
Abstract
Rationale
This review attempts to summarize the current status in relation to the use of positron emission tomography (PET) imaging in the assessment of synaptic concentrations of endogenous mediators in the living brain.
Objectives
Although PET radioligands are now available for more than 40 CNS targets, at the initiation of the Innovative Medicines Initiative (IMI) “Novel Methods leading to New Medications in Depression and Schizophrenia” (NEWMEDS) in 2009, PET radioligands sensitive to an endogenous neurotransmitter were only validated for dopamine. NEWMEDS work-package 5, “Cross-species and neurochemical imaging (PET) methods for drug discovery”, commenced with a focus on developing methods enabling assessment of changes in extracellular concentrations of serotonin and noradrenaline in the brain.
Results
Sharing the workload across institutions, we utilized in vitro techniques with cells and tissues, in vivo receptor binding and microdialysis techniques in rodents, and in vivo PET imaging in non-human primates and humans. Here, we discuss these efforts and review other recently published reports on the use of radioligands to assess changes in endogenous levels of dopamine, serotonin, noradrenaline, γ-aminobutyric acid, glutamate, acetylcholine, and opioid peptides. The emphasis is on assessment of the availability of appropriate translational tools (PET radioligands, pharmacological challenge agents) and on studies in non-human primates and human subjects, as well as current challenges and future directions.
Conclusions
PET imaging directed at investigating changes in endogenous neurochemicals, including the work done in NEWMEDS, have highlighted an opportunity to further extend the capability and application of this technology in drug development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brain imaging with positron emission tomography (PET) is now widely used in both academic and industry-driven research for a range of applications in neuroscience research and drug discovery. PET radioligands are available for many G-protein-coupled receptors (GPCRs) and neurotransmitter transporters, as well as some enzymes and ion channels, and have been used to provide information about the concentration, distribution, and occupancy of specific drug targets in the central nervous system (CNS) (for reviews see Grimwood and Hartig 2009; Halldin et al. 2001; Jones et al. 2012; Lee and Farde 2006; Zimmer and Luxen 2012). Molecular imaging with PET provides high sensitivity when compared to other methods such as single photon emission computed tomography (SPECT), and with the recent development of a new generation of PET systems, also fairly high spatial resolution. The current model of the high-resolution research tomograph (ECAT HRRT; Siemens CTI, Knoxville, TN, USA) allows for spatial image resolution of 1.5 mm when employing point spread function reconstruction and allows for reliable imaging of small brain structures (Varrone et al. 2009). Moreover, the development of PET systems dedicated to the imaging of rodents has provided opportunities for both forward- and back-translation across species and increased confidence in the validity of animal models (Nagy et al. 2013).
PET imaging can be employed in the drug development process in several different ways. First, radiolabeling and administration of a microgram dose of the radiolabeled drug can confirm brain exposure using PET (Bergstrom et al. 2003). Second, target engagement can be confirmed in a receptor occupancy study in which radioligand and drug interact at the same target. Target engagement has now been demonstrated for many different drug mechanisms and has provided significant progress towards establishing a better understanding of relationships between drug exposure levels and drug target occupancy, informing dose selection for studies aiming to demonstrate therapeutic efficacy in patients (for review see Grimwood and Hartig 2009). One potential confounding factor of target occupancy determinations is that endogenous ligands may compete directly with radioligand binding. However, this potential interaction raises the opportunity for a third PET application, to investigate drug-induced changes in neurotransmitter release, providing insight on receptor function beyond occupancy as well as modes of drug action.
Although PET radioligands are now available for more than 40 CNS targets, at the initiation of the Innovative Medicines Initiative (IMI) “Novel Methods leading to New Medications in Depression and Schizophrenia” (NEWMEDS) in 2009, PET radioligands sensitive to an endogenous neurotransmitter were limited to dopamine. Spurred by the success of developing novel radioligands optimized towards assessment of neurotransmitter release, such as the dopamine D2 receptor agonists [11C]NPA (Narendran et al. 2004) and [11C]MNPA (Seneca et al. 2006), NEWMEDS work-package 5, “Cross-species and neurochemical imaging (PET) methods for drug discovery”, commenced with a focus on developing methods enabling assessment of changes in extracellular concentrations of serotonin and noradrenaline in the brain. Here, we discuss these efforts and also review other recently published reports on the use of radioligands to assess changes in endogenous levels of dopamine, serotonin, noradrenaline, γ-aminobutyric acid (GABA), glutamate, acetylcholine (ACh), and opioid peptides, also including detailed summary tables of the PET studies referred to (Tables 1, 2, 3, and 4). Focus has been placed on assessing the availability of appropriate tools (PET radioligand, pharmacological challenge agent) and studies in non-human primates and human subjects, as well as current challenges and future directions.
Current state of the art: imaging neurotransmitter changes using PET
Dopamine
The dopamine system has historically been one of the most extensively studied neurotransmitter systems of the brain. The results of these investigations have been fruitful in terms of basic science and therapeutic applications. Studies on the dopamine system have over time seen considerable advancements of new technologies and methodological developments. Dopamine was also the first neurotransmitter for which changes in extracellular concentrations could be evaluated in the living human brain using PET and SPECT (Farde et al. 1992; Laruelle et al. 1995; Volkow et al. 1994).
The effects of dopamine are mediated through five receptor subtypes, divided into two families, the D1-like receptors (D1 and D5) and the D2-like receptors (D2, D3, and D4) (for review see Vallone et al. 2000). Several dopamine D2-like receptor radioligands have now been tested for sensitivity to endogenous dopamine in experiments conducted using 3H-labeled radioligands for in vitro or in vivo binding measurements in rodents, and 11C- or 18F-labeled radioligands for in vivo PET measurements in animals or humans. In these studies, extracellular dopamine concentrations in the brain have typically been elevated using amphetamine or methylphenidate, or reduced using reserpine and/or alpha-methyl-para-tyrosine (AMPT), respectively.
Initially, the butyrophenone derivatives [3H]spiperone (Leysen et al. 1978) and [3H]- and [11C]N-methyl-spiperone ([3H]/[11C]NMSP) (Lyon et al. 1986; Wagner et al. 1983) were found not to display changes in binding consistent with competition by endogenous dopamine, possibly because these radioligands also bind to intracellular or internalized receptors (Chugani et al. 1988; for review see Laruelle 2000). The substituted benzamide derivatives [11C]raclopride (Ehrin et al. 1985) and [123I]IBZM (Kung et al. 1988) have, however, in a large number of animal and human studies, consistently been shown to display changes in binding which are consistent with dopamine competition (for review see Laruelle 2000). In general, clinically applicable doses of amphetamine have reduced [11C]raclopride binding in the human striatum by up to 20 %, which is a sufficient effect size to be robustly measured using PET and SPECT (Breier et al. 1997; Dewey et al. 1993; for review see Laruelle 2000).
Two early observations greatly stimulated the use of the developed methodology for further research applications. First, an enhanced amphetamine effect on [11C]raclopride binding was observed in patients with schizophrenia (Breier et al. 1997; Laruelle et al. 1996). Second, videogame playing, employed as a rewarding stimulus, was shown to cause decreases in [11C]raclopride binding (Egerton et al. 2009; Koepp et al. 1998). Several studies have now shown that dopamine release induced by pharmacological and cognitive/behavioral interventions can be investigated using dopamine D2-like receptor radioligands (for reviews see Egerton et al. 2009; Laruelle 2000), and the methodology has provided increased understanding of the role of synaptic dopamine in drug actions (Brody et al. 2004), normal neuropsychology (for review see Egerton et al. 2009), and the pathophysiology of addiction (Volkow et al. 1997), Parkinson’s disease (Piccini et al. 2003), and schizophrenia (Laruelle et al. 1996).
More recently, antagonist radioligands with much higher dopamine D2 receptor affinity compared to raclopride, such as [11C]FLB 457 and [11C]/[18F]fallypride, have been used to investigate extrastriatal brain regions, where the density of dopamine D2 receptors is much lower than in the striatum (Halldin et al. 1995; Mukherjee et al. 2004; Mukherjee et al. 1995). PET studies evaluating the dopamine sensitivity of these radioligands are summarized in Table 1. Explorative studies in monkeys initially indicated that relatively high intravenous doses of amphetamine reduced cortical radioligand binding to a lesser extent than striatal [11C]raclopride binding (Chou et al. 2000; Okauchi et al. 2001) (Table 1). However, follow-up investigations employing amphetamine challenge have confirmed that the high-affinity (<1 nM) antagonist radioligands may allow for monitoring of changes in cortical synaptic dopamine concentrations in monkeys and human subjects (Aalto et al. 2009; Cropley et al. 2008; Mukherjee et al. 2005; Narendran et al. 2009; Narendran et al. 2013; Narendran et al. 2014; Riccardi et al. 2006; Sandiego et al. 2015; Slifstein et al. 2010; Slifstein et al. 2004). Moreover, modulators of endogenous dopamine, including methylphenidate, ketamine, and MK801, have been shown to reduce radioligand binding (Aalto et al. 2005; Montgomery et al. 2007; Tsukada et al. 2005b). However, AMPT (Cropley et al. 2008; Frankle et al. 2010; Riccardi et al. 2008) and ketamine (Vernaleken et al. 2013) administration in humans and nicotine administration in monkeys (Tsukada et al. 2005a) have not been found to affect extra-striatal radioligand binding (Table 1). This development of methodology has provided new opportunities for assessment of extra-striatal dopamine pathways and may facilitate the understanding of the treatment and pathophysiology of psychiatric diseases, including schizophrenia (Slifstein et al. 2015; Woodward et al. 2011) and smoking (Wing et al. 2014).
The published reports on dopamine also include important validation aspects with regard to PET measurement of neurotransmitter release in general. First, release-dependent changes in dopamine D2-like receptor radioligand binding were only observed when endogenous dopamine was available for release. Amphetamine-induced changes in radioligand binding were attenuated by pre-administration of inhibitors of dopamine transport (GBR12909), synthesis (AMPT), or storage (reserpine) (Innis et al. 1992; Laruelle et al. 1997; Villemagne et al. 1999). Second, the reductions in radioligand binding were shown to be proportional to drug-induced increases in dopamine concentrations measured in the extracellular fluid with microdialysis (Breier et al. 1997; Endres et al. 1997; Laruelle et al. 1997; Narendran et al. 2014; Tsukada et al. 1999a). Third, changes in radioligand binding were found to mainly reflect modulation of dopamine in the synaptic cleft, instead of inherently reflecting changes in the extracellular fluid dopamine concentration across drugs with different modes of action. Relative reductions in [11C]raclopride binding in the striatum of monkeys have been shown to be similar after administration of direct dopamine enhancers (GBR12909 and methamphetamine) or indirect dopamine modulators (benztropine and ketanserin), while microdialysis-measured increases in dopamine concentrations were much higher for the direct dopamine enhancers (500–1200 %) in comparison to the indirect dopamine modulators (140–160 %) (Tsukada et al. 1999a). Schiffer and colleagues demonstrated similar results when comparing methylphenidate and amphetamine in a combined microdialysis and PET study in rodents (Schiffer et al. 2006). Differences in endogenous ligand assessment might be expected between microdialysis and PET, since microdialysis provides a direct method of measurement, giving absolute values, and PET is an indirect method for displaying changes. In addition, changes in neurotransmitter levels measured with microdialysis might appear to be relatively larger than when measured using PET since some protocols include uptake blockers in the microdialysis probes. Nevertheless, together these studies provide support for the notion that this approach might be applied to other neurotransmitters.
