
Abstract
Heart failure is a clinical syndrome where cardiac output is not sufficient to sustain adequate perfusion and normal bodily functions, initially during exercise and in more severe forms also at rest. The two most frequent forms are heart failure of ischemic origin and of non-ischemic origin. In heart failure of ischemic origin, reduced coronary blood flow is causal to cardiac contractile dysfunction, and this is true for stunned and hibernating myocardium, coronary microembolization, myocardial infarction and post-infarct remodeling, possibly also for the takotsubo syndrome. The most frequent form of non-ischemic heart failure is dilated cardiomyopathy, caused by genetic mutations, myocarditis, toxic agents or sustained tachyarrhythmias, where alterations in coronary blood flow result from and contribute to cardiac contractile dysfunction. Hypertrophic cardiomyopathy is caused by genetic mutations but can also result from increased pressure and volume overload (hypertension, valve disease). Heart failure with preserved ejection fraction is characterized by pronounced coronary microvascular dysfunction, the causal contribution of which is however not clear. The present review characterizes the alterations of coronary blood flow which are causes or consequences of heart failure in its different manifestations. Apart from any potentially accompanying coronary atherosclerosis, all heart failure entities share common features of impaired coronary blood flow, but to a different extent: enhanced extravascular compression, impaired nitric oxide-mediated, endothelium-dependent vasodilation and enhanced vasoconstriction to mediators of neurohumoral activation. Impaired coronary blood flow contributes to the progression of heart failure and is thus a valid target for established and novel treatment regimens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
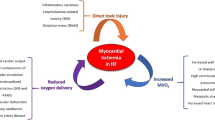
Heart failure and atherosclerosis are frequent and frequently co-exist, as they develop not only from more or less specific genetic predispositions but also from life style-related risk factors and comorbidities, such as physical inactivity, obesity [201] and metabolic syndrome, diabetes, hypertension, but also from environmental pollution [187]. The co-existence of predisposing risk factors and comorbidities, coronary atherosclerosis and coronary microvascular dysfunction is particularly obvious in patients who have heart failure with preserved ejection fraction. The interaction between coronary atherosclerosis and heart failure is complex. Coronary atherosclerosis on the one hand can induce myocardial ischemia and infarction which then causes heart failure. On the other hand, genetic mutations can cause heart failure, and coronary blood flow even in the absence of coronary atherosclerosis is then impaired as a consequence of heart failure. Then, both heart failure and impaired coronary blood flow impact on each other—any form of heart failure predisposes to myocardial ischemia through increased extravascular compression and increased coronary vasoconstriction in response to neurohumoral activation, and any form of myocardial ischemia further impairs left ventricular (LV) function (Fig. 1). The coronary circulation in heart failure is characterized by morphological alterations (arteriolar hypertrophy, capillary rarefication) and functional abnormalities, such as impaired endothelium-dependent and metabolic vasodilation, enhanced vasoconstriction to mediators of neurohumoral activation, and increased extravascular compression. A positive interaction between heart failure and impaired coronary vascular function, as evidenced by reduced coronary dilator reserve in heart failure, predisposes to poor clinical outcome. Comprehensive review articles on the coronary circulation in more general [12, 45, 74, 183], the coronary microcirculation in more particular, [40, 170, 273] and on the coronary circulation in specific forms of heart failure, e.g., hypertrophy, [10, 26, 31] heart failure of hypertensive origin [263] or heart failure with preserved ejection fraction [175, 216, 239] already exist. The present review attempts a comprehensive analysis of the common features of coronary blood flow impairment in the entire spectrum of heart failure syndromes and the cause-and-consequence relationships between heart failure and coronary blood flow. More specifically, this review identifies the common grounds of impaired coronary dilator reserve in all heart failure as well as the more specific defects of the coronary circulation in the different heart failure entities.

Impairment of coronary blood flow in heart failure of ischemic origin, of non-ischemic origin and of a pathogenesis with ischemic and non-ischemic contributions
Heart failure of ischemic origin
A reduction in cardiac contractile function is the earliest and most obvious manifestation of each critical reduction in coronary blood flow, whether reversible or not. The critical threshold of coronary blood flow is 8–10 µl per g of myocardial tissue and cardiac cycle [91].
Stunned and hibernating myocardium
Stunned and hibernating myocardium are characterized by reversible contractile dysfunction during the recovery from an episode of myocardial ischemia (stunning) or during still ongoing more moderate myocardial ischemia (hibernation) [94]. Heart failure can develop from such reversible contractile dysfunction acutely (stunning) or more chronically (hibernation), provided the respective coronary perfusion territory, the severity and the duration of coronary blood flow reduction are large enough [94].
Stunned myocardium Almost by definition, stunned myocardium is reperfused, i.e., in chronically instrumented conscious dogs, there is typically an initial reactive hyperemia followed by a normalization of myocardial blood flow over several hours during which some transmural redistribution of blood flow at the expense of subendocardial layers remains [99, 100]. Depending on the severity and duration of the preceding myocardial ischemia, full recovery of regional contractile function occurs over several hours to days [100, 140, 236]. When reperfusion occurs through a residual stenosis in chronically instrumented conscious pigs, coronary perfusion abnormalities and contractile dysfunction persist longer and may induce hibernation [238]. Whereas the myocardial contractile dysfunction of stunned myocardium is caused by increased reactive oxygen species formation and impaired excitation–contraction coupling, [94] there is also a coronary vascular stunning component, with an impaired reactive hyperemia response after brief coronary occlusion [247] and impaired vasodilator responses to intravenous adenosine or papaverine, [19] and a particularly impaired endothelium-dependent coronary vasodilator response to acetylcholine [47] in anesthetized dogs and pigs [141]. Whereas the Gregg phenomenon (an increase in contractile function in response to increased coronary blood flow) is not operative in normal myocardium, [207] the coronary autoregulation in stunned myocardium appears blunted, predisposing it to a Gregg effect, i.e., there is increased regional contractile function in anesthetized dogs to intravenous dipyridamole or papaverine [223] and in anesthetized pigs to intracoronary adenosine [208].
Most importantly, stunning contributes to contractile dysfunction following non-transmural myocardial infarction, i.e., there is both an irreversible and a reversible component of contractile dysfunction. In anesthetized dogs with 2 h coronary occlusion, regional myocardial blood flow recovered to 50% of baseline after 2 h reperfusion and regional contractile function recovered back to about 40% of baseline within 2 weeks. [50] In conscious dogs, which were otherwise healthy and without coronary atherosclerosis, 1 h coronary occlusion induced severe regional contractile dysfunction which recovered back to > 50% within 4 weeks, but there was no recovery after 3 h coronary occlusion. [129] In anesthetized dogs, the coronary dilator response to intracoronary acetylcholine was severely impaired at 30 min reperfusion in the myocardium surviving 1 h coronary occlusion, particularly in its subendocardial layers (Fig. 2) [47].

Increment in coronary blood flow in response to intracoronary acetylcholine (in % of dilator response at baseline) at 30 min reperfusion following either 15 min or 60 min coronary occlusion in anesthetized dogs, in reversibly (TTC-positive) and irreversibly (TTC-negative) injured myocardium. Endo: flow to subendocardial layers; Mid: flow to midmyocardial layers; Epi: flow to subepicardial layers. TTC, triphenyl tetrazolium chloride. From [47] by permission
Stunning in the clinic Pure stunning, i.e., fully reversible contractile dysfunction following an episode of myocardial ischemia, occurs clinically following percutaneous coronary intervention (PCI) [102, 161, 211] or a protocol of exercise-induced ischemia [4, 61, 135] but rarely poses a clinical problem, notably does not cause heart failure per se. [87] However, stunning may contribute to other myocardial ischemia-related heart failure scenarios, e.g., recovery from myocardial infarction (see above [24, 210, 240]) or from cardioplegic ischemic cardiac arrest. Unfortunately, sequential measurements of coronary blood flow and its relation to contractile function during the recovery from myocardial infarction or cardioplegia are not available. There is also vascular stunning, a reduced coronary vasodilator response to dipyridamole in patients after PCI, [252] but its functional importance is not really clear.
Hibernating myocardium Different from stunning with its transient nature, hibernation is a sustained state of regional myocardial contractile dysfunction which may indeed cause chronic heart failure. By definition, hibernating myocardium has reduced blood flow and its contractile dysfunction recovers after revascularization [22, 88, 94, 98, 118, 185, 186]. Hibernation was originally regarded as an adaptive response of the myocardium to ischemia, in that contractile function was downregulated to match the decrease in myocardial blood flow such that the myocardium could retain its viability and contractile function recover after revascularization [186]. Indeed, evidence for such perfusion–contraction matching not only during brief episodes of myocardial ischemia [14] was provided in a number of experimental studies in anesthetized and chronically instrumented conscious dogs and pigs, and the adaptive nature of such perfusion–contraction matching was supported by the recovery of metabolic perturbations during the progression from early to more sustained ischemia over several hours [94, 196]. The idea of an adaptive downregulation in response to reduced blood flow in hibernating myocardium was challenged since in some experimental studies, in chronically instrumented conscious pigs with coronary stenosis, contractile function was reduced but myocardial blood flow was not [212, 213]. A heated debate on whether hibernating myocardium was an adaptation to persistent ischemia or a result of repetitive stunning followed, but resolved by elegant experiments of Canty and colleagues who demonstrated in chronically instrumented conscious pigs with coronary stenosis, that indeed there is a progression from repetitive stunning to hibernation where myocardial blood flow and coronary reserve are reduced [55, 57]. When such chronic hibernating myocardium with reduced regional contractile function and blood flow affects both the left anterior descending and the left circumflex coronary arteries in pigs, a typical situation of compensated heart failure develops [56]. Hibernation characterized not only contractile function and metabolism distal to a chronic coronary stenosis, but also the coronary circulation which developed atrophy of larger (> 75 µm diameter) and hypertrophy of smaller (< 75 µm diameter) microvessels distal to the stenosis [148]. Induction of angiogenesis by endothelial nitric oxide synthase transfection in a pig model of hibernation, conversely, improved blood flow and contractile reserve [125]. Revascularization of chronically hibernating myocardium quickly normalizes adenosine-recruitable coronary reserve but recovery of contractile function is more delayed [171].
Hibernating myocardium in the clinic In patients with chronic coronary artery disease and contractile dysfunction, there is solid evidence from studies using positron emission tomography (PET) that myocardial blood flow in the hibernating regions is reduced [88, 98, 258] but higher than in regions which did not recover contractile function after revascularization [41, 276]. Dipyridamole-recruited coronary reserve is more reduced in patients with coronary artery disease and LV dysfunction than in those without LV dysfunction [256]. The viability of hibernating myocardium which is then an indication for revascularization is best assessed by a combination of imaging of decreased myocardial blood flow and increased glucose uptake by PET [69].
Whereas the prognostic benefit from optimal medical therapy vs. that from revascularization in patients with stable coronary artery disease and angina is contentious, [18, 137] it is particularly the group of patients with coronary artery disease and ischemic heart failure who benefit from coronary revascularization. In the STICH trial, 1212 patients with chronic coronary artery disease and a LV ejection fraction of ≤ 35% were randomized to medical treatment of surgical revascularization, and those with revascularization had better outcome in mortality, cardiovascular mortality and hospitalization for heart failure, [104, 259] notwithstanding some critical considerations on the value of viability testing in this trial [5]. Also, in the otherwise neutral large ISCHEMIA trial, in 5179 patients with stable coronary artery disease and angina, it was the subgroup of 398 patients with a history of heart failure or LV ejection fraction ≥ 35 but < 45% who had a worse 4-year outcome than patients without heart failure or LV dysfunction. Of note, however, this subgroup of patients had a better outcome in terms of all-cause mortality, cardiovascular mortality or hospitalization for heart failure with coronary revascularization by PCI or coronary artery bypass graft surgery than with medical therapy [131]. Although pre-specified, this was a subgroup analysis only and must be considered hypothesis-generating at this point. However, it does support the notion that coronary revascularization is of particular benefit for patients with heart failure of ischemic origin, supporting the concept of hibernating myocardium [94].
Coronary microembolization
Coronary microembolization occurs spontaneously or iatrogenically during PCI when atherothrombotic particulate debris and soluble vasoconstrictor, thrombogenic and inflammatory substances are released from erosion or rupture of an atherosclerotic plaque [117]. Spontaneous coronary microembolization may be clinically silent and become only apparent by chance in elevated serum troponin concentrations. Direct evidence for coronary microembolization is achieved only when it occurs clinically as an acute coronary syndrome or during PCI [117]. Repetitive, also repetitive clinically silent coronary microembolization may ultimately result in diffuse ischemic cardiomyopathy [117].
In animal experiments, coronary microembolization of inert particles was historically used to induce acute heart failure and cardiogenic shock [2]. Franciosa et al. then introduced the intracoronary embolization of glass beads of 400–600 µm in diameter into conscious dogs as a model of chronic heart failure, [62] which was subsequently further refined by Sabbah et al. who used repeated intracoronary injections of polystyrene microspheres of 70–110 µm in diameter to induce a stable situation of chronic heart failure in conscious dogs [199]. This heart failure model is characterized by LV hypertrophy, patchy myocardial fibrosis, and neurohumoral activation, [198, 199] and such model was also replicated in sheep [107] and pigs [228]. The microembolization-induced heart failure model has the advantage of reasonable stability such that therapeutic strategies can be studied. Using this model, different treatment regimens, including metoprolol, enalapril [198] and cell therapy [228] were tested. With a more limited repetitive coronary microembolization using microspheres of 115 µm in diameter in conscious dogs, a situation of heart failure with preserved ejection fraction, no reduction in end-systolic elastance and in ventricular relaxation but with intravascular volume expansion, neurohumoral activation and elevated LV end-diastolic pressure was induced [81]. Somewhat surprisingly, most of these studies which intentionally impaired coronary blood flow to induce heart failure did not report coronary blood flow at baseline before and after repetitive coronary microembolization and established heart failure. More acutely, coronary microembolization is typically characterized by elevated baseline coronary blood flow through reactive hyperemia in the coronary vasculature around the microembolized vascular territory and reduced adenosine-recruitable coronary blood flow through physical obstruction of some microvessels, acting jointly to reduce the amplitude of coronary reserve; [217] the same elevation of baseline coronary blood flow and reduction of coronary reserve is seen in patients with peri-interventional coronary microembolization [84]. In one study with repetitive coronary microembolization in dogs, the coronary vasodilator response to intravenous acetylcholine was depressed before heart failure had developed, and adenosine-recruitable coronary reserve was decreased with established heart failure [120].
Myocardial infarction and post-infarct remodeling
Myocardial infarction results from sustained and severe impairment of coronary blood flow after rupture or erosion of an epicardial coronary atherosclerotic plaque and/or coronary microvascular obstruction and manifests in injury to the myocardium and the coronary microcirculation; reperfusion is mandatory to salvage myocardium from impending infarction but inflicts additional injury to the myocardium and the coronary microcirculation [93]. Heart failure can result from myocardial infarction acutely in the form of cardiogenic shock or more chronically as a consequence of LV remodeling [97]. Since myocardial infarction affects a particular coronary perfusion territory, distinction is needed between blood flow to the infarcted and to the non-infarcted remote myocardium.
The infarct region The coronary circulation experiences massive injury during myocardial ischemia and in the following reperfusion, including increased vascular permeability and edema formation, platelet and leukocyte plugging and ultimately capillary destruction and intra-myocardial hemorrhage [16, 93]. In its extreme form, this coronary microvascular injury manifests during reperfusion following myocardial ischemia in the form of coronary microvascular obstruction and a no-reflow phenomenon, in both experimental animals and patients with reperfused acute myocardial infarction [92]. In experimental studies, coronary microvascular obstruction is best quantified by lack of endothelial staining with thioflavin, and in preclinical and clinical studies, it is quantified as an increased microvascular resistance by measurement of perfusion pressure and coronary blood flow or visualized by magnetic resonance imaging (MRI) (see Figs. 2 and 3 in [92]). In the further time course after acute myocardial infarction, not only the myocardium remodels and, if the infarcted region is large enough, eventually develops heart failure, [97, 142, 168, 180] but also the culprit coronary circulation remodels. Following the microvascular injury and destruction during immediate reperfusion, there is infarct healing with coronary angiogenesis and myocardial revascularization, and the disruption of angiogenesis contributes to the development of post-myocardial infarct heart failure in mice [215]. The post-infarct myocardial revascularization is dependent on angiogenic factors, notably vascular endothelial growth factor (VEGF), [15, 193] which in turn is increased by paracrine mechanisms involving cardiomyocyte alpha 1 receptor activation [279] and beta blockade in rats, [193] and nitric oxide in mice, which again is promoted by statins [128] or cell therapeutic approaches [112, 127]. Stimulation of angiogenesis in experimental animals improves LV function and attenuates the development of heart failure. [15, 128, 193, 215, 266] There appears to be a positive feed-back vicious cycle between heart failure following myocardial infarction and an inflammatory dysregulation of the bone marrow niche to mobilize cells for myocardial or coronary vascular repair in mice and also humans [101].

