motif) and SK (serine/threonine kinase). One previously established cardiomyopathy associated mutation in OBSCN was marked in red.
Since our initial assumption was that these genes were linked to HCM in our patient group, we wondered if there was any evidence from previously published studies that would support an association between the putative genes and HCM. Therefore, we then searched public database, GTEx, ZFIN, MGD and PubMed, for their expression and animal model data (see Methods and Materials). Tissue expression profiles showed that TTN, RYR2, OBSCN, CMYA5, NES, CAP1 and CFLAR displayed relatively high expression in the cardiovascular system (Fig. 2b). Animal model data are summarized in Table 3. TTN, ranking the first place according to the ToppGene analysis, was previously found to be a rare causative gene for HCM29,30. Interestingly, we found TTN significant compared to control in our analysis, despite its high mutation rate. One identified titin mutation, p.E10320X, was located in the PEVK region, which was previously confirmed to contribute to the elastic properties of the cardiac ventricle and thus probably lead to cardiomyopathy with diastolic dysfunction31.
CMYA5
Although CMYA5 was named a “cardiomyopathy associated” gene, a search showed that the OMIM database did not map the locus of myospryn to a susceptibility region for cardiovascular disease. Myospryn was originally coined on purely hypothetical grounds: a putative link to cardiomyopathy was suggested due to its coexpression with known cardiomyopathy-related genes32. It was supported by a recent report showing an association of a myospryn polymorphism (p.K2906N) with left ventricular wall thickening and diastolic dysfunction in hypertensive patients33. Furthermore, knockdown studies of myospryn transcripts in the zebrafish model showed cardiovascular abnormalities, including mild structural abnormalities, pericardial edema and ventricular hypoplasia34. These data suggest that myospryn could be involved in proper heart development and function. In line with this, the putative HCM associated gene CMYA5 was statistically significant in our analysis, further supporting its involvement in HCM. Moreover, in the patient carrying the mutation p.D857H in ACTN2, we also found one LoF mutation, p.S2404X in CMYA5, which lost three highly conserved domains: two fibronectin 3 repeats (FN3) and one SPRY (SPIa/ryanodine receptor) domain. The patient was diagnosed at the age of 50 years and presented the neutral septum hypertrophy. The LoF mutation was predicted to abolish most of the C-terminal part of myospryn, the region that was reported to contain the binding sites for desmin, α-actinin and titin34,35. Previous studies showed that the myospryn colocalizes with desmin, predominantly at intercalated disks and at the Z-line costamere connection level of the sarcolemma in adult mouse heart muscle36. Complete loss of the desmin binding site will probably lead to a disruption of intermediate filaments. Thus, we suspect that the combined effects of the two mutations in ACTN2 and CMYA5 contribute to the development of HCM. Three more damaging mutations, p.S2813L, p.K1822I and p.D3051E, were also found in CMYA5, which may disrupt its interactions with other proteins, but their exact roles need further confirmation (Fig. 2c).
RYR2
The cardiac ryanodine receptor encoded by RYR2 is found in the cardiac sarcoplasmic reticulum, which is the major source of calcium required for cardiac muscle excitation-contraction coupling37. Mutations in RYR2 are associated with stress-induced polymorphic ventricular tachycardia38,39 and arrhythmogenic right ventricular dysplasia40. Although the role of the cardiac ryanodine receptor in heart disease seems quite clear, little is known about whether there is an association between RYR2 and HCM, though the mutation p.T1107M was previously reported in one family in Japan17,41. RYR2 was on the top of our list of putative HCM causative genes provided by TADA and ToppGene and we found two novel missense mutations and one splicing mutation in three patients (Fig. 2c). Surprisingly, all of them presented the apical hypertrophy, with a mean onset age of 55.3 ± 13.4 years. Both of the two missense mutations, p.E3809G and p.R929H, have been predicted by SIFT and Polyphen2 to be damaging mutations. Additionally, these are two highly conserved residues, suggesting that changes at this amino acid could be detrimental. The novel splicing mutation c.7966-2A>T has also been predicted to disrupt several important domains, probably leading to an abnormal protein or degradation of an unfinished protein product. The patient with the splicing mutation showed impaired heart function with low LVEF (54%). None of the three mutations has been reported to be associated with any other kind of cardiac disease, indicating they may be specific to HCM. Further evaluation of these mutations may shed light on the role of RYR2 in the molecular pathogenesis of HCM.
