
Abstract
Aneurysmal subarachnoid hemorrhage (aSAH) causes a robust inflammatory response which leads worse brain injury and poor outcomes. We investigated if stimulation of nicotinic acetylcholine α7 receptors (α7-AChR) (receptors shown to have anti-inflammatory effects) would reduce inflammation and improve outcomes. To investigate the level of peripheral inflammation after aSAH, inflammatory markers were measured in plasma samples collected in a cohort of aSAH patients. To study the effect of α7-AChR stimulation, SAH was induced in adult mice which were then treated with a α7-AChR agonist, galantamine, or vehicle. A battery of motor and cognitive tests were performed 24 h after subarachnoid hemorrhage. Mice were euthanized and tissue collected for analysis of markers of inflammation or activation of α7-AChR-mediated transduction cascades. A separate cohort of mice was allowed to survive for 28 days to assess long-term neurological deficits and histological outcome. Microglia cell culture subjected to hemoglobin toxicity was used to assess the effects of α7-AChR agonism. Analysis of eighty-two patient plasma samples confirmed enhanced systemic inflammation after aSAH. α7-AChR agonism reduced neuroinflammation at 24 h after SAH in male and female mice, which was associated with improved outcomes. This coincided with JAK2/STAT3 and IRAK-M activity modulations and a robust improvement in neurological/cognitive status that was effectively reversed by interfering with various components of these signaling pathways. Pharmacologic inhibition partially reversed the α7-AChR agonist’s benefits, supporting α7-AChR as a target of the agonist’s therapeutic effect. The cell culture experiment showed that α7-AChR agonism is directly beneficial to microglia. Our results demonstrate that activation of α7-AChR represents an attractive target for treatment of SAH. Our findings suggest that α7-AChR agonists, and specifically galantamine, might provide therapeutic benefit to aSAH patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) affects about thirty thousand Americans every year and has a mortality rate as high as 67% [1]. As the average age of individuals affected by aSAH is 45–55 years [2], the socio-economic burden is large. Despite advances in management of aSAH, many survivors develop significant neurological and cognitive impairments [3,4,5].
Both experimental and clinical studies have shown that aSAH induces an intensive inflammatory reaction both peripherally and centrally [6, 7]. This excessive inflammation has been hypothesized to be a main driver of secondary brain injury after aSAH leading to worse clinical outcomes [8, 9]. Several lines of evidence suggest that acetylcholine α7 receptors (α7-AChR) in the brain modulate cerebral inflammation. For example, in animal models of aSAH, increased parasympathetic tone, either directly through stimulation of the sphenopalatine ganglion or by activation of perivascular α7-AChR, improves outcome [10, 11]. Furthermore, acetylcholine levels in cerebrospinal fluid after aSAH are significantly lower in the acute phase of illness, suggesting that reduced Ach receptor activity may play a role in SAH injury [12].
α7-AChR agonists have been reported to offer benefit after a number of hemorrhagic insults to the brain including intracerebral hemorrhage [13], traumatic brain injury [14], and SAH [15]. Specifically, agonism of α7-AChR has been observed to provide anti-inflammatory and anti-apoptotic mechanisms which can improve outcome from experimental hemorrhagic stroke. Studies have indicated that α7-AChR signaling can produce anti-inflammatory effects via JAK2 [13, 16] or IRAK-M pathways [17]. JAK2 signaling negatively regulates inflammation via STAT phosphorylation. On the other hand, IRAK-M negatively regulates TLR4-mediated inflammation via inhibition of MyD88. Since TLR4 has been implicated as a major contributor to poor outcome after SAH [18, 19], we hypothesized that α7-AChR agonism will reduce neuroinflammation (via JAK2/STAT3 and IRAK-M/MyD88) following SAH in mice, thereby improving functional outcomes.
Materials and Methods
Human plasma samples (n = 59 aSAH patients, n = 23 unruptured aneurysm patients) from patients enrolled in an observational study approved by the Institutional Review Board (HSC-MS-12-0637) were used. Plasma samples were collected within 24 h after SAH and the cytokines were measured using a multiplex human immunoassay kit (Millipore) following established protocols [20].
Animal experiments were approved by the local Animal Welfare Committee, conducted in compliance with the National Institutes of Health Guidelines for the Use of Animals in Neuroscience Research, and are reported in compliance with the ARRIVE guidelines.
Study Design
A total of 104 male C57BL/6J mice (28–36 g), 18 female C57BL/6J mice (25–30 g), and 6 P0 C57BL/6J pups were used. Animals, housed in a temperature-controlled room with a 12-h light-dark cycle, had complete access to food and water. Mice were electronically randomized into groups before surgery. SigmaPlot 11.0 was used to estimate all sample sizes using data from previous experiments and preliminary data with α = 0.05 and β = 0.2. All investigators performing functional assessment, measurement of outcomes, and/or data analysis were blinded to the experimental groups. A flowchart of each experiment is provided in the Supplemental Material.
Male and female mice were analyzed in separate groups in order to test if there were any significant differences between outcome for the SAH + Vehicle or the SAH + α7-AChR agonist (galantamine) groups. The mice were allocated into three main groups: sham, SAH + vehicle, and SAH + intervention. The sham group (which undergoes all surgical procedures but does not experience SAH) is used to assess the injury caused by SAH alone (i.e., sham outcome is compared to SAH + vehicle outcome). To assess only the effects of the interventions (galantamine, MLA, HY-13277, and LY2784544), the solution used for dissolving the drugs (called vehicle) was given to SAH mice; comparison of the outcomes from SAH + vehicle mice and SAH + intervention mice allows for the effects of the invention alone to be determined.