Mechanisms of impairment of coronary blood flow in heart failure: mechanical extravascular compression by left ventricular pressure (LVP), attenuated metabolic and nitric oxide (NO)-mediated endothelium-dependent coronary vasodilation secondary to increased formation of reactive oxygen species (ROS) formation, increased vasoconstriction to mediators of neurohumoral activation (norepinephrine, angiotensin, endothelin)
The remote region In experimental studies, alterations in coronary blood flow were also seen in the non-infarcted remote myocardium. In pigs with left circumflex coronary artery occlusion, cardiac output 2–3 weeks later was reduced and there was neurohumoral activation with increased plasma norepinephrine, epinephrine, angiotensin, and endothelin, reflecting LV dysfunction [77]. In this model, exercise-induced coronary vasodilation was preserved but attenuated, [77] and increased activation of ATP-dependent K channels, [147] maintenance of nitric oxide-mediated endothelium-dependent vasodilation [78] and attenuated vasoconstrictor impact of angiotensin [145] and endothelin [146] contributed to such adaptation of the remote coronary circulation in post-infarct left ventricular dysfunction [43]. These studies used systemic blockers to address the mediator mechanisms; it is therefore unclear, in which cellular compartment (myocardial [277] vs. vascular) the activation of ATP-dependent K channels occurs.
Coronary microvascular obstruction in the clinic Microvascular obstruction occurs in many patients with successfully reperfused myocardial obstruction, ranging from 5 to 70% depending on the method and parameter and the time of its assessment. [92] Not only infarct size but also the extent of coronary microvascular obstruction on MRI is a major determinant of cardiogenic shock [190]. However, coronary microvascular obstruction after successful reperfusion by PCI also predicts the long-term development of LV dysfunction [20, 134, 250] and clinical outcome in terms of mortality and hospitalization for heart failure [29, 39, 119, 191, 255, 268]. Infusion of bone marrow-derived or circulating progenitor cells into the infarct-related coronary artery in patients with reperfused myocardial infarction increased adenosine-recruitable coronary reserve on follow-up in the TOPCARE-AMI and REPAIR-AMI trials, [9, 52, 53] and this effect was associated with improved LV function and clinical outcome [8, 203]. Unfortunately, the clinical value of such autologous cell therapy approaches in patients with acute myocardial infarction remains uncertain, given the lack of a positive large prospective clinical outcome trial [139].
Clinically, in patients with uncomplicated reperfused acute myocardial infarction, adenosine-recruitable coronary velocity reserve (Doppler) is decreased immediately after PCI in the culprit and the non-culprit coronary artery as compared to propensity-matched controls. [38] The impairment in coronary reserve of the non-culprit coronary arteries as measured by PET is more severe in patients with coronary artery disease and heart failure than in those without heart failure [253]. Patients with myocardial infarction in the absence of significant obstructive coronary artery disease (MINOCA) have milder impairment of coronary blood flow and coronary reserve than those with classical myocardial infarction and obstructive coronary artery disease [149] and better outcome on follow-up, including the development of heart failure; [173] however, the specific role of coronary blood flow impairment for heart failure development in MINOCA is not clear at present.
Heart failure of non-ischemic origin
Dilative cardiomyopathy
Dilated cardiomyopathy in humans arises from genetic mutations in sarcomeric or mitochondrial proteins, [195] myocarditis [249] or toxic agents, such as ethanol [59] or chemotherapy, [83, 241] and from sustained tachyarrhythmias [49, 105]. Pacing-induced heart failure in experimental animals does not only mimic the clinical syndrome of tachycardia-induced cardiomyopathy but is also considered as a model of dilated cardiomyopathy, which mimics the features of ventricular dilatation and dysfunction, systemic congestion, exercise intolerance and dyspnea, neurohumoral activation, cardiomyocyte loss and hypertrophy of remaining cardiomyocytes, fibrosis and apoptosis [90]. In conscious pigs with chronic supraventricular pacing, there is capillary rarefication, reduced myocardial blood flow, and adenosine-recruitable coronary reserve particularly in the LV subendocardium [106, 220, 221]. Reduced baseline myocardial blood flow and adenosine-recruitable coronary reserve were also seen in conscious dogs with chronic right ventricular pacing, but there was no evidence for capillary rarefication [209]. In early stages of pacing-induced heart failure, despite neurohumoral activation and increased plasma concentrations of vasoconstrictor substances (norepinephrine, angiotensin, endothelin), [123, 162] nitric oxide formation may be increased and act to preserve coronary blood flow [162, 200]. Also, ATP-dependent K-channel activation may contribute to attenuate decreases in myocardial blood flow in dogs with pacing-induced heart failure [110, 244, 269]. While endothelium-dependent coronary vasodilation is still preserved, however, adenosine-recruitable coronary vascular reserve is already reduced through increased extravascular compression [242]. In an early state of pacing-induced heart failure, the vasoconstrictor effect of angiotensin was attenuated and the bradykinin-dependent vasodilator effect of the ACE inhibitor enalapril enhanced, supporting the notion of an increased nitric oxide formation [163]. Conscious dogs with chronic left ventricular pacing and established heart failure then had decreased epicardial coronary dilation and coronary blood flow response to acetylcholine and less coronary vascular nitrite formation in response to acetylcholine ex vivo, suggesting a defect in endothelial nitric oxide formation [227, 265]. The defect of endothelial nitric oxide formation in dogs with pacing-induced heart failure also impaired the cholinergic coronary vasodilation as part of the Bezold-Jarisch or carotid chemoreflex [278]. The reduced nitric oxide formation in established pacing-induced heart failure in dogs also induced a switch in cardiac substrate utilization from free fatty acid to glucose uptake. [189] The attenuation of nitric oxide-mediated, endothelium-dependent coronary vasodilation in pacing-induced heart failure is secondary to nitric oxide inactivation by reactive oxygen species [157] and NADPH oxidase activity [231, 275]. It is currently unclear in which cellular compartment (vascular or myocardial) the responsible NADPH oxidase activation occurs and where the increased reactive oxygen species formation originates; [154] this distinction, however is important to decide whether the impaired coronary vasomotion is a consequence of heart failure (myocardial origin) or a bystander (vascular origin) induced by the conditions leading to heart failure, e.g., sustained rapid pacing (Fig. 3). In any event, increased endothelial nitric oxide synthase activity [231, 248] by statins preserves endothelium-dependent coronary vasodilation in pacing-induced heart failure. Pacing-induced heart failure, [221] endothelium-dependent coronary vasodilation, [251] and endothelial nitric oxide formation [71] recover after termination of chronic pacing over several weeks. The relatively fast recovery of the pacing-induced heart failure after cessation of pacing is a disadvantage for the study of treatment regimens in this model, but it does mimic the clinical syndrome of tachycardic cardiomyopathy particularly well [90]. In dilated cardiomyopathy of tachycardic origin, the impairment of the coronary circulation plays a particularly prominent role since tachycardia increases myocardial oxygen consumption and decreases diastolic duration, thereby increasing the susceptibility to myocardial ischemia [14, 89]. In conscious dogs [160, 245] and pigs [123] with chronic rapid pacing, the exercise-induced increases in cardiac output but also in regional myocardial blood flow to the left and right ventricle, skeletal muscle blood flow and renal blood flow were attenuated (Fig. 4). The decrease in myocardial blood flow at baseline and during exercise in dogs with chronic pacing-induced heart failure was associated with a proportionate decrease in myocardial oxygen consumption and occurred in the absence of myocardial ischemia (net lactate production) [245]. The metabolic coronary vasodilation during pacing-induced tachycardia in dogs with established pacing-induced heart failure depends on nitric oxide formation, [229] and nitric oxide formation may inhibit myocardial oxygen consumption in the failing heart [243].

Attenuated increases in regional myocardial blood flow of the left (LV) and right (RV) ventricle in chronically instrumented conscious dogs with pacing-induced heart failure during treadmill exercise. Endo: flow to subendocardial layers; Mid: flow to midmyocardial layers; Epi: flow to subepicardial layers. Trans: flow to the entire transmural region. Data from [160]
The calcium antagonist amlodipine, [124] but not the angiotensin AT1 receptor antagonist valsartan [33] improved myocardial blood flow during exercise in pacing-induced heart failure. Not only extravascular compression by increased left ventricular end-diastolic pressure but also increased plasma vasoconstrictor concentrations from neurohumoral activation limit coronary blood flow in heart failure. The muscle metaboreflex-induced sympathetic activation during exercise in dogs with pacing-induced heart failure induced coronary vasoconstriction, [7] which was abrogated by alpha1-adenoceptor blockade with prazosin; [34] prazosin also attenuated resting coronary vasomotor tone in dogs with pacing-induced heart failure [232]. Endothelin-A receptor blockade also increased coronary blood flow during exercise in dogs with pacing-induced heart failure [103]. Apparently, coronary vasomotion in established pacing-induced heart failure at rest and during exercise is characterized by reduced nitric oxide-mediated, endothelium-dependent vasodilation and enhanced vasoconstriction by norepinephrine and endothelin.
In clinical dilated cardiomyopathy, impaired endothelium-dependent coronary vasodilation of the epicardial coronary arteries and of the microcirculation in response to intracoronary acetylcholine was demonstrated by angiography and Doppler velocity flow measurements (Table 1); [27, 138, 246] an impaired adenosine-recruitable coronary reserve was only apparent in patients with chronic, [27, 246] but not with acute onset—idiopathic dilated cardiopathy [138].
Decreased coronary reserve, as recruited by intravenous dipyridamole, was confirmed for patients with chronic idiopathic dilated cardiomyopathy using PET, [159, 224, 254] and decreased coronary reserve [159] and the spatial heterogeneity of myocardial blood flow [214] were associated with poor prognosis (mortality, heart failure progression). On MRI of patients with dilated cardiomyopathy, there was evidence for an increased extracellular matrix [111, 158] in association with reduced myocardial blood flow at rest [111] and with reduced angiographic coronary vasodilator response to intracoronary acetylcholine [158]. Somewhat surprisingly, patients with dilated cardiomyopathy had no reduction, but a modest increase in myocardial blood flow at rest, but again a decrease in adenosine-recruitable coronary reserve in MRI perfusion imaging [76]. The decrease in adenosine-recruitable coronary reserve was, however, not sufficient to induce a myocardial oxygen deficiency, supporting the non-ischemic nature of idiopathic dilated cardiomyopathy [36]. Apart from endothelial dysfunction as evidenced by the impaired coronary dilator response to acetylcholine, there is also neurohumoral activation in patients with dilated cardiomyopathy, [63] and antagonism of neurohumoral activation is an essential part of all medical treatment of heart failure [75, 108]. However, to which extent attenuated coronary vasoconstriction, as evidenced in the above experimental studies, contributes to the treatment success in patients with heart failure is unclear, given the systemic effects of such treatment on heart rate, blood pressure and ventricular function which all impact on coronary blood flow. Collectively, the clinical imaging data in patients with idiopathic dilated cardiomyopathy confirm a depression of endothelium-dependent coronary vasodilation and a reduction of coronary vasodilator reserve.
Hypertrophic cardiomyopathy
Hypertrophy of the myocardium develops as an adaptive response to pressure or volume overload or can be the manifestation of a genetic disease [167, 264]. In both scenarios, the hypertrophy may decompensate into heart failure, with or without preserved ejection fraction [26]. Remodeling and dysfunction of the coronary microcirculation are typically involved in hypertrophic cardiomyopathy, even in the absence of atherosclerosis [26].
In experimental animals, LV hypertrophy is morphologically not only characterized by increased cardiomyocyte cross-sectional area, but also by decreased capillary density and increased intercapillary distances [10, 17]. For myocardial blood flow and its distribution, it is important to distinguish between scenarios where the coronary circulation is also exposed to pressure or volume overload (supravalvular aortic stenosis/banding, hypertension) or where coronary perfusion pressure is reduced (aortic valve stenosis). In chronically instrumented conscious dogs with banding of the ascending aorta at 6–8 weeks of age, LV myocardial blood flow as assessed by the microsphere technique was increased after hypertrophy had developed after several months and even further increased when hypertrophy had decompensated to failure, as defined by increased LV end-diastolic pressure > 18 mmHg [172]. Using the same model, again increased myocardial blood flow commensurate with the increased myocardial work was seen, and there was no depletion of myocardial energy-rich phosphates, not even when fractional shortening was decreased in dogs with decompensated hypertrophy [65]. This model is, however, characterized by decreased adenosine-recruitable coronary reserve [17, 109]. During exercise, increases in myocardial blood flow were greater in dogs with LV hypertrophy commensurate with their greater increase in myocardial oxygen consumption. For the increase in coronary blood flow during exercise, dogs with a hypertrophied heart used a greater activation of ATP-dependent K-channels than normal dogs; [144] however, the subendocardial were less than the subepicardial blood flow increases, reflecting potential susceptibility to ischemia [11, 44]. The relative underperfusion of subendocardial layers of hypertrophied myocardium during exercise was attributed to increased extravascular compression, [44] but not to a deficit in nitric oxide bioavailability [46]. Coronary blood flow returned to normal after regression of hypertrophy [109]. Different from the above studies which used supravalvular aortic banding, experimental aortic valve stenosis in young dogs also resulted in LV hypertrophy several months later, but a more substantial reduction of adenosine-recruitable coronary reserve and a subnormal increase in blood flow during pacing-induced tachycardia particularly in the subendocardium [3]. Likewise, in chronically instrumented conscious dogs with renal hypertension, LV hypertrophy developed and coronary autoregulation was impaired such that at the lower range of coronary autoregulation (40–70 mmHg), myocardial blood flow was reduced to a greater extent than in normal dogs, particularly in subendocardial layers [80]. In pigs with corticosterone-induced hypertension, LV hypertrophy developed over 12 weeks, and the dobutamine stress-recruited perfusion reserve on MRI was reduced as compared to normal pigs [192]. Volume overload by severe experimental mitral regurgitation in dogs also induced LV hypertrophy after several months [28, 274]. Myocardial blood flow at rest and its increases during pacing and intravenous adenosine were, however, not different between dogs without or with mitral regurgitation [28, 274]. Nevertheless, energy-rich phosphates [274] and contractile function [28] were impaired in these dog studies with chronic mitral regurgitation, thus excluding a role of coronary blood flow in these impairments.
Also, in patients with hypertrophic cardiomyopathy, but absence of valve disease or hypertension, there are structural alterations in the coronary circulation; at autopsy, remodeling of intramural coronary arteries (< 1500 µm in diameter) with intimal and medial hypertrophy and narrowed lumen were seen in the majority of cases [136]. Small vessel disease of intramural coronary arteries (20–1000 µm in diameter) was also evident in the autopsy of patients with hypertrophic cardiomyopathy of various origin, including hypertension, with significant luminal narrowing which correlated to measures of hypertrophy and presence of fibrosis [233]. In young patients with hypertrophic cardiomyopathy and sudden cardiac death, there was morphological evidence of small vessel coronary disease and patchy myocardial scars, supporting the occurrence of ischemia in the natural history of hypertrophic cardiomyopathy [13]. Patients with hypertrophic cardiomyopathy in the absence of other cardiovascular disease, notably coronary atherosclerosis, had normal myocardial blood flow at rest but decreased coronary reserve in response to intracoronary adenosine in Doppler flow measurements [122] or to intravenous dipyridamole on PET [25, 30, 85, 164, 165]. Patients with chest pain had a more pronounced impairment of coronary reserve, [25] and the decrease in coronary reserve was related to poor clinical outcome. [30, 164] Intravenous infusion of the ACE inhibitor perindopilat in type 2 diabetic patients with LV hypertrophy improved the dipyridamole-recruitable coronary reserve acutely [85]. Patients with a genotype-positive sarcomeric mutation and hypertrophic cardiomyopathy had greater reduction in coronary reserve than genotype-negative patients, and they also had more fibrosis on gadolinium contrast MRI [165]. Multiparametric MRI appears to be of particular value in hypertrophic cardiomyopathy, as it can not only determine the severity of left ventricular hypertrophy and contractile dysfunction, but also the attenuation of coronary reserve and the extent of fibrosis [179]. As in the experimental studies, coronary blood flow is particularly impaired in patients with aortic stenosis when LV hypertrophy is associated with reduced coronary perfusion pressure [272]. On PET, the decrease in dipyridamole-recruitable coronary reserve was related to the severity of aortic stenosis and more pronounced in subendocardial than in subepicardial layers [188]. The magnitude of coronary reserve reduction was related to greater hypertrophy and left ventricular dysfunction and also to plasma hs-troponin T concentration as an injury marker, [282] and it was a marker of worse prognosis on follow-up [280]. The impairment of coronary reserve was reversible on transcatheter or surgical aortic valve replacement with regression of hypertrophy on follow-up [133, 282].
Hypertension not only induces LV hypertrophy but is also a major pathogenetic risk factor for coronary atherosclerosis; however, an impairment of dipyridamole-recruitable coronary vasodilator reserve is evident also in the absence of coronary artery disease [166, 204, 205, 225, 226]. The reduction in coronary reserve appeared to be specifically pronounced with hypertension as compared to other pathogenesis of left ventricular hypertrophy, [226] and a greater reduction in coronary reserve was associated with ST segment depression in Holter monitoring [205]. Episodes of ST segment depression corresponded to a greater reduction in subendocardial than subepicardial dipyridamole-recruitable coronary reserve in patients with hypertensive hypertrophy in PET [194]. Attenuation of coronary dilator reserve in patients with heart failure of hypertensive origin predicts worse clinical outcome on follow-up [281]. Chronic ACE inhibition with enalapril improved coronary reserve and reduced exercise-induced ST segment depression in a small group of hypertensive patients [151].