OBSCN
OBSCN is the gene encoding obscurin, the third giant protein of the contractile apparatus of striated muscles. We have only recently learned of OBSCN’s linkage to cardiomyopathies, owing to technical challenges posed by the large size of its coding sequence (approximately 170 kb). Although obscurin has been found to be indispensable in myofibrillogenesis and hypertrophic growth from small interfering RNA-mediated gene silencing, the role of OBSCN in HCM is still unclear42. Nevertheless, up-regulation of different OBSCN gene products, including full length obscurin and several smaller MLCK variants, has been reported to occur in mice with myocardial hypertrophy induced by aortic constriction43. In addition, phenotype data from ZFIN have shown abnormal heart contraction and abnormal heart structure in a zebrafish knockout model. Only one sequence variation has been detected through linkage analysis in the OBSCN gene in the region encoding the site of interaction for the Z-disc region of titin (Ig58/59), specifically, an R4344Q variant in the Ig58 domain of obscurin44. In vitro studies have demonstrated that this variant results in decreased binding of obscurin to titin as well as mis-localization of obscurin to the Z-disc. Now WES enabled us to sequence all the exons of OBSCN and six rare damaging mutations were identified in six patients, four of which were LoF mutations (Fig. 2c). The patient carrying the mutation p.A1088fs had an early onset age of 35 years. The six patients showed various types of septal morphology. The present study provided additional evidence for the involvement of OBSCN in the development of HCM, even though further functional experiment still need to be conducted to unveil the specific pathogenic mechanism.
MYH15
MYH15, a member of the conventional myosin, presumably possesses a muscle contraction role45,46. Even at low levels of expression, the protein seems to play a role in heart development. A recent study showed that one common SNP, rs3900940 (p.T1125A) in MYH15, was associated with an elevated risk for heart disease47. Two LoF mutations and two missense mutations were found in four patients in our study (Fig. 2c). One patient with a splice variant in MYH15, also carrying a titin mutation p.L9683P, presented the reverse curvature hypertrophy, while all the other three patients showed the apical hypertrophy.
Other putative genes have also been investigated to understand their functions. NEB has been found to be involved in heart development through a zebrafish knockout model and to regulate length maintenance in rat cardiac myocytes48, whereas CFLAR mainly functions as a regulator of apoptosis and contributes to regulating cardiac hypertrophy in response to pressure overload49,50. Both of the two patients carrying LoF mutations in CFLAR exhibited the apical hypertrophy. Decreased LVEF (53%) was found in the patient carrying the mutation p.S135X in CFLAR (Table 3). Taken together, it is likely that these genes act in a causative role or at least in a modifying role in the pathogenesis of HCM.
Protein interaction networks
Knowledge of molecular-level interaction between proteins has enabled the development of protein–protein interaction (PPI) networks enriched for known HCM disease genes and putative HCM associated genes. The underlying connections were explored using experimentally verified interaction data from StringDB51 and Biogrid52 and PPI networks were formed to summarize these links (Fig. 3). We found that this combined network encompasses three broad functional modules. The first component (15 proteins) forms a highly interconnected set of sarcomere function associated proteins, including the eight common sarcomere genes that are important in HCM pathogenesis, two putative genes in our study and five known rare HCM disease genes, whereas the second component (16 proteins) contains both calcium signaling functions (RYR2, CALM3, JPH2, PLN, SRI, CALR3 and CASQ2) and substrate and energy metabolic pathways (or metabolism pathway) (AGK, COX15, GLA, FXN, PRKAG2, MRPL3, LAMP2, TAZ and SLC25A4).

Protein–protein interaction network created by known HCM associated genes (blue circle) and putative HCM associated genes (red circle) reveals functional enrichment of genes involved in sarcomere function, calcium signaling and substrate and energy metabolic pathway.
The network also includes 19 intermediate genes (white circle). Solid line indicates direct interaction and dotted line indicates indirect interaction.
Pathway analysis showed that the sarcomere function proteins constitute the most important part of the network, consistent with previous findings about the pathogenesis of HCM5,53. The putative genes identified in our study, including TTN and NEB, are among the central part of this component. Both the calcium signaling and the metabolism pathway also play important roles in heart development5. RYR2 is involved in the calcium signaling pathway. The other putative genes are scattered over the network, suggesting the involvement of other potential processes in the pathogenesis.
