
Abstract
The objective of this work was to identify genetic variants in Mexican patients diagnosed with hypertrophic cardiomyopathy (HCM). According to world literature, the genes mainly involved are MHY7 and MYBPC3, although variants have been found in more than 50 genes related to heart disease and sudden death, and to our knowledge there are no studies in the Mexican population. These variants are reported and classified in the ClinVar (PubMed) database and only some of them are recognized in the Online Mendelian Information in Men (OMIM). The present study included 37 patients, with 14 sporadic cases and 6 familial cases, with a total of 21 index cases. Next-generation sequencing was performed on a predesigned panel of 168 genes associated with heart disease and sudden death. The sequencing analysis revealed twelve (57%) pathogenic or probably pathogenic variants, 9 of them were familial cases, managing to identify pathogenic variants in relatives without symptoms of the disease. At the molecular level, nine of the 12 variants (75%) were single nucleotide changes, 2 (17%) deletions, and 1 (8%) splice site alteration. The genes involved were MYH7 (25%), MYBPC3 (25%) and ACADVL, KCNE1, TNNI3, TPM1, SLC22A5, TNNT2 (8%). In conclusion; we found five variants that were not previously reported in public databases. It is important to follow up on the reclassification of variants, especially those of uncertain significance in patients with symptoms of the condition. All patients included in the study and their relatives received family genetic counseling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary hypertrophic cardiomyopathy (HCM) is considered a priority health problem in Mexico (INEGI 2020; Gobierno de México 2022) and globally (Antzelevitch 2007; Cheng et al. 2021), which in some cases begins with sudden death. Approximately 50% of HCM cases are caused by variations in genes that code for sarcomere proteins (Marian and Roberts 1995; Kimura et al. 1997; McKenna and Monserrat Iglesias 2000). More than 8000 gene variants have been identified in more than 50 genes associated with heart disease and sudden death (Coppini et al. 2014; Herrera-Rodriguez et al. 2020), most of them reported in the ClinVar database of the National Center of Biotechnology Information of PubMed (Sayers et al. 2021), and to the best of our knowledge, there have been no reports in the Mexican population.
Most cases are inherited in an autosomal dominant manner, which is why they affect both sexes equally, reaching genealogies with repetition of the disease, with incomplete penetrance and variable expressivity (Antzelevitch 2007; Maron et al. 2022). In familial cases, there are only 25 recognized variants in the Online Mendelian Inheritance in Man (OMIM) (Amberger et al. 2015; Herrera-Rodriguez et al. 2020) and less than 5% of cases have more than one variant with the severity of the phenotype due to gene dose effect (Wang et al. 2014; Rafael et al. 2017).
The genes most frequently associated with HCM are MYH7 and MYBPC3, in 15–25% of cases. Others such as TNNT2 and TNNI3 are found with frequencies lower than 5% (Ross et al. 2017; Herrera-Rodriguez et al. 2020). The types of gene variants that can be found are pathogenic and probably pathogenic (PV, PPV) variants that increase or probably increase the predisposition to the disease, benign or probably benign (BV, PBV) variants, which are not associated with the disease, and variants of uncertain significance (VUS), in which it is unknown whether or not it can contribute to the development of the disease (Alyousfi et al. 2021; Sayers et al. 2021; Richards et al. 2015).
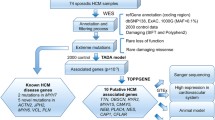
The objective of this study is to identify genetic variants in Mexican patients with a previous clinical diagnosis of HCM through next-generation sequencing (NGS) with a panel of 168 genes associated with heart disease and sudden death.
Material and methods
Patients
Male and female patients, of any age, with a previous diagnosis of HCM were recruited. The diagnosis was established by specialists in cardiology from the Mexican Institute of Social Security, based on the guidelines of the American College of Cardiology (Ommen et al. 2020). All patients agreed to participate in the study and signed a written informed consent; in minors, the consent was signed by one of their parents. In patients with a positive molecular result for any PV or PPV, their relatives were invited to participate, exploring their family history with suspected HCM or sudden death in the family, before 60 years of age. Patients who reported a family history were taken as family cases and patients without a family history were considered sporadic.