Experiment 1
Male mice (n = 12/group) were allocated into sham, SAH + vehicle, SAH + galantamine (Tocris Bioscience), or SAH + galantamine + MLA (methyllycaconitine citrate, α7-AChR antagonist, Tocris Bioscience), to test treatment effects on functional performance and inflammation. MLA was chosen as the antagonist of α7-AChR since it is 40-fold more selective for α7-AChR than its other cholinergic receptors (α4β2 or α6β2). Sample sizes were calculated using data from previous neurobehavioral experiments [21, 22]. Female mice (assigned to sham, SAH + vehicle, or SAH + galantamine, n = 6/group) were used to test treatment effects on behavior and inflammation. A cohort of male mice (n = 6/group), assigned to SAH + vehicle or SAH + galantamine, were euthanized 6 h after SAH for measurement of cytokines. The sample sizes for the female and 6 h male cohorts were calculated using previous and preliminary data from ELISAs [23].
Experiment 2
To study the long-term beneficial effects of galantamine treatment, mice were allocated into sham (n = 10), SAH + vehicle (n = 12), or SAH + galantamine (n = 12) and allowed to survive for 28 days post-SAH. Vehicle or galantamine was administered twice daily on days 0–2 post-SAH. Sample sizes were estimated using previous data from water maze [24].
Experiment 3
To determine the anti-inflammatory mechanism of galantamine, mice (n = 6/group) were allocated into sham, SAH + vehicle, SAH + galantamine, SAH + galantamine + MLA, SAH + galantamine + HY-13277 (IRAK-M inhibitor, MedChemExpress), or SAH + galantamine + LY2784544 (JAK2 inhibitor, Selleckchem). Mice were used to examine functional performance and protein expressions of the MyD88/Nf-κB-p65 and JAK2/STAT3 pathways. HY-13277 and LY2784544 were chosen as antagonists for IRAK-M and JAK2, respectively. HY-13277 has no known off-targets but there is a possibility that it interacts with other IRAKs. LY2784544 is 8-fold, 20-fold, and 14-fold more selective for JAK2 than JAK1, JAK3, and FMS-like tyrosine kinase 3 [25], respectively. Sample sizes were estimated using previous data from Western blots [16].
BV2 mouse microglial cells (ATCC) were used to assess the effect of galantamine on cytokines and protein expressions. Cells were grown in Dulbecco’s modified eagle’s medium (DMEM, GIBCO) supplemented with 10% fetal bovine serum at 37 °C in 95% air and 5% CO2 on tissue-culture plates. When the cells reached 75% confluency, MLA (10 μM) or saline (10 μL) was added to the wells. Thirty minutes later, galantamine (10 μM) [26] or saline (0.8 μL) was added, followed by hemoglobin (50 nM) [27] or saline (10 μL) 1 h later. Twenty-four hours after hemoglobin administration, the media was removed and stored at − 80 °C and cells were prepared for Western blot.
Subarachnoid Hemorrhage Model
Subarachnoid hemorrhage was induced in mice via endovascular perforation following established protocols [22]. Perforation was confirmed by respiratory distress [22, 28, 29]. Perforation was also confirmed by assessing mice during the first 6 h after recovery for functional deficits. Vehicle (normal saline for experiments 1 and 2, 50% ethanol for experiments 3 and 4), galantamine (0.5 mg/kg in normal saline (experiments 1 and 2), or 50% ethanol (experiment 3)), MLA (0.1 mg/kg in normal saline (experiments 1 and 2) or 50% ethanol (experiment 3)), HY-13277 (0.1 mg/kg in 50% ethanol), and LY2787544 (0.1 mg/kg in 50% ethanol) were intranasally administered 1 and 8 h post-SAH (BID on days 0–2 for experiment 2). Briefly, alternating each nostril, 5 μL was given for a total of 30 μL; there was 1 min between administrations.
Mice were subjected to functional testing before euthanasia. Mice allocated for histology (experiment 2) were transcardially perfused with PBS followed by 4% paraformaldehyde, and brains were carefully removed and stored in paraformaldehyde. The remaining mice, used for Western blot or ELISA, had blood collected before transcardial perfusion with PBS, followed by removal of the brains which were snap frozen. For experiments 1 and 3, immediately following removal of the brains, the brains were observed for blood clot(s) near the circle of Willis to confirm SAH.
Neurobehavioral Performance
All animals underwent functional performance evaluation using a 24-point neuroscore, beam walking, T-maze, and rotarod 1 day post-surgery. The neuroscore examines sensorimotor function using 8 sub-tests [21]. Beam walking assesses the animal’s ability to traverse a round rod [30]. T-maze was used to detect working memory deficits; spontaneous alterations were recorded for 10 individual trials. Rotarod assesses the animal’s motor skills. Briefly, two trials of constant 5 rpm were used to train mice. Then, mice were subjected to 2–3 trials of a 5 rpm starting speed with an 8 rpm acceleration and then 2–3 trials of a 5 rpm starting speed with a 16 rpm acceleration. Each trial was a total of 2 min and at least 2 min rest time were given between trials.
Experiment 2
All mice underwent assessment of functional performance using the neuroscore, beam walking, T-maze, and rotarod on days 1–3, 5, and 7 post-SAH. Neuroscore and rotarod were also performed on day 24. On days 25–27, water maze was performed before euthanasia on day 28. On day 25, water maze visible platform training was performed; then 3 h later, the mice began the hidden platform memory block 1. On day 26, mice were subjected to memory blocks 2 and 3 separated by 3 h. On day 27, mice performed memory block 4, and then 3 h later, the probe test was performed [31].
ELISA
Plasma was obtained by spinning down mouse whole blood. Brain concentrations of interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) used 50 μL of the supernatant after protein extraction (see Western blot section below). Enzyme-linked immunosorbent assay (ELISA) kits were used to assess the concentrations of IL-1β (MLB00C) and TNF-α (MTA00B) in plasma and brain samples in duplicates following the manufacturer’s instructions (R&D Systems).