Heart failure of ischemic and non-ischemic origin
Heart failure with preserved ejection fraction
Heart failure with preserved ejection fraction is characterized by typical heart failure symptoms with mostly diastolic LV dysfunction but preserved ejection fraction. It is typically associated with comorbidities, such as obesity, diabetes and hypertension [182]. Experimental models of heart failure with preserved ejection fraction are available. With a more limited coronary microembolization than in the creation of heart failure with reduced ejection fraction, dogs developed heart failure with preserved ejection fraction [81]. In pigs with corticosterone-induced hypertension, heart failure with preserved ejection fraction developed and was characterized by decreased coronary reserve, [192] but no alteration in capillary density [152]. Pigs with chronic aortic banding developed LV hypertrophy with both systolic and diastolic dysfunction but still had preserved ejection fraction; [51] in this model, the increment in coronary blood flow for a given increase in myocardial oxygen consumption during pressure load was attenuated, suggesting impaired metabolic coronary vasodilation [51]. In a pig model with multiple comorbidities (diabetes, hyperlipidemia, renal hypertension), there was LV hypertrophy and fibrosis, but ejection fraction was preserved; [219] in this model there was increased nitric oxide synthase uncoupling, associated with increased reactive oxygen species formation and decreased nitric oxide bioavailability. Accordingly, the ex vivo coronary vasodilator responses to bradykinin were reduced [219]. In a mouse model of heart failure with preserved ejection fraction, secondary to a combination of hypertension through systemic nitric oxide synthase inhibition and a diet-induced obesity and metabolic syndrome, [206] there was an increased expression of inducible nitric oxide synthase with a resulting substantial increase in circulating nitric oxide which induced nitrosylation of proteins, including proteins of the unfolded protein response which serve to control protein quality. In this model, coronary endothelial function was impaired and coronary reserve was reduced [206].
In patients with heart failure and preserved ejection fraction, there is LV hypertrophy, fibrosis and microvascular coronary rarefication even in the absence of epicardial coronary stenosis at autopsy [150]. In the myocardium of these patients, there are an increased expression of inflammatory proteins as well as increased reactive oxygen species and decreased nitrite/nitrate concentrations secondary to increased vascular expression of NADPH oxidase and uncoupling of endothelial nitric oxide synthase [64]. Consistently, patients with heart failure and preserved ejection fraction have reduced coronary reserve in the absence of coronary artery disease [42, 114, 197, 222, 235] on Doppler angiography, [42, 197] PET [222, 235] or MRI [113, 114, 197]. Almost all patients with heart failure and preserved ejection fraction have either coronary artery disease on angiography, coronary microvascular dysfunction (increased minimal resistance on Doppler) and vasomotor dysfunction (impaired dilator response to acetylcholine) or both; [197] however, half of these patients have in fact epicardial coronary artery disease. The reduction in coronary reserve predicts adverse events on follow-up in these patients [113]. Collectively, coronary vascular dysfunction is a hallmark of heart failure with preserved ejection fraction, predisposing to myocardial ischemia. However, the causality of impaired coronary blood flow for the development of this heart failure entity is not established, as the typically predisposing comorbidities (obesity, diabetes, hypertension) each and in combination predispose also to coronary atherosclerosis such that heart failure with preserved ejection fraction and impaired coronary blood flow may develop in parallel from a common systemic inflammatory activation [175, 216].
Takotsubo
Takotsubo cardiomyopathy is a clinical syndrome which is typically precipitated by extreme stress situation with an excessive catecholamine release [267] and characterized by features of both, myocardial infarction and heart failure [126]. Patients experience pain, ST segment alterations in their ECG and increased plasma troponin concentrations, mimicking acute myocardial infarction, yet their coronary circulation is not obstructed on angiography. Severe LV dysfunction with characteristic apical dyskinesis (“ballooning”) reflects the cardiomyopathy [70, 132, 177]. The takotsubo syndrome typically affects postmenopausal women in stress situations and it is reversible. The pathophysiology of the takotsubo syndrome is not fully clear, but coronary vascular dysfunction is definitely involved [260]. Using myocardial contrast echocardiography, a perfusion deficit in the dysfunctional region was identified [1, 67] which partially recovered during intravenous adenosine challenge along with an improvement of regional contractile function, [67] somewhat reminiscent of the Gregg effect seen in experimental studies of stunned myocardium [208]. Both, perfusion and contractile function recovered completely within 1-month follow-up [67]. On angiography, thrombolysis in myocardial infarction (TIMI) flow in patients with takotsubo was similarly impaired as in ST segment elevation myocardial infarction (STEMI) patients with microvascular obstruction on reperfusion [37]. Reduced myocardial blood flow reflecting coronary microvascular dysfunction was also demonstrated using single photon emission computed tomography [202, 270] and PET [32, 58, 121, 270] along with alterations in myocardial substrate metabolism suggestive of stunning/hibernation [58, 121, 202] and signs of inflammation [48, 267]. Endothelial dysfunction with focal or diffuse coronary vasoconstriction in response to intracoronary acetylcholine was seen a significant proportion of takotsubo patients [202]. While the pathophysiology of the takotsubo syndrome is not fully clear, the predominance of postmenopausal women being affected and the characteristic severe stress situations precipitating this syndrome suggest an interaction of estrogen deficiency possibly contributing to microvascular endothelial dysfunction [230] and increased responsiveness of the myocardium and coronary vasculature to catecholamines, which may be reflective of a more sparse sympathetic innervation of apical than basal myocardium [115] with a resulting catecholamine hypersensitivity [176]. Both, beta-adrenoceptor-mediated catecholamine toxicity on cardiomyocytes [132] and increased alpha-adrenoceptor-mediated vasoconstriction [95] may then induce a situation of transient ischemic dysfunction with subsequent stunning [132, 177]. However, at present, it is not fully clear whether reduced coronary blood flow is causal for the takotsubo syndrome; the only suggestive evidence originates from the observation that recruitment of dilator reserve with adenosine improves regional contractile function [67].
Cardio-oncology
Patients with a cancer history have more coronary ischemic events [234] and a higher incidence of myocardial infarction [153] than those without. They also have a higher incidence of plaque erosion which is, in turn, associated with coronary microembolization, [117] and they have worse clinical outcome [234]. Cancer therapy not only induces toxic or inflammatory injury to cardiomyocytes [83, 241] but also to the vasculature, including the coronary vasculature [82, 178, 271]. Not only anti-angiogenic therapies, but also conventional chemotherapy or radiation therapy promotes reduced nitric oxide availability and endothelial dysfunction, predisposes to vasoconstriction and can ultimately precipitate angina or myocardial infarction. In a pig model of anthracycline cardiotoxicity, coronary arterial structural damage and reduced coronary reserve in response to papaverine became apparent before a myocardial contractile defect, whereas more microvascular structural alterations were only seen when also LV dysfunction had developed [66]. Whereas this study suggested that anthracycline chemotherapy-induced coronary vascular injury might contribute to LV dysfunction, the vascular and myocardial contribution to cardiac toxicity from chemotherapy and radiation therapy are clinically more difficult to dissect. Patients with pre-existing coronary artery disease have an increased risk to develop heart failure from anthracycline [54, 60, 184]. Thus, the contribution of an impaired coronary blood flow to the development of cancer therapy-induced heart failure is not really clear.
The right ventricle in heart failure
The right ventricle is equally involved as the LV when the conditions causing heart failure affect the entire heart, such as genetic mutations, myocarditis, tachyarrhythmias or toxic agents, or when ischemia also affects right ventricular perfusion territories. The right ventricle may be less involved in failure when pressure or volume overload (hypertension, aortic valve disease) affects primarily the LV. The right ventricle, however, is more affected in pulmonary hypertension. The failing right ventricle has only recently received more attention, [79, 130, 261] and the coronary circulation in right ventricular failure has received little attention at all. Yet, there are some special considerations to the coronary circulation in the right ventricle, [35] since coronary perfusion pressure is above right ventricular pressure throughout the cardiac cycle such that extravascular compression and diastolic duration during tachycardia are of lesser importance than in the LV. Also, the thinner wall of the right ventricle may receive some retrograde perfusion through Thebesian veins. On the other hand, coronary autoregulation is less pronounced and alpha-adrenergic coronary vasoconstriction during sympathetic activation more pronounced in the right than the LV. Nevertheless, on the aggregate, the susceptibility to ischemia is less in the right than in the LV. However, in acute right ventricular pressure overload by acute pulmonary banding in dogs, there is increased alpha-adrenergic coronary vasoconstriction, increased extravascular compression and subendocardial ischemia [72, 73]. With chronic right ventricular pressure overload by chronic pulmonary stenosis, adenosine-recruitable coronary vasodilator reserve in the hypertrophied right ventricle is reduced particularly in the subendocardium [155] which impairs metabolic vasodilation during exercise [23, 156]. Patients with chronic pulmonary hypertension have reduced right coronary artery blood flow in proportion to right ventricular hypertrophy [257] and reduced adenosine-recruitable coronary reserve on MRI [262]. A recent NIH consensus workshop recommended directions for future research on the genetic, molecular and cellular processes in right heart failure, [130] but further research on the coronary circulation in right heart failure is also warranted. Arrhythmogenic right ventricular cardiomyopathy is a relatively infrequent form of human heart failure, caused by genetic mutations mostly in desmosomal proteins and characterized morphologically by diffuse fibrosis and inflammatory infiltration [68]. No specific alteration in coronary blood flow has been reported, but as in other heart failure entities, adenosine-recruitable coronary reserve on PET is reduced [174].
Conclusions and directions for future research
Heart failure is almost invariably associated with coronary vascular dysfunction, not only in the frequent presence but also in the absence of coronary atherosclerosis. Cause-and-consequence relationships between heart failure and impaired coronary blood flow are complex. In stunning and hibernation, coronary microembolization, myocardial infarction and post-infarct remodeling, heart failure is clearly a consequence of myocardial ischemia without or with reperfusion—these are heart failure syndromes of ischemic origin. Vice versa, in all forms of heart failure, including hypertrophic and dilated cardiomyopathy with underlying genetic mutations and in the absence of coronary artery disease, increased extravascular compression and coronary vasoconstriction by the mediators of neurohumoral activation (norepinephrine, angiotensin, and endothelin) are clearly a consequence of heart failure. The invariably impaired endothelium-dependent coronary dilation as well as eventual morphological alterations of the coronary circulation could be a consequence of heart failure but also a consequence of the underlying conditions inducing heart failure (e.g., in pressure or volume overload). In some forms of heart failure, both ischemic and non-ischemic causes contribute to heart failure. In takotsubo cardiomyopathy, the causal contribution of coronary vascular and myocardial disturbances to the heart failure syndrome is not clear. In heart failure with preserved ejection fraction, the underlying comorbidities with the resulting systemic inflammatory state may cause both impairment of the coronary circulation and the myocardium in parallel. In aortic stenosis, there is both reduced coronary perfusion pressure causing ischemia and pressure overload causing LV hypertrophy.
In any form of heart failure, there is a vicious cycle between the impairment of myocardial contractile function and the impairment of the coronary circulation in that myocardial ischemia worsens heart failure and vice versa (Fig. 5), and it is reflected by the prediction of poor clinical outcome from heart failure by the reduction of coronary dilator reserve [116, 237].

Vicious cycle between heart failure and impairment of coronary blood flow by common features of all heart failure entities: increased extravascular compression, reduced endothelium-dependent vasodilation, enhanced vasoconstriction to neurohumoral mediators and (to a variable extent) vascular remodeling and rarefication
Obviously, therapeutic restoration of coronary blood flow is of pivotal importance in all forms of heart failure for which ischemia is causal. For reversible ischemia and hibernating myocardium, the jury is still out in which clinical condition reperfusion by optimal medical therapy or by interventional/surgical revascularization is better. For irreversible ischemia and myocardial infarction, prevention of coronary microvascular obstruction is of pivotal importance. Unfortunately, interventional approaches using protection devices to attenuate coronary microvascular obstruction are of limited value and recommended only in cases of large atherothrombotic burden on angiography [117]. Also, pharmacological approaches to attenuate coronary microvascular obstruction, i.e., by use of adenosine, nitroprusside or calcium antagonists have been of limited clinical value [92, 96]. Currently, there is no evidence at all for clinical benefit from stimulation of angiogenesis through growth factor transfection or cell therapy. In heart failure of non-ischemic origin, there is no evidence that improvement of coronary blood flow specifically provides clinical benefit. Nevertheless, the above common features of coronary blood flow impairment in all forms of heart failure render them a valid target also for all established treatment strategies (statins, ACE inhibitors, AT1 blockers), but also a potential caveat (beta blockers: increased diastolic duration vs. increased vasoconstriction [86, 95]) and a worthwhile target in the study of novel treatment options, e.g., neprilysin or sodium glucose transporter 2 (SGLT2) inhibition. [6, 21, 143, 169, 181, 218].