Genetic study
The DNA was extracted from a peripheral blood sample of the patients. Subsequently, NGS (Rubio et al. 2020) using a hybridization-based protocol, and sequenced using Illumina technology was performed with a predesigned genetic panel named Invitae Arrhythmia and Cardiomyopathy Comprehensive panel, of 168 genes associated with cardiomyopathies and sudden death (Table 1). These genes were selected using oligonucleotide primers designed to capture exons, the 10–20 bases flanking intronic sequences, and certain noncoding regions of interest (Agilent Technologies, Santa Clara, CA; Roche, Pleasanton, CA; Integrated DNA Technologies, Coralville, AI). The selected gene regions were sequenced with an average coverage of 350⨉ (50 ⨉ minimum). The GRCh37 reference genome database was used for single nucleotide variants (SNVs), small and large insertions/deletions (indels), structural variants, and intragenic copy number variants (Truty et al. 2019). Clinically significant variants not meeting strict NGS quality metrics were confirmed using an orthogonal method (Lincoln et al. 2019). Enrichment and analysis focus on the coding sequence of the indicated transcripts, 20 bp of flanking intronic sequence, and other specific genomic regions demonstrated to be causative of disease at the time of assay design. Markers across the X and Y chromosomes are analyzed for quality control purposes and may detect deviations from the expected sex chromosome complement. Detected variants were interpreted using Sherloc (semiquantitative, hierarchical evidence-based rules for locus interpretation), (Nykamp et al. 2017), using a point-based system incorporating the American College of Medical Genetics and Association of Molecular Pathology (ACMG–AMP) joint consensus statement guidelines (Ommen et al. 2020; Richards et al. 2015) and classified as: PV, PPV, BV, PBV, and VUS (den Dunnen and Antonarakis 2014; Richards 2015). Rare variants were defined as those with a minor allelic filtering frequency [MAF] < 1.0e − 4 based on a public data set.
Results
A total of 37 samples were analyzed. Of these, 14 were sporadic cases and 6 familial cases (7 index cases and 16 relatives), with a total of 21 index cases. The age range of the patients was from 7 to 83 years; 24 (65%) were women and 13 (35%) men.
Twelve (57%) 12 PV and PPV / 21 index cases, were detected in patients with an established diagnosis of HCM. Genes with gene variants were MYH7 (25%), MYBPC3 (25%), ACADVL, KCNE1, TNNI3, TPM1, SLC22A5, and TNNT2 (1 each, 8%), of which 9 (75%) were SNVs, 2 (17%) deletions, and 1 (8%) splicing site alteration.
Family cases
Eight PV and PPV were detected in families with a history of HCM. The genealogical trees of the families are presented (Fig. 1) and the gene variants found, indicating whether or not they are reported in the ClinVar database, their variant number, and their probable consequence at the molecular level (Table 2) (den Dunnen and Antonarakis 2014).


Pedigree and gene variants of the families studied
Family 1
The son (IV-2) is the index case; he has an established diagnosis of HCM, the mother (III-1) and the daughters (IV-1 and IV-3) have symptoms. In all of them, an SNV was found in the MYH7 gene, in a heterozygous state, with a change in codon 1357 from cytosine (C) to thymine (T), which causes a change in amino acid 453 from arginine (Arg) to cysteine (Cys), considered as PV. The other daughter (IV-4) has no symptoms and was negative for PV. This family has a history of two cases of sudden death (II-1 and II-3).
Family 2
Cases II-1 and III-1 have an established diagnosis of HCM. They have a history of two sudden deaths in different generations (I-2 and III-3). In both patients, a variant in TNNT2 was found, in the heterozygous state, with a change in codon 275 from guanine (G) to alanine (A) and a change in amino acid 92 from Arg to glutamine (Gln), considered as PV.