Although these putative genes are in fact not significantly more connected to each other in the network than a random set of genes (due to the high interconnectedness of the global PPI network), viewing them in the context of the central network may highlight new genes and pathways to study, since these are promising HCM candidates. In addition to the known and putative disease genes, we also included 19 intermediate genes (white circle) that connect the known genes and putative genes. We found that the intermediate genes are mostly transcription factors and protein kinases. These novel genes, suggested by PPI networks, can also be explored to reveal further functions and pathways. For example, SAMD3 has been suggested to play a role in cardiac remodeling54, while FAS, connected to TCAP and CFLAR, is involved in the apoptotic pathway, a process that has recently been shown to play a major role in cardiac disease55. It is likely that sequencing studies of patients will identify novel candidates for PPI networks, creating a reiterative process by which networks and genetics mutually inform.
More significantly, the most connected intermediate gene, ubiquitin C (UBC), which belongs to the ubiquitin family, links three major pathways of the network. It can signal for protein degradation through the ubiquitin-proteasome system (UPS). Both increases and decreases in UPS function are regularly observed in animal models of HCM, cardiac hypertrophy and heart failure, suggesting that regulation of UPS contributes to important adaptations in cardiac disease56. Recent investigations suggest that UPS is involved in degrading mutant proteins in HCM and that UPS impairment may play an important role in the pathophysiology of HCM as well. It has been shown that truncated cMyBP-Cs resulting from human MYBPC3 mutations are unstable, not well incorporated into the sarcomere and finally degraded by UPS. Continuous degradation of mutant cMyBP-C proteins also leads to UPS impairment57. However, the mechanism by which UPS is impaired has not been elucidated yet. In addition to MYBPC3, the expression of missense and truncating FHL1 mutations as well as missense ANKRD1 mutations is markedly regulated by UPS after gene transfer in cardiac myocytes or in rat engineered heart tissue58,59. Hence, other disease genes also need to be underlined, even if mutations are rarely found in isolated sporadic cases of HCM. Furthermore, UPS has also been proposed to be established as a target in the therapy of cardiac diseases60.
Taken together, these findings demonstrate that the PPI network reveals underlying interaction webs between putative genes and known HCM disease genes, further supporting their connections with HCM.
Discussion
Next generation sequencing technologies have proved to be robust and cost-effective and are revolutionizing the way human disease is studied. In particular, the WES approach not only is capable of expanding our knowledge of novel mutations of established genes linked to a specific disorder, but also helps uncover genetic modifiers that contribute to a disease phenotype61. Consequently, WES has been widely used to detect genetic factors related to a range of diseases in various medical fields, particularly diseases that exhibit broad genetic or phenotypic heterogeneity62.
To explore possible pathogenic mechanisms responsible for the development of sHCM, we applied WES on 74 selected sHCM cases, who had not been found to carry damaging mutations in eight common sarcomere genes using the Sanger sequencing screening. With respect to the clinical features of our HCM cohort, two notable findings stood out. First, nearly half of our cases (44.6%) had apical hypertrophy, whereas epidemiological studies have shown that the usual prevalence of apical HCM is 5% to 20%63. This indicates that most apical HCM cases cannot be explained by mutations in the eight sarcomere genes and is in accordance with published literature on the genetic background of apical HCM64,65. Second, the proportion of obstructive HCM patients (55.4%) was relatively high, further substantiating findings by Gruner et al. that genotype-negative patients showed increased prevalence of left ventricular outflow tract (LVOT) obstruction22. Therefore, our study clearly demonstrates the value of WES in its ability to unveil previously inaccessible genetic properties of apical HCM and obstructive HCM.
Since a large number of mutations were uncovered in the 74 exomes through WES, we focused instead on the extreme mutations. Still, two mutations in one sarcomere gene MYH7 were present in two patients. It turned out that these were false negative errors in the Sanger sequencing method. As previously studies have suggested66,67, possible explanations are improper PCR primers that lead to imbalanced amplification of the two alleles due to overlapping unknown DNA variants and difficulty of automated software to correctly call heterozygous sites. Fortunately, the WES approach was able to resequence the samples and acquire definitive results. A further five mutations in five known rare HCM disease genes were also identified, leaving the rest 90% of the HCM cohort genetically unexplained. Next, analysis via the TADA program and the ToppGene program revealed several novel and promising putative HCM associated genes with strong statistical support, including two previously suspected rare HCM risk genes, TTN and RYR2.