References
Abdelmoneim SS, Mankad SV, Bernier M, Dhoble A, Hagen ME, Ness SA, Chandrasekaran K, Pellikka PA, Oh JK, Mulvagh SL (2009) Microvascular function in Takotsubo cardiomyopathy with contrast echocardiography: prospective evaluation and review of literature. J Am Soc Echocardiogr 22:1249–1255. https://doi.org/10.1016/j.echo.2009.07.012
Agress CM, Rosenberg MJ, Jacobs HI, Binder MJ, Schneiderman A, Clark WG (1952) Protracted shock in the closed-chest dog following coronary embolization with graded microspheres. Am J Physiol 170:536–549. https://doi.org/10.1152/ajplegacy.1952.170.3.536
Alyono D, Anderson RW, Parrish DG, Dai XZ, Bache RJ (1986) Alterations of myocardial blood flow associated with experimental canine left ventricular hypertrophy secondary to valvular aortic stenosis. Circ Res 58:47–57. https://doi.org/10.1161/01.res.58.1.47
Ambrosio G, Betocchi S, Pace L, Losi MA, Perrone-Filardi P, Soricelli A, Piscione F, Taube J, Squame F, Salvatore M, Weiss JL, Chiariello M (1996) Prolonged impairment of regional contractile function after resolution of exercise-induced angina. Evidence of myocardial stunning in patients with coronary artery disease. Circulation 94:2455–2464. https://doi.org/10.1161/01.cir.94.10.2455
Anavekar NS, Chareonthaitawee P, Narula J, Gersh BJ (2016) Revascularization in patients with severe left ventricular dysfunction: Is the assessment of viability still viable? J Am Coll Cardiol 67:2874–2887. https://doi.org/10.1016/j.jacc.2016.03.571
Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Bohm M, Brunner-La Rocca HP, Choi DJ, Chopra V, Chuquiure-Valenzuela E, Giannetti N, Gomez-Mesa JE, Janssens S, Januzzi JL, Gonzalez-Juanatey JR, Merkely B, Nicholls SJ, Perrone SV, Pina IL, Ponikowski P, Senni M, Sim D, Spinar J, Squire I, Taddei S, Tsutsui H, Verma S, Vinereanu D, Zhang J, Carson P, Lam CSP, Marx N, Zeller C, Sattar N, Jamal W, Schnaidt S, Schnee JM, Brueckmann M, Pocock SJ, Zannad F, Packer M, Investigators EM-PT (2021) Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med 385:1451–1461. https://doi.org/10.1056/NEJMoa2107038
Ansorge EJ, Augsutyniak RA, Perinot ML, Hammond RL, Kim J-K, Sala-Mercado JA, Rodriguez J, Rossi NF, O’Leary DS (2005) Altered muscle metaboreflex control of coronary blood flow and ventricular function in heart failure. Am J Physiol Heart Circ Physiol 288:H1381–H1388. https://doi.org/10.1152/ajpheart.00985.2004
Assmus B, Honold J, Schachinger V, Britten MB, Fischer-Rasokat U, Lehmann R, Teupe C, Pistorius K, Martin H, Abolmaali ND, Tonn T, Dimmeler S, Zeiher AM (2006) Transcoronary transplantation of progenitor cells after myocardial infarction. N Engl J Med 355:1222–1232. https://doi.org/10.1056/NEJMoa051779
Assmus B, Schächinger V, Teupe C, Britten M, Lehmann R, Döbert N, Grünwald F, Aicher A, Urbich C, Martin H, Hoelzer D, Dimmeler S, Zeiher AM (2002) Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction (TOPCARE-AMI). Circulation 106:3009–3017. https://doi.org/10.1161/01.cir.0000043246.74879.cd
Bache RJ (1988) Effects of hypertrophy on the coronary circulation. Prog Cardiovasc Dis 30:403–440. https://doi.org/10.1016/0033-0620(88)90005-9
Bache RJ, Dai XZ (1990) Myocardial oxygen consumption during exercise in the presence of left ventricular hypertrophy secondary to supravalvular aortic stenosis. J Am Coll Cardiol 15:1157–1164. https://doi.org/10.1016/0735-1097(90)90258-q
Bassenge E, Heusch G (1990) Endothelial and neuro-humoral control of coronary blood flow in health and disease. Rev Physiol Biochem Pharmacol 116:77–165. https://doi.org/10.1007/3540528806_4
Basso C, Thiene G, Corrado D, Buja G, Melacini P, Nava A (2000) Hypertrophic cardiomyopathy and sudden death in the young: pathologic evidence of myocardial ischemia. Hum Pathol 31:988–998. https://doi.org/10.1053/hupa.2000.16659
Bertero E, Heusch G, Münzel T, Maack C (2021) A pathophysiological compass to personalize antianginal drug treatment. Nat Rev Cardiol 18:838–852. https://doi.org/10.1038/s41569-021-00573-w
Besnier M, Galaup A, Nicol L, Henry JP, Coquerel D, Gueret A, Mulder P, Brakenhielm E, Thuillez C, Germain S, Richard V, Ouvrard-Pascaud A (2014) Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. Faseb J 28:3351–3361. https://doi.org/10.1096/fj.13-245753
Betgem RP, de Waard GA, Nijveldt R, Beek AM, Escaned J, van Royen N (2015) Intramyocardial haemorrhage after acute myocardial infarction. Nat Rev Cardiol 12:156–167. https://doi.org/10.1038/nrcardio.2014.188
Bishop SP, Powell PC, Hasebe N, Shen YT, Patrick TA, Hittinger L, Vatner SF (1996) Coronary vascular morphology in pressure-overload left ventricular hypertrophy. J Mol Cell Cardiol 28:141–154. https://doi.org/10.1006/jmcc.1996.0014
Boden WE, O’Rourke RA, Teo KK, Hartigan PM, Maron DJ, Kostuk WJ, Knudtson M, Dada M, Casperson P, Harris CL, Chaitman BR, Shaw L, Gosselin G, Nawaz S, Title LM, Gau G, Blaustein AS, Booth DC, Bates ER, Spertus JA, Berman DS, Mancini GB, Weintraub WS (2007) Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med 356:1503–1516. https://doi.org/10.1056/NEJMoa070829
Bolli R, Triana JF, Jeroudi MO (1990) Prolonged impairment of coronary vasodilation after reversible ischemia. Circ Res 67:332–343. https://doi.org/10.1161/01.res.67.2.332
Borlotti A, Jerosch-Herold M, Liu D, Viliani D, Bracco A, Alkhalil M, De Maria GL, Ox AMISI, Channon KM, Banning AP, Choudhury RP, Neubauer S, Kharbanda RK, Dall’Armellina E (2019) Acute microvascular impairment post-reperfused STEMI is reversible and has additional clinical predictive value: a CMR OxAMI Study. J Am Coll Cardiol Cardiovasc Imaging 12:1783–1793. https://doi.org/10.1016/j.jcmg.2018.10.028
Braunwald E (2021) SGLT2 inhibitors: the statins of the 21st century. Eur Heart J. https://doi.org/10.1093/eurheartj/ehab765
Bristow JD, McFalls EO, Anselone CG, Pantely GA (1987) Coronary vasodilator reserve persists despite tachycardia and myocardial ischemia. Am J Physiol Heart Circ Physiol 253:H422–H431. https://doi.org/10.1152/ajpheart.1987.253.2.H422
Cai Z, van Duin RWB, Stam K, Uitterdijk A, van der Velden J, Vonk Noordegraaf A, Duncker DJ, Merkus D (2019) Right ventricular oxygen delivery as a determinant of right ventricular functional reserve during exercise in juvenile swine with chronic pulmonary hypertension. Am J Physiol Heart Circ Physiol 317:H840–H850. https://doi.org/10.1152/ajpheart.00130.2019
Calabretta R, Castello A, Linguanti F, Tutino F, Ciaccio A, Giglioli C, Sciagra R (2018) Prediction of functional recovery after primary PCI using the estimate of myocardial salvage in gated SPECT early after acute myocardial infarction. Eur J Nucl Med Mol Imaging 45:530–537. https://doi.org/10.1007/s00259-017-3891-1
Camici P, Chiriatti G, Lorenzoni R, Bellina RC, Gistri R, Italiani G, Parodi O, Salvadori PA, Nista N, Papi L et al (1991) Coronary vasodilation is impaired in both hypertrophied and nonhypertrophied myocardium of patients with hypertrophic cardiomyopathy: a study with nitrogen-13 ammonia and positron emission tomography. J Am Coll Cardiol 17:879–886. https://doi.org/10.1016/0735-1097(91)90869-b
Camici PG, Tschope C, Di Carli MF, Rimoldi O, Van Linthout S (2020) Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc Res 116:806–816. https://doi.org/10.1093/cvr/cvaa023
Canetti M, Akhter MW, Lerman A, Karaalp IS, Zell JA, Singh H, Mehra A, Elkayam U (2003) Evaluation of myocardial blood flow reserve in patients with chronic congestive heart failure due to idiopathic dilated cardiomyopathy. Am J Cardiol 92:1246–1249. https://doi.org/10.1016/j.amjcard.2003.08.002
Carabello BA, Nakano K, Ishihara K, Kanazawa S, Biederman RW, Spann JF Jr (1991) Coronary blood flow in dogs with contractile dysfunction due to experimental volume overload. Circulation 83:1063–1075. https://doi.org/10.1161/01.cir.83.3.1063
Carrick D, Haig C, Ahmed N, Rauhalammi S, Clerfond G, Carberry J, Mordi I, McEntegart M, Petrie MC, Eteiba H, Hood S, Watkins S, Lindsay MM, Mahrous A, Welsh P, Sattar N, Ford I, Oldroyd KG, Radjenovic A, Berry C (2016) Temporal evolution of myocardial hemorrhage and edema in patients after acute ST-segment elevation myocardial infarction: pathophysiological insights and clinical implications. J Am Heart Assoc 5:e002834. https://doi.org/10.1161/JAHA.115.002834
Cecchi F, Olivotto I, Lorenzoni R, Chiriatti G, Camici PG (2003) Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med 349:1027–1035. https://doi.org/10.1056/NEJMoa025050
Cecchi F, Sgalambro A, Baldi M, Sotgia B, Antoniucci D, Camici PG, Sciagra R, Olivotto I (2009) Microvascular dysfunction, myocardial ischemia, and progression to heart failure in patients with hypertrophic cardiomyopathy. J Cardiovasc Transl Res 2:452–461. https://doi.org/10.1007/s12265-009-9142-5
Christensen TE, Ahtarovski KA, Bang LE, Holmvang L, Soholm H, Ghotbi AA, Andersson H, Vejlstrup N, Ihlemann N, Engstrom T, Kjaer A, Hasbak P (2015) Basal hyperaemia is the primary abnormality of perfusion in Takotsubo cardiomyopathy: a quantitative cardiac perfusion positron emission tomography study. Eur Heart J Cardiovasc Imaging 16:1162–1169. https://doi.org/10.1093/ehjci/jev065
Clair MJ, Krombach S, Coker ML, Heslin TL, Kribbs SB, de Casparo M, Spinale FG (1998) Angiotensin AT1 angiotensin II receptor inhibition in pacing-induced heart failure: effects on left ventricular performance and regional blood flow patterns. J Mol Cell Cardiol 30:2355–2364. https://doi.org/10.1016/s1071-9164(98)90237-8
Coutsos M, Sala-Mercado JA, Ichinose M, Li Z, Dawe EJ, O’Leary DS (2013) Muscle metaboreflex-induced coronary vasoconstriction limits ventricular contractility during dynamic exercise in heart failure. Am J Physiol Heart Circ Physiol 304:H1029–H1037. https://doi.org/10.1152/ajpheart.00879.2012
Crystal GJ, Pagel PS (2018) Right ventricular perfusion: physiology and clinical implications. Anesthesiology 128:202–218. https://doi.org/10.1097/ALN.0000000000001891
Dass S, Holloway CJ, Cochlin LE, Rider OJ, Mahmod M, Robson M, Sever E, Clarke K, Watkins H, Ashrafian H, Karamitsos TD, Neubauer S (2015) No evidence of myocardial oxygen deprivation in nonischemic heart failure. Circ Heart Fail 8:1088–1093. https://doi.org/10.1161/CIRCHEARTFAILURE.114.002169
De Caterina AR, Leone AM, Galiuto L, Basile E, Fedele E, Paraggio L, De Maria GL, Porto I, Niccoli G, Burzotta F, Trani C, Rebuzzi AG, Crea F (2013) Angiographic assessment of myocardial perfusion in Tako-Tsubo syndrome. Int J Cardiol 168:4717–4722. https://doi.org/10.1016/j.ijcard.2013.07.172
de Waard GA, Hollander MR, Teunissen PF, Jansen MF, Eerenberg ES, Beek AM, Marques KM, van de Ven PM, Garrelds IM, Danser AH, Duncker DJ, van Royen N (2016) Changes in coronary blood flow after acute myocardial infarction: insights from a patient study and an experimental porcine model. J Am Coll Cardiol Cardiovasc Interv 9:602–613. https://doi.org/10.1016/j.jcin.2016.01.001
de Waha S, Patel MR, Granger CB, Ohman EM, Maehara A, Eitel I, Ben-Yehuda O, Jenkins P, Thiele H, Stone GW (2017) Relationship between microvascular obstruction and adverse events following primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: an individual patient data pooled analysis from seven randomized trials. Eur Heart J 38:3502–3510. https://doi.org/10.1093/eurheartj/ehx414
Del Buono MG, Montone RA, Camilli M, Carbone S, Narula J, Lavie CJ, Niccoli G, Crea F (2021) Coronary microvascular dysfunction across the spectrum of cardiovascular diseases: JACC state-of-the-art review. J Am Coll Cardiol 78:1352–1371. https://doi.org/10.1016/j.jacc.2021.07.042
Depré C, Vanoverschelde J-LJ, Melin JA, Borgers M, Bol A, Ausma J, Dion R, Wijns W (1995) Structural and metabolic correlates of the reversibility of chronic left ventricular ischemic dysfunction in humans. Am J Physiol Heart Circ Physiol 268:H1265–H1275. https://doi.org/10.1152/ajpheart.1995.268.3.H1265
Dryer K, Gajjar M, Narang N, Lee M, Paul J, Shah AP, Nathan S, Butler J, Davidson CJ, Fearon WF, Shah SJ, Blair JEA (2018) Coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol 314:H1033–H1042. https://doi.org/10.1152/ajpheart.00680.2017
Duncker DJ, de Beer V, Merkus D (2008) Alterations in vasomotor control of coronary resistance vessels in remodelled myocardium of swine with a recent myocardial infarction. Med Biol Eng Comput 46:485–497. https://doi.org/10.1007/s11517-008-0315-1
Duncker DJ, Ishibashi Y, Bache RJ (1998) Effect of treadmill exercise on transmural distribution of blood flow in hypertrophied left ventricle. Am J Physiol Heart Circ Physiol 275:H1274–H1282. https://doi.org/10.1152/ajpheart.1998.275.4.H1274
Duncker DJ, Koller A, Merkus D, Canty JM Jr (2015) Regulation of coronary blood flow in health and ischemic heart disease. Prog Cardiovasc Dis 57:409–422. https://doi.org/10.1016/j.pcad.2014.12.002
Duncker DJ, Traverse JH, Ishibashi Y, Bache RJ (1999) Effect of NO on transmural distribution of blood flow in hypertrophied left ventricle during exercise. Am J Physiol Heart Circ Physiol 276:H1305–H1312. https://doi.org/10.1152/ajpheart.1999.276.4.H1305
Ehring T, Krajcar M, Baumgart D, Kompa S, Hümmelgen M, Heusch G (1995) Cholinergic and α-adrenergic coronary vasomotion with increasing ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 268:H886–H894. https://doi.org/10.1152/ajpheart.1995.268.2.H886
Eitel I, von Knobelsdorff-Brenkenhoff F, Bernhardt P, Carbone I, Muellerleile K, Aldrovandi A, Francone M, Desch S, Gutberlet M, Strohm O, Schuler G, Schulz-Menger J, Thiele H, Friedrich MG (2011) Clinical characteristics and cardiovascular magnetic resonance findings in stress (Takotsubo) cardiomyopathy. JAMA 306:277–286. https://doi.org/10.1001/jama.2011.992
Ellis ER, Josephson ME (2013) What about tachycardia-induced cardiomyopathy? Arrhythm Electrophysiol Rev 2:82–90. https://doi.org/10.15420/aer.2013.2.2.82
Ellis SG, Henschke CI, Sandor T, Wynne J, Braunwald E, Kloner RA (1983) Time course of functional and biochemical recovery of myocardium salvaged by reperfusion. J Am Coll Cardiol 1:1047–1055. https://doi.org/10.1016/s0735-1097(83)80107-7
Emter CA, Tharp DL, Ivey JR, Ganjam VK, Bowles DK (2011) Low-intensity interval exercise training attenuates coronary vascular dysfunction and preserves Ca(2)(+)-sensitive K(+) current in miniature swine with LV hypertrophy. Am J Physiol Heart Circ Physiol 301:H1687-1694. https://doi.org/10.1152/ajpheart.00610.2011
Erbs S, Linke A, Adams V, Lenk K, Thiele H, Diederich K-W, Emmrich F, Kluge R, Kendziorra K, Sabri O, Schuler G, Hambrecht R (2005) Transplantation of blood-derived progenitor cells after recanalization of chronic coronary artery occlusion. First randomized and placebo-controlled study. Circ Res 97:756–762. https://doi.org/10.1161/01.RES.0000185811.71306.8b
Erbs S, Linke A, Schachinger V, Assmus B, Thiele H, Diederich KW, Hoffmann C, Dimmeler S, Tonn T, Hambrecht R, Zeiher AM, Schuler G (2007) Restoration of microvascular function in the infarct-related artery by intracoronary transplantation of bone marrow progenitor cells in patients with acute myocardial infarction: the Doppler Substudy of the Reinfusion of Enriched Progenitor Cells and Infarct Remodeling in Acute Myocardial Infarction (REPAIR-AMI) trial. Circulation 116:366–374. https://doi.org/10.1161/CIRCULATIONAHA.106.671545
Ezaz G, Long JB, Gross CP, Chen J (2014) Risk prediction model for heart failure and cardiomyopathy after adjuvant trastuzumab therapy for breast cancer. J Am Heart Assoc 3:e000472. https://doi.org/10.1161/JAHA.113.000472
Fallavollita JA, Canty JM (1999) Differential 18F–2-deoxyglucose uptake in viable dysfunctional myocardium with normal resting perfusion. Circulation 99:2798–2805. https://doi.org/10.1161/01.cir.99.21.2798
