
Abstract
The genetic diversity of Patellifolia patellaris has been investigated to generate information required for the organisation of a systematic genetic resources conservation action combining the best elements of the ex situ and in situ conservation concept. To this end, ten occurrences of the species were sampled on the Iberian Peninsula in Portugal and Spain. A total of 271 individuals was analysed using 24 microsatellite markers. The factorial analysis separated the material into two occurrences from Portugal, two occurrences sampled east of Gibraltar and six occurrences from the eastern Spanish coast. The pairwise genetic distance between occurrences and the complementary compositional differentiation among occurrences were calculated. The genetic distance values were used to construct an Unweighted Pair Group Method with Arithmetic Mean tree, which can be separated into four equidistant clusters. Two clusters are located in Portugal and two further clusters in Spain. The factorial analysis as well as the genetic distance and differentiation analysis indicate that the two occurrences from Portugal are clearly genetically different from the Spanish group of occurrences. Both occurrences have low population sizes and are therefore highly endangered. In situ conservation actions are urgently required for both occurrences. Further investigations are needed to organise better informed conservation actions for P. patellaris, namely to study genetic relationships between occurrences on the Spanish mainland and occurrences distributed on the Canary Islands, Madeira and Cape Verde Islands as well as in Morocco.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Species of the genus Patellifolia A. J. Scott, Ford-Lloyd and J. T. Williams (syn. Beta section Procumbentes) belong to the tertiary gene pool of cultivated beets and are resistant to Curly Top Virus, Beet Necrotic Yellow Vein Virus, and the leaf spot disease, Cercospora beticola (see review paper of McGrath et al. 2011). Patellifolia patellaris (Moq.) A. J. Scott, Ford-Lloyd and J. T. Williams occurrences that are heavily infested with beet rust, Uromyces betae, (e.g.: at Faro del Cabo de Gata or Balerma, Almeria, Spain) as well as healthy occurrences (Calnegre, Murcia, Spain) can be observed in the distribution area of the species which indicates the presence of variation for this trait. Of particular economic importance is the resistance to the Beet Cyst Nematode (Heterodera schachtii) which was introgressed from Patellifolia procumbens (Smith) A. J. Scott, Ford-Lloyd & J. T. Williams and Patellifolia webbiana (Moq.) A. J. Scott, Ford-Lloyd and J. T. Williams into Beta vulgaris ssp. vulgaris and is being used in sugar beet production today (Thurau et al. 2011). Genetic resources of Patellifolia species will gain even more importance with the expansion of sugar beet production to new regions and ecological conditions (Hassan 2008). For that reason, the interest in Patellifolia genetic resources is increasing.
A sound taxonomic classification of plant material is at the core of any research project. One objective of the present study was to measure genetic diversity within the genus using genetic markers. This may also be used to solve pending taxonomic questions (ongoing study). Ideally, breeding research projects should rely on a small, well-described collection of genetic resources representing the genetic diversity of all species. Strikingly, not even the taxonomy of the genus Patellifolia has been settled. Although the genus Patellifolia consists of only three recognized species, several taxonomic problems still need to be solved. Wagner et al. (1989) doubted that P. procumbens and P. webbiana are distinct diploid species. Szota (1964, 1971; cited in Jassem 1992) observed that the diploid species hybridize spontaneously, form fertile offspring, and therefore could be treated as distinct variants of the same species. P. patellaris was considered to be a tetraploid self-pollinating species and thus readily distinguishable from the diploid cross-pollinating species until a flow cytometric pattern that did not fully agree with this assumption was detected (Beta patellaris = P. patellaris, Marrakesh, leg. L. Frese, 13.04.2007 (GAT-Herbarium of the Leibniz Institute of Plant Genetics and Crop Plant Research); O. Schrader, JKI, pers. communication). Later on, Giménez and Cueto (2009) described P. patellaris distributed in East Andalucia as a species composed of tetraploid as well as diploid plants. Patellifolia species are mainly found on the Canary Islands, Madeira and Cape Verde Islands, in Morocco as well as on the Iberian Peninsula. The species occur in ruderal habitats, such as roadsides, in abandoned agricultural fields, and in rocky or sandy areas mainly in dry coastal regions.
If the geographical distribution of genetic diversity is known, the information can be used to organize a systematic genetic resources conservation programme that combines the best elements of the ex situ and in situ conservation concept (Maxted et al. 2015). A carefully selected set of genebank accessions representing the genetic diversity of the genus will facilitate the efficient use of genetic resources in breeding research and crop breeding programmes.
Sustainable use of plant genetic resources implies the establishment of genetic reserves for selected occurrences. Unlike ex situ accessions preserved in genebanks, occurrences growing in their natural habitat can continuously adapt to changing growing conditions and may, as a result of long-term evolutionary processes, generate novel genetic traits of interest to plant breeding. Consequently, there is a need to identify a number of sites within the distribution area of species suited for the establishment of genetic reserves. The respective populations would altogether represent the collection of distinct alleles as well as their distinct genic associations that allow the species to reproduce, to regenerate genetic variation and to adapt over time. The collection would be composed of Most Appropriate Wild Populations (MAWP), a term introduced by Kell et al. (2016). Their ex situ conservation complement should ideally be the Most Appropriate Accession (MAA) (Engels and Maggioni 2012) i.e. the quasi genetic snapshot of the respective MAWP. The task of identifying of MAWP is quite similar to the delineation of provenances (Kleinschmit et al. 2004) in forest tree seed production. The delineation of seed provenances starts with the assessment of the population range using genetic markers. A population, or a reproductively coherent collection of plants, can be established based on two criteria. First, matings within the collection are distinctively more frequent than matings with individuals from outside the collection and secondly, offspring with one parent from the collection and the other from outside the group are less likely to survive to adulthood or to stay within the collection than offspring with both parents from the collection (Kleinschmit et al. 2004). As long as the location and limits of distinct P. patellaris populations are unknown, the term occurrence is used instead of the term population in this paper.