Family 3
Patient II-2 is the index case, with an established diagnosis of HCM. A variant was found in MYBPC3, with a change in codon 1457 in a splicing site cataloged as PPV. Daughter III-4 was also heterozygous for PPV, with no clinical symptoms. Daughters III-5 and III-6 were also studied and were negative for PPV. There is no history of sudden death or major cardiac events in previous generations.
Family 4
Patient II-1 is the index case with an established diagnosis of HCM. A PV was found in MYBPC, with a change at codon 772 from G to A, causing a change at amino acid 258 from glutamic acid (Glu) to lysine (Lys). Daughter III-3 was also heterozygous for PV without having an established diagnosis of HCM. Her twin sister (III-2) has no symptoms and it was not possible to perform the molecular study on her. Apparently, there is no history in previous generations.
Family 5
This is a very large family where the index case was patient II-9 with an established diagnosis of HCM. A PV was found in MYH7, in the heterozygous state, with a change in codon 1063 from G to A, which leads to a change in amino acid 355 from alanine (Ala) to Threonine (Thr). Siblings II-8 and II-10 were also heterozygous for PV, without presenting symptoms of the disease. The daughters of patient II-8 (III-4, III-5, and III-6) were studied and were negative for PV. This family has a history of sudden death (III-3) and other family members with symptoms did not agree to be analyzed.
Family 6
The index case in this family is patient II-2 with an established diagnosis of HCM. This patient was double heterozygous for two PVs, the first in MYH7 with a change in codon 4135 from G to A, which modifies amino acid 1379 from Ala to Thr, and the second in ACADVL with a change in codon 481 from G to A, with change in amino acid 161 of Ala for Thr. In son III-3, he presented symptoms of heart disease, without having an established diagnosis of HCM, and he also turned out to be double heterozygous for the same PVs in MYH7 and ACADVL. Son III-4 does not present symptoms of heart disease and was heterozygous but for a different PV located in KCNE1, which is caused by a deletion of the entire coding sequence, without presenting the other PV in MYH7 and ACADVL that his mother and brother have. This family has a history of heart disease in case II-2.
Sporadic cases
PV and PPV were found in 4 cases (29%) of the 14 patients with no history of HCM in the family, of which 3 PV were found in MYBPC3, TNNI3, and SLC22A5 and one PPV in TPM1 (Table 2). The rest of the sporadic cases were negative for PV and PPV.
Genetic counselling
All HCM patients included in this study and their relatives were referred to a geneticist for genetic counselling, regardless of the type of variant found. The classification of genetic variants may change as the databases are fed back with results from new studies. It is important to monitor these variants, especially those VUS found in patients with severe symptoms of the disease.
Discussion
PV and PPV were identified in 57% (12/21) of the patients analyzed with an established diagnosis of HCM. The genes involved are similar to those previously reported in the literature MYH7 (25%) and MYBPC3 (25%) (Amberger et al. 2015; Chiou et al. 2015; Herrera-Rodriguez et al. 2020) and TNNI3, TPM1, and TNNT2 (8%), (García-Castro 2009; Herrera-Rodriguez et al. 2020). Of the 12 PV and PPV found, 7 are reported in ClinVar (Sayers et al. 2021) (Table 2) and the other 5 are not found in this database, but they were designated as PV and PPV by the ACMG–AMP variant classification criteria (Nykamp et al. 2017). The PVs in ACADVL, KCNE1, and SLC22A5 are not included in the OMIM within the 25 most frequent variants in HCM (Amberger et al. 2015). At the molecular level, we found 9 (75%) SNVs that lead to changes in the amino acid sequence of the protein and prevent its correct functioning (Amberger et al. 2015), 2 deletions (17%), and 1 alteration in the splicing site (8%). Presence of PV and PPV in these genes makes it possible to improve the follow-up of carrier patients, offering genetic counseling to the family depending on their mode of inheritance. As well as early management of relatives who did not present symptoms of the disease.