In our dataset, several novel variants in the known HCM disease genes were present in a small minority of patients, while most patients were left with unknown genetic basis. After analyzing the putative HCM associated genes, we found many novel and rare variants that were more enriched in our patient group. Most of the ten putative genes were examined for connections with cardiovascular diseases. Despite the fact that gene expression cannot fully explain the pathogenicity, tissue expression profiles of the putative genes were explored. Databases of animal models were also mined to help elucidate gene function. In addition, genotype–phenotype associations between gene mutations and septal morphology evaluated in this study were examined. Our study generated interesting findings that may serve as valuable clues for future studies to pursue. Moreover, we established the PPI network link between putative HCM associated genes and known HCM disease genes through direct or indirect interaction. The network contained helpful information for understanding the role of these proteins in the development of HCM and we found genes were convergent on three functional pathways: sarcomere function, calcium signaling and substrate and energy metabolic pathway. Consistent with results by other studies, we also found the important role of UPS from the PPI56,60.
Additionally, published studies on the function of the other candidate genes that were not at the top of the list but exhibited statistical significance also suggest they may be responsible for increased susceptibility to HCM. For instance, DNAH11 encodes a ciliary outer dynein arm protein and is a member of the dynein heavy chain family. McGrath et al. reported that Dnahc11-null embryos showed abnormalities in asymmetric calcium signaling at the embryonic node in mice68. As a microtubule-dependent motor ATPase, combined with its role in proper left-right patterning, both are predicted to be crucial in the HCM pathway. KCNA7 has been shown to be associated with inherited cardiac disorders69. CACNA1G, a type of voltage-gated calcium channel involved in a variety of calcium-dependent processes, including muscle contraction, has recently been found to play an important role in the heart70. Furthermore, several genome-wide association studies identified a significant association of SNP rs2106261 in the ZFHX3 gene with atrial fibrillation in participants both in the European population and the Chinese Han population71,72. Recent knockdown studies of ZFHX3 transcripts in a zebrafish model also showed abnormal cardiac looping, indicating the role of ZFHX3 in heart development73. As with top putative HCM associated genes, experimental data about these genes have yet to be fully investigated to determine their pathogenic relevance.
Although studies in the past decades have generated a wealth of information that may help lead to the identification of remaining causative genes for HCM, our knowledge concerning the genetic basis and disease development remains very limited. Considerable phenotypic heterogeneity exists between patients with the same mutation, suggesting the complexity underlying the phenotype of HCM. Therefore, efforts on discovering novel HCM disease genes and validating existing results are necessary for a more complete understanding of the pathogenic mechanism. The present study represents a new approach to identifying promising candidate genes associated with HCM. We believe future studies using WES with larger samples of sHCM patients are required to validate what has been found so far and possibly generate more variants to further advance our understanding of the genetics underlying HCM. However, additional functional studies are also needed to determine which of the predicted genes are real and relevant to HCM and the exact roles they play.
Materials and Methods
Subjects
The study included seventy-four unrelated Chinese patients diagnosed with HCM, who were identified at, or referred to, ZhongDa Hospital, Southeast University, Nanjing or Nanjing General Hospital of Nanjing Military Command between September 2000 and July 2012. All the patients were screened for variants in the coding regions of eight common sarcomere genes, MYH7, MYBPC3, TNNT2, TNNI3, TPM1, ACTC1, MYL2 and MYL3, through Sanger sequencing, which generated negative findings (Supplement Table S1). Informed consent, blood samples and clinical evaluations were obtained from all of the HCM patients, under protocols approved by the Ethics Committee of Southeast University. Genetic counseling was provided to all participants. Only one case per family was included in the present analysis. The methods in the study were performed in accordance with the relevant guidelines and regulations.
Clinic evaluations
Patients with HCM were diagnosed based on medical history, physical examination, electrocardiogram and echocardiogram showing maximum left ventricular wall thickness (MLVWT) ≥13 mm in at least one myocardial segment, or MLVWT exceeding two SDs corrected for age, size and gender, in the absence of other diseases that could explain the hypertrophy74.