In an attempt to increase the sensitivity of radioligand binding to endogenous dopamine, relative to that of antagonist radioligands such as [11C]raclopride, agonist radioligands were developed for dopamine D2-like receptors, such as [11C]NPA (Hwang et al. 2000), [11C]MNPA (Finnema et al. 2005), [11C]PHNO (Wilson et al. 2005), and [18F]MCL-524 (Finnema et al. 2014e; for review see Finnema et al. 2010a). Agonist radioligands preferentially bind to the high-affinity, G-protein-coupled state of receptors (Sibley et al. 1982). Agonist radioligand binding is thereby proposed to be more susceptible to the endogenous agonist than the binding of antagonist radioligands, which do not discriminate between the low- and high-affinity states of the receptor. Indeed, these agonist radioligands were shown to be more sensitive to drug-induced changes in extracellular dopamine concentrations than [11C]raclopride, both in anesthetized animals (Gallezot et al. 2014b; Ginovart et al. 2006; Narendran et al. 2004; Seneca et al. 2006; Skinbjerg et al. 2010) and in awake human subjects (Caravaggio et al. 2014; Narendran et al. 2010; Shotbolt et al. 2012). However, there is a growing body of evidence which challenges the existence of two distinct affinity states in vivo (for reviews see Finnema et al. 2010a; Skinbjerg et al. 2012), and the exact mechanism of the increased sensitivity of agonist radioligands to dopamine therefore warrants further evaluation.
In contrast to the dopamine D2-like receptor radioligands, dopamine D1-like receptor antagonist radioligands such as [11C]SCH-23390 (DeJesus et al. 1987; Halldin et al. 1986) and [11C]NNC-112 (Halldin et al. 1998) have not appeared to be sensitive to acute changes in extracellular dopamine concentrations (Abi-Dargham et al. 1999; Chou et al. 1999; Tsukada et al. 2001). A number of hypotheses have been proposed to explain this difference, including that (1) dopamine D1-like receptors are predominantly located extra-synaptically (Hersch et al. 1995; Levey et al. 1993), (2) dopamine has significantly lower affinity to D1 receptors than D2 receptors (Marcellino et al. 2012), and (3) dopamine D1 receptors exist predominantly in the low-affinity state (Richfield et al. 1989). Recently developed partial dopamine D1 receptor agonist radioligands, such as (R)-(+)-[11C]SKF 82957 and (S)-[11C]N-methyl-NNC 01-0259, were also found not to be sensitive to amphetamine-induced changes in dopamine release (Finnema et al. 2013a; Palner et al. 2010).
The described studies on PET-measured dopamine release illustrate the potential of the methodology but also provide caution for the interpretation of changes in radioligand binding. The PET outcome measuring binding potential (BP) represents the ratio of B max/K D, in which B max is the receptor density and K D the equilibrium dissociation rate constant. Changes in BP are typically interpreted according to a competition model, e.g., reflecting a change in K D, but several observations suggest a contribution of dopamine receptor internalization as well (see for reviews Ginovart 2005; Laruelle 2000). First, a temporal discrepancy has been observed between the amphetamine-induced dopamine pulse (∼2 h) and the prolonged effect on radioligand binding (∼24 h) (Cardenas et al. 2004; Ginovart et al. 2006; Houston et al. 2004; Laruelle et al. 1997; Narendran et al. 2007). Second, a change in cellular environment consequent to receptor internalization affects radioligand binding properties differently for radioligands originating from diverse chemical classes (e.g., butyrophenones vs. benzamides) (Guo et al. 2010; Sun et al. 2003). Third, amphetamine has been shown to decrease [11C]raclopride B max values in vivo in cat and ex vivo in rat (Ginovart et al. 2004; Sun et al. 2003). These observations together suggest a contribution of receptor internalization to the observed changes in radioligand binding, possibly affecting both B max and K D. Skinbjerg et al. (2010) indeed confirmed that the long-lasting effect of amphetamine on radioligand binding was caused by internalization of the D2 receptors. Acute amphetamine administration decreased radioligand binding to the same extent in wild-type and arrestin 3 knockout mice. However, 4 h post-amphetamine administration, radioligand binding was still reduced in wild-type mice but recovered to baseline in arrestin 3 knockout mice, which lack the capacity to internalize D2 receptors (Skinbjerg et al. 2010). This study supports that amphetamine-induced changes in radioligand binding mainly reflect changes in extracellular dopamine concentration initially, but after a prolonged time-interval also reflect internalization.
Serotonin
Fourteen mammalian serotonin (5-HT) receptor subtypes (assigned to seven families, 5-HT1–7) have been characterized, and 13 of these have been identified in the human brain. Assessment of extracellular serotonin with PET imaging has the potential to provide important insights into the pathophysiology and treatment of common CNS diseases such as anxiety and mood disorders, as well as sleep and eating behaviors (for review see Millan et al. 2008). Several radioligands have been developed for PET imaging of the serotonin system, specifically for the 5-HT1A, 5-HT1B, 5-HT2A, 5-HT4, and 5-HT6 receptor subtypes and the neuronal serotonin transporter (SERT) (for review see Paterson et al. 2013). Most of these radioligands have been investigated for their sensitivity to extracellular serotonin levels. Typically, extracellular concentrations of serotonin have been elevated using serotonin releasers, e.g., fenfluramine, or SERT inhibitors (SSRIs), such as fluoxetine and the racemate citalopram, or reduced using tryptophan depletion. Paterson et al. previously reviewed these studies in great detail (Paterson et al. 2010), so only more recent work has been summarized in Table 2.
The 5-HT1A receptor distribution has been extensively characterized and high levels are found in the cortex, hippocampus, and raphe nuclei (Pazos et al. 1987a). The 5-HT1A receptors function as somatodendritic autoreceptors in the raphe nuclei and as postsynaptic receptors in other brain regions (for review see Millan et al. 2008). PET studies performed with the high-affinity (∼2 nM) 5-HT1A receptor antagonist [11C]WAY-100635 (Pike et al. 1995; Pike et al. 1996) have demonstrated that fenfluramine-induced increases in synaptic serotonin concentration decreased radioligand binding in the hippocampus of rats (Hume et al. 2001), although this was not reported by others (Maeda et al. 2001). Tryptophan depletion and infusion did not affect [11C]WAY-100635 binding in human subjects (Rabiner et al. 2002). The 5-HT1A receptor antagonist [18F]MPPF (Shiue et al. 1997), which has lower affinity when compared with [11C]WAY-100635, has been evaluated in a series of studies. Fluoxetine-induced elevation of serotonin concentrations has been demonstrated to decrease [18F]MPPF binding in the raphe nuclei of cats (Aznavour et al. 2006) and humans (Sibon et al. 2008), while citalopram and fenfluramine did not affect [18F]MPPF binding in rats (Moulin-Sallanon et al. 2009) and monkeys (Udo de Haes et al. 2006), respectively. These results suggest limited sensitivity of [18F]MPPF to serotonin, which is consistent with tryptophan depletion and infusion studies also showing no effect on [18F]MPPF binding in humans (Praschak-Rieder et al. 2004; Udo de Haes et al. 2002). Interestingly, it has been demonstrated that sleep may cause an elevation in [18F]MPPF binding when compared to wakefulness, but this study was performed in narcolepsy cataplexy patients and confirmation is required in healthy subjects (Derry et al. 2006). Similar to [18F]MPPF, [18F]FPWAY is a structural analogue of [11C]WAY100635 with moderate affinity (<10 nM) to the 5-HT1A receptor (Lang et al. 1999). Administration of paroxetine in monkeys induced 8–27 % decreases in [18F]FPWAY binding in the raphe nuclei and 10–20 % increases in cortical [18F]FPWAY binding (Giovacchini et al. 2005). Although these results were encouraging, so far no follow-up human study has been reported.
In summary, serotonin-dependent decreases in 5-HT1A receptor antagonist radioligand binding have primarily been observed in the raphe nuclei and have been small or absent in serotonin projection regions. These regional differences may be partly caused by SSRI-induced increases in extracellular serotonin being larger in the raphe nuclei than in serotonin projection regions, as shown with microdialysis (Fuller 1994). In addition, it has been proposed that there may be differences in the regional responses to increased synaptic serotonin. 5-HT1A autoreceptors in the raphe nuclei have been suggested to undergo internalization, while postsynaptic receptors in the serotonin projection regions may not internalize (Aznavour et al. 2006; Riad et al. 2004). The raphe nuclei are, however, small structures of the brain stem, and reliable determination of 5-HT1A receptor binding has proven challenging because of limitations in spatial resolution. This consideration may have limited the application of the use of 5-HT1A receptor radioligands for measurement of acute changes in extracellular serotonin in patient populations.
The 5-HT2A receptor is the main excitatory GPCR for serotonin and is widely distributed in the brain, with highest density in the cerebral cortex (Pazos et al. 1987b). Several 5-HT2A receptor antagonist radioligands have been developed for PET, e.g., [18F]altanserin (Lemaire et al. 1991) and [11C]MDL 100907 (Lundkvist et al. 1996). The 5-HT2A receptor antagonist radioligands have in general shown no serotonin sensitivity in PET studies on rats (Hirani et al. 2003), baboons (Staley et al. 2001), or humans (Matusch et al. 2007; Meyer et al. 1999; Pinborg et al. 2004), but Yatham et al. found a paradoxical decrease in cortical [18F]setoperone binding following tryptophan depletion (Yatham et al. 2001). As exceptions, clomipramine and dexfenfluramine have been found to decrease cortical [18F]altanserin binding in human subjects (Larisch et al. 2003; Quednow et al. 2012). These two studies, however, may be inconclusive as clomipramine and the main metabolite of dexfenfluramine, nordexfenfluramine, might also have binding affinity for the 5-HT2A receptor, which may have contributed to the observed reductions in binding (Paterson et al. 2010; Rothman et al. 2000).
Another approach has made use of radioligands targeting the SERT, e.g., [11C]DASB (Houle et al. 2000) and [11C]MADAM (Halldin et al. 2005), which were originally used for the determination of SERT occupancy by inhibitors of serotonin reuptake (tricyclic antidepressants and SSRI drugs). This approach to demonstrate changes in endogenous serotonin has been more challenging as most serotonin challenge studies utilize drugs that directly act on the SERT. However, several attempts have been reported using non-selective MAO inhibitors, 5-hydroxytryptophan (5-HTP), or tryptophan depletion. After elevation of extracellular serotonin levels, decreases in [11C]DASB binding have been consistently shown in the brain of rats (Lundquist et al. 2007), cats (Ginovart et al. 2003), and non-human primates (NHPs) (Lundquist et al. 2007; Yamamoto et al. 2007). Milak and colleagues, however, reported that tryptophan depletion paradoxically decreased binding potential values, possibly related to protein kinase-C-dependent SERT sequestration being less inhibited by serotonin and resulting in increased SERT internalization (Milak et al. 2005). In two reported human studies, tryptophan depletion did, however, not affect [11C]DASB binding (Praschak-Rieder et al. 2005; Talbot et al. 2005).