Fallavollita JA, Canty JMJ (2002) Ischemic cardiomyopathy in pigs with two-vessel occlusion and viable, chronically dysfunctional myocardium. Am J Physiol Heart Circ Physiol 282:H1370–H1379. https://doi.org/10.1152/ajpheart.00138.2001
Fallavollita JA, Malm BJ, Canty JMJ (2003) Hibernating myocardium retains metabolic and contractile reserve despite regional reductions in flow, function, and oxygen consumption at rest. Circ Res 92:48–55. https://doi.org/10.1161/01.res.0000049104.57549.03
Feola M, Chauvie S, Rosso GL, Biggi A, Ribichini F, Bobbio M (2008) Reversible impairment of coronary flow reserve in Takotsubo cardiomyopathy: a myocardial PET study. J Nucl Cardiol 15:811–817. https://doi.org/10.1007/BF03007363
Fernandez-Sola J (2015) Cardiovascular risks and benefits of moderate and heavy alcohol consumption. Nat Rev Cardiol 12:576–587. https://doi.org/10.1038/nrcardio.2015.91
Fogarassy G, Vathy-Fogarassy A, Kenessey I, Kasler M, Forster T (2019) Risk prediction model for long-term heart failure incidence after epirubicin chemotherapy for breast cancer—a real-world data-based, nationwide classification analysis. Int J Cardiol 285:47–52. https://doi.org/10.1016/j.ijcard.2019.03.013
Fragasso G, Benti R, Sciammarella M, Rossetti E, Savi A, Gerundini P, Chierchia S (1991) Symptom-limited exercise testing causes sustained diastolic dysfunction in patients with coronary disease and low effort tolerance. J Am Coll Cardiol 17:1251–1255. https://doi.org/10.1016/s0735-1097(10)80131-7
Franciosa JA, Heckel R, Limas C, Cohn JN (1980) Progressive myocardial dysfunction associated with increased vascular resistance. Am J Physiol Heart Circ Physiol 239:H477–H482. https://doi.org/10.1152/ajpheart.1980.239.4.H477
Francis GS, Benedict C, Johnstone DE (1990) Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. Circulation 82:1724–1729. https://doi.org/10.1161/01.cir.82.5.1724
Franssen C, Chen S, Unger A, Korkmaz I, De Keulenaer GW, Tschöpe C, Leite-Moreira AF, Musters R, Niessen HWM, Linke WA, Paulus WJ, Hamdani N (2016) Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. J Am Coll Cardiol Heart Failure 4:312–324. https://doi.org/10.1016/j.jchf.2015.10.007
Gaasch WH, Zile MR, Hoshino PK, Apstein CS, Blaustein AS (1989) Stress-shortening relations and myocardial blood flow in compensated and failing canine hearts with pressure-overload hypertrophy. Circulation 79:872–883. https://doi.org/10.1161/01.cir.79.4.872
Galan-Arriola C, Vilchez-Tschischke JP, Lobo M, Lopez GJ, de Molina-Iracheta A, Perez-Martinez C, Villena-Gutierrez R, Macias A, Diaz-Rengifo IA, Oliver E, Fuster V, Sanchez-Gonzalez J, Ibanez B (2021) Coronary microcirculation damage in anthracycline cardiotoxicity. Cardiovasc Res. https://doi.org/10.1093/cvr/cvab053:10.1093/cvr/cvab053
Galiuto L, De Caterina AR, Porfidia A, Parraggio L, Barchetta S, Locorotondo G, Rebuzzi AG, Crea F (2010) Reversible coronary microvascular dysfunction: a common pathogenetic mechanism in apical ballooning or Tako-Tsubo syndrome. Eur Heart J 31:1319–1327. https://doi.org/10.1093/eurheartj/ehq039
Gandjbakhch E, Redheuil A, Pousset F, Charron P, Frank R (2018) Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. J Am Coll Cardiol 72:784–804. https://doi.org/10.1016/j.jacc.2018.05.065
Gewirtz H, Dilsizian V (2017) Myocardial viability: survival mechanisms and molecular imaging targets in acute and chronic ischemia. Circ Res 120:1197–1212. https://doi.org/10.1161/CIRCRESAHA.116.307898
Ghadri JR, Wittstein IS, Prasad A, Sharkey S, Dote K, Akashi YJ, Cammann VL, Crea F, Galiuto L, Desmet W, Yoshida T, Manfredini R, Eitel I, Kosuge M, Nef HM, Deshmukh A, Lerman A, Bossone E, Citro R, Ueyama T, Corrado D, Kurisu S, Ruschitzka F, Winchester D, Lyon AR, Omerovic E, Bax JJ, Meimoun P, Tarantini G, Rihal C, Migliore F, Horowitz JD, Shimokawa H, Luscher TF, Templin C (2018) International expert consensus document on Takotsubo syndrome (Part I): clinical characteristics, diagnostic criteria, and pathophysiology. Eur Heart J 39:2032–2046. https://doi.org/10.1093/eurheartj/ehy076
Gill RM, Braz JC, Jin N, Etgen GJ, Shen W (2007) Restoration of impaired endothelium-dependent coronary vasodilation in failing heart: role of eNOS phosphorylation and CGMP/cGK-I signaling. Am J Physiol Heart Circ Physiol 292:H2782–H2790. https://doi.org/10.1152/ajpheart.00831.2006
Gold FL, Bache RJ (1982) Transmural right ventricular blood flow during acute pulmonary artery hypertension in the sedated dog. Evidence for subendocardial ischemia despite residual vasodilator reserve. Circ Res 51:196–204. https://doi.org/10.1161/01.res.51.2.196
Gold FL, Horwitz LD, Bache RJ (1984) Adrenergic coronary vasoconstriction in acute right ventricular hypertension. Cardiovasc Res 18:447–454. https://doi.org/10.1093/cvr/18.7.447
Goodwill AG, Dick GM, Kiel AM, Tune JD (2017) Regulation of coronary blood flow. Compr Physiol 7:321–382. https://doi.org/10.1002/cphy.c160016
Group M-HS (1999) Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised intervention trial in congestive heart failure. Lancet 353:2001–2007. https://doi.org/10.1016/S0140-6736(99)04440-2
Gulati A, Ismail TF, Ali A, Hsu LY, Goncalves C, Ismail NA, Krishnathasan K, Davendralingam N, Ferreira P, Halliday BP, Jones DA, Wage R, Newsome S, Gatehouse P, Firmin D, Jabbour A, Assomull RG, Mathur A, Pennell DJ, Arai AE, Prasad SK (2019) Microvascular dysfunction in dilated cardiomyopathy: a quantitative stress perfusion cardiovascular magnetic resonance study. J Am Coll Cardiol Cardiovasc Imaging 12:1699–1708. https://doi.org/10.1016/j.jcmg.2018.10.032
Haitsma DB, Bac D, Raja N, Boomsma F, Verdouw PD, Duncker DJ (2001) Minimal impairment of myocardial blood flow responses to exercise in the remodeled left ventricle early after myocardial infarction, despite significant hemodynamic and neurohumoral alterations. Cardiovasc Res 52:417–428. https://doi.org/10.1016/s0008-6363(01)00426-6
Haitsma DB, Merkus D, Vermeulen J, Verdouw PD, Duncker DJ (2002) Nitric oxide production is maintained in exercising swine with chronic left ventricular dysfunction. Am J Physiol Heart Circ Physiol 282:H2198–H2209. https://doi.org/10.1152/ajpheart.00834.2001
Harjola VP, Mebazaa A, Celutkiene J, Bettex D, Bueno H, Chioncel O, Crespo-Leiro MG, Falk V, Filippatos G, Gibbs S, Leite-Moreira A, Lassus J, Masip J, Mueller C, Mullens W, Naeije R, Nordegraaf AV, Parissis J, Riley JP, Ristic A, Rosano G, Rudiger A, Ruschitzka F, Seferovic P, Sztrymf B, Vieillard-Baron A, Yilmaz MB, Konstantinides S (2016) Contemporary management of acute right ventricular failure: a statement from the Heart Failure Association and the Working Group on Pulmonary Circulation and Right Ventricular Function of the European Society of Cardiology. Eur J Heart Fail 18:226–241. https://doi.org/10.1002/ejhf.478
Harrison DG, Florentine MS, Brooks LA, Cooper SM, Marcus ML (1988) The effect of hypertension and left ventricular hypertrophy on the lower range of coronary autoregulation. Circulation 77:1108–1115. https://doi.org/10.1161/01.cir.77.5.1108
He K-L, Dickstein M, Sabbah HN, Yi G-H, Gu A, Maurer M, Wei C-M, Wang J, Burkhoff D (2004) Mechanisms of heart failure with well preserved ejection fraction in dogs following limited coronary microembolization. Cardiovasc Res 64:72–83. https://doi.org/10.1016/j.cardiores.2004.06.007
Herrmann J (2020) Vascular toxic effects of cancer therapies. Nat Rev Cardiol 17:503–522. https://doi.org/10.1038/s41569-020-0347-2
Herrmann J (2020) Adverse cardiac effects of cancer therapies: cardiotoxicity and arrhythmia. Nat Rev Cardiol 17:474–502. https://doi.org/10.1038/s41569-020-0348-1
Herrmann J, Haude M, Lerman A, Schulz R, Volbracht L, Ge J, Schmermund A, Wieneke H, von Birgelen C, Eggebrecht H, Baumgart D, Heusch G, Erbel R (2001) Abnormal coronary flow velocity reserve following coronary intervention is associated with cardiac marker elevation. Circulation 103:2339–2345. https://doi.org/10.1161/01.cir.103.19.2339
Hesse B, Meyer C, Nielsen FS, Sato A, Hove JD, Holm S, Bang LE, Kofoed KF, Svendsen TL, Parving HH, Opie LH (2004) Myocardial perfusion in type 2 diabetes with left ventricular hypertrophy: normalisation by acute angiotensin-converting enzyme inhibition. Eur J Nucl Med Mol Imaging 31:362–368. https://doi.org/10.1007/s00259-003-1388-6
Heusch G (1990) α-Adrenergic mechanisms in myocardial ischemia. Circulation 81:1–13. https://doi.org/10.1161/01.cir.81.1.1
Heusch G (1998) Stunning—great paradigmatic, but little clinical importance. Basic Res Cardiol 93:164–166. https://doi.org/10.1007/s003950050081
Heusch G (1998) Hibernating myocardium. Physiol Rev 78:1055–1085. https://doi.org/10.1152/physrev.1998.78.4.1055
Heusch G (2008) Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol 153:1589–1601. https://doi.org/10.1038/sj.bjp.0707673
Heusch G (2011) Heart rate and heart failure. Circ J 75:229–236. https://doi.org/10.1253/circj.cj-10-0925
Heusch G (2019) Myocardial ischemia: lack of coronary blood flow, myocardial oxygen supply-demand imbalance, or what? Am J Physiol Heart Circ Physiol 316:H1439–H1446. https://doi.org/10.1152/ajpheart.00139.2019
Heusch G (2019) Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Res Cardiol 114:45. https://doi.org/10.1007/s00395-019-0756-8
Heusch G (2020) Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol 17:773–789. https://doi.org/10.1038/s41569-020-0403-y
Heusch G (2021) Myocardial stunning and hibernation revisited. Nat Rev Cardiol 18:522–536. https://doi.org/10.1038/s41569-021-00506-7
Heusch G, Baumgart D, Camici P, Chilian W, Gregorini L, Hess O, Indolfi C, Rimoldi O (2000) α-Adrenergic coronary vasoconstriction and myocardial ischemia in humans. Circulation 101:689–694. https://doi.org/10.1161/01.cir.101.6.689
Heusch G, Kleinbongard P, Boese D, Levkau B, Haude M, Schulz R, Erbel R (2009) Coronary microembolization: from bedside to bench and back to bedside. Circulation 120:1822–1836. https://doi.org/10.1161/CIRCULATIONAHA.109.888784
Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, Opie L (2014) Cardiovascular remodeling in coronary artery disease and heart failure. Lancet 383:1933–1943. https://doi.org/10.1016/S0140-6736(14)60107-0
Heusch G, Schulz R, Rahimtoola SH (2005) Myocardial hibernation: a delicate balance. Am J Physiol Heart Circ Physiol 288:H984–H999. https://doi.org/10.1152/ajpheart.01109.2004
Heyndrickx GR, Baig H, Nellens P, Leusen I, Fishbein MC, Vatner SF (1978) Depression of regional blood flow and wall thickening after brief coronary occlusions. Am J Physiol Heart Circ Physiol 234:H653–H659. https://doi.org/10.1152/ajpheart.1978.234.6.H653
Heyndrickx GR, Millard RW, McRitchie RJ, Maroko PR, Vatner SF (1975) Regional myocardial functional and electrophysiological alterations after brief coronary artery occlusion in conscious dogs. J Clin Invest 56:978–985. https://doi.org/10.1172/JCI108178
Hoffmann J, Luxan G, Abplanalp WT, Glaser SF, Rasper T, Fischer A, Muhly-Reinholz M, Potente M, Assmus B, John D, Zeiher AM, Dimmeler S (2021) Post-myocardial infarction heart failure dysregulates the bone vascular niche. Nat Commun 12:3964. https://doi.org/10.1038/s41467-021-24045-4
Hoole SP, Heck PM, White PA, Read PA, Khan SN, West NE, O’Sullivan M, Dutka DP (2010) Stunning and cumulative left ventricular dysfunction occurs late after coronary balloon occlusion in humans insights from simultaneous coronary and left ventricular hemodynamic assessment. J Am Coll Cardiol Cardiovasc Interv 3:412–418. https://doi.org/10.1016/j.jcin.2009.12.014
Hou M, Chen Y, Traverse JH, Li Y, Barsoum M, Bache RJ (2004) ET-A receptor activity restrains coronary blood flow in the failing heart. J Cardiovasc Pharmacol 43:764–769. https://doi.org/10.1097/00005344-200406000-00005
Howlett JG, Stebbins A, Petrie MC, Jhund PS, Castelvecchio S, Cherniavsky A, Sueta CA, Roy A, Pina IL, Wurm R, Drazner MH, Andersson B, Batlle C, Senni M, Chrzanowski L, Merkely B, Carson P, Desvigne-Nickens PM, Lee KL, Velazquez EJ, Al-Khalidi HR, Investigators S (2019) CABG Improves outcomes in patients with ischemic cardiomyopathy: 10-Year follow-up of the STICH trial. J Am Coll Cardiol Heart Failure 7:878–887. https://doi.org/10.1016/j.jchf.2019.04.018
Huizar JF, Ellenbogen KA, Tan AY, Kaszala K (2019) Arrhythmia-induced cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol 73:2328–2344. https://doi.org/10.1016/j.jacc.2019.02.045
Huo Y, Kassab GS (2015) Remodeling of left circumflex coronary arterial tree in pacing-induced heart failure. J Appl Physiol 119:404–411. https://doi.org/10.1152/japplphysiol.00262.2015
Ikram H, Rogers SJ, Charles CJ, Sands J, Richards AM, Bridgman PG, Gooneratne R (1997) An ovine model of acute myocardial infarction and chronic left ventricular dysfunction. Angiology 48:679–688. https://doi.org/10.1177/000331979704800803
Investigators TS (1991) Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 325:293–302. https://doi.org/10.1056/NEJM199108013250501
Ishihara K, Zile MR, Nagatsu M, Nakano K, Tomita M, Kanazawa S, Clamp L, DeFreyte G, Carabello BA (1992) Coronary blood flow after the regression of pressure-overload left ventricular hypertrophy. Circ Res 71:1472–1481. https://doi.org/10.1161/01.RES.71.6.1472
Jameel MN, Xiong Q, Mansoor A, Bache RJ, Zhang J (2016) ATP sensitive K(+) channels are critical for maintaining myocardial perfusion and high energy phosphates in the failing heart. J Mol Cell Cardiol 92:116–121. https://doi.org/10.1016/j.yjmcc.2016.02.005
Jerosch-Herold M, Sheridan DC, Kushner JD, Nauman D, Burgess D, Dutton D, Alharethi R, Li D, Hershberger RE (2008) Cardiac magnetic resonance imaging of myocardial contrast uptake and blood flow in patients affected with idiopathic or familial dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 295:H1234–H1242. https://doi.org/10.1152/ajpheart.00429.2008
Kainuma S, Miyagawa S, Fukushima S, Pearson J, Chen YC, Saito A, Harada A, Shiozaki M, Iseoka H, Watabe T, Watabe H, Horitsugi G, Ishibashi M, Ikeda H, Tsuchimochi H, Sonobe T, Fujii Y, Naito H, Umetani K, Shimizu T, Okano T, Kobayashi E, Daimon T, Ueno T, Kuratani T, Toda K, Takakura N, Hatazawa J, Shirai M, Sawa Y (2015) Cell-sheet therapy with omentopexy promotes arteriogenesis and improves coronary circulation physiology in failing heart. Mol Ther 23:374–386. https://doi.org/10.1038/mt.2014.225
Kato S, Fukui K, Kodama S, Azuma M, Nakayama N, Iwasawa T, Kimura K, Tamura K, Utsunomiya D (2021) Cardiovascular magnetic resonance assessment of coronary flow reserve improves risk stratification in heart failure with preserved ejection fraction. J Cardiovasc Magn Reson 23:112. https://doi.org/10.1186/s12968-021-00807-3
Kato S, Saito N, Kirigaya H, Gyotoku D, Iinuma N, Kusakawa Y, Iguchi K, Nakachi T, Fukui K, Futaki M, Iwasawa T, Kimura K, Umemura S (2016) Impairment of coronary flow reserve evaluated by phase contrast cine-magnetic resonance imaging in patients with heart failure with preserved ejection fraction. J Am Heart Assoc 5:e002649. https://doi.org/10.1161/JAHA.115.002649
Kawano H, Okada R, Yano K (2003) Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels 18:32–39. https://doi.org/10.1007/s003800300005
Kelshiker MA, Seligman H, Howard JP, Rahman H, Foley M, Nowbar AN, Rajkumar CA, Shun-Shin MJ, Ahmad Y, Sen S, Al-Lamee R, Petraco R (2021) Coronary flow reserve and cardiovascular outcomes: a systematic review and meta-analysis. Eur Heart J. https://doi.org/10.1093/eurheartj/ehab775
Kleinbongard P, Heusch G (2021) A fresh look at coronary microembolization. Nat Rev Cardiol. https://doi.org/10.1038/s41569-021-00632-2
Kloner RA, Przyklenk K (1991) Hibernation and stunning of the myocardium. N Engl J Med 325:1877–1879. https://doi.org/10.1056/NEJM199112263252610