Particularly in the context of in situ conservation management, a set of P. patellaris populations with a good long-term survival prognosis is to be identified. The long-term survival is largely determined by the environmental conditions at a growing site. Some disturbance of the site can be an advantage to this ruderal species, such as the building of touristic infrastructure or roadside cleaning, which promotes long-term population survival depending on extent and type of human disturbance. The possible or actual impact of human activities on selected growing sites can be assessed during collecting and later be re-assessed during monitoring. In addition to the assessment of the site-specific conditions and the recommendation of sites best suited to conserve P. patellaris in situ, the selection of MAWP is to be based on genetic criteria.
For this purpose, we need to quantify genetic diversity and understand how it is distributed over the landscape. To this end, a rich repertoire of statistical tools has been developed which can be applied to analyse molecular genetic data and measure genetic diversity and differentiation (Manel et al. 2003). However, some of the more frequently used measures such as F st and its derivatives suffer from widespread misinterpretation, as pointed out by Gregorius and Gillet (2015). An alternative concept for analysing genetic variation has been suggested by Gillet and Gregorius (2008) but may not yet have reached the mainstream research.
Microsatellite markers have been widely used to study genetic diversity in Beta vulgaris ssp. maritima (inter alia Fievet et al. 2007; Stevanato et al. 2013). A comprehensive analysis of genetic diversity of P. patellaris has so far not been possible, as only few polymorphic SSR markers were available in the past. Nachtigall et al. (2016) published a set of 25 new microsatellite markers, allowing studies on the relationships between the three Patellifolia species and of the spatial distribution of genetic diversity within the species. The set complements a set of 6 SSR markers used by El Bahloul and Gaboun (2013) to investigate genetic diversity and differentiation of P. patellaris occurrences distributed along the coastline of Morocco.
The aim of this study was to assess the distribution of genetic diversity of P. patellaris in the Iberian Peninsula in order to establish a representative network of genetic reserves for effective in situ conservation of the MAWP.
Materials and methods
Sampling
Potential collecting sites of P. patellaris were identified using the International Database for Beta (IDBB), the genebank information system (GBIS) of the Institute for Plant Genetics and Crop Plant Research as well as the Genetic Resources Information Network (GRIN) of the USDA/ARS. These information systems contain passport data on historical and living ex situ accessions collected in the Iberian-Macaronesian region over the past three decades. The global biodiversity information facility (GBIF) database including chorological data of wild plant taxa was also consulted. Portugal and Spain proved to be a large geographic gap in Patellifolia genebank holdings. In the region of Lisbon, Algarve, Málaga, Almeria, Murcia, Alicante, Castellón and Tarragona, potential collecting sites were identified with the help of local experts. Regional governments and national authorities (as applicable) were asked for collecting permissions in protected areas.
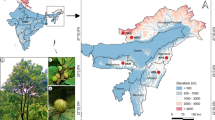
After visiting and confirming the presence of the populations recorded in the databases, samples were collected in ten locations along the distribution range in Spain and Portugal (Fig. 1), representing most of the known localities. The occurrence in Cabo Espichel (Setúbal, Portugal), discovered in the 1980s, was no longer found. Each collection site was described using a subset of the “Core descriptors for in situ conservation of crop wild relatives v.1.” of Thormann et al. (2013). The complete set of passport data is given in Supplementary file 1.

Map of the sampled Patellifolia patellaris sites in west and southwest Portugal and southeast and east Spain. The identifiers of samples taken in Spain are composed of the abbreviation of the site location, sampling date and a time stamp e.g.: BAL (Balerma, Playa de Balerma)—210415 (21 April 2015)—0900 (09.00 h). The map of sampling sites was created with OpenStreetMap. ©OpenStreetMap contributors
To support taxonomic revision of the genus Patellifolia (ongoing study), herbarium specimens of all occurrences were sent to the Herbarium of the Instituto de Investigação Científica Tropical (LISC), University of Lisbon, Portugal. At each collection site, a leaf sample of 0.5–1 g fresh weight was taken from up to 40 individual plants. When possible, plants were sampled at about 2 m distance from each other to minimize kinship. Each occurrence was geo-referenced with a GPS. The leaves of each individual were torn into small pieces, separately placed in tea bags and stored in a large zipped plastic bag or airtight plastic box with 20 g of silica gel/500 mg fresh leaf in order to desiccate the material. Several photos per site were taken to illustrate passport data records as well as to support the planned taxonomic review. In general, an overview of the site and population as well as a close-up showing details of the leaves, flowers and seeds were photographed. The picture files were stored at the Centre of Ecology, Evolution and Environmental Changes, Lisbon. The in situ and ex situ passport data were annotated to spread sheet files by the field teams, and the individual data sets were edited and stored in a standardized table. The ten occurrences of P. patellaris sampled on the coastline of the Iberian Peninsula are listed in Table 1.
Microsatellite marker analysis
Genomic DNA was prepared from dried (20 mg) leaf material after vigorous homogenization in a mixer-mill disruptor according to the modified CTAB protocol (Saghai-Maroof et al. 1984). Except for a single sample from occurrence AZO2403151630, the DNA-quality was sufficiently high for analysing the individuals at 24 microsatellite loci suitable for P. patellaris genotyping (Nachtigall et al. 2016). In total, 272 individuals were genetically analysed. DNA amplification was carried out in a total volume of 10 µl. The PCR mix contained 25 ng template DNA, 1.5 mM MgCl2, 200 µM of each dNTP, 0.25 µM of each primer and 0.5 U Taq DNA polymerase. A touch-down PCR profile was generally used as described by Nachtigall et al. (2016). The microsatellite analysis was conducted using a capillary electrophoresis GenomeLab™ GeXP Genetic Analysis System (Beckman Coulter). Fragment sizes were determined and documented within a database developed by Enders (2010). The analysis of a probe was once repeated in case of absence of the expected fragment. If the fragment remained absent, the allele was recorded as a null allele. Individual no. 40 from population STE2104151800 was excluded from further analysis, as it showed null alleles for 22 out of the 24 loci. The remaining 271 individuals were statistically analysed. The ALLELE procedure of SAS (version 9.3) was used to compile fragment sizes for all 24 markers. In case of a 1 nt deviation from a marker-specific frequent fragment size, a manual binning procedure was performed in order to avoid overestimation of genetic diversity (Amos et al. 2007). The binned data was used for the statistical analysis, while the raw data was archived in a separate database. The microsatellite markers used in the present study were all derived from diploid P. procumbens. Statistical procedures for diploid species were therefore employed in subsequent analyses (Nachtigall et al. 2016).