Histological Assessment
Brains were vibratome sectioned at −2 from bregma (30 μm thick) and stored free-floating at 4 °C. For experiment 2, 30-μm-thick brain sections, taken every mm from 1 to −3 from bregma, were used to measure white matter area and ventricle volume after H&E staining. ImageJ was used to determine the white matter area and area of the ventricles in each slice. The total white matter area is the sum of the white matter areas in each slice. The ventricle volume is determined by summing all ventricle areas and multiplying by the thickness (1 mm) of each slice. For experiment 2, sections were stained with primary antibodies against Iba1 (1:2000, ab5076, Abcam) at 4 °C overnight followed by secondary antibody (anti-rabbit Alexa Fluor 488 (1:200, 111-545-003, Jackson ImmunoResearch)) at room temperature for 1 h. Stained sections were imaged at 6 different locations and the number of Iba1-positive cells were recorded. The cell counts are presented as the number of positive stained cells per square millimeter.
Western Blot
Frozen brains were homogenized and sonicated in Lysis buffer (Biorad) and protein concentrations were measured. Cells were sonicated in lysis buffer and protein concentrations were measured. Western blot was performed, as previously described [32], using primary antibodies for IRAK-M (ab8116, Abcam), MyD88 (4283S, Cell Signaling Technology), p-JAK2 (3776, Cell Signaling Technology), JAK2 (3230, Cell Signaling Technology), p-STAT3 (ab476315, Abcam), p-NF-κB p65 (3033T, Cell Signaling Technology), NF-κB p65 (8242T, Cell Signaling Technology), IL-10 (BioLegend), and β-actin (sc-47778, Santa Cruz Biotechnology). Signals were normalized to β-actin for each lane and then, within each gel, all lanes were normalized to the sham values [2–3 per membrane].
Statistical Analysis
Data is presented as individual data points with the mean and standard deviation shown. All tests were two sided, all data was assessed for normality and homoscedasticity, and p < 0.05 was considered statistically significant. Data for cell counts, ELISA, Western blot, white matter area, ventricle volume, rotarod, and water maze probe trial were analyzed using one-way ANOVAs with Tukey post hoc. Sex differences for the functional data for sham, SAH + vehicle, and SAH + galantamine groups were analyzed using unpaired t tests. Human cytokine data was analyzed using Mann-Whitney U tests. Neuroscore, beam walking, and T-maze were analyzed using Kruskal-Wallis with Dunn’s post hoc. Water maze memory trials were analyzed using two-way ANOVA with Tukey post hoc. GraphPad Prism 6 (La Jolla, CA, USA) and SigmaPlot 11.0 (SysStat, Germany) were all used for analyzing and graphing data. Data is available upon reasonable request.
Results
For experiments 1 and 3, 0/18 sham male, 1/18 SAH + vehicle male, 2/18 SAH + galantamine male, 2/18 SAH + galantamine + MLA male, 1/6 SAH + galantamine + HY-13277 male, 0/6 SAH + galantamine + LY2784544 male, 0/6 sham female, 0/6 SAH + vehicle female, and 0/6 SAH + galantamine female mice died before neurobehavioral testing 1 day post-SAH. No mice (SAH + vehicle or SAH + galantamine) died for the 6 h endpoint (experiment 1). For experiment 2, 0/10 sham, 3/12 SAH + vehicle, and 5/12 SAH + galantamine mice died. One SAH + vehicle mice died within 2 days and the other 2 mice died from delayed cerebral ischemia. Three SAH + galantamine mice died within 2 days and the other 2 SAH + galantamine mice died from delayed cerebral ischemia. Details of the mortalities are reported in the Supplement Material. Samples from mice which died within 24 h were excluded from statistical analysis. Complete statistical reports are available in the Supplemental Material.
aSAH Leads to Significant Elevation of Plasma Cytokines in Patients
In humans with aSAH, a number of cytokines and chemokines, including the pro-inflammatory cytokine IL-1β, are significantly elevated within 24 h compared to unruptured aneurysm controls (Fig. 1).

Cytokines and chemokines are elevated 24 h after SAH. (a) In aSAH patients, 40 cytokines and chemokines were measured. (b) In aSAH patients, several cytokines and chemokines which were significantly different (after multiple comparison adjustment) between aSAH and unruptured aneurysm (UA) patients. n = 23 UA, n = 59 aSAH. *p < 0.05
Galantamine Reduces Neuroinflammation After SAH
Six hours following SAH, galantamine-treated mice had a significantly lower concentration of IL-1β in the brain than vehicle-treated mice (p = 0.0149). At 6 h post-SAH, there was no difference between vehicle- and galantamine-treated SAH mice for the levels of brain TNF-α, plasma IL-1β, nor plasma TNF-α (Fig. 2a). Twenty-four hours after SAH, vehicle-treated male mice had significantly elevated levels of IL-1β and TNF-α in both their plasma and brain compared to sham controls. Galantamine treatment decreased the levels of brain IL-1β (p = 0.0070) and brain TNF-α (p = 0.0140). For the plasma levels of IL-1β and TNF-α, galantamine-treated mice had levels similar to those observed in uninjured shams. When an antagonist of α7-AChR (MLA) was coadministered with galantamine, the reduction of IL-1β in the brain by galantamine alone was reversed (Fig. 2b). In female mice, galantamine treatment significantly reduced the SAH-elevated levels of brain IL-1β, plasma IL-1β, and brain TNF-α (brain IL-1β: p = 0.0117; brain TNF-α: p = 0.0009; plasma IL-1β: p = 0.0003), but did not alter plasma TNF-α (Fig. 2c).