Previous investigations in other populations report 54.2, 60.6 and 43.8% in the United States, France, and Japan, respectively (Richard et al. 2003; Van Driest et al. 2004; Otsuka et al. 2012). This percentage can be explained as our population was clinically selected and a history of severe heart disease and sudden death was considered in family cases.
Genes with variants encode or are associated with sarcomeric proteins and the change found causes that had some effect, or absence of protein formation, or they are related to ion transport processes associated with HCM. The MYH7 gene (OMIM 160760) is located on chromosome 14 at position q11.2 and codes for the heavy chain of β-myosin, involved in cardiac muscle contraction (Perrot et al. 2005; O’Leary et al. 2016). In families 1, 5, and 6, PVs were found in this gene, all with a single nucleotide change and previously reported (Burns et al. 2017, Nykamp et al. 2017, Salazar-Mendiguchia et al. 2020). In this gene, the gene variant that changes Arg to Cys at position 453 has been reported to have a more aggressive phenotype, due to a change in amino acid charge, compared with other reported variants (Epstein et al. 1992; Frisso et al. 2009).
The MYBPC3 gene (OMIM 6000958) (Amberger et al. 2015) is located at 11p11.2 and codes for myosin-binding protein C. The molecular consequence of the deletion found in this gene is the formation of a premature termination codon, which results in an absent or altered protein. This variant has been previously reported in 0.003% of HCM cases (O’Leary et al. 2016; Walsh et al. 2017; Nykamp et al. 2017). The alteration in the splicing site occurs at the border between an exon and an intron and can lead to the loss of exons or the inclusion of introns that also alter the protein sequence (Amberger et al. 2015).
The TNNI3 gene (OMIM 191044) is located at 19q13.42 and codes for type 3 troponin I related to cardiac muscle contraction (Amberger et al. 2015; Walsh et al. 2017; Herrera-Rodriguez et al. 2020). On the other hand, TPM1 (OMIM 191010) is located at 15q22.2 and encodes for tropomyosin 1. In the case of TNN2 (OMIM 191045), it is located at 1q32.1 and encodes the cardiac isoform of troponin T type 2. These proteins are located in the thin filaments and regulate muscle contraction in response to changes in intracellular calcium ion concentration (O'Leary et al. 2016). Variants in these three genes have been associated with a family history of sudden death and other prognoses (Anan et al. 1998; Karibe et al. 2001; Rani et al. 2012; Renaudin et al. 2018).
The SLC22A5 gene (OMIM 603377), located at 5q31.1, is a member of the organic cation transporter family and is expressed in the kidney, skeletal muscle, heart, and placenta (Amberger et al. 2015; Mutlu-Albayrak et al. 2015). Some variants in SLC22A5 cause primary systemic carnitine deficiency, skeletal myopathy, or cardiomyopathy (O’Leary et al. 2016), due to a defect in the carnitine transporter. Patients present with hypoketotic hypoglycemia, HCM, and sudden death in children and adults (Frigeni et al. 2017). In our patient, the clinical history does not show any symptoms of primary carnitine deficiency.
An interesting case was found in family 6 where one of the members presented a deletion in KCNE1 (OMIM 176261), located at 21q22.12 and belonging to the KCNE family of potassium channels (Chen, et al. 2003; Amberger et al. 2015). This gene codes for a transmembrane protein that, together with the KVLQT1 gene product, forms the delayed rectifier potassium channel (Avalos Prado et al. 2021). There are few reports of deletions in KCNE1 and those have been identified in patients with long QT syndrome (Splawski et al. 2000). Experimentally, a deletion in KCNE1 has been found to increase susceptibility to atrial fibrillation in mice (Avalos Prado et al. 2021). This family member did not present symptoms of HCM but was included because his mother and brother were double heterozygotes for PV, in MYH7 and ACADVL, which were not present in this patient.