Data analysis
The processes of adaptation act upon whole genotypes at all developmental stages of plants and not on single isolated alleles in the gene pool. Thus, if we do not analyse the compositional differentiation at higher levels of genic integration, we miss information that may help explaining interactions between genotypes and selective forces (Gillet and Gregorius 2008). To this end, Gregorius et al. (2003) developed the measure Δ of genetic distance between populations. So far, the measure Δ has only been used in forest tree population genetics (Gregorius and Kownatzki 2005; Gillet and Gregorius 2008) and, except for one single case (Frese et al. 2013), not yet for the analysis of crop species and their wild allies. In this paper, measures of compositional differentiation as described in detail by Gillet (2013a, b) are given preference.
The software package DifferInt can be applied to calculate compositional differentiation among occurrences. The measures calculated by DifferInt are based on the minimality principle, which states that the distance between two occurrences equals the minimum extent to which one of the occurrences must be altered in order to make it match the other. Two forms of dissimilarity are important. The first—elementary genic difference d0 between individuals—establishes the dissimilarity between two individuals as the proportion of alleles of one individual’s genotype that must be changed in order to make this individual match the other individual’s genotype at all loci. The second establishes the frequency difference between two occurrences as the proportion of individuals in (either) one of the two occurrences whose genetic types must be altered in order to make the composition of genetic types in this occurrence match the composition in the other occurrence, that is, the genetic distance d0. Simultaneous minimization of the extent of alteration with respect to both elementary genic and frequency differences yields the measure of pair wise occurrence distance Δ. In order to analyse more than two occurrences, the pair wise genetic distances Δ and d0 are used to derive compositional differentiation measures for higher number of occurrences. The complementary compositional differentiation ΔSD establishes the mean pair wise occurrence distance Δ between each occurrence and the entity consisting of all other occurrences (the complement). The dispersive compositional differentiation \(\bar{\Delta }\) establishes the mean pair wise occurrence distance over all pairs of occurrences. Another unique and important feature of Δ is that it allows the measurement of compositional differentiation at all three levels of genic integration, i.e. the gene-pool, the single-locus genotypes and multi-locus genotypes. A comprehensive description of DifferInt, the underlying concept for measuring and analysing genetic variation, and an application case study is given by Gillet (2013a, b) and Gillet and Gregorius (2008).
In the present study we follow the terminology of Gregorius and Gillet (2015), where the meaning of conceptual classes of biological variation, e.g. diversity, compositional differentiation, and partitioning of variation was reviewed.
Community
Any collection of individuals that are connected and delineated by specified ecological or reproductive factors (e.g. a population in the sense of a coherent reproductive community or an occurrence).
Occurrence
In the context of this study, the term replaces the commonly used term population. An occurrence is a collection of individuals at a specific site that may or may not form a population.
Trait
In the context of the present P. patellaris study, this term describes an SSR marker which is a qualitative trait. The alleles of an SSR locus of an individual can be of the same (A1A1) or different (A1A2) type. These are called single locus types. Accordingly, the association of several single loci is termed multilocus type.
Difference
Assessment of the qualitative and quantitative difference between the types of two individuals (e.g. alleles at a locus).
Diversity
Assessment of the effective number of types in a community.
Compositional differentiation
Assessment of differences between occurrences within a community for the distribution of the trait types of a specific trait.
Partitioning of variation
Assessment of the partitioning of the total (community) variation into variation within and between occurrences.
Statistical parameters
The whole dataset used for the calculation of descriptive genetic parameters and for visualising the variation pattern included all the 271 individuals. The numbers of SSR alleles and polymorphism information content (PIC) were calculated for each locus over all individuals. The deviation from Hardy–Weinberg Proportions (HWP) was assessed for each individual occurrence using the χ2 test. The ALLELE procedure of SAS (version 9.3) was used for all calculations. The factorial analysis was conducted with the DARwin software (Perrier and Jacquemound-Collet 2006) to illustrate the variation patterns.
DifferInt was used for statistical analysis of the genetic data. This computer programme is applicable to a set of co-dominant marker data without null alleles. The measure is free of assumptions such as the existence of HWP in panmictic populations. Null alleles can be included in the analysis, but then results must be interpreted as phenotypic differences (Gillet 2013b; Frese et al. 2013). Null alleles for microsatellites are caused by a different genetic phenomenon than genetic polymorphism, in that mutations in the flanking regions of microsatellites prevent the primer from annealing to template DNA during the PCR-amplification of the microsatellite locus. For this reason, all individuals having a homozygous null allele at any of the loci were removed, reducing the number of entries in the data set from 271 to 195. The smaller data set was then used to calculate the pair wise genetic distance Δ between occurrences and the complementary compositional differentiation ΔSD. The genetic distances were further used to construct a UPGMA tree. To this end, the matrix of pairwise genetic distances was loaded into DARwin.
Conservation biologists need to understand the processes leading to local adaptation. Selective forces act upon individuals and determine their ability to reproduce. This determines the chance of individuals to pass specific non-random combinations of alleles (single locus gene association) or non-random combinations of single locus genotypes (multilocus gene association) to the next generation. DifferInt calculates measures for compositional differentiation at three levels of genic integration, i.e. gene pool, single locus and multilocus level. Different patterns of associations at the gene pool and single locus level, or single locus and multilocus level provide information of forces creating these association patterns. The permutation analysis is used to test the significance of observed differences between two levels of association (Gillet 2013a, b).