In conclusion, at the start of NEWMEDS, no established methodology existed for assessment of changes in extracellular serotonin concentrations in the human brain. We therefore initiated the validation of the use of the novel 5-HT1B receptor radioligand [11C]AZ10419369 for evaluation of changes in extracellular serotonin. The characterization of [11C]AZ10419369 binding in NHPs and humans was initiated at the start of NEWMEDS (Pierson et al. 2008; Varnäs et al. 2011), allowing for a quick cross-species evaluation of serotonin sensitivity. The 5-HT1B receptor was considered a promising target as serotonin binds with quite high affinity to this receptor (∼1 nM) (Millan et al. 2002; for review see Paterson et al. 2010), and a relatively large proportion of the receptors may be in the high affinity state in vitro (Granas et al. 2001). In addition, 5-HT1B receptors are expressed at relatively high density in the raphe nuclei and in serotonergic projection areas (Bonaventure et al. 1997), allowing for detailed examination of regional effects.
The serotonin sensitivity of [11C]AZ10419369 binding was initially explored in three monkeys using the potent serotonin releaser (±)-fenfluramine. Fenfluramine reduced the specific binding of [11C]AZ10419369 in a dose-dependent manner with a regional average of 27 % after 1.0 mg/kg and 50 % after 5.0 mg/kg (Finnema et al. 2010b). This study showed for the first time a major decrease in radioligand receptor binding measured with PET in the NHP brain after administration of a serotonin releasing agent. Importantly, these observations have now been confirmed in four other studies using the 5-HT1B receptor radioligands [11C]P943 or [11C]AZ10419369 (Cosgrove et al. 2011; Finnema et al. 2012c; Ridler et al. 2011; Yamanaka et al. 2014) (Table 2). These studies indicate that 5-HT1B receptor radioligands are promising tools for the measurement of serotonin release and provide a good rationale for further development of the methodology.
It is not easily feasible to continue the fenfluramine studies in human subjects since fenfluramine products were withdrawn from the market in 1997 following reports of valvular heart disease and pulmonary hypertension in chronic users (Connolly et al. 1997). The serotonin sensitivity of [11C]AZ10419369 in humans was therefore assessed using the SSRI escitalopram which has been shown devoid of significant affinity for the 5-HT1B receptor (Maier et al. 2009; Sanchez et al. 2003). In pilot PET studies in monkeys, we confirmed that a high dose of escitalopram (2.0 mg/kg, i.v.) decreased [11C]AZ10419369 binding by 11 % in serotonin projection areas and by 25 % in the raphe nuclei (Nord et al. 2013). In healthy human subjects, administration of a single dose of escitalopram (20 mg, p.o.), a daily dose commonly used in the treatment of depression, tended to decrease [11C]AZ10419369 binding in the raphe nuclei but increased radioligand binding by 5 % in serotonergic projection areas (p < 0.05) (Table 2). In a consecutive test-retest study in human subjects, [11C]AZ10419369 binding was found to be very reproducible with an absolute mean difference in BP ND of less than 3 % in serotonergic projection areas between morning and afternoon measurements (Nord et al. 2014). These studies suggested that a single clinically relevant dose of escitalopram may decrease extracellular serotonin concentrations in serotonergic projection areas in the human brain. Since, hypothetically, desensitization of inhibitory serotonin autoreceptors will cause the serotonin concentration in projection areas to increase over time with chronic administration, these findings might aid in understanding the mechanism of the delayed onset of the clinical effects of SSRIs.
Concomitantly, other investigators also examined the effects of SSRI-induced changes in extracellular serotonin using radioligand binding. The binding of [11C]P943 was reduced by 9–30 % in serotonergic projection areas in monkeys after a large acute dose of citalopram (4 mg/kg, i.v.) (Ridler et al. 2011), but to date there have been no follow-up studies reported in human subjects. Kumar and colleagues reported the development of [11C]CUMI-101 which was demonstrated to be an agonist to human 5-HT1A receptors expressed on Chinese hamster ovary cells (Kumar et al. 2007). However, more recently, [11C]CUMI-101 was shown to be an 5-HT1A receptor antagonist in binding assays using native rat, NHP, or human brain tissue (Hendry et al. 2011; Shrestha et al. 2014). Nevertheless, [11C]CUMI-101 was found to be sensitive to serotonin, and citalopram (4 mg/kg, i.v.) reduced its binding by 30 % in serotonergic projection areas in monkeys (Milak et al. 2011). Selvaraj and colleagues demonstrated that citalopram (10 mg, i.v.) increased the cortical binding of this radioligand by 7 % in human subjects (Selvaraj et al. 2012), but this finding was not replicated in another study (Pinborg et al. 2012). Other recent initiatives include work with the 5-HT4 receptor antagonist [11C]SB207145 and the 5-HT2A receptor agonist [11C]Cimbi-36. Binding of [11C]SB207145 was not found to be sensitive to acute changes in extracellular serotonin following pindolol and acute citalopram administration (Marner et al. 2010), but treatment with fluoxetine (40 mg/day) decreased [11C]SB207145 binding by 5 % in a 3-week placebo-controlled study in healthy human subjects (Haahr et al. 2014). [11C]Cimbi-36 is a recently developed agonist radioligand for the 5-HT2A receptor (Ettrup et al. 2014; Ettrup et al. 2011; Finnema et al. 2014d), and preliminary evaluation of the serotonin sensitivity of [11C]Cimbi-36 in monkeys demonstrated that fenfluramine (5 mg/kg) reduced cortical BP ND values by ∼60 % (Finnema et al. 2012b) (Table 2). These results warrant further evaluation of [11C]Cimbi-36 in humans to confirm the potential of agonist radioligands for PET measurement of changes in extracellular neurotransmitter concentrations.
In conclusion, the recently developed radioligands [11C]AZ10419369, [11C]P943, and [11C]CUMI-101 were shown to be highly sensitive to SSRI-induced changes in serotonin concentrations in NHPs (Milak et al. 2011; Nord et al. 2013; Ridler et al. 2011). Studies using [11C]AZ10419369 and [11C]CUMI-101 in human subjects (Nord et al. 2013; Pinborg et al. 2012; Selvaraj et al. 2012) suggest that single doses of SSRIs do not increase extracellular serotonin concentrations in serotonergic projection regions, as assessed with PET. Of interest is that the cross-species differential effect of SSRIs on cortical extracellular serotonin concentrations may be related to the SSRI dose. In a recent preliminary study, we determined SERT occupancy after single doses of citalopram and escitalopram and concluded that the previously applied doses of citalopram and escitalopram nearly saturated SERT in the monkey brain (Finnema et al. 2014a). Similarly, brain microdialysis experiments in rodents have demonstrated that a high dose of citalopram (10 mg/kg, i.p.) significantly increased serotonin concentrations in the cortex but that a lower dose (1 mg/kg, i.p.) did not have the same effect (Invernizzi et al. 1992). Consideration of SERT occupancy is therefore of importance when comparing changes in extracellular serotonin across species.
A second observation was that a single dose of an SSRI was associated with possible reductions in extracellular serotonin in the projection regions of the human brain (Nord et al. 2013; Selvaraj et al. 2012). These results are very similar to a previous monkey study in which paroxetine induced a decrease in [18F]FPWAY binding in the raphe nuclei while binding in the cortex was increased (Giovacchini et al. 2005). These results appear in contrast to the majority of brain microdialysis experiments in rodents. However, a number of microdialysis studies have shown that SSRI-induced increases in extracellular serotonin in the raphe nuclei may lead to activation of 5-HT1A autoreceptors and decreased neuronal firing and serotonin release in the projection areas (Bel and Artigas 1992; Hervas and Artigas 1998; Invernizzi et al. 1992). The effect of an SSRI on extracellular serotonin in the projection regions depends on the balance between local SERT occupancy, promoting increased extracellular serotonin concentrations, and the consequent decrease in neuronal firing and serotonin release leading to decreased extracellular serotonin concentrations. It may thus be hypothesized that overall, a single clinically relevant dose of an SSRI results in a decrease in the extracellular concentration of serotonin in the projection regions. These studies may thus provide further understanding of the mode of action of SSRIs, and the developed radioligands appear useful for applications in the clinical development of drug treatments intended to modulate endogenous serotonin. However, to date, no methodology has been sufficiently validated for assessment of acute changes in extracellular serotonin in the human brain. Other serotonin challenge paradigms than SSRI administration should be considered for combination with these new 5-HT1A and 5-HT1B receptor radioligands.
Noradrenaline
Receptors for noradrenaline (and adrenaline) are divided into three main classes, i.e., α1-, α2-, and β-adrenoceptors (ARs), each of which is comprised of three subtypes. There are three α2-AR subtypes in humans and other mammalians: the α2A-, α2B-, and α2C-ARs. Of these, the α2A-AR subtype has the most widespread distribution both in the CNS and in peripheral tissues, and has wide-ranging physiological functions that include a role as a presynaptic and auto-inhibitory regulator of neurotransmitter release and many postsynaptic effects. The α2B-subtype does not have a significant presence in the CNS. The α2C-subtype is considered to have a predominantly modulatory function, at least to some extent with a presynaptic auto- and heteroreceptor localization (Ihalainen and Tanila 2002; Sallinen et al. 1997). The lack of major physiological functions in peripheral tissues, together with a neuromodulatory heteroreceptor role, makes the α2C-AR subtype an appealing potential CNS drug target (Scheinin et al. 2001).
There is no subtype-selective radioligand for PET imaging of CNS ARs available for clinical use. Most experience has been gained from attempts to develop PET radioligands for α2-ARs, whereas α1-ARs and β-ARs have received less attention. Two 11C-labeled sertindole analogues exhibited high in vitro selectivity for α1-ARs, but PET imaging in cynomolgus monkeys revealed poor brain uptake and a high degree of non-displaceable binding (Airaksinen et al. 2013). [18F](Fluoromethoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)propan-2-ol ([18F]FPTC) showed specific binding to β-ARs in vitro, but binding was non-displaceable in vivo in rats (Mirfeizi et al. 2014).
The recently developed PET radioligand candidates [11C]MBF and [11C]JP-1302 are indeed selective for the α2C-AR subtype, but have poor access to their targets in the CNS due to the effect of efflux transporters, which became evident in a study performed with p-glycoprotein and breast cancer resistance protein (P-gp/Bcrp) knockout mice compared to wild-type mice (Kawamura et al. 2010). The α2-AR radioligands [11C]MK-912 (Shiue et al. 1998), [O-methyl-11C]RS-15385-197(Hume et al. 2000), [11C]R107474 (Van der Mey et al. 2006), [11C]yohimbine (Jakobsen et al. 2006), and [11C]mirtazapine (Marthi et al. 2002) do not differentiate between the α2-AR subtypes. Brain uptake of [O-methyl-11C]RS-15385-197 was minimal in humans during a 90-min PET measurement (Hume et al. 2000).
In a PET study on pigs, based on a decreased volume of distribution of [11C]yohimbine, a high dose (10 mg/kg, i.v.) of amphetamine led to approximately 5–30 % binding reductions in different brain regions known to express α2-ARs (Table 3). These data suggested that [11C]yohimbine binding could be used as a surrogate marker of noradrenaline release in vivo (Landau et al. 2012). This finding has so far not been extended to alternative challenge protocols or to human subjects. The test-retest reliability of [11C]yohimbine has also yet to be confirmed. After i.v. administration of [11C]yohimbine, there were no radioactive metabolites observed in plasma (at least in pigs), which should facilitate quantification of [11C]yohimbine binding. However, because of the wide distribution of the α2A-AR subtype in the CNS, no suitable reference region could be identified in pigs (Jakobsen et al. 2006). Very recently, first in human studies with [11C]yohimbine were reported. For quantitative analysis in humans, a metabolite-corrected arterial input function may be required, although the corpus callosum was explored as a potential reference region (Nahimi et al. 2015). Phan et al. (2015) recently demonstrated that amphetamine also reduced [11C]yohimbine binding in rats (Phan et al. 2015).