The ACADVL gene (OMIM 609575) is located at 17p13.1 and codes for the very long chain of acyl-CoA dehydrogenase. Deficiency of this enzyme causes an inborn error in mitochondrial fatty acid β-oxidation that causes severe cardiomyopathy and/or sudden death during the neonatal period. This condition is rare and is inherited in an autosomal recessive manner. To our knowledge, there is only one report of a variant in ACADVL in a patient with HCM, caused by frameshift duplication (Kim et al. 2018). Due to the difference in PV present in this family, we suggest performing molecular studies in the other members, to confirm whether the PV in KCNE1 is de novo and whether there are other heterozygotes for PV in ACADVL and MYH7.
Of the sporadic cases, a 7-year-old patient with severe symptoms of HCM stands out in whom only 2 VUS were found in ACTC1 (OMIM 102540) and EYA4 (OMIM 603550). The parents were negative for these VUS, which proves that they are de novo variants, and they do not report a history of HCM in the family. It is important to continue analyzing the presence of VUS to define its clinical importance in HCM since the meaning of these can change as more variants are categorized (Burke et al. 2022). It is suggested to perform a microarray study on the patient to see if these variants come from some de novo chromosomal rearrangement.
There is a great need to include the identification of gene variants to support the diagnosis of HCM, as the diagnosis is currently only made based on the patient’s symptoms and the results of imaging studies (Ommen et al. 2020). The identification of PV and PPV allows the detection of carriers in the family even before expressing symptoms of the disease. With this methodology, it is possible to distinguish double heterozygous patients, with two or more variants in different genes, where the clinical manifestations are more severe. Similarly, compound heterozygotes that present genetic variants in both alleles of the same gene, where the clinical phenotype leads to death in a few months (Rafael et al. 2017; Carrier 2021), could be identified. Currently, there are gene therapy proposals for compound heterozygotes for MYBPC3 (Carrier 2021).
Limitations
In this manuscript, we focus only on reporting the gene variants observed in our patients with HCM. To establish a prevalence of each of them in the Mexican population, it is necessary to increase the sample size of the population studied, as has been done in other populations (Erdmann et al. 2003; Richard et al. 2003; Van Driest et al. 2003; García-Castro et al. 2009; Otsuka et al. 2012; Saposnik et al. 2014).
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Consent for publication
Consent for publication was obtained for every individual person’s data included in the study.
Data availability
The empirical data generated in this research is available upon request to the corresponding author, except for the personal data of the patients or any other data that could identify them.
Change history
08 October 2023
The co-corresponding author has been updated.
20 October 2023
A Correction to this paper has been published: https://doi.org/10.1007/s00438-023-02069-3
References
Alyousfi D, Baralle D, Collins A (2021) Essentiality-specific pathogenicity prioritization gene score to improve filtering of disease sequence data. Brief Bioinform 22(2):1782–1789
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A (2015) OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res 43:D789-798