Results
Descriptive genetic parameters
Within the total data set of 13,008 data points, 260 (2%) homozygous null alleles were observed (Table 2). The number of alleles per marker loci ranged between 3 (JKIPat17) and 14 (JKIPat11), the PIC between 0.0505 (JKIPat18) and 0.7943 (JKIPat11). Out of the 240 combinations (10 occurrences × 24 loci), only 13 did not deviate significantly from Hardy–Weinberg Proportion.
Phenotypic structure
The results of the factorial analysis are shown in Fig. 2. The first two axes explain 35.93% of the total variation. Three groups can be recognized: (A) the two occurrences PS and FM from Portugal, (B) PCA and BAL sampled east of Gibraltar and (C) a group consisting of CGO, STE, AZO, COL, MOR and CNE plants from the eastern Spanish coast. Although some occurrences within groups overlap (A) PS/FM, (B) PCA/BAL, (C) STE/AZO/COL/MOR), the individual occurrences can be distinguished. The Portuguese occurrences PS and FM are most distant from the Spanish occurrences. This is in accordance with the fact that the Portuguese occurrences are also geographically distant from the Spanish ones. In addition, morphological variation of P. patellaris observed in the field agrees well with these findings (Fig. 3).

Plot of the first two components derived from principal component analysis performed with DARwin. SSR data including null alleles of a total of 271 individuals from ten occurrences are shown. AZO La Azohía, Playa de la Azohía, Murcia; BAL Balerma, Playa de Balerma, Almería; CGO La Cizaña, campo de Golf, Málaga; CNE Parque regional de Calnegre, Puntas de Calnegre, Murcia; COL Isla de Columbretes, Alicante; FM Setúbal, Serra da Arrábida, Fojo dos Morcegos, Lisboa; MOR Moraira, Cap de Moraira, Cova de les Cendres, Alicante; PCA Peñoncillo de Calaceite, Málaga; PS Sagres, Ponta de Sagres, Algarve; STE Rambla de San Telmo, Antigua carretera de entrada a Almería desde Aguadulce, Almería. The geographical sequences from the western (south of Lisbon) to the eastern (Isla de Columbretes) coastline of the Iberian Peninsula: FM, PS, CGO, PCA, BAL, STE, CNE, AZO, MOR and COL

Patellifolia patellaris in (left) west Portugal at Setúbal, Serra da Arrábida, Fojo dos Morcegos, Population ID FM (photo by M. C. Duarte, 19 April 2015); and (right) Spain at Balerma, Playa de Balerma, Population ID BAL2104150900 (photo by M. L. Rubio Teso, 21 April 2015)
Analysis of genetic diversity
Within the whole data set, i.e. 271 individuals analysed with 24 markers, 169 distinct alleles (excluding null alleles) were counted. On average, seven microsatellite alleles per locus were observed. Within the reduced data set of 195 individuals, 149 distinct alleles, 156 distinct single locus types (Fig. 4) and 134 distinct multilocus types were identified. None of the distinct multilocus genotypes was shared between the occurrences. Within occurrences, some individual multilocus genotypes were found to be duplicated between two and eight times.

Number of distinct alleles per locus as well as single locus genotypes are shown on the y-axis. Data based on DifferInt output files. Numbers on the x-axis refer to marker loci (JKIPat01, JKIPat02, etc.). JKIPat09 and JKIPat16 have not been used in this study. The number of alleles and single locus types ranged from 2 to 13 and 3 to 16, respectively
The pairwise genetic distance Δ between occurrences was calculated. Table 3a, b and c show the genetic distances for each level of gene association. The lowest distances exist between COL and MOR at each level 0.1846, 0.1846 and 0.1840 (gene pool, single locus and multilocus) and the highest distance between AZO and FM (0.6128, 0.6302 and 0.637). The UPGMA tree illustrates the genetic relationships among the ten occurrences at the gene pool level (Table 3a; Fig. 5). At the 0.2 genetic distance level, the set can be separated into four equidistant clusters, two located in Portugal (cluster I: FM, cluster II: PS) and two in Spain (cluster III: BAL and PCA as well as a larger cluster IV: CGO, AZO, STE, CNE, COL and MOR).

Cluster analysis using the unweighted pair-group method using arithmetic averages (UPGMA) of ten occurrences. The dendrogram is based on the pairwise genetic distance values Δ presented in Table 3a. It should be noted that the FM group consists of four plants, only. The bar on the left side indicates a genetic distance of 0.1 on a 0–1 scale
The measure ΔSD was used to quantify differences among occurrences. ΔSD is the mean genetic distances ΔSD(j) between each occurrence j and its complement, which is composed of all occurrences except for the jth one. The distance ΔSD(j) is maximal if the jth occurrence is completely distinct from its complement and equals 0 if the jth occurrence exactly represents its complement. Table 3 shows the components of the communities’ differentiation for each level of association. The Portuguese occurrences PS and FM, systematically showed the highest ΔSD values at all three levels, whereas CNE and MOR had the lowest values.
The mean ΔSD within the collection of ten occurrences at the gene pool and mean single locus level of genic association is presented for all 24 loci in Table 4. A small value indicates low level of compositional differentiation (e.g. JKIPat18 with ΔSD = 0.026 at the gene pool level) while high values indicate the opposite (e.g. JKIPat11 with ΔSD = 0.82 at the gene pool level). A small value does not necessarily indicate low levels of diversity since each of the occurrences within a community may be equally diverse and hence not different from each other. The ΔSD-values of all loci are comparable between both levels of genic association and 7 loci even show identical values (JKIPat01, JKIPat08, JKIPat10, JKIPat18, JKIPat19, JKIPat20, and JKIPat23).
The differentiation within a set of occurrences can be illustrated by snail diagrams. Each of the occurrences is represented by a sector, and the radius of the sector equals the contribution ΔSD(j) of a single occurrence to differentiation. The sectors are arranged by radius, with the largest contribution placed to the right of 12 o’clock, the second largest to its right followed by the rest in diminishing order. The measure (ΔSD) equals 1 if all occurrences are genetically disjoint and 0 if all occurrences show identical frequency distributions for the genetic types.