IL-1β and TNF-α concentrations following SAH in mice. (a) Six hours post-SAH in male mice, galantamine reduces IL-1β in the brain but not brain TNF-α, plasma IL-1β, nor plasma TNF-α. One plasma sample and one brain sample from mice in the SAH + galantamine group had undetectable levels of TNF-α. For statistical purposes, these samples were assigned the minimal detectable concentration (0.36 pg/mL) as indicated by the manufacturer. (b) Twenty-four hours after SAH in male mice, galantamine reduces the amount of IL-1β in the plasma and brain, although neither reached statistical significance. Co-administration of MLA with galantamine increased IL-1β in the brain. Galantamine reduces brain TNF-α but not plasma TNF-α. (c) In female mice, galantamine reduces plasma IL-1β, brain IL-1β, and brain TNF-α, but not plasma TNF-α 24 h after SAH. One plasma sample from a sham mouse had an undetectable amount of IL-1β. One plasma sample from a sham mouse had an undetectable amount of TNF-α. For TNF-α in the brain, one sham and one SAH + galantamine mice had undetectable levels. For statistical purposes, these samples were assigned the minimal detectable concentrations (TNF-α: 0.36 pg/mL, IL-1β: 0.46 pg/mL) as indicated by the manufacturer. n = 5–6/group. *p < 0.05 vs Sham, #p < 0.05 vs SAH + vehicle, †p < 0.05 vs SAH + galantamine
Galantamine Improves Functional Performance
One day after SAH, vehicle-treated male mice had significant functional deficits (p < 0.05 vs Sham). Galantamine-treated mice performed significantly better than vehicle-treated SAH mice on neuroscore and rotarod (p < 0.05 vs SAH + vehicle) (Fig. 3a). Notably, galantamine treatment improved the performances of SAH mice to levels comparable to those observed in sham mice in all of the behaviors tested (p > 0.05 vs Sham). Galantamine’s improvement of behavioral deficits caused by SAH was also observed in female mice (Fig. 3b). Interestingly, there was a sex difference in the protective effect of galantamine; female SAH + galantamine mice had greater behavioral improvement compared to male SAH + galantamine mice in neuroscore and T-maze (Fig. 3c).

Galantamine improves neurobehavioral performance on sensory, motor, and working memory tests 24 h post-SAH. (a) In male mice, galantamine improves performance on the neuroscore and rotarod tests. Co-administration of MLA reverses galantamine’s protection. *p < 0.05 vs Sham, #p < 0.05 vs SAH + vehicle, †p < 0.05 vs SAH + galantamine. (b) In female mice, galantamine improves performance on the beam walking, rotarod, and T-maze tests. *p < 0.05 vs Sham, #p < 0.05 vs SAH + vehicle. (c) Females treated with galantamine after SAH have significantly better behavioral performances than their male counter-parts on the neuroscore, beam walking, and T-maze tests. Males: n = 5–6/group, females: n = 5–6/group. ‡p < 0.05 between the male and female mouse for the specific injury group
To determine if acute treatment with galantamine offers lasting improvement in SAH outcome, long-term functional performance was assessed. Galantamine-treated mice had significantly improved memory (Fig. 4c, d) and sensorimotor function (Fig. 4e, f) 24 days post-SAH. Swimming speed did not differ across groups (Fig. 3). Histological analysis of brains from mice surviving 28 days after SAH showed that galantamine-treated mice had less white matter damage than vehicle-treated mice (p < 0.0001) and ventricle volumes were reduced to the sham level (p = 0.0136 vs SAH + vehicle) (Fig. 4g–i).

Galantamine prevents long-term memory, sensory, and motor deficits while preserving white matter integrity in SAH mice. (a) Water maze working memory test on days 24–27 post-SAH. n = 6–10/group. *p < 0.05 Sham vs SAH + vehicle at indicated time-point. #p < 0.05 SAH + vehicle vs SAH + galantamine at indicated time-point. (b) Water maze probe test on day 27 post-SAH. Dotted line indicates random chance for being in the target quadrant (25%). n = 6–10/group. *p < 0.05 vs Sham, #p < 0.05 vs SAH + vehicle. (c) Sensory and motor functional testing on day 24 after SAH was evaluated using the neuroscore and rotarod. n = 6–10/group. *p < 0.05 vs Sham, #p < 0.05 vs SAH + vehicle. (d) Representative H&E stained brains from mice euthanized 28 days post-SAH. H&E staining was used to quantify the white matter area (internal capsule and corpus callosum) and ventricle volume. n = 6/group. *p < 0.05 vs Sham, #p < 0.05 vs SAH + vehicle
Beneficial Effects of Galantamine Occur Through α7-AChR Signaling
When the α7-AChR antagonist MLA is co-administered with galantamine, the beneficial effects of galantamine on brain IL-1β (Fig. 2) and functional outcome (Fig. 3) were lost.
Anti-inflammatory Effects of α7-AChR Stimulation
Since the α7-AChR is expressed on immune cells, we probed the signaling pathways downstream of α7-AChR to determine whether galantamine acts via JAK2/STAT3 or IRAK-M/MyD88 signaling.
When the antagonists of the α7-AChR/JAK2/STAT3 and α7-AChR/IRAK-M/MyD88 pathways are coadministered with galantamine, the functional benefits of galantamine treatment are significantly reversed (Fig. 5a). Within immune cells, α7-AChR activation may provide anti-inflammatory effects through JAK2/STAT3 signaling leading to increased anti-inflammatory IL-10 production, or by inhibiting the pro-inflammatory TLR4/MyD88/NF-κB-p65 signaling pathway.

Role of galantamine in the MyD88/NF-κB-p65 and JAK2/STAT3 signaling pathways to attenuate neuroinflammation 24 h after SAH in mice. (a) Behavioral performance of SAH mice. (b, c) Mouse protein expressions of IRAK-M, p-JAK2, MyD88, p-STAT3, p-NF-κB-p65/NF-κB-p65, and IL-10. n = 5–6/group. *p < 0.05 vs Sham, #p < 0.05 vs SAH + vehicle, †p < 0.05 vs SAH + galantamine. Galan: galantamine. HY: HY-13277. LY: LY2784544
HY-13277 (IRAK-M antagonist) was used to attenuate the beneficial effects of galantamine on the TLR4/MyD88 signaling pathway. Treatment with galantamine did not have any effect on TLR4 expression (Fig, 6). MyD88 and phospho p-65 (NF-κB) were elevated in the brain following SAH, and galantamine reduced both MyD88 and phosphorylated p65 (NF-κB) levels to those of sham (p < 0.05 SAH + vehicle vs SAH + galantamine). Galantamine also increased expression of IRAK-M (p < 0.05 vs Sham and SAH + vehicle). MLA (α7-AChR antagonist) prevented galantamine’s effects on these three proteins. HY-13277 was able to block galantamine’s benefit on IRAK-M and MyD88 expressions, and partially counteracted galantamine’s reduction of phospho p65 (NF-κB) (Fig. 5b, c).