Whole exome sequencing and identification and annotation of SNPs and InDels
DNA was extracted with 8 mL peripheral blood through the salting out method and stored at −20 °C until use. WES was performed on DNA samples of the HCM patients at Beijing Genomics Institute (BGI) using the Agilent SureSelect Exon Capture kit (48-Mb) (Agilent, Santa Clara, CA) and the Illumina HiSeq 2000 sequencer (Illumina, San Diego, CA). For quality control, low quality reads were filtered and 3′/5′ adapters were trimmed using the Trim Galore program. Illumina clean reads were aligned using the BWA program on the human reference genome build hg19 and quality scores were recalibrated and realigned to reference using the GATK software package. Following the exclusion of duplicate reads, insertion-deletions (InDels) and single-nucleotide polymorphisms (SNPs) were called using Sequence Alignment/Map tools (SAMtools)75. SNPs were detected and genotyped with the GATK Unified Genotyper in single-sample mode (with parameters -im ALL -mbq 20 -mmq 20 -mm42 3 -deletions 0.05). Variants were filtered with the GATK Variant Filtration module (with filters “QUAL<50.0 & QD<5.0 & HRun>10 & DP<4” and parameters –cluster 3 -window 10). Indels were detected with GATK InDel GenotyperV2 (with parameters -im ALL) and filtered with a custom python module that removed sites with amax_cons_av≥1.9 (maximum average number of mismatches across reads supporting the InDel) or max_cons_nqs_av_mm ≥0.2 (maximum average mismatch rate in the 5-bp NQS window around the InDel, across InDel-supporting reads). In-house developed bioinformatics tools with RefSeq (hg19, from UCSC) and UCSC annotation (http://www.ncbi.nlm.nih.gov/refseq/) were applied to annotate the variants, such as locations (exonic, intronic and intergenic region, etc.) and effects on protein coding (synonymous, missense, nonsense, frameshift, etc.).
Identification of extreme mutations
We defined our set of candidate variants for further analysis mainly based on allele frequency and function. The frequency filter used allele frequency estimates from dbSNP (v138) (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 Genome (http://www.ncbi.nlm.nih.gov/Ftp/), ExAC (http://exac.broadinstitute.org/) and 2,000 in-house exome data (from the Beijing Genomics Institute, Shenzhen, China), with a 0.1% cut-off to remove possible common variants. For the functional filter, synonymous and non-frameshift mutations were eliminated due to their low possibility to contribute to disorders. LoF mutations, such as nonsense/splicing mutations and frameshift InDels, were directly considered damaging. For missense mutations, prediction of in silico pathogenicity was performed using polymorphism phenotyping v2 (Polyphen2) and sorts intolerant from tolerant (SIFT)76,77. A mutation was predicted to be deleterious if it was classified as ‘damaging’ by SIFT and ‘possibly damaging’ or ‘probably damaging’ by Polyphen2. Genes harboring rare LoF/deleterious SNPs, which we refer to as extreme mutations, were used for candidate genes prioritization (Fig. 1).
Prioritization of candidate genes
One recently developed program, Transmission and De novo Association (TADA)15 can accurately predict risk genes on the basis of allele frequencies, gene-specific penetrance and mutation rate. The TADA P-value (PTADA) for the likelihood of each gene contributing to the corresponding disorders was calculated with default parameters. For each gene, the mutation rate was estimated using a probability model taking the gene length and base content into account. Another program, ToppGene Suite28 (http://toppgene.cchmc.org) was an online candidate gene prioritization tool based on functional similarity between a set of genes known to be associated with the disease of interest and a set of candidate genes that has been potentially linked with the disease. The training set for ranking HCM candidate genes was a set of known HCM disease genes (Table S1). A set of genes with PTADA ≤ 0.001 (P-value generated by the TADA program) were used as the test set on ToppGene.
Tissue expression profiles and animal model data
The Genotype-Tissue Expression database (GTEx, http://www.gtexportal.org/) was used to investigate the expression of candidate genes in multiple human tissues. The zebrafish information network78 (ZFIN, http://zfin.org/) and the mouse genome database79 (MGD, http://www.informatics.jax.org/) databases, as well as PubMed, were used to investigate the phenotype of candidate genes in zebrafish and mice.
Protein interaction network
StringDB51, Biogrid52 (http://www.thebiogrid.org) were used to integrate the experimentally verified interaction data for protein interaction network. Only medium- and high-confidence experimental interactions are shown, although these may not always represent local interactions. Only one intermediate gene that is known to interact between two genes is included. Functional modules were manually grouped and labeled using Cytoscape 2.880 (www.cytoscape.org).