Anan R, Shono H, Kisanuki A, Arima S, Nakao S, Tanaka H (1998) Patients with familial hypertrophic cardiomyopathy caused by a Phe110Ile missense mutation in the cardiac troponin T gene have variable cardiac morphologies and a favorable prognosis. Circulation 98(5):391–397
Antzelevitch C (2007) Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 4:964–972
Burke W, Parens E, Chung WK, Berger SM, Appelbaum PS (2022) The challenge of genetic variants of uncertain clinical significance : a narrative review. Ann Intern Med 175(7):994–1000
Burns C, Bagnall RD, Lam L, Semsarian C, Ingles J (2017) Multiple gene variants in hypertrophic cardiomyopathy in the era of next-generation sequencing. Circ Cardiovasc Genet. https://doi.org/10.1161/CIRCGENETICS.116.001666
Carrier L (2021) Targeting the population for gene therapy with MYBPC3. J Mol Cell Cardiol 150:101–108
Chen S, Zhang L, Bryant RM, Vincent GM, Flippin M, Lee JC et al (2003) KCNQ1 mutations in patients with a family history of lethal cardiac arrhythmias and sudden death. Clin Genet 63:273–282
Cheng Z, Fang T, Huang J, Guo Y, Alam M, Qian H (2021) Hypertrophic cardiomyopathy: from phenotype and pathogenesis to treatment. Front Cardiovasc Med 8:722340
Chiou KR, Chu CT, Charng MJ (2015) Detection of mutations in symptomatic patients with hypertrophic cardiomyopathy in Taiwan. J Cardiol 65(3):250–256
Coppini R, Ho CY, Ashley E, Day S, Ferrantini C, Girolami F, Tomberli B, Bardi S, Torricelli F, Cecchi F, Mugelli A, Poggesi C, Tardiff J, Olivotto I (2014) Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J Am Coll Cardiol 64(24):2589–2600
den Dunnen J, Antonarakis S (2014) Nomenclature for the description of human sequence variations. Hum Genet 109(1):121–124
Epstein ND, Fananapazir L, Lin HJ, Mulvihill J, White R, Lalouel JM, Lifton RP, Nienhuis AW, Leppert M (1992) Evidence of genetic heterogeneity in five kindreds with familial hypertrophic cardiomyopathy. Circulation 85(2):635–647
Erdmann J, Daehmlow S, Wischke S, Senyuva M, Werner U, Raible J, Tanis N, Dyachenko S, Hummel M, Hetzer R, Regitz-Zagrosek V (2003) Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet 64(4):339–349
Frigeni M, Balakrishnan B, Yin X, Calderon FRO, Mao R, Pasquali M, Longo N (2017) Functional and molecular studies in primary carnitine deficiency. Hum Mutat 38(12):1684–1699
Frisso G, Limongelli G, Pacileo G, Del Giudice A, Forgione L, Calabro P, Iacomino M, Detta N, Di Fonzo LM, Maddaloni V, Calabro R, Salvatore F (2009) A child cohort study from southern Italy enlarges the genetic spectrum of hypertrophic cardiomyopathy. Clin Genet 76(1):91–101
García-Castro M, Coto E, Reguero JR, Berrazueta JR, Alvarez V, Alonso B, Sainz R, Martín M, Morís C (2009) Mutations in sarcomeric genes MYH7, MYBPC3, TNNT2, TNNI3, and TPM1 in patients with hypertrophic cardiomyopathy. Rev Esp Cardiol 62:48–56
Gobierno de México (2022). Problemas prioritarios de salud del Instituto Mexicano del Seguro Social: https://www.imss.gob.mx/sites/all/statics/profesionalesSalud/investigacionSalud/temas_prioritarios_2022.pdf
Herrera-Rodriguez DL, Totomoch-Serra A, Rosas-Madrigal S, Luna-Limon C, Marroquin-Ramirez D, Carnevale A, Rosendo-Gutierrez R, Villarreal-Molina MT, Marquez-Murillo MF (2020) Genes frequently associated with sudden death in primary hypertrophic cardiomyopathy. Arch Cardiol Mex 90(1):58–68
INEGI, I. N. d. E. y. G. (2020). "Características de las defunciones registradas en México durante 2018. Consultado el 4 de septiembre 2020."