Of the evaluated radioligand candidates for α2-AR, [11C]mirtazapine has also been tested in humans (Munk and Smith 2011). However, its non-selectivity and binding to 5-HT2, 5-HT3, and histamine H1 receptors (de Boer 1996) precludes an assessment of its suitability as a radioligand for α2-ARs and for monitoring noradrenaline release. While [11C]mirtazapine binding has been reported to be significantly reduced in non-responders to antidepressant treatments (Smith et al. 2009), the receptor and neurotransmitter implications of this finding are at most speculative because of this lack of selectivity. It can be concluded that there was no methodology for measurement of endogenous synaptic noradrenaline in vivo in the human brain before the initiation of NEWMEDS.
Labeling of the subtype-selective α2C-AR antagonist ORM-13070 (1-[(S)-1-(2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl]-4-(3-methoxymethylpyridin-2-yl)-piperazine) with 11C at high radiochemical purity (>99 %) and specific radioactivity (640 ± 390 GBq/μmol) and the preliminary characterization of the radioligand candidate in rats and α2-AR subtype knockout mice (Arponen et al. 2014) paved the way for a metabolism and dosimetry study in healthy human males (Luoto et al. 2014). This study revealed the presence of two radioactive metabolites of the radioligand in human blood, in agreement with a previous investigation in rats (Arponen et al. 2014), but indicated that reference tissue methods would still be applicable for clinical imaging. The molecular identity of these metabolites has not been determined, but it was concluded that they probably represent small volatile molecules not detectable with the employed HPLC-mass spectroscopy approaches and unlikely to interfere with receptor binding (Arponen et al. 2014; Luoto et al. 2014). Also, from a radiation safety viewpoint, [11C]ORM-13070 appeared suitable for repeated measurements in humans (Luoto et al. 2014).
The test-retest reliability of [11C]ORM-13070 PET was investigated by imaging six healthy male subjects twice (Lehto et al. 2015b). The bound/free ratio of radioligand uptake relative to non-specific uptake into the cerebellum during the time interval of 5–30 min was largest in the dorsal striatum: 0.77 in the putamen and 0.58 in the caudate nucleus. Absolute test-retest variability in the bound/free ratio of the radioligand was 4.3 % in the putamen and also <10 % in the caudate nucleus and thalamus. The pattern of [11C]ORM-13070 binding, as determined by PET, was in agreement with receptor density results previously derived from postmortem autoradiography (Fagerholm et al. 2008). PET data analysis results obtained with a compartmental model fit, the simplified reference tissue model, and a graphical reference tissue analysis method were convergent with the tissue ratio method (Lehto et al. 2015b). The results of this study supported the use of [11C]ORM-13070 PET in the quantitative assessment of α2C-ARs in the human brain in vivo.
The suitability of [11C]ORM-13070 for measurement of amphetamine-evoked changes in extracellular noradrenaline levels was explored ex vivo in rat brain sections and in vivo with PET imaging in monkeys; rat striatal microdialysis experiments confirmed amphetamine-induced elevations in noradrenaline and dopamine concentrations (Finnema et al. 2014b). After injection of [11C]ORM-13070 in rats, mean striatal specific binding ratios, determined using the cerebellum as a reference region, were 1.4 ± 0.3 after saline pre-treatment and 1.2 ± 0.2 after amphetamine administration (0.3 mg/kg, s.c.). Injection of [11C]ORM-13070 in NHPs resulted in mean striatal binding potential (BP ND) estimates of 0.65 ± 0.12 at baseline. Intravenous administration of amphetamine (0.5 and 1.0 mg/kg) reduced BP ND values by 31–50 % in monkeys (Table 3). Amphetamine (0.3 mg/kg, s.c.) increased extracellular noradrenaline (by 400 %) and dopamine (by 270 %) in the rat striatum. Together, these results indicated that [11C]ORM-13070 may be a useful tool for evaluation of synaptic noradrenaline concentrations in vivo (Finnema et al. 2014b).
A limitation of the use of amphetamine in the animal validation studies was the non-selective elevation of noradrenaline and dopamine. Although the relative amphetamine-evoked increase in noradrenaline concentrations was greater than the dopamine increase, dopamine also binds to α2C-ARs, albeit with 5-fold lower affinity than noradrenaline (Ruuskanen et al. 2005). It could thus not be ruled out that the amphetamine-induced decrease in [11C]ORM-13070 binding was partly related to increased dopamine. In an attempt to selectively elevate synaptic noradrenaline concentrations, we performed experiments with the potent noradrenaline transporter (NET) inhibitor atomoxetine. In preliminary PET studies in two monkeys, atomoxetine did not influence the striatal binding of [11C]ORM-13070 consistently (Finnema et al. 2014b). This was not surprising since atomoxetine increased extracellular noradrenaline to a smaller extent than amphetamine, as shown by microdialysis.
A subsequent study explored the use of [11C]ORM-13070 to monitor α2C-AR occupancy and the tracer’s capacity to reflect changes in extracellular concentrations of endogenous noradrenaline in the human brain. The subtype non-selective α2-AR antagonist atipamezole was administered at different doses to eight healthy volunteer subjects. Using the bound/free ratio during 5–30 min after [11C]ORM-13070 injection, the maximal extent of inhibition of striatal [11C]ORM-13070 uptake achieved by atipamezole was 78 % (95 % CI 69–87 %) in the caudate nucleus and 65 % (53–77 %) in the putamen. The EC50 estimates of atipamezole (1.6 and 2.5 ng/ml, respectively) were in agreement with the drug’s α2C-AR affinity. This provided strong support for the use of [11C]ORM-13070 to monitor drug occupancy of α2C-ARs in the living human brain. Three of the four noradrenaline challenges that were explored, each challenge in four subjects, were associated with small, approximately 10–16 % average reductions in [11C]ORM-13070 binding (atomoxetine, ketamine, and the cold pressor test; p < 0.05 for all), but the fourth investigated challenge, insulin-induced hypoglycemia, did not have any evident effect on radioligand uptake (Table 3). This suggested that [11C]ORM-13070 has potential as a tool for in vivo monitoring of synaptic concentrations of noradrenaline in the human brain (Lehto et al. 2015a) and provided a rationale for a larger study aimed to confirm these findings. The results of the follow-up study are featured in this issue (Lehto et al. 2015c). This study showed significant effects on [11C]ORM-13070 binding in the dorsal striatum in response to ketamine infusion (up to 24 % average reduction in B/F), and also in response to a combination of oral atomoxetine and cold stimulation (up to 23 % average reduction in B/F). These results provide evidence in favor of [11C]ORM-13070 binding in the dorsal striatum being sensitive to increased extracellular concentrations of noradrenaline in healthy human subjects.
GABA
GABA is the principal inhibitory neurotransmitter in the mammalian CNS. Heteropentameric GABAA receptors in the CNS mainly contain two α, two β, and a single γ subunit (McKernan and Whiting 1996). Compounds that bind to the GABAA benzodiazepine (BZD) binding site are allosteric ligands; they exhibit no intrinsic activity of their own, but potentiate or inhibit the effects of GABA at receptors that contain α1, α2, α3, or α5 subunits (Dawson et al. 2005). Changes in synaptic GABA levels have been assessed in healthy control subjects with the GABAA receptor BZD site radioligands [11C]flumazenil and [11C]Ro15-4513 (Frankle et al. 2012; Frankle et al. 2009; Stokes et al. 2014). Synaptic GABA levels were reported to be increased following oral administration of tiagabine, which acts by blocking the GABA transporter 1 (GAT1). Maximal effects observed for [11C]flumazenil were a ∼15 % increase in the BP ND for the cortex and medial temporal lobe after administration of 0.21 mg/kg tiagabine (Frankle et al. 2009) (Table 3). Although the observed changes in [11C]flumazenil binding were relatively small, they appeared to be reproducible (Frankle et al. 2012). More recently, Stokes et al. demonstrated that oral tiagabine administration (0.18 mg/kg) resulted in significant reductions in α1-subunit specific [11C]Ro15-4513 binding in the hippocampus, parahippocampus, amygdala, and anterior cingulate (Stokes et al. 2014). The maximal effects were greater than those seen with [11C]flumazenil, with tiagabine causing a 61 % reduction in the α1-subunit specific [11C]Ro15-4513 binding (V S) (Stokes et al. 2014) (Table 3). The proposed underlying mechanism is that increased extracellular GABA levels evoked by tiagabine result in changes in the affinity of BZD receptor ligands produced by GABAA receptor activation, known as the “GABA shift” (Kemp et al. 1987). The observed differences in sensitivity between the two radioligands can therefore be explained by [11C]flumazenil acting as a weak partial agonist in vivo (Higgitt et al. 1986; Miller et al. 1988) and [11C]Ro15-4513 being a partial inverse agonist at the BZD site, respectively. Within NEWMEDS, we have attempted to measure tiagabine-induced changes in 3H- and 11C-labeled flumazenil and Ro15-4513 binding to brains of rodents and anesthetized NHPs, but did not observe significant changes in radioligand binding (Finnema et al. 2012a). Our ongoing efforts in this area are to investigate whether a novel BZD site full agonist, [11C]RO6899880, provides a more sensitive approach for measuring changes in synaptic GABA concentrations (Finnema et al. 2013b, 2014c; Stepanov et al. 2013).
Glutamate
l-Glutamate is an important mediator of excitatory neurotransmission in the CNS. A large and diverse array of receptors, comprising both ligand-gated ion channels (ionotropic) and GPCRs (metabotropic), mediates the cellular effects of glutamate (Kew and Kemp 2005). With growing knowledge and understanding on the physiological roles of the glutamate receptor subtypes, interest has developed in their potential as promising CNS therapeutic targets in a large number of diseases. This has provided a powerful stimulus for the development of new tools, including PET radioligands, to assist translational drug discovery and also enabling further insight into glutamatergic neurotransmission in the human brain.
Ionotropic glutamate receptors are comprised of N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors; these gate monovalent and/or divalent cations and have widespread distributions in the brain (Watkins and Jane 2006). Several NMDA receptor radioligands have been evaluated for their utility as PET imaging agents, but no suitable AMPA or kainate receptor radioligands have so far been developed. The suitability of [11C]ketamine, which binds to the intra-channel phencyclidine (PCP) site of NMDA receptors, as a PET ligand was first established in NHPs (Hartvig et al. 1994). A subsequent study in humans demonstrated that (S)-[N-methyl-11C]ketamine produced a radioactivity distribution in the brain consistent with the known NMDA receptor expression pattern, and that its binding could be displaced by unlabeled enantiomers of ketamine (Hartvig et al. 1995). In patients with medial temporal lobe epilepsy, (S)-[N-methyl-11C]ketamine demonstrated decreased binding in the temporal lobes of ictal onset, in comparison with the contralateral lobes (Kumlien et al. 1999). Since endogenous glutamate concentrations may change in relation to seizure activity (Carlson et al. 1992), these binding differences may be explained by glutamate-dependent changes to the opening state of the NMDA receptor ion channel, thereby regulating radioligand binding to the PCP binding site, but they might also be a consequence of neuronal loss or reduced regional blood flow (Kumlien et al. 1999). There is also some evidence, albeit mixed, from 1H-MRS imaging in rats (Kim et al. 2011) and humans (Rowland et al. 2005; Stone et al. 2012), indicating that ketamine can elevate brain glutamate or related metabolite levels, suggesting that ketamine may also be a useful pharmacological challenge tool for examining glutamate release.