Sanger sequencing
For the case data, all mutations described and analyzed here were validated by standard PCR combined with Sanger sequencing81.
Additional Information
How to cite this article: Xu, J. et al. Investigation of Pathogenic Genes in Chinese sporadic Hypertrophic Cardiomyopathy Patients by Whole Exome Sequencing. Sci. Rep. 5, 16609; doi: 10.1038/srep16609 (2015).
References
Maron, B. J. et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 92, 785–789 (1995).
Zou, Y. et al. Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults. Am J Med 116, 14–18 (2004).
Gersh, B. J. et al. ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines developed in collaboration with the American Association for Thoracic Surgery, American Society of echocardiography, American Society of nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions and Society of Thoracic Surgeons. J Am Coll Cardiol 58, e212–e260 (2011).
Maron, B. J. Hypertrophic cardiomyopathy: a systematic review. JAMA 287, 1308–1320 (2002).
Frey, N., Luedde, M. & Katus, H. A. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol 9, 91–100 (2012).
Bos, J. M., Ommen, S. R. & Ackerman, M. J. Genetics of hypertrophic cardiomyopathy: one, two, or more diseases? Curr Opin Cardiol 22, 193–199 (2007).
Lopes, L. R. et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet 50, 228–239 (2013).
Keren, A., Syrris, P. & McKenna, W. J. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med 5, 158–168 (2008).
Millat, G. et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J Med Genet 53, 261–267 (2010).
Nunez, L. et al. Somatic MYH7, MYBPC3, TPM1, TNNT2 and TNNI3 mutations in sporadic hypertrophic cardiomyopathy. Circ J 77, 2358–2365 (2013).
Bamshad, M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12, 745–755 (2011).
Hou, H. et al. MagicViewer: integrated solution for next-generation sequencing data visualization and genetic variation detection and annotation. Nucleic Acids Res 38, W732–W736 (2010).
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714 (2008).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
He, X. et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. Plos Genet 9, e1003671 (2013).
Wolny, M. et al. Cardiomyopathy mutations in the tail of beta-cardiac myosin modify the coiled-coil structure and affect integration into thick filaments in muscle sarcomeres in adult cardiomyocytes. J Biol Chem 288, 31952–31962 (2013).
Chiu, C. et al. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: a genome-wide analysis. J Am Coll Cardiol 55, 1127–1135 (2010).
Vasile, V. C. et al. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol Genet Metab 87, 169–174 (2006).
Girolami, F. et al. Novel alpha-actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: a massively parallel sequencing study. Circ Cardiovasc Genet 7, 741–750 (2014).
Janssen, M. E., Liu, H., Volkmann, N. & Hanein, D. The C-terminal tail domain of metavinculin, vinculin’s splice variant, severs actin filaments. J Cell Biol 197, 585–593 (2012).
Theis, J. L. et al. Echocardiographic-determined septal morphology in Z-disc hypertrophic cardiomyopathy. Biochem Biophys Res Commun 351, 896–902 (2006).
Gruner, C. et al. Toronto hypertrophic cardiomyopathy genotype score for prediction of a positive genotype in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 6, 19–26 (2013).
Binder, J. et al. Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc 81, 459–467 (2006).
Matsushita, Y. et al. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J Hum Genet 52, 543–548 (2007).
Landstrom, A. P., Adekola, B. A., Bos, J. M., Ommen, S. R. & Ackerman, M. J. PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. Am Heart J 161, 165–171 (2011).
Smeazzetto, S., Saponaro, A., Young, H. S., Moncelli, M. R. & Thiel, G. Structure-function relation of phospholamban: modulation of channel activity as a potential regulator of SERCA activity. Plos One 8, e52744 (2013).
Carniel, E. et al. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 112, 54–59 (2005).
Chen, J., Bardes, E. E., Aronow, B. J. & Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–W311 (2009).
Herman, D. S. et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366, 619–628 (2012).
Satoh, M. et al. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun 262, 411–417 (1999).
Granzier, H. L. et al. Truncation of titin’s elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circ Res 105, 557–564 (2009).
Walker, M. G. Pharmaceutical target identification by gene expression analysis. Mini Rev Med Chem 1, 197–205 (2001).
Nakagami, H. et al. Gene polymorphism of myospryn (cardiomyopathy-associated 5) is associated with left ventricular wall thickness in patients with hypertension. Hypertens Res 30, 1239–1246 (2007).