Klug G, Mayr A, Schenk S, Esterhammer R, Schocke M, Nocker M, Jaschke W, Pachinger O, Metzler B (2012) Prognostic value at 5 years of microvascular obstruction after acute myocardial infarction assessed by cardiovascular magnetic resonance. J Cardiovasc Magn Reson 14:46. https://doi.org/10.1186/1532-429X-14-46
Knecht M, Burkhoff D, Yi G-H, Popilskis S, Homma S, Packer M, Wang J (1997) Coronary endothelial dysfunction precedes heart failure and reduction of coronary reserve in awake dogs. J Mol Cell Cardiol 29:217–227. https://doi.org/10.1006/jmcc.1996.0266
Kobylecka M, Budnik M, Kochanowski J, Piatkowski R, Chojnowski M, Fronczewska-Wieniawska K, Mazurek T, Maczewska J, Peller M, Opolski G, Krolicki L (2018) Takotsubo cardiomyopathy: FDG myocardial uptake pattern in fasting patients. Comparison of PET/CT, SPECT, and ECHO results. J Nucl Cardiol 25:1260–1270. https://doi.org/10.1007/s12350-016-0775-x
Krams R, Kofflard MJM, Duncker DJ, von Birgelen C, Carlier S, Kliffen M, Ten Cate FJ, Serruys PW (1998) Decreased coronary flow reserve in hypertrophic cardiomyopathy is related to remodeling of the coronary microcirculation. Circulation 97:230–233. https://doi.org/10.1161/01.cir.97.3.230
Kribbs SB, Merritt WM, Clair MJ, Krombach RS, Houck WV, Dodd MG, Mukherjee R, Spinale FG (1998) Amlodipine monotherapy, angiotensin-converting enzyme inhibition, and combination therapy with pacing-induced heart failure. Hypertension 31:755–765. https://doi.org/10.1161/01.hyp.31.3.755
Krombach RS, Clair MJ, Hendrick JW, Mukherjee R, Houck WV, Hebbar L, Kribbs SB, Dodd MG, Spinale FG (1999) Amlodipine therapy in congestive heart failure: hemodynamic and neurohormonal effects at rest and after treadmill exercise. Am J Cardiol 84:3L-15L. https://doi.org/10.1016/s0002-9149(99)00359-8
Kupatt C, Hinkel R, von Bruhl ML, Pohl T, Horstkotte J, Raake P, El AC, Thein E, Dimmeler S, Feron O, Boekstegers P (2007) Endothelial nitric oxide synthase overexpression provides a functionally relevant angiogenic switch in hibernating pig myocardium. J Am Coll Cardiol 49:1575–1584. https://doi.org/10.1016/j.jacc.2006.11.047
Kurisu S, Sato H, Kawagoe T, Ishihara M, Shimatani Y, Nishioka K, Kono Y, Umemura T, Nakamura S (2002) Tako-tsubo-like left ventricular dysfunction with ST-segment elevation: a novel cardiac syndrome mimicking acute myocardial infarction. Am Heart J 143:448–455. https://doi.org/10.1067/mhj.2002.120403
Lancaster J, Juneman E, Hagerty T, Do R, Hicks M, Meltzer K, Standley P, Gaballa M, Kellar R, Goldman S, Thai H (2010) Viable fibroblast matrix patch induces angiogenesis and increases myocardial blood flow in heart failure after myocardial infarction. Tissue Eng Part A 16:3065–3073. https://doi.org/10.1089/ten.TEA.2009.0589
Landmesser U, Engberding N, Bahlmann FH, Schaefer A, Wiencke A, Heineke A, Spiekermann S, Hilfiker-Kleiner D, Templin C, Kotlarz D, Mueller M, Fuchs M, Hornig B, Haller H, Drexler H (2004) Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation 110:1933–1939. https://doi.org/10.1161/01.CIR.0000143232.67642.7A
Lavallee M, Cox D, Patrick TA, Vatner SF (1983) Salvage of myocardial function by coronary artery reperfusion 1, 2 and 3 hours after occlusion in conscious dogs. Circ Res 53:235–247. https://doi.org/10.1161/01.res.53.2.235
Leopold JA, Kawut SM, Aldred MA, Archer SL, Benza RL, Bristow MR, Brittain EL, Chesler N, DeMan FS, Erzurum SC, Gladwin MT, Hassoun PM, Hemnes AR, Lahm T, Lima JAC, Loscalzo J, Maron BA, Rosa LM, Newman JH, Redline S, Rich S, Rischard F, Sugeng L, Tang WHW, Tedford RJ, Tsai EJ, Ventetuolo CE, Zhou Y, Aggarwal NR, Xiao L (2021) Diagnosis and treatment of right heart failure in pulmonary vascular diseases: a national heart, lung, and blood institute workshop. Circ Heart Fail 14:e007975. https://doi.org/10.1161/CIRCHEARTFAILURE.120.007975
Lopes RD, Alexander KP, Stevens SR, Reynolds HR, Stone GW, Pina IL, Rockhold FW, Elghamaz A, Lopez-Sendon JL, Farsky PS, Chernyavskiy AM, Diaz A, Phaneuf D, DeBelder MA, Ma Y-t, Guzman LA, Khouri M, Sionis A, Hausenloy DJ, Doerr R, Selvanayagam JK, Maggioni AP, Hochman JS, Maron DJ (2020) Initial invasive versus conservative management of stable ischemic heart disease patients with a history of heart failure or left ventricular dysfunction: insights from the ISCHEMIA Trial. Circulation 142:1725–1735. https://doi.org/10.1161/CIRCULATIONAHA.120.050304
Lyon AR, Citro R, Schneider B, Morel O, Ghadri JR, Templin C, Omerovic E (2021) Pathophysiology of Takotsubo syndrome: JACC state-of-the-art review. J Am Coll Cardiol 77:902–921. https://doi.org/10.1016/j.jacc.2020.10.060
Mahmod M, Francis JM, Pal N, Lewis A, Dass S, De Silva R, Petrou M, Sayeed R, Westaby S, Robson MD, Ashrafian H, Neubauer S, Karamitsos TD (2014) Myocardial perfusion and oxygenation are impaired during stress in severe aortic stenosis and correlate with impaired energetics and subclinical left ventricular dysfunction. J Cardiovasc Magn Reson 16:29. https://doi.org/10.1186/1532-429X-16-29
Mangion K, Carrick D, Clerfond G, Rush C, McComb C, Oldroyd KG, Petrie MC, Eteiba H, Lindsay M, McEntegart M, Hood S, Watkins S, Davie A, Auger DA, Zhong X, Epstein FH, Haig CE, Berry C (2019) Predictors of segmental myocardial functional recovery in patients after an acute ST-elevation myocardial infarction. Eur J Radiol 112:121–129. https://doi.org/10.1016/j.ejrad.2019.01.010
Maranta F, Tondi L, Agricola E, Margonato A, Rimoldi O, Camici PG (2015) Ivabradine reduces myocardial stunning in patients with exercise-inducible ischaemia. Basic Res Cardiol 110:55. https://doi.org/10.1007/s00395-015-0511-8
Maron BJ, Wolfson JK, Epstein SE, Roberts WC (1986) Intramural (“small vessel”) coronary artery disease in hypertrophic cardiomyopathy. J Am Coll Cardiol 8:545–557. https://doi.org/10.1016/s0735-1097(86)80181-4
Maron DJ, Hochman JS, Reynolds HR, Bangalore S, O’Brien SM, Boden WE, Chaitman BR, Senior R, Lopez-Sendon J, Alexander KP, Lopes RD, Shaw LJ, Berger JS, Newman JD, Sidhu MS, Goodman SG, Ruzyllo W, Gosselin G, Maggioni AP, White HD, Bhargava B, Min JK, Mancini GBJ, Berman DS, Picard MH, Kwong RY, Ali ZA, Mark DB, Spertus JA, Krishnan MN, Elghamaz A, Moorthy N, Hueb WA, Demkow M, Mavromatis K, Bockeria O, Peteiro J, Miller TD, Szwed H, Doerr R, Keltai M, Selvanayagam JB, Steg PG, Held C, Kohsaka S, Mavromichalis S, Kirby R, Jeffries NO, Harrell FE Jr, Rockhold FW, Broderick S, Ferguson TB Jr, Williams DO, Harrington RA, Stone GW, Rosenberg Y, Group IR (2020) Initial invasive or conservative strategy for stable coronary disease. N Engl J Med 382:1395–1407. https://doi.org/10.1056/NEJMoa1915922
Mathier MA, Rose GA, Fifer MA, Miyamoto MI, Dinsmore RE, Castano HH, Dec GW, Palacios IF, Semigran MJ (1998) Coronary endothelial dysfunction in patients with acute-onset idiopathic dilated cardiomyopathy. J Am Coll Cardiol 32:216–224. https://doi.org/10.1016/s0735-1097(98)00209-5
Mathur A, Fernandez-Aviles F, Dimmeler S, Hauskeller C, Janssens S, Menasche P, Wojakowski W, Martin JF, Zeiher A, Investigators B (2017) The consensus of the Task Force of the European Society of Cardiology concerning the clinical investigation of the use of autologous adult stem cells for the treatment of acute myocardial infarction and heart failure: update 2016. Eur Heart J 38:2930–2935. https://doi.org/10.1093/eurheartj/ehw640
Matsuzaki M, Gallagher KP, Kemper WS, White F, Ross J Jr (1983) Sustained regional dysfunction produced by prolonged coronary stenosis: gradual recovery after reperfusion. Circulation 68:170–182. https://doi.org/10.1161/01.CIR.68.1.170
McFalls EO, Duncker DJ, Ward H, Fashingbauer P (1995) Impaired endothelium-dependent vasodilation of coronary resistance vessels in severely stunned porcine myocardium. Basic Res Cardiol 90:498–509. https://doi.org/10.1007/BF00788543
McKay RG, Pfeffer MA, Pasternak RC, Markis JE, Come PC, Nakao S, Alderman JD, Ferguson JJ, Safian RD, Grossman W (1986) Left ventricular remodeling after myocardial infarction: a corollary to infarct expansion. Circulation 74:693–702. https://doi.org/10.1161/01.cir.74.4.693
McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Belohlavek J, Bohm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukat A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O’Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjostrand M, Langkilde AM, Committees D-HT, Investigators (2019) Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 381:1995–2008. https://doi.org/10.1056/NEJMoa1911303
Melchert PJ, Duncker DJ, Traverse JH, Bache RJ (1999) Role of K+ATP channels and adenosine in regulation of coronary blood flow in the hypertrophied left ventricle. Am J Physiol Heart Circ Physiol 277:H617–H625. https://doi.org/10.1152/ajpheart.1999.277.2.H617
Merkus D, Haitsma DB, Sorop O, Boomsma F, de Beer VJ, Lamers JM, Verdouw PD, Duncker DJ (2006) Coronary vasoconstrictor influence of angiotensin II is reduced in remodeled myocardium after myocardial infarction. Am J Physiol Heart Circ Physiol 291:H2082–H2089. https://doi.org/10.1152/ajpheart.00861.2005
Merkus D, Houweling B, van den Meiracker AH, Boomsma F, Duncker DJ (2005) Contribution of endothelin to coronary vasomotor tone is abolished after myocardial infarction. Am J Physiol Heart Circ Physiol 288:H871–H880. https://doi.org/10.1152/ajpheart.00429.2004
Merkus D, Houweling B, van Vliet M, Duncker DJ (2005) Contribution of K+ATP channels to coronary vasomotor tone regulation is enhanced in exercising swine with a recent myocardial infarction. Am J Physiol Heart Circ Physiol 288:H1306–H1313. https://doi.org/10.1152/ajpheart.00631.2004
Mills I, Fallon JT, Wrenn D, Sasken H, Gray W, Bier J, Levine D, Berman S, Gilson M, Gewirtz H (1994) Adaptive responses of coronary circulation and myocardium to chronic reduction in perfusion pressure and flow. Am J Physiol Heart Circ Physiol 266:H447–H457. https://doi.org/10.1152/ajpheart.1994.266.2.H447
Mochula A, Mochula OV, Maltseva AN, Vorobyeva DA, Ryabov VV, Zavadovsky KV (2021) Dynamic SPECT with assessment myocardial blood flow and coronary flow reserve in MINOCA patients: comparison with cardiac magnetic resonance. Eur Heart J Cardiovasc Imaging. https://doi.org/10.1093/ehjci/jeaa356.339
Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM (2015) Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 131:550–559. https://doi.org/10.1161/CIRCULATIONAHA.114.009625
Motz W, Strauer BE (1996) Improvement of coronary flow reserve after long-term therapy with enalapril. Hypertension 27:1031–1038. https://doi.org/10.1161/01.hyp.27.5.1031
Muhlfeld C, Rajces A, Manninger M, Alogna A, Wierich MC, Scherr D, Post H, Schipke J (2020) A transmural gradient of myocardial remodeling in early-stage heart failure with preserved ejection fraction in the pig. J Anat 236:531–539. https://doi.org/10.1111/joa.13117
Mulder FI, Horvath-Puho E, van Es N, Pedersen L, Buller HR, Botker HE, Sorensen HT (2021) Arterial thromboembolism in cancer patients. A Danish population-based cohort study. J Am Coll Cardiol Cardiooncol 3:205–218. https://doi.org/10.1016/j.jaccao.2021.02.007
Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, Vanhoutte D, Heymans S, Grieve DJ, Cave AC, Brewer AC, Zhang M, Shah AM (2014) Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol 63:2734–2741. https://doi.org/10.1016/j.jacc.2014.02.572
Murray PA, Vatner SF (1981) Reduction of maximal coronary vasodilator capacity in conscious dogs with severe right ventricular hypertrophy. Circ Res 48:25–33. https://doi.org/10.1161/01.res.48.1.25
Murray PA, Vatner SF (1981) Abnormal coronary vascular response to exercise in dogs with severe right ventricular hypertrophy. J Clin Invest 67:1314–1323. https://doi.org/10.1172/jci110160
Nakamura R, Egashira K, Arimura K, Machida Y, Ide T, Tsutsui H, Shimokawa H, Takeshita A (2001) Increased inactivation of nitric oxide is involved in impaired coronary flow reserve in heart failure. Am J Physiol Heart Circ Physiol 281:H2619–H2625. https://doi.org/10.1152/ajpheart.2001.281.6.H2619
Nakayama M, Yamamuro M, Takashio S, Uemura T, Nakayama N, Hirakawa K, Oda S, Utsunomiya D, Kaikita K, Hokimoto S, Yamashita Y, Morita Y, Kimura K, Tamura K, Tsujita K (2018) Late gadolinium enhancement on cardiac magnetic resonance imaging is associated with coronary endothelial dysfunction in patients with dilated cardiomyopathy. Heart Vessels 33:393–402. https://doi.org/10.1007/s00380-017-1069-1
Neglia D, Michelassi C, Trivieri MG, Sambuceti G, Giorgetti A, Pratali L, Gallopin M, Salvadori P, Sorace O, Carpeggiani C, Poddighe R, L’Abbate A, Parodi O (2002) Prognostic role of myocardial blood flow impairment in idiopathic left ventricular dysfunction. Circulation 105:186–193. https://doi.org/10.1161/hc0202.102119
Neumann T, Heusch G (1997) Myocardial, skeletal muscle, and renal blood flow during exercise in conscious dogs with heart failure. Am J Physiol Heart Circ Physiol 273:H2452–H2457. https://doi.org/10.1152/ajpheart.1997.273.5.H2452
Nienaber CA, Brunken RC, Sherman CT, Yeatman LA, Gambhir SS, Krivokapich J, Demer LL, Ratib O, Child JS, Phelps ME, Schelbert HR (1991) Metabolic and functional recovery of ischemic human myocardium after coronary angioplasty. J Am Coll Cardiol 18:966–978. https://doi.org/10.1016/0735-1097(91)90755-x
Niitsuma T, Saito T, Tamagawa K, Saitoh S, Mitsugi M, Sato M, Maehara K, Maruyama Y (1999) Augmented basal nitric oxide production contributes to maintenance of coronary blood flow in dogs with pacing-induced heart failure. Jpn Heart J 40:629–644. https://doi.org/10.1536/jhj.40.629
Oikawa Y, Maehara K, Saito T, Tamagawa K, Maruyama Y (2001) Attenuation of angiotensin II-mediated coronary vasoconstriction and vasodilatory action of angiotensin-converting enzyme inhibitor in pacing-induced heart failure in dogs. J Am Coll Cardiol 38:1188–1194. https://doi.org/10.1016/s0735-1097(01)01494-2
Olivotto I, Cecchi F, Gistri R, Lorenzoni R, Chiriatti G, Girolami F, Torricelli F, Camici PG (2006) Relevance of coronary microvascular flow impairment to long-term remodeling and systolic dysfunction in hypertrophic cardiomyopathy. J Am Coll Cardiol 47:1043–1048. https://doi.org/10.1016/j.jacc.2005.10.050
Olivotto I, Girolami F, Sciagra R, Ackerman MJ, Sotgia B, Bos JM, Nistri S, Sgalambro A, Grifoni C, Torricelli F, Camici PG, Cecchi F (2011) Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. J Am Coll Cardiol 58:839–848. https://doi.org/10.1016/j.jacc.2011.05.018
Olsen MH, Wachtell K, Meyer C, Hove JD, Palmieri V, Dige-Petersen H, Rokkedal J, Hesse B, Ibsen H (2004) Association between vascular dysfunction and reduced myocardial flow reserve in patients with hypertension: a LIFE substudy. J Hum Hypertens 18:445–452. https://doi.org/10.1038/sj.jhh.1001716
Ommen SR, Semsarian C (2021) Hypertrophic cardiomyopathy: a practical approach to guideline directed management. Lancet 398:2102–2108. https://doi.org/10.1016/S0140-6736(21)01205-8
Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA (2006) Controversies in ventricular remodelling. Lancet 367:356–367. https://doi.org/10.1016/S0140-6736(06)68074-4
Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, Januzzi J, Verma S, Tsutsui H, Brueckmann M, Jamal W, Kimura K, Schnee J, Zeller C, Cotton D, Bocchi E, Bohm M, Choi DJ, Chopra V, Chuquiure E, Giannetti N, Janssens S, Zhang J, Gonzalez Juanatey JR, Kaul S, Brunner-La Rocca HP, Merkely B, Nicholls SJ, Perrone S, Pina I, Ponikowski P, Sattar N, Senni M, Seronde MF, Spinar J, Squire I, Taddei S, Wanner C, Zannad F, Investigators EM-RT (2020) Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med 383:1413–1424. https://doi.org/10.1056/NEJMoa2022190