The radius of the dotted circle in a snail diagram equals the weighted mean of the sector radii and marks the community differentiation. The smaller the weighted mean, the less differentiated the community is. It is important to comprehend that the magnitude of the weighted means is largely independent of the diversity observed within the community (Gillet 2013a). For example, the diversity of allelic types of JKIPat02 (11 types) is more than double of JKIPat20 (4 types). The snail diagram for locus JKIPat02 (Fig. 6, left) starts with the largest contribution (BAL), continues clock-wise and ends with the smallest contribution to the differentiation of the community of ten occurrences (CNE). For locus JKIPat20 (Fig. 6, right), STE, PCA, CGO and BAL differ most from the complement while PS represents best the complement. The weighted mean of the sector radii is ΔSD = 0.626 for JKIPat02 and ΔSD = 0.653 for JKIPat20. The degree of mean differentiation among the ten occurrences is almost similar for both markers although JKIPat20 is less variable. The contribution of individual occurrences to differentiation in turn depends on the marker chosen. CNE, for instance, represents the complement in the case of JKIPat02 best. The short radii lengths below mean of PCA, MOR, FM, CGO, PS and CNE indicate that these occurrences are more suited to conserve the total genetic diversity of JKIPat02 contained in the community of ten occurrences than the remaining four occurrences (BAL, STE, COL and AZO).

Diversity and differentiation are two different and interdependent measures as is exemplified by the comparison of differentiation snails of markers JKIPat02 (left) and JKIPat20 (right). The snails refer to the gene pool level of integration
The individual marker loci not only differ in terms of mean differentiation values. JKIPat18 having 6 alleles sticks out by showing the lowest mean values and overall short radii length of the individual sectors (data not shown). This finding agrees with the low PIC-value (see Table 2). In future studies, with a different set of material, JKIPat18 should be tested, and if the variation within the set of the plants used is still low, the marker could be excluded from routine analysis. However, a low PIC-value does not necessarily indicate that the marker locus is not suited for differentiation analysis, as is exemplified by JKIPat08 with a PIC-value of 0.2139 and ΔSD = 0.357. The radii lengths of occurrences FM and PS differ considerably from the remaining eight occurrences (snail not shown). The marker loci JKIPat06, JKIPat21, JKIPat23 and JKIPat26 show a similar pattern as JKIPat08 and therefore would be a sufficiently large set of markers suitable to distinguish Portuguese and Spanish occurrences sampled on the mainland.
To understand whether the observed distances are not just one of the possible random arrangements of genes within occurrences, 10,000 data sets were generated by random permutations of the genes at each locus within all occurrences (Table 5). The permutation analysis tests the hypothesis that forces within populations which create gene association in individuals at a given level of integration do this independently of the allelic type at each locus: if less than 1% of permutations yield distances equal or greater than the observed distances, the results are considered as non-random effects. Similarly the hypothesis can be tested that the forces which associate individuals with occurrences do this independently of their genetic types at a given level of integration (Gillet 2013a). According to the results of the permutation analysis (Table 5), the genic associations within occurrences differ significantly from random patterns at the level of single locus genotypes, while associations in multilocus genotypes are random. The distribution of genotypes between occurrences is non-random at all levels of associations.
The differentiation of occurrences was calculated considering all 24 markers (Fig. 7). Mean ΔSD (dotted circle) only slightly increases with each higher integration level from 0.402 to 0.417 and 0.424. The levels of gene association therefore have little effect on the differentiation among occurrences. The result of the permutation analysis (Table 5, all individual genotypes between occurrences) supports this assumption.

The snails visualise the complementary compositional differentiation over all 24 marker loci. From left to right a gene pool, b mean single locus and c multilocus level for all levels of genetic integration
Discussion
Genetic investigations of Patellifolia with microsatellite markers are just beginning and knowledge on genome organization is still scanty. Here, we report the use of microsatellite markers developed by Nachtigall et al. (2016) in population genetic analysis. This study aims at starting effective planning of genetic resources conservation actions for P. patellaris.
Twenty-four markers were used for analysis. Fu et al. (2014) showed in cultivated celery that 29 SSR markers yield a sufficiently high resolution to distinguish 99% of the investigated genotypes. Interestingly, only two distinct fragments per marker were generally observed, indicating that P. patellaris may be an allopolyploid species with P. procumbens as one of its ancestors. Consequently, it was assumed that the employed microsatellite markers are specific to the P. procumbens-derived part of the P. patellaris genome. For each individual marker, the allele frequency, the PIC-values, and the ΔSD-values were calculated. Duplications within the set of 134 multilocus genotypes were observed, but this does not necessarily indicate identity of the genotypes.
Although the first and second axis of the factorial analysis explain only 35.93% of the total variation in the set of 271 plants, the phenotypic variation pattern shown in Fig. 2 is generally consistent with our expectations. Three groups of individuals can be recognized that correlate with the geographic origin of the samples. The results of the genetic analysis using a subset of 195 individuals support these findings.
Regarding the establishment of a knowledge basis for informed in situ and ex situ conservation action, FM and PS occurrences should clearly be selected based on the results of the factorial and UPGMA analyses and the ΔSD values. They are not only clearly genetically different from the Spanish group of occurrences. FM is morphologically distinct from the other nine occurrences (Fig. 3) and in addition grows, as well as PS, on a limestone cliff, an unusual site for P. patellaris. Hence, with FM a unique adaptive potential shall be conserved for future needs. Both occurrences have a very low population size of five and about 50 individuals (Supplement 1), respectively, and are threatened populations. Seed samples of both populations have already been donated to the Portuguese national genebank. In situ conservation actions are urgently required for both occurrences. Based on the factorial and UPGMA analyses, additional candidates for the network of MAWP should be chosen from the PCA-BAL and CGO-STE-CNE-AZO-MOR-COL clusters. If the capacities for the establishment and management of a genetic reserves network are limited, occurrences with the lowest ΔSD values of each cluster, i.e. BAL and MOR, respectively, should be the first choice. The establishment of genetic reserves for MAWP should not be misunderstood as a license to negligent conservation actions for other occurrences in the distribution area. The number of individuals of PCA, CGO, AZO, MOR and COL is less than 40 per occurrence (Supplement 1). These occurrences seem to be threatened and should at least be monitored to prevent accidental loss.