Somewhat more promising progress has been made towards PET imaging of metabotropic glutamate receptor subtypes, particularly with some non-competitive antagonists of the mGluR5 subtype (Sobrio 2013). Application of one of these radioligands, the negative allosteric modulator [11C]ABP688, has provided initial indirect evidence to support sensitivity to changes in glutamate release. [11C]ABP688 binding appears to be sensitive to increased glutamate release, most likely via an allosteric interaction, following N-acetylcysteine administration (Miyake et al. 2011; Sandiego et al. 2013) and sub-anesthetic ketamine challenge (DeLorenzo et al. 2014), although N-acetylcysteine and MK-801 did not affect [11C]ABP688 binding in rats (Wyckhuys et al. 2013; Table 4). [18F]FPEB (Wong et al. 2013; Zhang et al. 2014) and [18F]SP203 (Kimura et al. 2012) have also been demonstrated as mGluR5 PET radioligands, but there have been no studies reported with them investigating endogenous glutamate changes.
Acetylcholine
Muscarinic receptor (mAChR: M1–M5) GPCRs and nicotinic ligand-gated ion channel receptors (nAChRs) mediate diverse effects of ACh in the CNS (for reviews see Gotti et al. 2009; Langmead et al. 2008). There has been a strong drive to identify and develop molecules with high receptor subtype selectivity, and as a result, many receptor-selective cholinergic ligands, including putative PET radioligands, have been developed (for review see Eckelman 2006), some of which have been shown to be sensitive to endogenous ACh (Table 4).
The M2 mAChR subtype selective agonist [18F]FP-TZTP has been shown sensitive to ACh in monkeys (Carson et al. 1998). Intravenous administration of physostigmine reduced radioligand binding across brain regions with significant reductions of 22–29 % in V T of cortical regions (Table 4). In a more recent study, it has been confirmed that physostigmine affects [18F]FP-TZTP binding in human subjects but the effect is influenced by age and APOE-ε4 genotype of the subjects (Cohen et al. 2006).
(+)N-[11C]methyl-3-piperidyl benzilate ([11C](+)3-MPB) is a subtype non-selective mAChR orthosteric antagonist which has been used to demonstrate mAChR occupancy by scopolamine in conscious monkey brain (Yamamoto et al. 2011). [11C](+)3-MPB was not sensitive to increased endogenous ACh levels following administration of an acetylcholinesterase (AChE) inhibitor, but two related compounds, [11C](+)3-EPB and [11C](+)3-PPB, with lower mAChR affinity, were sensitive to elevations of endogenous ACh provoked by administration of donepezil (250 μg/kg), leading to significantly reduced binding (e.g., for [11C](+)3-PPB ∼19 % in striatum and ∼52 % in frontal cortex) in monkey brain (Nishiyama et al. 2001; Tsukada et al. 2004) (Table 4). Another subtype non-selective mAChR radioligand, [11C]NMPYB, was found not to be sensitive to ACh in PET studies on rats (Ma et al. 2004).
Considerable efforts have been devoted to the development of subtype-selective mAChR PET radioligands, primarily to assist drug development, but also to provide more detailed insight into the specific roles of the different receptor subtypes in interactions between cholinergic mechanisms and other endogenous mediators in the CNS. Progress has been slow but some leads have emerged and some potential radioligands are in the early stages of evaluation and validation in animal studies, for example, the M1 selective mAChR agonist radioligand [11C]AF150(S) (Buiter et al. 2013). In rat brain, regional [11C]AF150(S) binding was consistent with the reported distribution of M1 receptors, although BP ND values were relatively low (<0.3; Buiter et al. 2013). Administration of the dopamine D2 receptor antagonist haloperidol (1 mg/kg, s.c.) reduced [11C]AF150(S) binding by 27 and 15 % in the striatum and hippocampus, respectively. Since haloperidol is known to elevate extracellular ACh levels at this dose, these findings suggest that [11C]AF150(S) may be sensitive to changes in extracellular concentrations of ACh, but further investigation is required (Buiter et al. 2013).
Developments in nAChR research have also stimulated the search for subtype-selective ligands, including the development of PET radioligands, particularly for the most abundant nAChR subtypes in mammalian brain, α4β2 and α7, due to their prospective therapeutic potential (for reviews see Jasinska et al. 2014; Mo et al. 2014). Many potential PET radioligands have been identified and investigated for imaging of brain nAChRs (Mo et al. 2014; Wu et al. 2010), but only the non-selective agonist [11C]nicotine and the α4β2 agonists 2-18F-fluoro-3-(2(S)-azetidinylmethoxy)pyridine (2-[18F]FA) and 6-[18F]FA have been available for studies in humans (for review see Horti et al. 2010). [11C]Nicotine has been used extensively to study brain nAChR changes in smokers and in different patient groups, but its utility is limited because of issues of cerebral blood flow dependence and relatively high non-specific binding (Nyback et al. 1994). 2-[18F]FA has also been used to show changes in α4β2 nicotinic receptor availability. For example, it has been demonstrated that cigarette smoking inhibits 2-[18F]FA binding in an exposure-related manner, such that smoking of one cigarette produced almost 90 % receptor occupancy (Brody et al. 2006). However, 2-[18F]FA has slow brain kinetics which is a limitation of a PET ligand, so new radioligands are being explored (Mo et al. 2014).
Investigations of the sensitivity of α4β2 nAChR PET radioligands for measuring ACh changes have not been reported in humans, but there are some indications from AChE inhibitor treatment studies in rats and NHPs which suggest that this might become feasible with improved radioligands (Table 4). Physostigmine infusion was shown to result in significant reductions in distribution volumes of 2-[18F]FA (Valette et al. 2005), [18F]-(−)-flubatine (Gallezot et al. 2014), and [18F]norchloro-fluoro-epibatidine ([18F]NFEP; Ding et al. 2000) in various brain regions of NHPs, as well as reducing [18F]nifene binding in rat brain (Hillmer et al. 2013). Similarly, donepezil and galantamine were shown to decrease [18F]NFEP binding in various regions of monkey brain (Gallezot et al. 2014) and [18F]nifene binding (Hillmer et al. 2013) in regions of rat brain, respectively (Table 4). Taken together, these studies demonstrate that α4β2 nAChR radioligands may be suitable for measuring ACh changes and future application of the methodology in human subjects may be anticipated.
Opioid peptides
The endogenous opioid systems of the brain contain four families of opioid peptides (endorphins, enkephalins, dynorphins, and endomorphins) which are thought to act through four subtypes of G-protein-coupled opioid receptors (μ-, κ-, δ-, and nociceptin-OR). Endorphins and enkephalins are thought to preferentially act on μ- and δ-ORs, dynorphins on κ-ORs, and endomorphins on μ-ORs (Gianoulakis 2009). Evidence for PET radioligand binding being sensitive to changes in endogenous opioid release originated from clinical PET studies. Acute somatic pain (Bencherif et al. 2002; Scott et al. 2007; Zubieta et al. 2001), inflammatory pain (Jones et al. 1994), affective responses (Zubieta et al. 2003), and placebo administration (Scott et al. 2008; Zubieta et al. 2005) were shown to modulate [11C]carfentanil or [11C]diprenorphine binding in a manner consistent with changes in endogenous opioid peptide concentrations. More recently, Colasanti and colleagues reported that a pharmacologically induced elevation in endogenous opioid release reduced [11C]carfentanil binding in several regions of the human brain, including the basal ganglia, frontal cortex, and thalamus (Colasanti et al. 2012). Oral administration of d-amphetamine, 0.5 mg/kg, 3 h before [11C]carfentanil injection, reduced BP ND values by 2–10 %. The results were confirmed in another group of subjects (Mick et al. 2014). However, Guterstam and colleagues observed no change in [11C]carfentanil binding when d-amphetamine, 0.3 mg/kg, was administered intravenously directly before injection of [11C]carfentanil (Guterstam et al. 2013). It has been hypothesized that this discrepancy may be related to delayed increases in extracellular opioid peptide concentrations following amphetamine-evoked monoamine release (Colasanti et al. 2012; Mick et al. 2014).
Summary of results of NEWMEDS work-package 5
The development and validation of PET radioligands requires significant resources and expertise and often coincides with drug development. Radioligand development specifically tailored toward measurement of neurotransmitter release has so far been limited. For NEWMEDS work-package 5 we collaborated across institutions to utilize radioligands that were already available from drug development programs, such as [11C]AZ10419369, [11C]ORM-13070, and [11C]Lu AE92686 (Fig. 1), and tested them for sensitivity to changes in extracellular concentrations of endogenous neurotransmitters. In addition, we initiated development of new radioligands tailored towards the measurement of neurotransmitter release, e.g., [11C]RO6899880 for GABA (Fig. 1). Sharing the workload across institutions, we utilized in vitro techniques in cell and tissue models, in vivo receptor binding and microdialysis techniques in rodents, and in vivo PET imaging in NHPs and humans (Fig. 2). Methods were validated for the measurement of changes in extracellular serotonin levels using the 5-HT1B receptor radioligand [11C]AZ10419369 in NHPs (Finnema et al. 2010b, 2012c; Nord et al. 2013) and humans (Nord et al. 2013, 2014). In addition, this cross-institution approach enabled us to validate measurement of endogenous noradrenaline using the α2C-AR antagonist [11C]ORM-13070 preclinically, using cross-species autoradiography (Arponen et al. 2014; Finnema et al. 2014b), rat microdialysis and PET imaging in NHPs (Finnema et al. 2014b), and clinically using PET (Lehto et al. 2015a, c) (Fig. 2). Work is ongoing to explore the utility of the novel GABAA agonist [11C]RO6899880 (Finnema et al. 2013b, 2014c; Stepanov et al. 2013) and the phosphodiesterase 10A (PDE10A) inhibitor [11C]LuAE92686 (Kehler et al. 2014) for the measurement of changes in concentrations of GABA and cyclic nucleotides, respectively, using cells, rodent in vivo receptor binding, and NHP PET imaging. Perhaps the most valuable aspect of being able to access multiple techniques was that different pharmacological challenge regimens could be explored in rodents, significantly improving the success of exploratory NHP studies, before progressing to humans.