Padro T, Manfrini O, Bugiardini R, Canty J, Cenko E, De Luca G, Duncker DJ, Eringa EC, Koller A, Tousoulis D, Trifunovic D, Vavlukis M, de Wit C, Badimon L (2020) ESC Working Group on Coronary Pathophysiology and Microcirculation position paper on ‘coronary microvascular dysfunction in cardiovascular disease.’ Cardiovasc Res 116:741–755. https://doi.org/10.1093/cvr/cvaa003
Page BJ, Banas MD, Suzuki G, Weil BR, Young RF, Fallavollita JA, Palka BA, Canty JM Jr (2015) Revascularization of chronic hibernating myocardium stimulates myocyte proliferation and partially reverses chronic adaptations to ischemia. J Am Coll Cardiol 65:684–697. https://doi.org/10.1016/j.jacc.2014.11.040
Parrish DG, Ring WS, Bache RJ (1985) Myocardial perfusion in compensated and failing hypertrophied left ventricle. Am J Physiol Heart Circ Physiol 249:H534-539. https://doi.org/10.1152/ajpheart.1985.249.3.H534
Pasupathy S, Lindahl B, Litwin P, Tavella R, Williams MJA, Air T, Zeitz C, Smilowitz NR, Reynolds HR, Eggers KM, Nordenskjold AM, Barr P, Jernberg T, Marfella R, Bainey K, Sodoon Alzuhairi K, Johnston N, Kerr A, Beltrame JF (2021) Survival in patients with suspected myocardial infarction with nonobstructive coronary arteries: a comprehensive systematic review and meta-analysis from the MINOCA Global Collaboration. Circ Cardiovasc Qual Outcomes 14:e007880. https://doi.org/10.1161/CIRCOUTCOMES.121.007880
Paul M, Rahbar K, Gerss J, Kies P, Schober O, Schafers K, Breithardt G, Schulze-Bahr E, Wichter T, Schafers M (2012) Microvascular dysfunction in nonfailing arrhythmogenic right ventricular cardiomyopathy. Eur J Nucl Med Mol Imaging 39:416–420. https://doi.org/10.1007/s00259-011-1985-8
Paulus WJ, Tschope C (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62:263–271. https://doi.org/10.1016/j.jacc.2013.02.092
Paur H, Wright PT, Sikkel MB, Tranter MH, Mansfield C, O’Gara P, Stuckey DJ, Nikolaev VO, Diakonov I, Pannell L, Gong H, Sun H, Peters NS, Petrou M, Zheng Z, Gorelik J, Lyon AR, Harding SE (2012) High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/Gi-dependent manner: a new model of takotsubo cardiomyopathy. Circulation 126:697–706. https://doi.org/10.1161/CIRCULATIONAHA.112.111591
Pelliccia F, Kaski JC, Crea F, Camici PG (2017) Pathophysiology of Takotsubo syndrome. Circulation 135:2426–2441. https://doi.org/10.1161/CIRCULATIONAHA.116.027121
Peretto G, Lazzeroni D, Sartorio CL, Camici PG (2017) Cardiotoxicity in oncology and coronary microcirculation: future challenges in theranostics. Front Biosci 22:1760–1773. https://doi.org/10.2741/4570
Petersen SE, Jerosch-Herold M, Hudsmith LE, Robson MD, Francis JM, Doll HA, Selvanayagam JB, Neubauer S, Watkins H (2007) Evidence for microvascular dysfunction in hypertrophic cardiomyopathy: new insights from multiparametric magnetic resonance imaging. Circulation 115:2418–2425. https://doi.org/10.1161/CIRCULATIONAHA.106.657023
Pfeffer MA, Braunwald E (1990) Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 81:1161–1172. https://doi.org/10.1161/01.cir.81.4.1161
Pfeffer MA, Claggett B, Lewis EF, Granger CB, Kober L, Maggioni AP, Mann DL, McMurray JJV, Rouleau JL, Solomon SD, Steg PG, Berwanger O, Cikes M, De Pasquale CG, East C, Fernandez A, Jering K, Landmesser U, Mehran R, Merkely B, Vaghaiwalla Mody F, Petrie MC, Petrov I, Schou M, Senni M, Sim D, van der Meer P, Lefkowitz M, Zhou Y, Gong J, Braunwald E, Investigators P-M, Committees (2021) Angiotensin receptor-neprilysin inhibition in acute myocardial infarction. N Engl J Med 385:1845–1855. https://doi.org/10.1056/NEJMoa2104508
Pieske B, Tschope C, de Boer RA, Fraser AG, Anker SD, Donal E, Edelmann F, Fu M, Guazzi M, Lam CSP, Lancellotti P, Melenovsky V, Morris DA, Nagel E, Pieske-Kraigher E, Ponikowski P, Solomon SD, Vasan RS, Rutten FH, Voors AA, Ruschitzka F, Paulus WJ, Seferovic P, Filippatos G (2019) How to diagnose heart failure with preserved ejection fraction: the HFA-PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur Heart J 40:3297–3317. https://doi.org/10.1093/eurheartj/ehz641
Pries AR, Badimon L, Bugiardini R, Camici PG, Dorobantu M, Duncker DJ, Escaned J, Koller A, Piek JJ, de Wit C (2015) Coronary vascular regulation, remodelling, and collateralization: mechanisms and clinical implications on behalf of the working group on coronary pathophysiology and microcirculation. Eur Heart J 36:3134–3146. https://doi.org/10.1093/eurheartj/ehv100
Qin A, Thompson CL, Silverman P (2015) Predictors of late-onset heart failure in breast cancer patients treated with doxorubicin. J Cancer Surviv 9:252–259. https://doi.org/10.1007/s11764-014-0408-9
Rahimtoola SH (1982) Coronary bypass surgery for chronic angina—1981. Circulation 65:225–241. https://doi.org/10.1161/01.cir.65.2.225
Rahimtoola SH (1985) A perspective on the three large multicenter randomized clinical trials of coronary bypass surgery for chronic stable angina. Circulation 72(Suppl V):V123–V135
Rajagopalan S, Landrigan PJ (2021) Pollution and the Heart. N Engl J Med 385:1881–1892. https://doi.org/10.1056/NEJMra2030281
Rajappan K, Rimoldi OE, Dutka DP, Ariff B, Pennell DJ, Sheridan DJ, Camici PG (2002) Mechanisms of coronary microcirculatory dysfunction in patients with aortic stenosis and angiographically normal coronary arteries. Circulation 105:470–476. https://doi.org/10.1161/hc0402.102931
Recchia FA, McConnell PI, Bernstein RD, Vogel TR, Xu X, Hintze TH (1998) Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ Res 83:969–979. https://doi.org/10.1161/01.res.83.10.969
Reinstadler SJ, Fuernau G, Eitel C, de Waha S, Desch S, Metzler B, Schuler G, Thiele H, Eitel I (2016) Shock index as a predictor of myocardial damage and clinical outcome in ST-elevation myocardial infarction. Circ J 80:924–930. https://doi.org/10.1253/circj.CJ-15-1135
Reinstadler SJ, Stiermaier T, Reindl M, Feistritzer HJ, Fuernau G, Eitel C, Desch S, Klug G, Thiele H, Metzler B, Eitel I (2019) Intramyocardial haemorrhage and prognosis after ST-elevation myocardial infarction. Eur Heart J Cardiovasc Imaging 20:138–146. https://doi.org/10.1093/ehjci/jey101
Reiter U, Reiter G, Manninger M, Adelsmayr G, Schipke J, Alogna A, Rajces A, Stalder AF, Greiser A, Muhlfeld C, Scherr D, Post H, Pieske B, Fuchsjager M (2016) Early-stage heart failure with preserved ejection fraction in the pig: a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson 18:63. https://doi.org/10.1186/s12968-016-0283-9
Rengo G, Cannavo A, Liccardo D, Zincarelli C, de Lucia C, Pagano G, Komici K, Parisi V, Scala O, Agresta A, Rapacciuolo A, Perrone Filardi P, Ferrara N, Koch WJ, Trimarco B, Femminella GD, Leosco D (2013) Vascular endothelial growth factor blockade prevents the beneficial effects of beta-blocker therapy on cardiac function, angiogenesis, and remodeling in heart failure. Circ Heart Fail 6:1259–1267. https://doi.org/10.1161/CIRCHEARTFAILURE.113.000329
Rimoldi O, Rosen SD, Camici PG (2014) The blunting of coronary flow reserve in hypertension with left ventricular hypertrophy is transmural and correlates with systolic blood pressure. J Hypertens 32:2465–2471. https://doi.org/10.1097/HJH.0000000000000338
Rosenbaum AN, Agre KE, Pereira NL (2020) Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol 17:286–297. https://doi.org/10.1038/s41569-019-0284-0
Ross J Jr (1991) Myocardial perfusion-contraction matching. Implications for coronary heart disease and hibernation. Circulation 83:1076–1083. https://doi.org/10.1161/01.cir.83.3.1076
Rush CJ, Berry C, Oldroyd KG, Rocchiccioli JP, Lindsay MM, Touyz RM, Murphy CL, Ford TJ, Sidik N, McEntegart MB, Lang NN, Jhund PS, Campbell RT, McMurray JJV, Petrie MC (2021) Prevalence of coronary artery disease and coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. JAMA Cardiol 6:130–1143. https://doi.org/10.1001/jamacardio.2021.1825
Sabbah HN, Shimoyama H, Kono T, Gupta RC, Sharov VG, Scicli G, Levine B, Goldstein S (1994) Effects of long-term monotherapy with enalapril, metoprolol, and digoxin on the progression of left ventricular dysfunction and dilation in dogs with reduced ejection fraction. Circulation 89:2852–2859. https://doi.org/10.1161/01.cir.89.6.2852
Sabbah HN, Stein PD, Kono T, Gheorghiade M, Levine TB, Jafri S, Hawkins ET, Goldstein S (1991) A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am J Physiol Heart Circ Physiol 29:H1379–H1384. https://doi.org/10.1152/ajpheart.1991.260.4.H1379
Saito T, Maehara K, Tamagawa K, Oikawa Y, Niitsuma T, Saitoh S, Maruyama Y (2002) Alterations of endothelium-dependent and -independent regulation of coronary blood flow during heart failure. Am J Physiol Heart Circ Physiol 282:H80-86. https://doi.org/10.1152/ajpheart.2002.282.1.H80
Samson WK, Yosten GLC, Remme CA (2022) A primer on obesity-related cardiomyopathy. Physiol Rev 102:1–6. https://doi.org/10.1152/physrev.00023.2021
Sato A, Aonuma K, Nozato T, Sekiguchi Y, Okazaki O, Kubota K, Hiroe M (2008) Stunned myocardium in transient left ventricular apical ballooning: a serial study of dual I-123 BMIPP and Tl-201 SPECT. J Nucl Cardiol 15:671–679. https://doi.org/10.1016/j.nuclcard.2008.03.010
Schachinger V, Erbs S, Elsasser A, Haberbosch W, Hambrecht R, Holschermann H, Yu J, Corti R, Mathey DG, Hamm CW, Suselbeck T, Assmus B, Tonn T, Dimmeler S, Zeiher AM (2006) Intracoronary bone marrow-derived progenitor cells in acute myocardial infarction. N Engl J Med 355:1210–1221. https://doi.org/10.1056/NEJMoa060186
Schafer S, Kelm M, Mingers S, Strauer BE (2002) Left ventricular remodeling impairs coronary flow reserve in hypertensive patients. J Hypertens 20:1431–1437. https://doi.org/10.1097/00004872-200207000-00031
Scheler S, Motz W, Strauer BE (1994) Mechanism of angina pectoris in patients with systemic hypertension and normal epicardial coronary arteries by arteriogram. Am J Cardiol 73:478–482. https://doi.org/10.1016/0002-9149(94)90678-5
Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, Hill TM, Mammen PPA, Huang J, Lee DI, Hahn VS, Sharma K, Kass DA, Lavandero S, Gillette TG, Hill JA (2019) Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568:351–356. https://doi.org/10.1038/s41586-019-1100-z
Schulz R, Guth BD, Heusch G (1991) No effect of coronary perfusion on regional myocardial function within the autoregulatory range in pigs: evidence against the Gregg phenomenon. Circulation 83:1390–1403. https://doi.org/10.1161/01.CIR.83.4.1390
Schulz R, Janssen F, Guth BD, Heusch G (1991) Effect of coronary hyperperfusion on regional myocardial function and oxygen consumption of stunned myocardium in pigs. Basic Res Cardiol 86:534–543. https://doi.org/10.1007/BF02190703
Shannon RP, Komamura K, Shen YT, Bishop SP, Vatner SF (1993) Impaired regional subendocardial coronary flow reserve in conscious dogs with pacing-induced heart failure. Am J Physiol Heart Circ Physiol 265:H801–H809. https://doi.org/10.1152/ajpheart.1993.265.3.H801
Sharif D, Matanis W, Sharif-Rasslan A, Rosenschein U (2016) Doppler echocardiographic myocardial stunning index predicts recovery of left ventricular systolic function after primary percutaneous coronary intervention. Echocardiography 33:1465–1471. https://doi.org/10.1111/echo.13305
Sheiban I, Tonni S, Benussi P, Martini A, Trevi GP (1993) Left ventricular dysfunction following transient ischaemia induced by transluminal coronary angioplasty. Beneficial effects of calcium antagonists against post-ischaemic myocardial stunning. Eur Heart J 14(Suppl. A):14–21. https://doi.org/10.1093/eurheartj/14.suppl_a.14
Shen Y-T, Kudej RK, Bishop SP, Vatner SF (1996) Inotropic reserve and histological appearance of hibernating myocardium in conscious pigs with ameroid-induced coronary stenosis. Basic Res Cardiol 91:479–485. https://doi.org/10.1007/BF00788729
Shen Y-T, Vatner SF (1995) Mechanism of impaired myocardial function during progressive coronary stenosis in conscious pigs. Hibernation versus stunning. Circ Res 76:479–488. https://doi.org/10.1161/01.res.76.3.479
Shikama N, Himi T, Yoshida K, Nakao M, Fujiwara M, Tamura T, Yamanouchi M, Nakagawa K, Kuwabara Y, Toyozaki T, Masuda Y (1999) Prognostic utility of myocardial blood flow assessed by N-13 ammonia positron emission tomography in patients with idiopathic dilated cardiomyopathy. Am J Cardiol 84:434–439. https://doi.org/10.1016/s0002-9149(99)00329-x
Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K (2005) Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 115:2108–2118. https://doi.org/10.1172/JCI24682
Sinha A, Rahman H, Webb A, Shah AM, Perera D (2021) Untangling the pathophysiologic link between coronary microvascular dysfunction and heart failure with preserved ejection fraction. Eur Heart J 42:4431–4441. https://doi.org/10.1093/eurheartj/ehab653
Skyschally A, Schulz R, Erbel R, Heusch G (2002) Reduced coronary and inotropic reserves with coronary microembolization. Am J Physiol Heart Circ Physiol 282:H611–H614. https://doi.org/10.1152/ajpheart.00797.2001
Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, Martinez F, Packer M, Pfeffer MA, Pieske B, Redfield MM, Rouleau JL, van Veldhuisen DJ, Zannad F, Zile MR, Desai AS, Claggett B, Jhund PS, Boytsov SA, Comin-Colet J, Cleland J, Dungen HD, Goncalvesova E, Katova T, Kerr Saraiva JF, Lelonek M, Merkely B, Senni M, Shah SJ, Zhou J, Rizkala AR, Gong J, Shi VC, Lefkowitz MP, Investigators P-H, Committees (2019) Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med 381:1609–1620. https://doi.org/10.1056/NEJMoa1908655
Sorop O, Heinonen I, van Kranenburg M, van de Wouw J, de Beer VJ, Nguyen ITN, Octavia Y, van Duin RWB, Stam K, van Geuns RJ, Wielopolski PA, Krestin GP, van den Meiracker AH, Verjans R, van Bilsen M, Danser AHJ, Paulus WJ, Cheng C, Linke WA, Joles JA, Verhaar MC, van der Velden J, Merkus D, Duncker DJ (2018) Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc Res 114:954–964. https://doi.org/10.1093/cvr/cvy038
Spinale FG, Grine RC, Tempel GE, Crawford FA, Zile MR (1992) Alterations in the myocardial capillary vasculature accompany tachycardia-induced cardiomyopathy. Basic Res Cardiol 87:65–79. https://doi.org/10.1007/BF00795391
Spinale FG, Tanaka R, Crawford FA, Zile MR (1992) Changes in myocardial blood flow during development of and recovery from tachycardia-induced cardiomyopathy. Circulation 85:717–729. https://doi.org/10.1161/01.cir.85.2.717
Srivaratharajah K, Coutinho T, deKemp R, Liu P, Haddad H, Stadnick E, Davies RA, Chih S, Dwivedi G, Guo A, Wells GA, Bernick J, Beanlands R, Mielniczuk LM (2016) Reduced myocardial flow in heart failure patients with preserved ejection fraction. Circ Heart Fail 9:e002562. https://doi.org/10.1161/CIRCHEARTFAILURE.115.002562
Stahl LD, Aversano TR, Becker LC (1986) Selective enhancement of function of stunned myocardium by increased flow. Circulation 74:843–851. https://doi.org/10.1161/01.CIR.74.4.843
Stolen KQ, Kemppainen J, Kalliokoski KK, Hallsten K, Luotolahti M, Karanko H, Lehikoinen P, Viljanen T, Salo T, Airaksinen KE, Nuutila P, Knuuti J (2004) Myocardial perfusion reserve and oxidative metabolism contribute to exercise capacity in patients with dilated cardiomyopathy. J Card Fail 10:132–140. https://doi.org/10.1016/j.cardfail.2003.08.009
Strauer BE (1984) The coronary circulation in hypertensive heart disease. Hypertension. https://doi.org/10.1161/01.hyp.6.6_pt_2.iii74
Strauer BE (1988) Regression of myocardial and coronary vascular hypertrophy in hypertensive heart disease. J Cardiovasc Pharmacol 12(Suppl 4):S45-54. https://doi.org/10.1097/00005344-198806124-00009
Sun D, Huang A, Zhao G, Bernstein R, Forfia P, Xu X, Koller A, Kaley G, Hintze TH (2000) Reduced NO-dependent arteriolar dilation during the development of cardiomyopathy. Am J Physiol Heart Circ Physiol 278:H461-468. https://doi.org/10.1152/ajpheart.2000.278.2.H461