The occurrences are distributed along the coastline from west (FM: south of Lisbon) to east (COL: Isla de Columbretes) in the following order: FM, PS, CGO, PCA, BAL, STE, CNE, AZO, MOR and COL. The results of the factorial analysis as well as the genetic distance analysis, visualised by the UPGMA tree, however do not fully agree with the spatial distribution pattern. Assuming that the distance between occurrences is a main factor influencing the pattern of genetic diversity of P. patellaris in the investigated area, we would have expected four groups consisting of pairs of spatially close occurrences (CGO/PCA, BAL/STE, CNE/AZO) separated by less than 50 km distance and four distinct single occurrences (FM, PS, MOR, COL) separated by more than 100 km from the next occurrence (Fig. 1).
The spatial distance between two adjacent collecting sites ranged between 30 and 180 km. P. patellaris is a predominantly self-pollinating species, and therefore pollen-mediated gene flow between adjacent populations is not very likely. Seed-mediated gene flow is possible, but the likelihood and extent has never been investigated. Until further investigations give evidence for the contrary, we assume that the sampled occurrences form a network of interrelated plant groups connected through gene flow.
The plants of FM were found 160 km north of PS and both form a separate single branch of clusters I and II (Fig. 5). MOR is separated from AZO by about 180 km distance. COL is isolated by the sea and was found about 130 km north of MOR. Instead of forming a cluster with CGO, PCA is genetically closer to BAL while CGO clusters into the group of occurrences distributed along the coastline north of PCA (cluster III). The geographically closest neighbour of BAL is STE. STE is included in cluster IV while BAL forms cluster III together with PCA. The results differ from those reported by El Bahloul and Gaboun (2013) who detected a clinal variation in P. patellaris distributed in Morocco.
Further investigations are required to understand whether the absence of a clear clinal variation pattern can be explained by independent colonisation events of the sites. Kadereit et al. (2006) supposed that the continental sites had been colonised from the Canary Islands. According to a hypothesis of Hansen (1968), the colonisation of the shore of Baltic Sea by Beta vulgaris subsp. maritima started with the unintended import of seeds in the ballast sand of sailing ships at the end of the 18th century. Seeds of P. patellaris may also have been distributed in a similar way from the islands of the Macaronesian archipelagos with increasing trade traffic in the period after the discovery of the islands by Spanish and Portuguese conquerors in the 15th century.
Genebank managers are primarily interested in capturing a maximum of distinct alleles during collecting missions. Marshall and Brown (1975) defined the aim of plant explorations as the collection of at least one copy of each variant that occurs in the target population with frequency greater than 0.05, whereby co-adaptive gene complexes were regarded as “variants”. Further theoretical considerations led to the recommendation to focus collecting efforts on a class of variants which are locally common and thus reflect distinct local adaptations. From plant breeders’ point of view, such a collecting strategy is productive, as it yields genetic diversity that can be introgressed or incorporated into the crop breeding pool. If the intention is to conserve and manage genetic diversity of a species within the natural habitat “in situ” or “on site”, management plans and decisions should be based on an understanding of mechanisms that help maintain the adaptability of a species. The availability of sufficient genetic diversity is a precondition for that. Genetic diversity can be shaped in space and time by forces such as founder effects, isolation by distance, habitat fragmentation, the amount of gene flow between adjacent subpopulations of a metapopulation, and selective forces that determine the reproductive success of individual genotypes. Prior to any in situ conservation planning, information on the main forces shaping genetic diversity of a species within the target area is to be investigated.
The permutation analysis (Table 5) indicates that gene associations in individuals within occurrences are dependent on the allelic type at each locus at the single locus genotype association level but not at the multilocus genotype association level. According to Gillet (2013a), this finding can be explained as the effect of self-pollination and inbreeding as well as selective forces being involved in differentiation. However, high percentages of heterozygosity for some loci indicate a certain degree of cross-pollination in P. patellaris.
Furthermore, a non-significant P value for gene integration into multilocus genotypes within occurrences indicates that the 24 loci are not linked. If individuals were associated with occurrences dependent of their genetic type at the gene pool and single locus level of association, directed selection would have contributed to differentiation. The permutation analysis indicates significant deviation from random association pattern for the gene pool and single locus association level. It can be concluded that selection on single loci within occurrences and directed genotypic selection are the two forces shaping genetic diversity of P. patellaris within the investigation area. The covariance for ΔSD-values at the mean single locus and multilocus level equals plus 1 (P = 0.0) and shows that the ranking of populations does not change with increasing integration level. The ten occurrences have therefore the same form of gene associations within populations which can be interpreted as selection acting on single loci and multilocus genotypes in a similar way.
As far as studies of differentiation among occurrences are concerned, there has been much debate within the research community (Ryman and Leimar 2009) about the appropriateness of the common usage of Wright’s fixation index F st or G st in population studies for measuring the level of population differentiation (Manel et al. 2003). Maras et al. (2006) also noticed that F st is inappropriate for establishing the genetic limits between occurrences or genebank accessions. In many publications Nei’s GST was used to partition total variation within sets of accessions into a “within groups” and “between groups” component and the mean GST-value was interpreted as differentiation between groups. In fact, as shown some time ago by Gregorius and Roberds (1986) and taken up by Jost (2008) in more detail, these measures do not assess differentiation in the sense of their not sharing genetic types, as already mentioned by Wright (1978). F st can achieve maximal values for a locus only when all populations are monomorphic (though not all for the same allele, in which case F st is undefined). F st can be very small at a locus not only when almost all populations are identical but also when the locus is highly polymorphic, as is typical for microsatellites (Gregorius et al. 2007). As reviewed by Gregorius and Gillet (2015), the notion of differentiation can in fact be approached from two perspectives, one relating to the partitioning of total diversity into components of diversity within and between occurrences, and the other perspective explicitly relating to differences in composition among occurrences, referred to as compositional differentiation. The perspective of partitioning of diversity is even more problematic, since it does not allow for studies of differentiation on principle (Gregorius 2016).