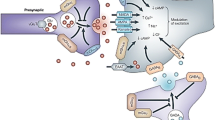
Translational techniques for measuring changes in endogenous neurotransmitter concentrations with radioligands. A series of experiments is illustrated for validation of measurement of changes in extracellular noradrenaline concentrations using the α2C-AR antagonist [11C]ORM-13070. From left to right: autoradiography of a coronal section of a human brain analyzed by incubation with [3H]ORM-13070 (unpublished results, Karolinska Institutet) and of a rat brain obtained after in vivo [11C]ORM-13070 binding (Arponen et al. 2014). Microdialysis studies demonstrated amphetamine-induced noradrenaline release in rat striatum (Finnema et al. 2014b). PET summation images after injection of [11C]ORM-13070 in a cynomolgus monkey or a human subject (Finnema et al. 2014b; Lehto et al. 2015a)

PET radioligands used in NEWMEDS work-package 5. PET summation images of 5-HT1B receptor radioligand [11C]AZ10419369, α2C-AR radioligand [11C]ORM-13070, PDE10A radioligand [11C]Lu AE92686, and GABAA receptor radioligand [11C]RO6899880 in monkey brain
Considerations and current challenges
Development and characterization of radioligands
Since PET imaging provides an indirect approach to assess changes in extracellular neurotransmitter levels, the need to have high-quality PET radioligands and pharmacological challenge tools is paramount. Several review papers have extensively discussed requirements for optimal radioligands for the CNS (Halldin et al. 2001; Pike 2009), and based on these criteria biomathematical modeling approaches have been described to predict the in vivo performance of radioligands based on in vitro and in silico assessments (Friden et al. 2014; Guo et al. 2009; Zhang et al. 2013). These approaches are promising and may possibly be further refined for radioligands tailored to the use of measuring changes in neurotransmitter concentrations. For instance, high affinity for the target (K D in the nanomolar range) and high selectivity towards the target versus other related proteins are considered important criteria for optimal CNS radioligands (Halldin et al. 2001; Pike 2009), but may need further consideration for radioligands tailored toward measurement of neurotransmitter release.
During PET measurements for the determination of drug occupancy, the receptor binding of the radioligand is typically assessed during approximate equilibrium and at tracer dose conditions, making the receptor occupancy estimate independent of the tracer’s K D. However, monitoring of changes in extracellular neurotransmitter concentrations is typically performed under rapidly changing conditions, and it has been suggested that the tracer’s K D, or more specifically, its dissociation rate, K off (corresponding to the PET rate constant k 4), is an important parameter allowing for rapid adjustments to changes in the concentration of the neurotransmitter (Endres and Carson 1998). Also, the rate constant k 2 has been proposed to be an important radioligand characteristic when the radioligand is intended to be used for measurement of neurotransmitter release during dynamic conditions (Endres and Carson 1998; Logan et al. 1991; Morris and Yoder 2007). The K off and k 2 may therefore need to be considered for tailored radioligand development. Experimental studies with high affinity dopamine D2 receptor radioligands suggest that a relative low K off rate does not limit a radioligand to be suitable for measurement of neurotransmitter release. A more systematic study may need to be conducted to further understand the relative importance of these parameters.
Target selectivity is another important parameter to be optimized for radioligands to be used for diagnostic purposes. However, when the radioligand is developed for measurement of neurotransmitter release, target selectivity might not be so critical since selectivity may be achieved by use of a well-characterized pharmacological challenge with high selectivity to a particular neurotransmitter. Differential affinities should also be considered. For example, WAY-100635, which was originally considered to be highly selective for 5-HT1A receptors, was subsequently shown to be a potent D4 receptor agonist, albeit with 10-fold lower affinity than for 5-HT1A (Chemel et al. 2006). Still, relative target distributions should also be considered, as for example in the case of WAY-100635 binding, the density of D4 receptors in vivo is considerably lower than that of 5-HT1A receptors. The importance of target selectivity is also exemplified by NMSP, which is selective for D2 receptors in the striatum and for 5-HT2A receptors in the cortex. However, although PET radioligand selectivity might not be an absolute prerequisite from a biological perspective, and such characterization may have been limited for older PET radioligands, more recent and future radioligands will likely be developed with the intention of high selectivity, which will facilitate the interpretation of the binding results, and this might be predicted for radioligands targeting allosteric binding sites.
In the selection of the radioligand to study changes in neurotransmitter release, it is also important to consider the nature of the interaction between the radioligand and the endogenous mediator. The current applications are primarily based on direct competition, e.g., with dopamine, serotonin, and noradrenaline. The effects of GABA and glutamate on tracer binding involve allosteric interactions and have so far been less well validated, and the reported changes in binding require further mechanistic understanding. In addition, it is worthwhile to consider the affinity of the neurotransmitter to the receptor targeted by the radioligand. Dopamine has higher affinity for the dopamine D2 receptor than the D1 receptor (Marcellino et al. 2012) and serotonin has relatively high affinity for the 5-HT1A, 5-HT1B, and 5-HT7 receptors (Paterson et al. 2010). The success of a radioligand for investigating changes in extracellular neurotransmitter levels may thus depend on the affinity of the neurotransmitter to the corresponding receptor.
Another criterion specifically related to the development of radioligands tailored towards measurement of neurotransmitter release is its intrinsic activity towards the target receptor. Agonist radioligands have been found to be more sensitive to changes in endogenous neurotransmitter release for dopamine, e.g., using dopamine D2-like receptor agonists, and for serotonin, using a 5-HT2A receptor agonist (see relevant sections above). Agonist radioligands preferentially bind to the high-affinity state of the receptors and may therefore be more susceptible to the endogenous agonist than antagonist radioligands, which have similar affinity for the low- and high-affinity states of the receptor. The existence of two distinct affinity states has, however, not yet been demonstrated in vivo (for reviews see Finnema et al. 2010a; Skinbjerg et al. 2012), and the exact mechanism of the increased sensitivity of agonist radioligands therefore warrants further evaluation.
Receptor internalization has been demonstrated to contribute to amphetamine-induced changes in dopamine D2 receptor radioligands. Since, for example, most GPCRs undergo internalization, it may be speculated that the internalization model also applies to other neurotransmitter systems discussed in this review. It is therefore important to understand effects of the physiological environment on the binding of a radioligand to a receptor. In vitro studies using native brain tissue or cell lines have provided understanding of the effect of cellular location on radioligand affinity to dopamine D2 receptors (Guo et al. 2010; Quelch et al. 2014b; Skinbjerg et al. 2009; Sun et al. 2003), SERT (Quelch et al. 2012), opioid receptors (Quelch et al. 2014a), and GABAA receptors (Quelch et al. 2015). Alterations in the ability to bind internalized receptors may thus be a characteristic of a radioligand which could be evaluated to understand if the radioligand will be sensitive to neurotransmitter release.
Quantification of PET signals
Quantification of drug-induced changes in extracellular neurotransmitter concentrations in the human brain is challenging since the magnitude of the observed change in radioligand binding has usually been quite small (typically <25 %) in comparison to the effects seen in drug occupancy studies (typically >50 %). The high cost of PET further limits the number of study subjects, thereby putting high demands on the reproducibility of the procedure. The use of reference region approaches is therefore highly desirable since the corresponding outcome parameters (BP P and BP ND) are more reproducible when compared to distribution volumes (V T) calculated using metabolite-corrected blood data. In particular, quantification of receptors with a broad regional brain distribution poses a methodological challenge as they lack a suitable reference region for the determination of non-specific binding. In addition, PET imaging studies are traditionally analyzed using the receptor occupancy model, but this assumes that the radioligand’s volume of distribution and fractional receptor occupancy are uniform across the brain. This characteristic makes it possible to use occupancy plots, a linear model for reliable quantification of receptor occupancy utilizing V T values (Cunningham et al. 2010). Importantly, this approach may be confounded when endogenous neurotransmitters are not released in equal fractions across different brain regions. Further development of quantitative methods dedicated towards measurement of neurotransmitter release is required. Recent progress include the development of methods aimed to detect relatively small changes in radioligand binding consequential of neurotransmitter release (Alpert et al. 2003; Friston et al. 1997; Watabe et al. 2000) and methods to uncover the temporal pattern of neurotransmitter release during a single PET measurement (Morris et al. 2005; Normandin et al. 2012).
Selection of pharmacological challenge
An additional important consideration for studying fluctuations in a certain neurotransmitter is to identify a pharmacological challenge which does not directly interfere with radioligand binding. Within NEWMEDS work-package 5, to allow for direct translation between methodologies and species, we aimed to evaluate the same pharmacological challenges across species. However, this was sometimes not feasible as not all compounds which were suitable for use in experimental animals were appropriate or available for human studies. Tables 1, 2, 3, and 4 include detailed information on pharmacological regimens that have been used in studies to investigate changes in extracellular neurotransmitter concentrations.
Other factors to consider
Other factors challenging or possibly confounding our studies included whether the interaction between the radioligand and the neurotransmitter was directly competitive, allosteric, or indirect. Endogenous mediators may not be released in equal amounts across different brain regions, the cell membrane transporters of the neurotransmitters may have different brain distributions than the target protein of the radioligand, and agonist-dependent receptor internalization may complicate the interpretation of the findings. In addition, the challenge itself might affect important physiological parameters such as blood pressure, cerebral blood flow, and peripheral clearance of the tracer, which in turn may affect radioligand uptake (Laruelle 2000). The use of anesthesia can also affect the release, re-uptake, and clearance of endogenous mediators in the brain and consequently the interpretation of the results of the pharmacological challenge. Dr. Tsukada and his colleagues (in Hamamatsu, Japan) have extensively investigated the neurotransmitter effects of anesthesia in NHPs by performing PET studies in awake and anesthetized experimental animals. They have shown that isoflurane anesthesia induced a decrease in the binding of [11C]raclopride binding to D2 receptors through increased extracellular levels of dopamine (Tsukada et al. 1999b) and that nicotine, in high tobacco-smoking related doses, did not release sufficient dopamine to displace [11C]raclopride in the striatum of awake NHPs, but did so when the animals were anesthetized with isoflurane (Tsukada et al. 2002). Similarly, in NEWMEDS, we determined using rat microdialysis that striatal extracellular dopamine and noradrenaline concentrations were significantly higher under isoflurane anesthesia compared to awake rats (Finnema et al. 2014b). Nevertheless, most of the findings of NEWMEDS work-package 5 are clearly independent of anesthesia-induced bias or changes in cerebral blood flow. As an example, the noradrenaline challenges monitored with [11C]ORM-13070 produced consistent effects across anesthetized rodents and monkeys and awake humans.
Future work
Assessment of molecular target engagement, accompanied by compound exposure levels at the site of action, is increasingly being recognized as critical information for effective decision making in drug discovery and development (Morgan et al. 2012; Van der Graaf and Danhof 1997). High-resolution imaging technology such as PET, which is amenable to preclinical-to-clinical translation, is playing a key role in addressing the need to investigate target occupancy as well as providing insight into pharmacodynamic actions related to the drug target. Pharmacological challenge approaches have proved useful in providing insight into direct or indirect drug effects on synaptic neurotransmission. This review attempts to summarize the current situation in relation to the use of PET imaging towards investigation of the interactions of endogenous mediators with their target receptors. The focus has been placed on assessing the availability of appropriate tools (PET radioligands, pharmacological challenge agents) and studies in NHPs and human subjects. Here, we have discussed in detail the progress made for PET measurement of dopamine, serotonin, noradrenaline, GABA, glutamate, ACh, and opioid peptides. In addition to the aforementioned investigation utilizing PDE10A radioligands to measure changes in cyclic nucleotides, ongoing activity towards the development of PET radioligands for GPCR histamine receptor subtypes H1 to H4 (for review see Funke et al. 2013) may soon provide radioligand tools which are sensitive to measuring endogenous histamine levels. The H1 receptor antagonist [11C]doxepin and the H3 receptor antagonists [11C]GSK189254 (Ashworth et al. 2010) and [11C]MK8278 (Van Laere et al. 2014) have been successfully characterized in human PET studies, but so far no reports exist in which these radioligands have been utilized for assessment of drug-induced changes in extracellular histamine levels.