The JAK-2 antagonist, LY2784544, reversed galatanmine’s functional benefits on neuroscore and rotarod (Fig. 5a). After SAH, p-JAK2 is elevated, but p-STAT3 and IL-10 levels were unchanged. Galantamine treatment further increases p-JAK2 levels (p < 0.05 vs SAH + vehicle) and also elevated p-STAT3 expression, leading to more IL-10 within the brain (p < 0.05 vs SAH + vehicle). Both MLA and LY2784544 prevented galantamine from increasing p-JAK2, p-STAT3, and IL-10 expressions. LY2784544 also prevented galantamine lowering of phosphorylated NF-κB p65 (Fig. 5b, c).
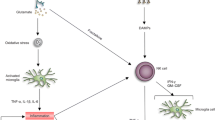
Microglia (BV2) cells were used to identify if galantamine had a direct effect on this cell type. Twenty-four hours after hemoglobin toxicity, IL-1β and TNF-α were significantly elevated in the media of BV2 cells (IL-1β: p = 0.0194; TNF-α: p < 0.0001). Galantamine treatment of the microglia cells partially attenuated the increase of IL-1β (Hb vs Hb + galantamine p = 0.0802), but had no effect on TNF-α. Antagonism of α7-AChR with MLA partially prevented galantamine’s protective effects (Fig. 6a, b). Protein expression from microglia shows that galantamine can provide benefit against the effects of hemoglobin toxicity in BV2 cells (Fig. 6c–i). Overall schematic of the pathway involved in galantamine’s protection against neuroinflammation is presented in Fig. 6j.

Galantamine reduces inflammation via MyD88/NF-κB-p65 and JAK2/STAT3 signaling in microglia. (a, b) Levels of IL-1β and TNF-α in the media from in vitro microglia subjected to hemoglobin toxicity. n = 6/group. (c–i) In vitro microglia protein expressions after hemoglobin toxicity. n = 6/group. *p < 0.05 vs Sham, #p < 0.05 vs SAH + vehicle, †p < 0.05 vs SAH + galantamine. Galan: galantamine. Hb: hemoglobin. (j) Schematic of galantamine’s anti-neuroinflammatory mode of action
Discussion
Agonism of α7-AChR has been suggested to have neuroprotective effects after brain hemorrhages via lower neuroinflammation. Herein, we investigated the therapeutic potential of galantamine, an FDA-approved cholinergic drug with α7-AChR agonist properties, for aSAH. Our findings show (1) an elevated inflammatory response following SAH in humans and mice, (2) agonism of α7-AChR can prevent neuroinflammation and improve functional outcomes in SAH mice, (3) α7-AChR is crucial for improving poor outcomes after SAH since α7-AChR null mice tend to have worse working memory after SAH, and (4) two major signaling pathways in microglia are directly affected and key in reducing neuroinflammation.
aSAH leads to a robust pro-inflammatory response as indicated by the elevation of numerous cytokines and chemokines (seen in this study and as reported by others [33,34,35,36]). Furthermore, a higher pro-inflammatory response leads to worsened delayed injury after SAH [36,37,38]. Previous clinical trials using drugs aimed at preventing inflammation have failed to offer significant benefit for aSAH [39]. One explanation for the failure of anti-inflammatory agents in improving SAH outcome is that the drugs only targeted inflammation. It is now accepted that injury following SAH does not just rely on a single pathological consequence, but rather multiple injury cascades, including inflammation and apoptosis [4, 5, 40]. α7-AChR has multiple downstream signaling pathways which are beneficial and can attenuate a variety of pathophysiological events [13,14,15,16, 41, 42]. In this study, we observed the potential benefit of α7-AChR agonism using galantamine, a cholinesterase inhibitor, and partial α7-AChR agonist, to improve outcome following SAH in mice through its anti-inflammatory effects. Intranasal administration of galantamine to mice after SAH was able to reduce neuroinflammation, leading to improved functional outcomes. To determine if the observed protection was via direct effects and signaling by microglia, we performed in vitro microglia cell culture experiments; we observed a direct anti-inflammatory effect by galantamine on microglia as assessed by measurement of inflammatory cytokines. Since galantamine is reported to have a number of mechanisms (i.e., agonism of α7-AChR and antagonism of acetylcholinesterase), we used α7-AChR knockout mice and observed that galantamine was unable to provide a therapeutic benefit for either neuroinflammation or functional outcome. Additionally, there is a worse trend for worse working memory (T-maze) after SAH in α7-AChR null mice compared to wild-type mice (Fig. 5). Although not powered to test for significant differences in functional outcome between α7-AChR knockout and wild-type SAH mice, a sample size estimation indicates that six to seven mice per group are needed, which suggests that there may be a significant differences in working memory (i.e., α7-AChR knockout mice have worse working memory than wild-type mice). This trend is a crucial piece of evidence that α7-AChR is a key modulator of brain injury after aSAH. Furthermore, the observed findings suggest that α7-AChR is the primary therapeutic target of galantamine for SAH injury.
Mechanisms of Therapeutic Benefit by α7-AChR Agonism
To identify the potential signaling cascades responsible for protection against secondary aSAH injury, we investigated two downstream signaling pathways. Our data indicates that neither signaling pathway was the key to the anti-inflammatory benefit offered by galantamine. Rather, each signaling pathway played a role in the overall protection provided by α7-AChR agonism; the combination of anti-inflammatory signaling (by JAK2/STAT3) and prevention of pro-inflammation (by IRAK-M/MyD88 and JAK2/STAT3) were needed to improve functional outcome.