In particular for the planning of in situ conservation programmes, it is essential to understand the adaptive processes that are shaping the distribution of genetic diversity in space and time. Conservationists aim at the maintenance of genetic diversity contained in the target species. The diversity is shared by occurrences located within the species’ distribution area and shaped by external forces such as geographic fragmentation, selection or random drift. An understanding of the compositional differentiation among occurrences is needed for planning conservation actions which can comprise re-sampling of the species to complement ex situ holdings or the identification of a set of occurrences representing the genetic diversity of a species found within a specified part of the species’ distribution area.
Further studies may include more occurrences and the other Patellifolia species from the Macaronesian archipelagos. Microsatellite markers are suited to calculate genetic distances among occurrences and to quantify genetic differentiation within this collection. Nuclear ribosomal DNA (nrDNA), internal transcribed spacer sequences (ITS) and/or chloroplast DNA markers are tools widely used to analyse and establish phylogenetic relationships between species (Romeiras et al. 2016) and between occurrences of the same species as well (Villain 2007). The latter two marker classes may help to explain why the geographic distribution pattern of occurrences does not fully agree with the observed genetic patterns. If two occurrences located close to each other show greater genetic distance than two occurrences more distantly located, this observation could be explained by independent colonisation of the sites. The existence of differences in nrDNA or cpDNA marker diversity in neighbouring occurrences would support this assumption. Romeiras et al. (2016) observed polymorphisms in nrDNA (ITS) and cpDNA gene (matK, trnH-psbA, trnL intron, rbcL) sequences of P. patellaris that are available for such kind of phylogeographic investigations. Until such work has been implemented, further priority setting for in situ conservation actions would be premature. However, the small number of P. patellaris plants found on the Iberian Peninsula indicates that the species is already under threat on the continental part of the distribution area. Species conservation agencies should therefore urgently start to monitor the demographic development of the species within the investigated regions.
Change history
31 August 2017
An erratum to this article has been published.
References
Amos W, Hoffman JI, Frodsham A, Zhang L, Best S, Hill AVS (2007) Automated binning of microsatellite alleles: problems and solutions. Mol Ecol Notes 7:10–14. doi:10.1111/j.1471-8286.2006.01560.x
El Bahloul Y, Gaboun F (2013) Genetic structure analysis for Moroccan wild beet germplasm. In: Maggioni L, Frese L, Lipman E (eds) Report of a Working Group on Beta and the World Beta Network. Fourth Joint Meeting, 20–22 June 2012, Cappelle-en-Pévèle, France. Bioversity International, Rome, Italy, p 5
Enders M (2010) Entwicklung und Anwendung molekularer und informatorischer Werkzeuge zum genetischen Monitoring bei Wildrüben. Martin-Luther-Universität Halle-Wittenberg Naturwissenschaftliche Fakultät III, Institut für Informatik, Diplomarbeit im Fach Bioinformatik, Halle, Germany
Engels J, Maggioni L (2012) AEGIS: a regionally based approach to PGR conservation. In: Maxted N, Dulloo ME, Ford-Lloyd BV, Frese L, Iriondo JM, Pinheiro de Carvalho MAA (eds) Agrobiodiversity conservation: securing the diversity of crop wild relatives and landraces. CAB International, Wallingford, pp 321–326
Fievet V, Touzet P, Arnaud J-F, Cuguen J (2007) Spatial analysis of nuclear and cytoplasmic DNA diversity in wild sea beet (Beta vulgaris ssp. maritima) populations: do marine currents shape the genetic structure? Mol Ecol 16:1847–1864
Fu N, Wang P-Y, Liu X-D, Shen H-L (2014) Use of EST-SSR markers for evaluating genetic diversity and fingerprinting celery (Apium graveolens L.) cultivars. Molecules 19:1939–1955. doi:10.3390/molecules19021939
Frese L, Enders M, Nachtigall M, Gillet, EM, Gregorius H-R (2013) Population differentiation in Beta vulgaris subsp. maritima measured with Delta In: Maggioni L, Frese L, Lipman E (eds) Report of a Working Group on Beta and the World Beta Network. Fourth Joint Meeting, 20-22 June 2012, Cappelle-en-Pévèle, France. Bioversity International, Rome, Italy. http://archive-ecpgr.cgiar.org/archive_up_to_phase_viii/-sugar_starch_fibre_crops/-beta/beta_meeting_2012/posters.html. Accessed 17 Nov 2016
Gillet EM (2013a) DifferInt: compositional differentiation among populations at three levels of genetic integration. Mol Ecol Res 13:953–964
Gillet EM (2013b) DifferInt Compositional differentiation among populations at three levels of genetic integration. User Manual. http://www.uni-goettingen.de/forstgenetik/differint. Accessed 17 Nov 2016
Gillet EM, Gregorius H-R (2008) Measuring differentiation among populations at different levels of genetic integration. BMC Genet 9:60
Giménez E, Cueto M (2009) Amaranthaceae. In Blanca G, Cabzudo B, Cueto M, Fernández López C, Morales Torres C (eds) Flora vascular de Andalucia oriental, vol 4. Consejería de Medio Ambiente, Junta de Andalucía, Sevilla, pp 157–158
Gregorius H-R (2016) Effective numbers in the partitioning of biological diversity. J Theor Biol 409:133–147
Gregorius H-R, Gillet EM (2015) Classifying measures of biological variation. PLoS ONE 10(3):e0115312. doi:10.1371/journal.pone.0115312
Gregorius H-R, Kownatzki D (2005) Spatiogenetic characteristics of beech stands with different degrees of authochthony. BMC Ecol 5:8. doi:10.1186/1472-6785-5-8