Karibe A, Tobacman LS, Strand J, Butters C, Back N, Bachinski LL, Arai AE, Ortiz A, Roberts R, Homsher E, Fananapazir L (2001) Hypertrophic cardiomyopathy caused by a novel alpha-tropomyosin mutation (V95A) is associated with mild cardiac phenotype, abnormal calcium binding to troponin, abnormal myosin cycling, and poor prognosis. Circulation 103(1):65–71
Kim YMK, G., Ko, H., Yoo, H. W., Lee, H. D. (2018) Treatable massive pericardial effusion and hypertrophic cardiomyopathy in an infant with a novel homozygous ACADVL mutation: a case report. Medicine (baltimore) 97(20):e10813
Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, Koyanagi T, Hwang TH, Choo JA, Chung KS, Hasegawa A, Nagai R, Okazaki O, Nakamura H, Matsuzaki M, Sakamoto T, Toshima H, Koga Y, Imaizumi T, Sasazuki T (1997) Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet 16(4):379–382
Lincoln SE, Truty R, Lin CF, Zook JM, Paul J, Ramey VH, Salit M, Rehm HL, Nussbaum RL, Lebo MS (2019) A rigorous interlaboratory examination of the need to confirm next-generation sequencing-detected variants with an orthogonal method in clinical genetic testing. J Mol Diagn 21(2):318–329
Marian AJ, Roberts R (1995) Recent advances in the molecular genetics of hypertrophic cardiomyopathy. Circulation 92(5):1336–1347
Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, Dearani JA, Rowin EJ, Maron MS, Sherrid MV (2022) Management of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol 79(4):390–414
McKenna WJ, Monserrat Iglesias L (2000) Identificación y tratamiento de los pacientes con miocardiopatía hipertrófica y riesgo de muerte súbita. Rev Esp Cardiol 53(1):123–130
Mutlu-Albayrak H, Bene J, Oflaz MB, Tanyalcin T, Caksen H, Melegh B (2015) Identification of SLC22A5 gene mutation in a family with carnitine uptake defect. Case Rep Genet 2015:259627
Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, Kobayashi Y, Patil N, Thusberg J, Westbrook M, Invitae Clinical Genomics Group, Topper S (2017) Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med 19:1105–1117
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O’Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD, Pruitt KD (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44(D1):D733-745
Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, Evanovich LL, Hung J, Joglar JA, Kantor P, Kimmelstiel C, Kittleson M, Link MS, Maron MS, Martinez MW, Miyake CY, Schaff HV, Semsarian C, Sorajja P (2020) 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol 76(25):3022–3055
Otsuka H, Arimura T, Abe T, Kawai H, Aizawa Y, Kubo T, Kitaoka H, Nakamura H, Nakamura K, Okamoto H, Ichida F, Ayusawa M, Nunoda S, Isobe M, Matsuzaki M, Doi YL, Fukuda K, Sasaoka T, Izumi T, Ashizawa N, Kimura A (2012) Prevalence and distribution of sarcomeric gene mutations in Japanese patients with familial hypertrophic cardiomyopathy. Circ J 76(2):453–461
Perrot A, Schmidt-Traub H, Hoffmann B, Prager M, Bit-Avragim N, Rudenko RI, Usupbaeva DA, Kabaeva Z, Imanov B, Mirrakhimov MM, Dietz R, Wycisk A, Tendera M, Gessner R, Osterziel KJ (2005) Prevalence of cardiac beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J Mol Med (Berl) 83(6):468–477
Prado PA, Hafner S, Comoglio Y, Wdziekonski B, Duranton C, Attali B, Barhanin J, Sandoz G (2021) KCNE1 is an auxiliary subunit of two distinct ion channel superfamilies. Cell 184(2):534-544 e511
Rafael JF, Cruz FF, Carvalho ACC, Gottlieb I, Cazelli JG, Siciliano AP, Dias GM (2017) Myosin-binding protein C compound heterozygous variant effect on the phenotypic expression of hypertrophic cardiomyopathy. Arq Bras Cardiol 108(4):354–360
Rani DS, Nallari P, Priyamvada S, Narasimhan C, Singh L, Thangaraj K (2012) High prevalence of Arginine to Glutamine substitution at 98, 141 and 162 positions in Troponin I (TNNI3) associated with hypertrophic cardiomyopathy among Indians. BMC Med Genet 13:69
Renaudin P, Janin A, Millat G, Chevalier P (2018) A novel missense mutation p.Gly162Glu of the gene MYL2 involved in hypertrophic cardiomyopathy: a pedigree analysis of a proband. Mol Diagn Ther 22(2):219–223
Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M (2003) Hypertrophic cardiomyopathy. Circulation 107(17):2227–2232