Tsoupri, E. & Capetanaki, Y. Μyospryn: a multifunctional desmin-associated protein. Histochem Cell Biol 140, 55–63 (2013).
Durham, J. T. et al. Myospryn is a direct transcriptional target for MEF2A that encodes a striated muscle, alpha-actinin-interacting, costamere-localized protein. J Biol Chem 281, 6841–6849 (2006).
Kouloumenta, A., Mavroidis, M. & Capetanaki, Y. Proper perinuclear localization of the TRIM-like protein myospryn requires its binding partner desmin. J Biol Chem 282, 35211–35221 (2007).
Tang, Y., Tian, X., Wang, R., Fill, M. & Chen, S. R. Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ Res 110, 968–977 (2012).
Laitinen, P. J. et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103, 485–490 (2001).
Priori, S. G. et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103, 196–200 (2001).
Tiso, N. et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 10, 189–194 (2001).
Noboru F. H. I. K. H. A novel missense mutation in cardiac ryanodine receptor gene as a possible cause of hypertrophic cardiomyopathy: Evidence from familial analysis. Circulation. 114, 165 (2006).
Borisov, A. B. et al. Essential role of obscurin in cardiac myofibrillogenesis and hypertrophic response: evidence from small interfering RNA-mediated gene silencing. Histochem Cell Biol 125, 227–238 (2006).
Borisov, A. B. et al. Rapid response of cardiac obscurin gene cluster to aortic stenosis: differential activation of Rho-GEF and MLCK and involvement in hypertrophic growth. Biochem Biophys Res Commun 310, 910–918 (2003).
Arimura, T. et al. Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem Biophys Res Commun 362, 281–287 (2007).
Park, J. S. et al. Isolation of a ventricle-specific promoter for the zebrafish ventricular myosin heavy chain (vmhc) gene and its regulation by GATA factors during embryonic heart development. Dev Dyn 238, 1574–1581 (2009).
Weiss, A. & Leinwand, L. A. The mammalian myosin heavy chain gene family. Annu Rev Cell Dev Biol 12, 417–439 (1996).
Bare, L. A. et al. Five common gene variants identify elevated genetic risk for coronary heart disease. Genet Med 9, 682–689 (2007).
McElhinny, A. S., Schwach, C., Valichnac, M., Mount-Patrick, S. & Gregorio, C. C. Nebulin regulates the assembly and lengths of the thin filaments in striated muscle. J Cell Biol 170, 947–957 (2005).
Giampietri, C. et al. c-Flip overexpression reduces cardiac hypertrophy in response to pressure overload. J Hypertens 26, 1008–1016 (2008).
Yeh, W. C. et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12, 633–642 (2000).
Jensen, L. J. et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 37, D412–D416 (2009).
Stark, C. et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34, D535–D539 (2006).
Keren, A., Syrris, P. & McKenna, W. J. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med 5, 158–168 (2008).
Bujak, M. et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 116, 2127–2138 (2007).
Harvey, P. A. & Leinwand, L. A. Cellular mechanisms of cardiomyopathy. The Journal of Cell Biology 194, 355–365 (2011).
Carrier, L., Schlossarek, S., Willis, M. S. & Eschenhagen, T. The ubiquitin-proteasome system and nonsense-mediated mRNA decay in hypertrophic cardiomyopathy. Cardiovasc Res 85, 330–338 (2010).
Sarikas, A. et al. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res 66, 33–44 (2005).
Friedrich, F. W. et al. Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum Mol Genet 21, 3237–3254 (2012).
Crocini, C. et al. Impact of ANKRD1 mutations associated with hypertrophic cardiomyopathy on contraction parameters of engineered heart tissue. Basic Res Cardiol 108, 349 (2013).
Schlossarek, S., Frey, N. & Carrier, L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J Mol Cell Cardiol 71, 25–31 (2014).
Sanders, S. J. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012).
Chae, J. H. et al. Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. J Med Genet 52, 208–216 (2015).
Kitaoka, H. et al. Comparison of prevalence of apical hypertrophic cardiomyopathy in Japan and the United States. Am J Cardiol 92, 1183–1186 (2003).
Gruner, C. et al. Sarcomere protein gene mutations in patients with apical hypertrophic cardiomyopathy. Circ Cardiovasc Genet 4, 288–295 (2011).
Arad, M. et al. Gene mutations in apical hypertrophic cardiomyopathy. Circulation 112, 2805–2811 (2005).