Gregorius H-R, Roberds JH (1986) Measurement of genetical differentiation among subpopulations. Theor Appl Genet 71:826–834
Gregorius H-R, Gillet EM, Ziehe M (2003) Measuring differences of trait distributions between populations. Biom J 45:959–973
Gregorius H-R, Degen B, König A (2007) Problems in the analysis of genetic differentiation among populations: a case study in Quercus robur. Silvae Genet 56:190–199
Hansen A (1968) Beta maritima L.: strand-bede. Botaniske Tidsskr 63:222–225
Hassan SF (2008) Development of sugar industry in Africa. Sugar Tech 10:197. doi:10.1007/s12355-008-0037-6
Szota Z (1964, 1971) cited by Jassem B (1992) Species relationship in the genus Beta as revealed by crossing experiments. In: Frese L (ed) International Beta Genetic Resources Network. A report on the 2nd international Beta genetic resources workshop held at the institute for crop science and plant breeding, Braunschweig, Germany, 24–28 June, 1991. International Crop Network Series No. 7. International Board for Plant Genetic Resources, Rome, pp 55–61
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kadereit G, Hohmann S, Kadereit JW (2006) A synopsis of Chenopodiaceae subfam. Betoideae and notes on the taxonomy of Beta. Willdenowia 36:9–19
Kell SP, Ford-Lloyd BV, Maxted N (2016) Europe’s crop wild relative diversity: from conservation planning to conservation action. In: Maxted N, Ehsan Dulloo M, Ford-Lloyd BV (eds) Enhancing crop genepool use: capturing wild relative and Landrace Diversity for crop improvement. CAB International, Wallingford, pp 125–134
Kleinschmit JRG, Kownatzki D, Gregorius H-R (2004) Adaptational characteristics of autochthonous populations—consequences for provenance delineation. For Ecol Manag 197:213–224
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197
Maras M, Sušnik S, Meglič V, Šuštar-Vozlič J (2006) Characterization and genetic diversity changes in the Slovenian common bean, Cesnjevec landrace. Acta Biol Crac Ser Bot 48(2):39–47
Marshall DR, Brown AHD (1975) Optimum sampling strategy. In: Frankel OH, Hawkes JG (eds) Crop genetic resources for today and tomorrow. Cambridge University Press, Cambridge, pp 379–396
Maxted N, Avagyan A, Frese L, Iriondo JM, Magos Brehm J, Singer A, Kell SP (2015) ECPGR concept for in situ conservation of crop wild relatives in Europe. Wild species in genetic reserves working group. European Cooperative Programme for Plant Genetic Resources. Rome, Italy
McGrath JM, Panella L, Frese L (2011) Beta. In: Kole C (ed) Wild crop relatives: genomic and breeding resources industrial crops. Springer, Heidelberg, pp 1–28
Nachtigall N, Bülow L, Schubert J, Frese L (2016) Development of SSR Markers for the Genus Patellifolia (Chenopodiaceae). Appl Plant Sci 4:8. doi:10.3732/apps.1600040
Perrier X, Jacquemound-Collet JP (2006) DARwin software http://darwin.cirad.fr/darwin. Accessed 10 Oct 2010
Romeiras MM, Vieira A, Silva DN, Moura M, Santos-Guerra A, Batista D, Duarte MC, Paulo OS (2016) Evolutionary and biogeographic insights on the macaronesian Beta-Patellifolia Species (Amaranthaceae) from a time-scaled molecular phylogeny. PLoS ONE 11(3):e0152456. doi:10.1371/journal.pone.0152456
Ryman N, Leimar O (2009) GST is still a useful measure of genetic differentiation: a comment on Jost’s D. Mol Ecol 18:2084–2087
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Stevanato P, Trebbi D, Biancardi E, Cacco G, McGrath JM, Saccomani M (2013) Evaluation of genetic diversity and root traits of sea beet accessions of the Adriatic Sea coast. Euphytica 189:135–146
Thormann I, Alercia A, Ehsan Dulloo M (2013) Core descriptors for in situ conservation of crop wild relatives v.1, Bioversity International, Rome, Italy
Thurau T, Ye W, Menkhaus J, Knecht K, Tang G, Cai D (2011) Plant Nematode Control. Sugar Tech 12:229–237. doi:10.1007/s12355-010-0056-y
Villain S (2007) Histoire evolutive de la section Beta. Mise en évidence des phénomènes d’hybridation et de spéciation au sein de la section dans le bassin méditerranéen. These pour obtenir le grade de docteur en sciences de l’Université de Lille 1. Spécialité: sciences de la vie. Présentée par Sarah Villain le 13 décembre 2007
Wagner H, Gimbel E-M, Wricke G (1989) Are Beta procumbens Chr. Sm. and B. webbiana Moq. different species? Plant Breed 102:17–21
Wright S (1978) Evolution and the genetics of populations, vol. 4: variability within and among natural populations. The University of Chicago Press, Chicago
Acknowledgements
Elisabeth Gillet and Hans-Rolf Gregorius critically read and edited the manuscript. We are very grateful for their suggestions and help. This research was co-funded by the European Cooperative Programme for Plant Genetic Resources (ECPGR), Rome, Italy (LOA 15/008).
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Plant Breeding: the Art of Bringing Science to Life. Highlights of the 20th EUCARPIA General Congress, Zurich, Switzerland, 29 August–1 September 2016
Edited by Roland Kölliker, Richard G. F. Visser, Achim Walter & Beat Boller
An erratum to this article is available at https://doi.org/10.1007/s10681-017-2003-4.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10681_2017_1942_MOESM1_ESM.xlsx
Description of Patellifolia patellaris sampled on the Iberian Peninsula in Portugal and Spain. The following data are provided: identifiers, designation and geographic coordinates of the sampling sites, population area and population size. Supplementary material 1 (XLSX 16 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Frese, L., Bülow, L., Nachtigall, M. et al. Genetic diversity of Patellifolia patellaris from the Iberian Peninsula, a crop wild relative of cultivated beets. Euphytica 213, 187 (2017). https://doi.org/10.1007/s10681-017-1942-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-1942-0