Neuropsychiatric disorders such as schizophrenia and depression present significant challenges to researchers striving to deliver improved therapeutics (Rizzo et al. 2013). The development of PET radioligands to enable dynamic monitoring of neurotransmission could be employed to determine changes in target expression and distribution within subjects, to measure phenotypic differences in target expression and distribution, to explore intervention-evoked changes in endogenous ligand levels within subjects, and to investigate phenotypic differences in brain neurotransmission over time. In addition to informing on the diagnosis and monitoring of disease states and their progression, PET radioligands that are sensitive to changes in endogenous mediators can be used to observe functional effects of drugs, thereby serving as biomarkers for target engagement in drug development. In order to achieve this, there is a need to develop more tailored PET radioligands and pharmacological challenge tools. Since a significant challenge of neuropsychiatric drug discovery is the availability of animal models with translational validity (O’Donnell 2013), the approaches reviewed here could also be used to test whether animal models show the same pathophysiology as seen in human patients, e.g., whether “schizophrenia models” show increased uptake of [18F]DOPA or increased amphetamine-induced reduced binding of [11C]raclopride, as is now well established for patients (Howes et al. 2012).
Finally, with recent technological advances in multimodality imaging, enabling simultaneous PET and functional magnetic resonance imaging (fMRI) determinations (for review see Duncan et al. 2014), there is potential to gain new insights into estimating basal receptor occupancy coupled to neurotransmitter levels. This has been successfully demonstrated by measuring neurovascular responses and dopamine receptor occupancy in the basal ganglia across a wide dynamic range using different specific activities of [11C]raclopride. Distinct relative cerebrovascular response magnitudes were observed between the putamen and the caudate nucleus, consistent with higher basal extracellular dopamine levels in the putamen (Sander et al. 2013). These applications are likely to further our understanding of psychiatric disorders and pave the way to novel effective therapeutics. Substantial progress has been made in the development of novel radiotracer tools and methodology, particularly over the past two decades. This has firmly established an important role for PET imaging in CNS research and drug discovery for target engagement analysis and confirmation of modes of action. PET imaging directed at investigating changes in endogenous neurochemicals, including the work done in NEWMEDS work-package 5, has highlighted an opportunity to further extend the capability and application of this technology in new directions.
References
Aalto S, Ihalainen J, Hirvonen J, Kajander J, Scheinin H, Tanila H, Nagren K, Vilkman H, Gustafsson LL, Syvalahti E, Hietala J (2005) Cortical glutamate-dopamine interaction and ketamine-induced psychotic symptoms in man. Psychopharmacology (Berl) 182:375–383
Aalto S, Hirvonen J, Kaasinen V, Hagelberg N, Kajander J, Nagren K, Seppala T, Rinne JO, Scheinin H, Hietala J (2009) The effects of d-amphetamine on extrastriatal dopamine D2/D3 receptors: a randomized, double-blind, placebo-controlled PET study with [11C]FLB 457 in healthy subjects. Eur J Nucl Med Mol Imaging 36:475–483
Abi-Dargham A, Simpson N, Kegeles L, Parsey R, Hwang DR, Anjilvel S, Zea-Ponce Y, Lombardo I, Van Heertum R, Mann JJ, Foged C, Halldin C, Laruelle M (1999) PET studies of binding competition between endogenous dopamine and the D1 radiotracer [11C]NNC 756. Synapse 32:93–109
Airaksinen AJ, Finnema SJ, Balle T, Varnäs K, Bang-Andersen B, Gulyas B, Farde L, Halldin C (2013) Radiosynthesis and evaluation of new alpha1-adrenoceptor antagonists as PET radioligands for brain imaging. Nucl Med Biol 40:747–754
Alpert NM, Badgaiyan RD, Livni E, Fischman AJ (2003) A novel method for noninvasive detection of neuromodulatory changes in specific neurotransmitter systems. Neuroimage 19:1049–1060
Arponen E, Helin S, Marjamaki P, Gronroos T, Holm P, Loyttyniemi E, Nagren K, Scheinin M, Haaparanta-Solin M, Sallinen J, Solin O (2014) A PET tracer for brain alpha2C adrenoceptors, 11C-ORM-13070: radiosynthesis and preclinical evaluation in rats and knockout mice. J Nucl Med 55:1171–1177
Ashworth S, Rabiner EA, Gunn RN, Plisson C, Wilson AA, Comley RA, Lai RY, Gee AD, Laruelle M, Cunningham VJ (2010) Evaluation of 11C-GSK189254 as a novel radioligand for the H3 receptor in humans using PET. J Nucl Med 51:1021–1029
Aznavour N, Rbah L, Riad M, Reilhac A, Costes N, Descarries L, Zimmer L (2006) A PET imaging study of 5-HT1A receptors in cat brain after acute and chronic fluoxetine treatment. Neuroimage 33:834–842
Bel N, Artigas F (1992) Fluvoxamine preferentially increases extracellular 5-hydroxytryptamine in the raphe nuclei: an in vivo microdialysis study. Eur J Pharmacol 229:101–103
Bencherif B, Fuchs PN, Sheth R, Dannals RF, Campbell JN, Frost JJ (2002) Pain activation of human supraspinal opioid pathways as demonstrated by [11C]-carfentanil and positron emission tomography (PET). Pain 99:589–598
Bergstrom M, Grahnen A, Langstrom B (2003) Positron emission tomography microdosing: a new concept with application in tracer and early clinical drug development. Eur J Clin Pharmacol 59:357–366
Bonaventure P, Schotte A, Cras P, Leysen JE (1997) Autoradiographic mapping of 5-HT1B- and 5-HT1D receptors in human brain using [3H]alniditan, a new radioligand. Recept Chann 5:225–230
Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D (1997) Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci U S A 94:2569–2574
Brody AL, Olmstead RE, London ED, Farahi J, Meyer JH, Grossman P, Lee GS, Huang J, Hahn EL, Mandelkern MA (2004) Smoking-induced ventral striatum dopamine release. Am J Psychiatry 161:1211–1218
Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D, Jou J, Allen V, Tiongson E, Chefer SI, Koren AO, Mukhin AG (2006) Cigarette smoking saturates brain alpha 4 beta 2 nicotinic acetylcholine receptors. Arch Gen Psychiatry 63:907–915
Buiter HJ, Windhorst AD, Huisman MC, Yaqub M, Knol DL, Fisher A, Lammertsma AA, Leysen JE (2013) [11C]AF150(S), an agonist PET ligand for M1 muscarinic acetylcholine receptors. EJNMMI Res 3:19
Caravaggio F, Nakajima S, Borlido C, Remington G, Gerretsen P, Wilson A, Houle S, Menon M, Mamo D, Graff-Guerrero A (2014) Estimating endogenous dopamine levels at D2 and D3 receptors in humans using the agonist radiotracer [11C]-(+)-PHNO. Neuropsychopharmacology 39:2769–2776
Cardenas L, Houle S, Kapur S, Busto UE (2004) Oral D-amphetamine causes prolonged displacement of [11C]raclopride as measured by PET. Synapse 51:27–31
Carlson H, Ronne-Engstrom E, Ungerstedt U, Hillered L (1992) Seizure related elevations of extracellular amino acids in human focal epilepsy. Neurosci Lett 140:30–32
Carson RE, Kiesewetter DO, Jagoda E, Der MG, Herscovitch P, Eckelman WC (1998) Muscarinic cholinergic receptor measurements with [18F]FP-TZTP: control and competition studies. J Cereb Blood Flow Metab 18:1130–1142
Chemel BR, Roth BL, Armbruster B, Watts VJ, Nichols DE (2006) WAY-100635 is a potent dopamine D4 receptor agonist. Psychopharmacology (Berl) 188:244–251
Chou YH, Karlsson P, Halldin C, Olsson H, Farde L (1999) A PET study of D1-like dopamine receptor ligand binding during altered endogenous dopamine levels in the primate brain. Psychopharmacology (Berl) 146:220–227
Chou YH, Halldin C, Farde L (2000) Effect of amphetamine on extrastriatal D2 dopamine receptor binding in the primate brain: a PET study. Synapse 38:138–143
Chugani DC, Ackermann RF, Phelps ME (1988) In vivo [3H]spiperone binding: evidence for accumulation in corpus striatum by agonist-mediated receptor internalization. J Cereb Blood Flow Metab 8:291–303
Cohen RM, Carson RE, Filbey F, Szczepanik J, Sunderland T (2006) Age and APOE-epsilon4 genotype influence the effect of physostigmine infusion on the in-vivo distribution volume of the muscarinic-2-receptor dependent tracer [18F]FP-TZTP. Synapse 60:86–92
Colasanti A, Searle GE, Long CJ, Hill SP, Reiley RR, Quelch D, Erritzoe D, Tziortzi AC, Reed LJ, Lingford-Hughes AR, Waldman AD, Schruers KR, Matthews PM, Gunn RN, Nutt DJ, Rabiner EA (2012) Endogenous opioid release in the human brain reward system induced by acute amphetamine administration. Biol Psychiatry 72:371–377
Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, Schaff HV (1997) Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 337:581–588
Cosgrove KP, Kloczynski T, Nabulsi N, Weinzimmer D, Lin SF, Staley JK, Bhagwagar Z, Carson RE (2011) Assessing the sensitivity of [11C]P943, a novel 5-HT1B radioligand, to endogenous serotonin release. Synapse 65:1113–1117
Cropley VL, Innis RB, Nathan PJ, Brown AK, Sangare JL, Lerner A, Ryu YH, Sprague KE, Pike VW, Fujita M (2008) Small effect of dopamine release and no effect of dopamine depletion on [18F]fallypride binding in healthy humans. Synapse 62:399–408
Cunningham VJ, Rabiner EA, Slifstein M, Laruelle M, Gunn RN (2010) Measuring drug occupancy in the absence of a reference region: the Lassen plot re-visited. J Cereb Blood Flow Metab 30:46–50
Dawson GR, Collinson N, Atack JR (2005) Development of subtype selective GABAA modulators. CNS Spectr 10:21–27
de Boer T (1996) The pharmacologic profile of mirtazapine. J Clin Psychiatry 57(Suppl 4):19–25
DeJesus OT, Van Moffaert GJ, Friedman AM (1987) Synthesis of [11C]SCH 23390 for dopamine D1 receptor studies. Int J Rad Appl Instrum A 38:345–348
DeLorenzo C, DellaGioia N, Bloch M, Sanacora G, Nabulsi N, Abdallah C, Yang J, Wen R, Mann JJ, Krystal JH, Parsey RV, Carson RE, Esterlis I (2014) In vivo ketamine-induced changes in [11C]ABP688 binding to metabotropic glutamate receptor subtype 5. Biol Psychiatry. doi:10.1016/j.biopsych.2014.06.024