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Ross SB, Bagnall RD, Ingles J, Van Tintelen JP, Semsarian C (2017) Burden of recurrent and ancestral mutations in families with hypertrophic cardiomyopathy. Circ Cardiovasc Genet 10(3):e001671
Rubio S, Pacheco-Orozco RA, Gómez A, Perdomo S, García-Robles R (2020) Secuenciación de nueva generación (NGS) de ADN: presente y futuro en la práctica clínica. Univ Med 61:49–63
Salazar-Mendiguchia J, Diez-Lopez C, Claver E, Cesar S, Campuzano O, Sarquella-Brugada G, Monserrat L (2020) Familial evaluation reveals a distinct genetic cause in a large Spanish family with neurofibromatosis 1 and hypertrophic cardiomyopathy. Gene 746:144658
Saposnik B, Binard S, Fenneteau O, Nurden A, Nurden P, Hurtaud-Roux MF, Schlegel N, M. Y. H. n. French, (2014) Mutation spectrum and genotype-phenotype correlations in a large French cohort of MYH9-related disorders. Mol Genet Genomic Med 2(4):297–312
Sayers EW, Beck J, Bolton EE, Bourexis D, Brister JR, Canese K, Comeau DC, Funk K, Kim S, Klimke W, Marchler-Bauer A, Landrum M, Lathrop S, Lu Z, Madden TL, O’Leary N, Phan L, Rangwala SH, Schneider VA, Skripchenko Y, Wang J, Ye J, Trawick BW, Pruitt KD, Sherry ST (2021) Database resources of the national center for biotechnology information. Nucleic Acids Res 49(D1):D10–D17
Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT (2000) Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102(10):1178–1185
Truty R, Paul J, Kennemer M, Lincoln SE, Olivares E, Nussbaum RL, Aradhya S (2019) Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet Med 21:114–123 (Epub 2018 Jun 12)
Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ (2003) Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 108(4):445–451
Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ (2004) Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol 44(9):1903–1910
Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, Exome Aggregation C, MacArthur DG, Farrall M, Cook SA, Watkins H (2017) Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 19(2):192–203
Wang J, Wang Y, Zou Y, Sun K, Wang Z, Ding H, Yuan J, Wei W, Hou Q, Wang H, Liu X, Zhang H, Ji Y, Zhou X, Sharma RK, Wang D, Ahmad F, Hui R, Song L (2014) Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy. Eur J Heart Fail 16(9):950–957
Acknowledgements
We thank the patients and relatives who agreed to participate in this study and the Invitae Corporation © for the support of the tests for the beneficiaries of Instituto Mexicano del Seguro Social.
Funding
Instituto Mexicano del Seguro Social in support of the English correction and the publication of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study’s conception and design. Material preparation was performed by CG-V and FG-S, data collection LGL-C, JCA-H, ENG-A, HL-Z, NEG-D, CG-V; and analysis Catalina G-V, FG-S. The first draft of the manuscript was written by CG-V and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
The study was conducted in accordance with the principles of the Declaration of Helsinki, approval was granted by the Ethics Committee of the National Commission for Health Research, of the Instituto Mexicano del Seguro Social: CONBIOÉTICA-09-CEI-009_20160601.
Consent to participate
Informed consent was obtained from all individual participants included in the study. When the participant was a minor, written informed consent was obtained from the parents. Additional informed consent was obtained from all individual participants to publish their results and family trees in family cases.
Additional information
Communicated by Shuhua Xu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised to update co-corresponding author of the article as Prof. Nancy Guzmán.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
García-Vielma, C., Lazalde-Córdova, L.G., Arzola-Hernández, J.C. et al. Identification of variants in genes associated with hypertrophic cardiomyopathy in Mexican patients. Mol Genet Genomics 298, 1289–1299 (2023). https://doi.org/10.1007/s00438-023-02048-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-023-02048-8