A major signaling pathway reported for α7-AChR is the JAK2 pathway [16]. SAH triggers a pro-inflammatory response in part through TLR4/MyD88/p65 (NF-κB) signaling. While we found no difference in TLR4 brain expression (Fig. 6), we observed that α7-AChR activation can inhibit MyD88 via IRAK-M and inhibit phosphorylation of p65 (NF-κB) via STAT3. Additionally, we found increased anti-inflammatory cytokine IL-10 via JAK2/STAT3 signaling. Our findings related to preventing neuroinflammation with α7-AChR agonists are supported by others [13,14,15,16, 23, 41, 42].
Microglia have been suggested to be a target for improving outcome after SAH in mice [43]. Specifically, deactivation of microglia resulted in less inflammation and neuronal survival [44]. In order to determine if there was a direct effect of α7-AChR agonism for neuroinflammation via microglia, we used in vitro microglia experiments and observed that galantamine was able to reduce inflammation in culture via IRAK-M/MyD88 and JAK2/STAT3 signaling pathways. Others have also shown anti-inflammatory properties of α7-AChR activation in immune cells [45, 46].
Implications for Human Use
Activation of α7-AChR has been shown to be a therapeutic target capable of preventing neuroinflammation, neuronal cell loss, blood-brain barrier disruption, and brain edema in numerous rodent models of brain injuries, including SAH [15], traumatic brain injury [14, 23], and intracerebral hemorrhage [13, 16, 41, 42]. In these studies, various agonists were used, but none have been translated into clinical practice. There is also clinical evidence that acetylcholinesterase inhibitors are beneficial in traumatic brain injury [47, 48]].Since galantamine is both a cholinesterase antagonist and an agonist for α7-AChR, and it is FDA approved for treatment of cognitive impairments associated with Alzheimer’s disease and dementia, galantamine can be quickly translated into the clinic for use to treat SAH.
Currently, a phase 2 clinical trial is underway to investigate the therapeutic benefit of galantamine for SAH (NCT02872857). This clinical trial will assess tolerability of galantamine, as well as examine if daily administration of galantamine to SAH patients can improve outcome at 90 days post-SAH.
Study Limitations
First, we did not investigate different α7-AChR agonists. However, several α7-AChR agonists are beneficial after brain hemorrhage. Second, we did not test different galantamine doses. Our dose was determined from the human dose [23] and was adjusted for intranasal administration. The third limitation is that α7-AChR agonism can also promote cell survival. [42] Herein, we only investigated the effects of α7-AChR agonism on neuroinflammation after SAH. Evidence from others [45, 46, 49,50,51] suggests that α7-AChR agonism can directly affect microglia and neurons, and there may be a synergistic effect when both these cell types are receiving α7-AChR activation [52]. Additionally, the contribution of anti-acetylcholinesterase activity was not studied here [53]. Inflammation has been suggested to lead to worse delayed injury and poor outcome [36,37,38, 54]. Herein, we chose to focus on the acute inflammation, but future studies will investigate the role of α7-AChR agonism on delayed injury. Another limitation is that we used only pharmacological inhibitors which usually have off-target effects. However, the drugs used are significantly more selective for the primary target than other targets (see methods). Future studies can investigate more selective inhibitors or protein knockdown. Finally, it was assumed that all mice had similar hematomas and SAH injury severities since neuroscore values are very comparable (for SAH + vehicle) among experiments 1, 2, and 3. SAH blood was not graded since mice have an inherently rapid clearance of the extravagated blood [55, 56]. Other methods which could provide insight into injury severity (e.g., MRI hours after SAH, ICP, or CBF monitoring during surgery) were not used.
References
Zacharia BE, Hickman ZL, Grobelny BT, DeRosa P, Kotchetkov I, Ducruet AF, et al. Epidemiology of aneurysmal subarachnoid hemorrhage. Neurosurgery Clinics of North America. 2010;21:221-233
de Rooij NK, Linn FH, van der Plas JA, Algra A, Rinkel GJ. Incidence of subarachnoid haemorrhage: A systematic review with emphasis on region, age, gender and time trends. Journal of Neurology Neurosurgery Psychiatry. 2007;78:1365-1372
Connolly ES, Jr., Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: A guideline for healthcare professionals from the american heart association/american stroke association. Stroke. 2012;43:1711-1737
Cossu G, Messerer M, Oddo M, Daniel RT. To look beyond vasospasm in aneurysmal subarachnoid haemorrhage. BioMed Research International. 2014;628597
Foreman B. The pathophysiology of delayed cerebral ischemia. Journal of Clinical Neurophysiology. 2016;33:174-182
Savarraj J, Parsha K, Hergenroeder G, Ahn S, Chang TR, Kim DH, et al. Early brain injury associated with systemic inflammation after subarachnoid hemorrhage. Neurocritical Care. 2018;28:203-211
Sehba FA, Bederson JB. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurological Research. 2006;28:381-398
Ahn SH, Savarraj JPJ, Parsha K, Hergenroeder GW, Chang TR, Kim DH, et al. Inflammation in delayed ischemia and functional outcomes after subarachnoid hemorrhage. Journal of Neuroinflammation. 2019;16:213
Friedrich V, Flores R, Muller A, Bi W, Peerschke EI, Sehba FA. Reduction of neutrophil activity decreases early microvascular injury after subarachnoid haemorrhage. Journal of Neuroinflammation. 2011;8:103
Si ML, Lee TJ. Alpha7-nicotinic acetylcholine receptors on cerebral perivascular sympathetic nerves mediate choline-induced nitrergic neurogenic vasodilation. Circulation Research. 2002;91:62-69
Yarnitsky D, Lorian A, Shalev A, Zhang ZD, Takahashi M, Agbaje-Williams M, et al. Reversal of cerebral vasospasm by sphenopalatine ganglion stimulation in a dog model of subarachnoid hemorrhage. Surgical Neurology. 2005;64:5–11; discussion 11
Kawamata T, Takeshita M, Ujiie H, Sato K, Izawa M, Kagawa M, et al. Acetylcholine determination of cerebrospinal fluid in aneurysmal subarachnoid hemorrhage. Surgical Neurology. 1994;41:399-404
Katsuki H, Matsumoto K. Nicotinic acetylcholine receptors in regulation of pathology of cerebrovascular disorders. In: Akaike A, Shimohama S, Misu Y, eds. Nicotinic acetylcholine receptor signaling in neuroprotection. Singapore: Springer
Copyright 2018, The Author(s). 2018:113–136.