Quinlan, A. R. & Marth, G. T. Primer-site SNPs mask mutations. Nat Methods 4, 192 (2007).
Stephens, M., Sloan, J. S., Robertson, P. D., Scheet, P. & Nickerson, D. A. Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat Genet 38, 375–381 (2006).
McGrath, J., Somlo, S., Makova, S., Tian, X. & Brueckner, M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell 114, 61–73 (2003).
Bardien-Kruger, S. et al. Characterisation of the human voltage-gated potassium channel gene, KCNA7, a candidate gene for inherited cardiac disorders and its exclusion as cause of progressive familial heart block I (PFHBI). Eur J Hum Genet 10, 36–43 (2002).
Le Quang, K. et al. T-type calcium current contributes to escape automaticity and governs the occurrence of lethal arrhythmias after atrioventricular block in mice. Circ Arrhythm Electrophysiol 6, 799–808 (2013).
Liu, Y. et al. Genetic polymorphisms in ZFHX3 are associated with atrial fibrillation in a Chinese Han population. PLOS ONE 9, e101318 (2014).
Benjamin, E. J. et al. Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet 41, 879–881 (2009).
Thomas Wieland, P. M. Pathophysiologische Mechanismen hereditärer Formen von Vorhofflimmern. (2014).
Dubourg, O. et al. Doppler echocardiography in familial hypertrophic cardiomyopathy: the French Cooperative Study. Echocardiography 12, 235–241 (1995).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. bioinformatics 25, 2078–2079 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081 (2009).
Sprague, J. et al. The Zebrafish Information Network: the zebrafish model organism database. Nucleic Acids Res 34, D581–D585 (2006).
Eppig, J. T. et al. The Mouse Genome Database (MGD): from genes to mice–a community resource for mouse biology. Nucleic Acids Res 33, D471–D475 (2005).
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L. & Ideker, T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 (2011).
Sanger, F. The Croonian Lecture, 1975. Nucleotide sequences in DNA. Proc R Soc Lond B Biol Sci 191, 317–333 (1975).
Anderson, S. L. et al. Nemaline myopathy in the Ashkenazi Jewish population is caused by a deletion in the nebulin gene. Hum Genet 115, 185–190 (2004).
Acknowledgements
We would like to express our gratitude to the patients for their participation in this study. Assistance in DNA sample collection from ZhongDa Hospital, Southeast University and Nanjing General Hospital of Nanjing Military Command is greatly appreciated. We are grateful to Dr. Jinyu Wu for help in data processing and statistical analysis. This study is supported by the National Natural Science Foundation of China (81270295), the Fundamental Research Funds for the Central Universities and the Graduate Innovation Program of Jiangsu Province (KYZZ_0065). Dr. Xiangdong Liu is a Fellow at the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine of Jiangsu Province, China.
Author information
Authors and Affiliations
Contributions
Conception and design: X.L., Q.D., J.X. and W.X. Development and Methodology: J.X. and Z.L. Acquisition and analysis of clinical sample: Q.D., J.X., X.R., Y.Z., X.S., M.D. and J.L. Analysis of sequencing data: J.X., Z.S. and Z.L. Statistical analysis and laboratory analysis: J.X., Z.L. and J.L. Drafting Manuscript: J.X. Review, revision of manuscript: X.L., Z.L. and Q.D. Study Supervision: X.L. and Q.D. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xu, J., Li, Z., Ren, X. et al. Investigation of Pathogenic Genes in Chinese sporadic Hypertrophic Cardiomyopathy Patients by Whole Exome Sequencing. Sci Rep 5, 16609 (2015). https://doi.org/10.1038/srep16609
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16609
- Springer Nature Limited
This article is cited by
-
Integration of transcriptomic data identifies key hallmark genes in hypertrophic cardiomyopathy
BMC Cardiovascular Disorders (2021)
-
Myospryn deficiency leads to impaired cardiac structure and function and schizophrenia-associated symptoms
Cell and Tissue Research (2021)
-
T-tubule remodeling in human hypertrophic cardiomyopathy
Journal of Muscle Research and Cell Motility (2021)
-
Double the trouble: giant proteins with dual kinase activity in the heart
Biophysical Reviews (2020)
-
Non-familial cardiomyopathies in Lebanon: exome sequencing results for five idiopathic cases
BMC Medical Genomics (2019)