Suzuki G, Young RF, Leiker MM, Suzuki T (2016) Heart-derived stem cells in miniature swine with coronary microembolization: Novel ischemic cardiomyopathy model to assess the efficacy of cell-based therapy. Stem Cells Int 2016:6940195. https://doi.org/10.1155/2016/6940195
Tada H, Egashira K, Yamamoto M, Usui M, Arai Y, Katsuda Y, Shimokawa H, Takeshita A (2001) Role of nitric oxide in regulation of coronary blood flow in response to increased metabolic demand in dogs with pacing-induced heart failure. Jpn Circ J 65:827–833. https://doi.org/10.1253/jcj.65.827
Taddei S, Virdis A, Ghiadoni L, Mattei P, Sudano I, Bernini G, Pinto S, Salvetti A (1996) Menopause is associated with endothelial dysfunction in women. Hypertension 28:576–582. https://doi.org/10.1161/01.hyp.28.4.576
Takayama T, Wada A, Tsutamoto T, Ohnishi M, Fujii M, Isono T, Horie M (2004) Contribution of vascular NAD(P)H oxidase to endothelial dysfunction in heart failure and the therapeutic effects of HMG-CoA reductase inhibitor. Circ J 68:1067–1075. https://doi.org/10.1253/circj.68.1067
Tamagawa K, Saito T, Oikawa Y, Maehara K, Yaoita H, Maruyama Y (2005) Alterations of alpha-adrenergic modulations of coronary microvascular tone in dogs with heart failure. J Card Fail 11:388–395. https://doi.org/10.1016/j.cardfail.2005.01.003
Tanaka M, Fujiwara H, Onodera T, Wu D-J, Matsuda M, Hamashima Y, Kawai C (1987) Quantitative analysis of narrowings of intramyocardial small arteries in normal hearts, hypertensive hearts, and hearts with hypertrophic cardiomyopathy. Circulation 75:1130–1139. https://doi.org/10.1161/01.cir.75.6.1130
Tanimura K, Otake H, Kawamori H, Toba T, Nagasawa A, Nakano N, Takahashi Y, Fukuyama Y, Kozuki A, Shite J, Iwasaki M, Kuroda K, Takaya T, Hirata K (2021) Morphological plaque characteristics and clinical outcomes in patients with acute coronary syndrome and a cancer history. J Am Heart Assoc 10:e020243. https://doi.org/10.1161/JAHA.120.020243
Taqueti VR, Solomon SD, Shah AM, Desai AS, Groarke JD, Osborne MT, Hainer J, Bibbo CF, Dorbala S, Blankstein R, Di Carli MF (2018) Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur Heart J 39:840–849. https://doi.org/10.1093/eurheartj/ehx721
Thaulow E, Guth BD, Heusch G, Gilpin E, Schulz R, Kröger K, Ross J Jr (1989) Characteristics of regional myocardial stunning after exercise in dogs with chronic coronary stenosis. Am J Physiol Heart Circ Physiol 257:H113–H119. https://doi.org/10.1152/ajpheart.1989.257.1.H113
Thomas M, Sperry BW, Peri-Okonny P, Malik AO, McGhie AI, Saeed IM, Chan PS, Spertus JA, Thompson RC, Bateman TM, Patel KK (2021) Relative prognostic significance of positron emission tomography myocardial perfusion imaging markers in cardiomyopathy. Circ Cardiovasc Imaging 14:e012426. https://doi.org/10.1161/CIRCIMAGING.121.012426
Thomas S, Fallavollita J, Borgers M, Canty J (2002) Dissociation of regional adaptations to ischemia and global myolysis in an accelerated swine model of chronic hibernating myocardium. Circ Res 91:970–977. https://doi.org/10.1161/01.RES.0000040396.79379.77
Tona F, Montisci R, Iop L, Civieri G (2021) Role of coronary microvascular dysfunction in heart failure with preserved ejection fraction. Rev Cardiovasc Med 22:97–104. https://doi.org/10.31083/j.rcm.2021.01.277
Topol EJ, Weiss JL, Brinker JA, Brin KP, Gottlieb SO, Becker LC, Bulkley BH, Chandra N, Flaherty JT, Gerstenblith G et al (1985) Regional wall motion improvement after coronary thrombolysis with recombinant tissue plasminogen activator: importance of coronary angioplasty. J Am Coll Cardiol 6:426–433. https://doi.org/10.1016/s0735-1097(85)80182-0
Totzeck M, Schuler M, Stuschke M, Heusch G, Rassaf T (2019) Cardio-oncology—strategies for management of cancer-therapy related cardiovascular disease. Int J Cardiol 280:163–175. https://doi.org/10.1016/j.ijcard.2019.01.038
Traverse JH, Chen Y-J, Crampton M, Voss S, Bache RJ (2001) Increased extravascular forces limit endothelium-dependent and -independent coronary vasodilation in congestive heart failure. Cardiovasc Res 52:454–461. https://doi.org/10.1016/s0008-6363(01)00392-3
Traverse JH, Chen Y, Hou M, Bache RJ (2002) Inhibition of NO production increases myocardial blood flow and oxygen consumption in congestive heart failure. Am J Physiol Heart Circ Physiol 282:H2278–H2283. https://doi.org/10.1152/ajpheart.00504.2001
Traverse JH, Chen Y, Hou M, Li Y, Bache RJ (2007) Effect of K+ATP channel and adenosine receptor blockade during rest and exercise in congestive heart failure. Circ Res 100:1643–1649. https://doi.org/10.1161/CIRCRESAHA.107.150219
Traverse JH, Melchert P, Pierpont GL, Jones B, Crampton M, Bache RJ (1999) Regulation of myocardial blood flow by oxygen consumption is maintained in the failing heart during exercise. Circ Res 84:401–408. https://doi.org/10.1161/01.res.84.4.401
Treasure CB, Vita JA, Cox DA, Fish RD, Gordon JB, Mudge GH, Colucci WS, Sutton MG, Selwyn AP, Alexander RW et al (1990) Endothelium-dependent dilation of the coronary microvasculature is impaired in dilated cardiomyopathy. Circulation 81:772–779. https://doi.org/10.1161/01.cir.81.3.772
Triana JF, Bolli R (1991) Decreased flow reserve in “stunned” myocardium after a 10-min coronary occlusion. Am J Physiol Heart Circ Physiol 261:H793–H804. https://doi.org/10.1152/ajpheart.1991.261.3.H793
Trochu J-N, Mital S, Zhang X, Xu X, Ochoa M, Liao JK, Recchia FA, Hintze T (2003) Preservation of NO production by statins in the treatment of heart failure. Cardiovasc Res 60:250–258. https://doi.org/10.1016/j.cardiores.2003.08.003
Tschope C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB, Hare JM, Heidecker B, Heymans S, Hubner N, Kelle S, Klingel K, Maatz H, Parwani AS, Spillmann F, Starling RC, Tsutsui H, Seferovic P, Van Linthout S (2021) Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol 18:169–193. https://doi.org/10.1038/s41569-020-00435-x
Ubachs JF, Engblom H, Koul S, Kanski M, Andersson P, van der Pals J, Carlsson M, Erlinge D, Arheden H (2013) Myocardium at risk can be determined by ex vivo T2-weighted magnetic resonance imaging even in the presence of gadolinium: comparison to myocardial perfusion single photon emission computed tomography. Eur Heart J Cardiovasc Imaging 14:261–268. https://doi.org/10.1093/ehjci/jes142
Ueno M, Kawashima S, Ikeoka K, Iwasaki T (1997) The delayed recovery of impaired endothelium dependent vasodilatory response after hemodynamic improvement in dogs with congestive heart failure. Jpn Circ J 61:936–942. https://doi.org/10.1253/jcj.61.936
Uren NG, Crake T, Lefroy DC, deSilva R, Davies GJ, Maseri A (1993) Delayed recovery of coronary resistive vessel function after coronary angioplasty. J Am Coll Cardiol 21:612–621. https://doi.org/10.1016/0735-1097(93)90092-f
van den Heuvel AF, Bax JJ, Blanksma PK, Vaalburg W, Crijns HJ, van Veldhuisen DJ (2002) Abnormalities in myocardial contractility, metabolism and perfusion reserve in non-stenotic coronary segments in heart failure patients. Cardiovasc Res 55:97–103. https://doi.org/10.1016/s0008-6363(02)00331-0
van den Heuvel AF, Blanksma PK, Siebelink HM, van Wijk LM, Boomsma F, Vaalburg W, Crijns HJ, van Veldhuisen DJ (2001) Impairment of myocardial blood flow reserve in patients with asymptomatic left ventricular dysfunction: effects of ACE-inhibition with perindopril. Int J Cardiovasc Imaging 17:353–359. https://doi.org/10.1023/a:1011971800052
van Kranenburg M, Magro M, Thiele H, de Waha S, Eitel I, Cochet A, Cottin Y, Atar D, Buser P, Wu E, Lee D, Bodi V, Klug G, Metzler B, Delewi R, Bernhardt P, Rottbauer W, Boersma E, Zijlstra F, van Geuns RJ (2014) Prognostic value of microvascular obstruction and infarct size, as measured by CMR in STEMI patients. JACC Cardiovasc Imaging 7:930–939. https://doi.org/10.1016/j.jcmg.2014.05.010
van Veldhuisen DJ, van den Heuvel AF, Blanksma PK, Crijns HJ (1998) Ischemia and left ventricular dysfunction: a reciprocal relation? J Cardiovasc Pharmacol 32(Suppl 1):S46-51. https://doi.org/10.1097/00005344-199800003-00008
van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM, Bronzwaer JG, Henkens IR, Gan CT, Boonstra A, Postmus PE, Vonk-Noordegraaf A (2008) Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J 29:120–127. https://doi.org/10.1093/eurheartj/ehm567
Vanoverschelde J-LJ, Wijns W, Depré C, Essamri B, Heyndrickx GR, Borgers M, Bol A, Melin JA (1993) Mechanisms of chronic regional postischemic dysfunction in humans. New insights from the study of noninfarcted collateral- dependent myocardium. Circulation 87:1513–1523. https://doi.org/10.1161/01.cir.87.5.1513
Velazquez EJ, Lee KL, Jones RH, Al-Khalidi HR, Hill JA, Panza JA, Michler RE, Bonow RO, Doenst T, Petrie MC, Oh JK, She L, Moore VL, Desvigne-Nickens P, Sopko G, Rouleau JL, Investigators S (2016) Coronary-artery bypass surgery in patients with ischemic cardiomyopathy. N Engl J Med 374:1511–1520. https://doi.org/10.1056/NEJMoa1602001
Vitale C, Rosano GM, Kaski JC (2016) Role of coronary microvascular dysfunction in takotsubo cardiomyopathy. Circ J 80:299–305. https://doi.org/10.1253/circj.CJ-15-1364
Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB (2006) Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 114:1883–1891. https://doi.org/10.1161/CIRCULATIONAHA.106.632208
Vogel-Claussen J, Skrok J, Shehata ML, Singh S, Sibley CT, Boyce DM, Lechtzin N, Girgis RE, Mathai SC, Goldstein TA, Zheng J, Lima JA, Bluemke DA, Hassoun PM (2011) Right and left ventricular myocardial perfusion reserves correlate with right ventricular function and pulmonary hemodynamics in patients with pulmonary arterial hypertension. Radiology 258:119–127. https://doi.org/10.1148/radiol.10100725
Vogt M, Strauer BE (1995) Systolic ventricular dysfunction and heart failure due to coronary microangiopathy in hypertensive heart disease. Am J Cardiol 76:48D-53D. https://doi.org/10.1016/S0002-9149(99)80492-5
Walsh R, Offerhaus JA, Tadros R, Bezzina CR (2021) Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. https://doi.org/10.1038/s41569-021-00608-2
Wang J, Seyedi N, Xu XB, Wolin MS, Hintze TH (1994) Defective endothelium-mediated control of coronary circulation in conscious dogs after heart failure. Am J Physiol Heart Circ Physiol 266:H670–H680. https://doi.org/10.1152/ajpheart.1994.266.2.H670
White CI, Jansen MA, McGregor K, Mylonas KJ, Richardson RV, Thomson A, Moran CM, Seckl JR, Walker BR, Chapman KE, Gray GA (2016) Cardiomyocyte and vascular smooth muscle-independent 11beta-hydroxysteroid dehydrogenase 1 amplifies infarct expansion, hypertrophy, and the development of heart failure after myocardial infarction in male mice. Endocrinology 157:346–357. https://doi.org/10.1210/en.2015-1630
Wittstein IS, Thiemann DR, Lima JA, Baughman KL, Schulman SP, Gerstenblith G, Wu KC, Rade JJ, Bivalacqua TJ, Champion HC (2005) Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 352:539–548. https://doi.org/10.1056/NEJMoa043046
Wu KC, Zerhouni EA, Judd RM, Lugo-Olivieri CH, Barouch LA, Schulman SP, Blumenthal RS, Lima JA (1998) Prognostic significance of microvascular obstruction by magnetic resonance imaging in patients with acute myocardial infarction. Circulation 97:765–772. https://doi.org/10.1161/01.cir.97.8.765
Yamamoto M, Egashira K, Arimura K-I, Tada H, Shimokawa H, Takeshita A (2000) Coronary vascular K+ATP channels contribute to the maintenance of myocardial perfusion in dogs with pacing-induced heart failure. Jpn Circ J 64:701–707. https://doi.org/10.1253/jcj.64.701
Yoshida T, Hibino T, Kako N, Murai S, Oguri M, Kato K, Yajima K, Ohte N, Yokoi K, Kimura G (2007) A pathophysiologic study of tako-tsubo cardiomyopathy with F-18 fluorodeoxyglucose positron emission tomography. Eur Heart J 28:2598–2604. https://doi.org/10.1093/eurheartj/ehm401
Zamorano J (2016) An ESC position paper on cardio-oncology. Eur Heart J 37:2739–2740. https://doi.org/10.1093/eurheartj/ehw359
Zelis JM, Tonino PAL, Pijls NHJ, De Bruyne B, Kirkeeide RL, Gould KL, Johnson NP (2020) Coronary microcirculation in aortic stenosis: pathophysiology, invasive assessment, and future directions. J Interv Cardiol 2020:4603169. https://doi.org/10.1155/2020/4603169
Zhang C, Rogers P, Merkus D, Muller-Delp J, Tiefenbacher C, Potter B, Knudson J, Rocic P, Chilian W (2011) Regulation of coronary microvascular resistance in health and disease. Compr Physiol 12:521–549. https://doi.org/10.1002/cphy.cp020412
Zhang J, Toher C, Erhard M, Zhang Y, Ugurbil K, Bache RJ, Lange T, Homans DC (1997) Relationships between myocardial bioenergetic and left ventricular function in hearts with volume-overload hypertrophy. Circulation 96:334–343. https://doi.org/10.1161/01.cir.96.1.334
Zhang P, Hou M, Li Y, Xu X, Barsoum M, Chen Y, Bache RJ (2009) NADPH oxidase contributes to coronary endothelial dysfunction in the failing heart. Am J Physiol Heart Circ Physiol 296:H840–H846. https://doi.org/10.1152/ajpheart.00519.2008
Zhang X, Schindler TH, Prior JO, Sayre J, Dahlbom M, Huang SC, Schelbert HR (2013) Blood flow, flow reserve, and glucose utilization in viable and nonviable myocardium in patients with ischemic cardiomyopathy. Eur J Nucl Med Mol Imaging 40:532–541. https://doi.org/10.1007/s00259-012-2311-9
Zhao G, Joca HC, Nelson MT, Lederer WJ (2020) ATP- and voltage-dependent electro-metabolic signaling regulates blood flow in heart. Proc Natl Acad Sci U S A 117:7461–7470. https://doi.org/10.1073/pnas.1922095117
Zhao G, Shen W, Zhang X, Smith CJ, Hintze TH (1996) Loss of nitric oxide production in the coronary circulation after the development of dilated cardiomyopathy: a specific defect in the neural regulation of coronary blood flow. Clin Exp Pharmacol Physiol 23:715–721. https://doi.org/10.1111/j.1440-1681.1996.tb01764.x
Zhao X, Balaji P, Pachon R, Beniamen DM, Vatner DE, Graham RM, Vatner SF (2015) Overexpression of cardiomyocyte alpha1a-adrenergic receptors attenuates postinfarct remodeling by inducing angiogenesis through heterocellular signaling. Arterioscler Thromb Vasc Biol 35:2451–2459. https://doi.org/10.1161/ATVBAHA.115.305919
Zhou W, Bajaj N, Gupta A, Sun YP, Divakaran S, Bibbo C, Hainer J, Taqueti V, Dorbala S, Blankstein R, Shah P, Kaneko T, Adler D, O’Gara P, Di Carli M (2021) Coronary microvascular dysfunction, left ventricular remodeling, and clinical outcomes in aortic stenosis. J Nucl Cardiol 28:579–588. https://doi.org/10.1007/s12350-019-01706-y
Zhou W, Brown JM, Bajaj NS, Chandra A, Divakaran S, Weber B, Bibbo CF, Hainer J, Taqueti VR, Dorbala S, Blankstein R, Adler D, O’Gara P, Di Carli MF (2020) Hypertensive coronary microvascular dysfunction: a subclinical marker of end organ damage and heart failure. Eur Heart J 41:2366–2375. https://doi.org/10.1093/eurheartj/ehaa191
Zhou W, Sun YP, Divakaran S, Bajaj NS, Gupta A, Chandra A, Morgan V, Barrett L, Martell L, Bibbo CF, Hainer J, Lewis EF, Taqueti VR, Dorbala S, Blankstein R, Slomka P, Shah PB, Kaneko T, Adler DS, O’Gara P, Di Carli MF (2021) Association of myocardial blood flow reserve with adverse left ventricular remodeling in patients with aortic stenosis: the microvascular disease in aortic stenosis (MIDAS) Study. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2021.3396
Acknowledgements
GH was supported by the German Research Foundation (SFB 1116 B8) and the European Union COST ACTION (CA 16225).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Frantz, Würzburg, Germany, served as guest editor for the manuscript and was responsible for all editorial decisions, including the selection of reviewers. The policy applies to all manuscripts with authors from the editor’s institution.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heusch, G. Coronary blood flow in heart failure: cause, consequence and bystander. Basic Res Cardiol 117, 1 (2022). https://doi.org/10.1007/s00395-022-00909-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-022-00909-8