Gatson JW, Simpkins JW, Uteshev VV. High therapeutic potential of positive allosteric modulation of alpha7 nachrs in a rat model of traumatic brain injury: Proof-of-concept. Brain Research Bull. 2015;112:35-41
Duris K, Manaenko A, Suzuki H, Rolland WB, Krafft PR, Zhang JH. Alpha7 nicotinic acetylcholine receptor agonist pnu-282987 attenuates early brain injury in a perforation model of subarachnoid hemorrhage in rats. Stroke. 2011;42:3530-3536
Krafft PR, McBride D, Rolland WB, Lekic T, Flores JJ, Zhang JH. Alpha7 nicotinic acetylcholine receptor stimulation attenuates neuroinflammation through jak2-stat3 activation in murine models of intracerebral hemorrhage. BioMed Research International. 2017;8134653
Maldifassi MC, Atienza G, Arnalich F, López-Collazo E, Cedillo JL, Martín-Sánchez C, et al. A new irak-m-mediated mechanism implicated in the anti-inflammatory effect of nicotine via α7 nicotinic receptors in human macrophages. PloS One. 2014;9:e108397
Akamatsu Y, Pagan VA, Hanafy KA. The role of tlr4 and ho-1 in neuroinflammation after subarachnoid hemorrhage. Journal of Neuroscience Research. 2020;98:549-556
Okada T, Suzuki H. Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural Regeneration Research. 2017;12:193-196
Savarraj JP, McGuire MF, Parsha K, Hergenroeder G, Bajgur S, Ahn S, et al. Disruption of thrombo-inflammatory response and activation of a distinct cytokine cluster after subarachnoid hemorrhage. Cytokine. 2018;111:334-341
Matsumura K, Kumar TP, Guddanti T, Yan Y, Blackburn SL, McBride DW. Neurobehavioral deficits after subarachnoid hemorrhage in mice: Sensitivity analysis and development of a new composite score. Journal of American Heart Association. 2019;8:e011699
Dienel A, Matsumura K, Veettil RA, Hong S-H, Kumar T. P, Yan Y, et al. Microthrombi correlates with infarction and delayed neurological deficits after subarachnoid hemorrhage in mice. Stroke. 2020;51:2249-2254
Zhao J, Hylin MJ, Kobori N, Hood KN, Moore AN, Dash PK. Post-injury administration of galantamine reduces traumatic brain injury pathology and improves outcome. Journal of Neurotrauma. 2018;35:362-374
Hill JL, Kobori N, Zhao J, Rozas NS, Hylin MJ, Moore AN, et al. Traumatic brain injury decreases amp-activated protein kinase activity and pharmacological enhancement of its activity improves cognitive outcome. Journal of Neurochemistry. 2016;139:106-119
Ma L, Clayton JR, Walgren RA, Zhao B, Evans RJ, Smith MC, et al. Discovery and characterization of ly2784544, a small-molecule tyrosine kinase inhibitor of jak2v617f. Blood Cancer Journal. 2013;3:e109
Liu Y, Zhang Y, Zheng X, Fang T, Yang X, Luo X, et al. Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. Journal of Neuroinflammation. 2018;15:112
Sankar SB, Donegan RK, Shah KJ, Reddi AR, Wood LB. Heme and hemoglobin suppress amyloid β-mediated inflammatory activation of mouse astrocytes. Journal of Biological Chemistry. 2018;293:11358-11373
Muroi C, Fujioka M, Okuchi K, Fandino J, Keller E, Sakamoto Y, et al. Filament perforation model for mouse subarachnoid hemorrhage: Surgical-technical considerations. British Journal of Neurosurgery. 2014;28:722-732
Kamii H, Tominaga T. Filament perforation subarachnoid hemorrhage mouse model. In: Chen J, Xu ZC, Xu XM, Zhang JH, eds. Animal models of acute neurological injury. Switerland: Springer; 2019.
McBride DW, Wang Y, Sherchan P, Tang J, Zhang JH. Correlation between subacute sensorimotor deficits and brain water content after surgical brain injury in rats. Behavioural Brain Research. 2015;290:161-171
Dash PK, Moore AN, Dixon CE. Spatial memory deficits, increased phosphorylation of the transcription factor creb, and induction of the ap-1 complex following experimental brain injury. Journal of Neuroscience. 1995;15:2030-2039
Wu G, McBride DW, Zhang JH. Axl activation attenuates neuroinflammation by inhibiting the tlr/traf/nf-kappab pathway after mcao in rats. Neurobiology Disease. 2018;110:59-67
Zeiler FA, Thelin EP, Czosnyka M, Hutchinson PJ, Menon DK, Helmy A. Cerebrospinal fluid and microdialysis cytokines in aneurysmal subarachnoid hemorrhage: A scoping systematic review. Front Neurology. 2017;8:379
Coulibaly AP, Provencio JJ. Aneurysmal subarachnoid hemorrhage: An overview of inflammation-induced cellular changes. Neurotherapeutics. 2020;17:436-445
Frontera JA, Provencio JJ, Sehba FA, McIntyre TM, Nowacki AS, Gordon E, et al. The role of platelet activation and inflammation in early brain injury following subarachnoid hemorrhage. Neurocritical Care. 2017;26:48-57
Schneider UC, Xu R, Vajkoczy P. Inflammatory events following subarachnoid hemorrhage (sah). Current neuropharmacology. 2018;16:1385-1395
Provencio JJ. Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: A review. Acta Neurochirurgica Supplement. 2013;115:233-238
Smithason S, Moore SK, Provencio JJ. Systemic administration of lps worsens delayed deterioration associated with vasospasm after subarachnoid hemorrhage through a myeloid cell-dependent mechanism. Neurocritical Care. 2012;16:327-334
de Oliveira Manoel AL, Macdonald RL. Neuroinflammation as a target for intervention in subarachnoid hemorrhage. Front Neurology. 2018;9:292
Macdonald RL. Delayed neurological deterioration after subarachnoid haemorrhage. Nature Reviews Neurology. 2014;10:44-58
Hijioka M, Matsushita H, Ishibashi H, Hisatsune A, Isohama Y, Katsuki H. Alpha7 nicotinic acetylcholine receptor agonist attenuates neuropathological changes associated with intracerebral hemorrhage in mice. Neuroscience. 2012;222:10-19
Krafft PR, Altay O, Rolland WB, Duris K, Lekic T, Tang J, et al. Alpha7 nicotinic acetylcholine receptor agonism confers neuroprotection through gsk-3beta inhibition in a mouse model of intracerebral hemorrhage. Stroke. 2012;43:844-850
Atangana E, Schneider UC, Blecharz K, Magrini S, Wagner J, Nieminen-Kelhä M, et al. Intravascular inflammation triggers intracerebral activated microglia and contributes to secondary brain injury after experimental subarachnoid hemorrhage (esah). Translational Stroke Research. 2017;8:144-156
Heinz R, Brandenburg S, Nieminen-Kelhä M, Kremenetskaia I, Boehm-Sturm P, Vajkoczy P, et al. Microglia as target for anti-inflammatory approaches to prevent secondary brain injury after subarachnoid hemorrhage (sah). Journal of Neuroinflammation. 2021;18:36
Hwang J, Hwang H, Lee HW, Suk K. Microglia signaling as a target of donepezil. Neuropharmacology. 2010;58:1122-1129
Tsoyi K, Jang HJ, Kim JW, Chang HK, Lee YS, Pae H-O, et al. Stimulation of alpha7 nicotinic acetylcholine receptor by nicotine attenuates inflammatory response in macrophages and improves survival in experimental model of sepsis through heme oxygenase-1 induction. Antioxidants Redox Signal. 2011;14:257-2070
Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E. An experimental model of closed head injury in mice: Pathophysiology, histopathology, and cognitive deficits. Journal of Neurotrauma. 1996;13:557-568
Yu TS, Kim A, Kernie SG. Donepezil rescues spatial learning and memory deficits following traumatic brain injury independent of its effects on neurogenesis. PloS One. 2015;10:e0118793
Bencherif M, Narla ST, Stachowiak MS. Alpha7 neuronal nicotinic receptor: A pluripotent target for diseases of the central nervous system. CNS Neurological Disorders Drug Targets. 2014;13:836-845
Takada-Takatori Y, Kume T, Ohgi Y, Fujii T, Niidome T, Sugimoto H, et al. Mechanisms of alpha7-nicotinic receptor up-regulation and sensitization to donepezil induced by chronic donepezil treatment. European Journal of Pharmacology. 2008;590:150-156
Takada-Takatori Y, Kume T, Ohgi Y, Izumi Y, Niidome T, Fujii T, et al. Mechanism of neuroprotection by donepezil pretreatment in rat cortical neurons chronically treated with donepezil. Journal of Neuroscience Research. 2008;86:3575-3583
Morioka N, Harano S, Tokuhara M, Idenoshita Y, Zhang FF, Hisaoka-Nakashima K, et al. Stimulation of α7 nicotinic acetylcholine receptor regulates glutamate transporter glast via basic fibroblast growth factor production in cultured cortical microglia. Brain Research. 2015;1625:111-120
Lilienfeld S. Galantamine--a novel cholinergic drug with a unique dual mode of action for the treatment of patients with alzheimer's disease. CNS Drug Reviews. 2002;8:159-176
Blecharz-Lang KG, Wagner J, Fries A, Nieminen-Kelhä M, Rösner J, Schneider UC, et al. Interleukin 6-mediated endothelial barrier disturbances can be attenuated by blockade of the il6 receptor expressed in brain microvascular endothelial cells. Translational Stroke Research. 2018;9:631-642
Lin CL, Calisaneller T, Ukita N, Dumont AS, Kassell NF, Lee KS. A murine model of subarachnoid hemorrhage-induced cerebral vasospasm. Journal of Neuroscience Methods. 2003;123:89-97
Required Author Forms
Disclosure Form provided by the authors are available with the online version of this article.
Disclosures
H.A.C. is involved in a clinical trial (NCT02872857). All other authors declare no conflicts of interest.
Funding
Funding support was provided by the Brain Aneurysm Foundation (D.W.M), a National Institute of Health K23 (S.L.B.), Center for Clinical and Translational Sciences Scholar Program (H.A.C.), and a seed grant provided by the Department of Neurosurgery at UTHealth (D.W.M).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
ESM 1
Supplementary file1 (PDF 959 KB)
ESM 2
Supplementary file2 (PDF 508 KB)
ESM 3
Supplementary file3 (PDF 959 KB)
ESM 4
Supplementary file4 (PDF 960 KB)
ESM 5
Supplementary file5 (PDF 973 KB)
ESM 6
Supplementary file6 (PDF 1217 KB)
ESM 7
Supplementary file7 (PDF 958 KB)
ESM 8
Supplementary file8 (PDF 949 KB)
ESM 9
Supplementary file9 (PDF 1353 KB)
ESM 10
Supplementary file10 (PDF 558 KB)
Rights and permissions
About this article

Cite this article
Dienel, A., Veettil, R.A., Matsumura, K. et al. α7-Acetylcholine Receptor Signaling Reduces Neuroinflammation After Subarachnoid Hemorrhage in Mice. Neurotherapeutics 18, 1891–1904 (2021). https://doi.org/10.1007/s13311-021-01052-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01052-3