Derry C, Benjamin C, Bladin P, le Bars D, Tochon-Danguy H, Berkovic SF, Zimmer L, Costes N, Mulligan R, Reutens D (2006) Increased serotonin receptor availability in human sleep: evidence from an [18F]MPPF PET study in narcolepsy. Neuroimage 30:341–348
Dewey SL, Smith GS, Logan J, Brodie JD, Fowler JS, Wolf AP (1993) Striatal binding of the PET ligand 11C-raclopride is altered by drugs that modify synaptic dopamine levels. Synapse 13:350–356
Ding YS, Logan J, Bermel R, Garza V, Rice O, Fowler JS, Volkow ND (2000) Dopamine receptor-mediated regulation of striatal cholinergic activity: positron emission tomography studies with norchloro[18F]fluoroepibatidine. J Neurochem 74:1514–1521
Duncan NW, Wiebking C, Munoz-Torres Z, Northoff G (2014) How to investigate neuro-biochemical relationships on a regional level in humans? Methodological considerations for combining functional with biochemical imaging. J Neurosci Methods 221:183–188
Eckelman WC (2006) Imaging of muscarinic receptors in the central nervous system. Curr Pharm Des 12:3901–3913
Egerton A, Mehta MA, Montgomery AJ, Lappin JM, Howes OD, Reeves SJ, Cunningham VJ, Grasby PM (2009) The dopaminergic basis of human behaviors: a review of molecular imaging studies. Neurosci Biobehav Rev 33:1109–1132
Ehrin E, Farde L, de Paulis T, Eriksson L, Greitz T, Johnstrom P, Litton JE, Nilsson JL, Sedvall G, Stone-Elander S et al (1985) Preparation of 11C-labelled Raclopride, a new potent dopamine receptor antagonist: preliminary PET studies of cerebral dopamine receptors in the monkey. Int J Appl Radiat Isot 36:269–273
Endres CJ, Carson RE (1998) Assessment of dynamic neurotransmitter changes with bolus or infusion delivery of neuroreceptor ligands. J Cereb Blood Flow Metab 18:1196–1210
Endres CJ, Kolachana BS, Saunders RC, Su T, Weinberger D, Breier A, Eckelman WC, Carson RE (1997) Kinetic modeling of [11C]raclopride: combined PET-microdialysis studies. J Cereb Blood Flow Metab 17:932–942
Ettrup A, Hansen M, Santini MA, Paine J, Gillings N, Palner M, Lehel S, Herth MM, Madsen J, Kristensen J, Begtrup M, Knudsen GM (2011) Radiosynthesis and in vivo evaluation of a series of substituted 11C-phenethylamines as 5-HT2A agonist PET tracers. Eur J Nucl Med Mol Imaging 38:681–693
Ettrup A, da Cunha-Bang S, McMahon B, Lehel S, Dyssegaard A, Skibsted AW, Jorgensen LM, Hansen M, Baandrup AO, Bache S, Svarer C, Kristensen JL, Gillings N, Madsen J, Knudsen GM (2014) Serotonin 2A receptor agonist binding in the human brain with [11C]Cimbi-36. J Cereb Blood Flow Metab 34:1188–1196
Fagerholm V, Rokka J, Nyman L, Sallinen J, Tiihonen J, Tupala E, Haaparanta M, Hietala J (2008) Autoradiographic characterization of alpha2C-adrenoceptors in the human striatum. Synapse 62:508–515
Farde L, Nordstrom AL, Wiesel FA, Pauli S, Halldin C, Sedvall G (1992) Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch Gen Psychiatry 49:538–544
Finnema SJ, Seneca N, Farde L, Shchukin E, Sovago J, Gulyas B, Wikstrom HV, Innis RB, Neumeyer JL, Halldin C (2005) A preliminary PET evaluation of the new dopamine D2 receptor agonist [11C]MNPA in cynomolgus monkey. Nucl Med Biol 32:353–360
Finnema SJ, Bang-Andersen B, Wikstrom HV, Halldin C (2010a) Current state of agonist radioligands for imaging of brain dopamine D2/D3 receptors in vivo with positron emission tomography. Curr Top Med Chem 10:1477–1498
Finnema SJ, Varrone A, Hwang TJ, Gulyas B, Pierson ME, Halldin C, Farde L (2010b) Fenfluramine-induced serotonin release decreases [11C]AZ10419369 binding to 5-HT1B-receptors in the primate brain. Synapse 64:573–577
Finnema SJ, Borroni E, Nakao R, Amini N, Takano A, Gobbi L, Honer M, Grimwood S, Farde L, Halldin C (2012a) Tiagabine does not modify [11C]Ro 15-4513 and [11C]flumazenil binding to a major extent in anesthetized non-human primates. J Cereb Blood Flow Metab 32:S65
Finnema SJ, Ettrup A, Stepanov V, Nakao R, Amini N, Varrone A, Knudsen G, Halldin C (2012b) Characterization of [11C]CIMBI-36 as a 5-HT2A receptor agonist PET radioligand in the non-human primate brain. J Cereb Blood Flow Metab 32:S64
Finnema SJ, Varrone A, Hwang TJ, Halldin C, Farde L (2012c) Confirmation of fenfluramine effect on 5-HT1B receptor binding of [11C]AZ10419369 using an equilibrium approach. J Cereb Blood Flow Metab 32:685–695
Finnema SJ, Bang-Andersen B, Jorgensen M, Christoffersen CT, Gulyas B, Wikstrom HV, Farde L, Halldin C (2013a) The dopamine D1 receptor agonist (S)-[11C]N-methyl-NNC 01-0259 is not sensitive to changes in dopamine concentration—a positron emission tomography examination in the monkey brain. Synapse 67:586–595
Finnema SJ, Stepanov V, Zonker B, Gobbi L, Honer M, Borroni E, Grimwood S, Halldin C (2013b) [11C]RO6899880, a novel agonist PET radioligand for the GABAA receptor. J Nucl Med 54:377
Finnema SJ, Halldin C, Bang-Andersen B, Bundgaard C, Farde L (2014a) Serotonin transporter occupancy by S-citalopram and R/S-citalopram in the nonhuman primate brain: a [11C]MADAM PET study. Neuroscience 2014, Washington DC, pp 426.403
Finnema SJ, Hughes ZA, Haaparanta-Solin M, Stepanov V, Nakao R, Varnäs K, Varrone A, Arponen E, Marjamaki P, Pohjanoksa K, Vuorilehto L, Babalola PA, Solin O, Grimwood S, Sallinen J, Farde L, Scheinin M, Halldin C (2014b) Amphetamine decreases alpha2C-adrenoceptor binding of [11C]ORM-13070—a PET study in the primate brain. Int J Neuropsychopharmacol 18(3). doi:10.1093/ijnp/pyu081
Finnema SJ, Stepanov V, Amini N, Martinsson S, Gobbi L, Honer M, Borroni E, Grimwood S, Farde L, Halldin C (2014c) Quantification of the full GABA-A receptor benzodiazepine (BZD) site agonist radioligand [11C]RO6899880 in the monkey brain. The 10th International Symposium on Functional NeuroReceptor Mapping of the Living Brain, Egmond aan Zee, The Netherlands. Available via http://www.vumc.nl/afdelingen-themas/50095/4720590/Program-abstract_book_NRM201.pdf?version=1. P-121:162
Finnema SJ, Stepanov V, Ettrup A, Nakao R, Amini N, Svedberg M, Lehmann C, Hansen M, Knudsen GM, Halldin C (2014d) Characterization of [11C]Cimbi-36 as an agonist PET radioligand for the 5-HT2A and 5-HT2C receptors in the nonhuman primate brain. Neuroimage 84:342–353
Finnema SJ, Stepanov V, Nakao R, Sromek AW, Zhang T, Neumeyer JL, George SR, Seeman P, Stabin MG, Jonsson C, Farde L, Halldin C (2014e) 18F-MCL-524, an 18F-labeled dopamine D2 and D3 receptor agonist sensitive to dopamine: a preliminary PET study. J Nucl Med 55:1164–1170
Frankle WG, Cho RY, Narendran R, Mason NS, Vora S, Litschge M, Price JC, Lewis DA, Mathis CA (2009) Tiagabine increases [11C]flumazenil binding in cortical brain regions in healthy control subjects. Neuropsychopharmacology 34:624–633
Frankle WG, Mason NS, Rabiner EA, Ridler K, May MA, Asmonga D, Chen CM, Kendro S, Cooper TB, Mathis CA, Narendran R (2010) No effect of dopamine depletion on the binding of the high-affinity D2/3 radiotracer [11C]FLB 457 in the human cortex. Synapse 64:879–885
Frankle WG, Cho RY, Mason NS, Chen CM, Himes M, Walker C, Lewis DA, Mathis CA, Narendran R (2012) [11C]flumazenil binding is increased in a dose-dependent manner with tiagabine-induced elevations in GABA levels. PLoS One 7:e32443
Friden M, Wennerberg M, Antonsson M, Sandberg-Ställ M, Farde L, Schou M (2014) Identification of positron emission tomography (PET) tracer candidates by prediction of the target-bound fraction in the brain. EJNMMI Res 4:50
Friston KJ, Malizia AL, Wilson S, Cunningham VJ, Jones T, Nutt DJ (1997) Analysis of dynamic radioligand displacement or “activation” studies. J Cereb Blood Flow Metab 17:80–93
Fuller RW (1994) Uptake inhibitors increase extracellular serotonin concentration measured by brain microdialysis. Life Sci 55:163–167
Funke U, Vugts DJ, Janssen B, Spaans A, Kruijer PS, Lammertsma AA, Perk LR, Windhorst AD (2013) 11C-labeled and 18F-labeled PET ligands for subtype-specific imaging of histamine receptors in the brain. J Labelled Comp Radiopharm 56:120–129
Gallezot JD, Esterlis I, Bois F, Zheng MQ, Lin SF, Kloczynski T, Krystal JH, Huang Y, Sabri O, Carson RE, Cosgrove KP (2014) Evaluation of the sensitivity of the novel alpha4beta2* nicotinic acetylcholine receptor PET radioligand 18F-(−)-NCFHEB to increases in synaptic acetylcholine levels in rhesus monkeys. Synapse 68:556–564
Gianoulakis C (2009) Endogenous opioids and addiction to alcohol and other drugs of abuse. Curr Top Med Chem 9:999–1015
Ginovart N (2005) Imaging the dopamine system with in vivo [11C]raclopride displacement studies: understanding the true mechanism. Mol Imaging Biol 7:45–52
Ginovart N, Wilson AA, Meyer JH, Hussey D, Houle S (2003) [11C]-DASB, a tool for in vivo measurement of SSRI-induced occupancy of the serotonin transporter: PET characterization and evaluation in cats. Synapse 47:123–133
Ginovart N, Wilson AA, Houle S, Kapur S (2004) Amphetamine pretreatment induces a change in both D2-receptor density and apparent affinity: a [11C]raclopride positron emission tomography study in cats. Biol Psychiatry 55:1188–1194
Ginovart N, Galineau L, Willeit M, Mizrahi R, Bloomfield PM, Seeman P, Houle S, Kapur S, Wilson AA (2006) Binding characteristics and sensitivity to endogenous dopamine of [11C]-(+)-PHNO, a new agonist radiotracer for imaging the high-affinity state of D2 receptors in vivo using positron emission tomography. J Neurochem 97:1089–1103
Giovacchini G, Lang L, Ma Y, Herscovitch P, Eckelman WC, Carson RE (2005) Differential effects of paroxetine on raphe and cortical 5-HT1A binding: a PET study in monkeys. Neuroimage 28:238–248
Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L, Zoli M (2009) Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol 78:703–711
Granas C, Nordquist J, Mohell N, Larhammar D (2001) Site-directed mutagenesis of the 5-HT1B receptor increases the affinity of 5-HT for the agonist low-affinity conformation and reduces the intrinsic activity of 5-HT. Eur J Pharmacol 421:69–76
Grimwood S, Hartig PR (2009) Target site occupancy: emerging generalizations from clinical and preclinical studies. Pharmacol Ther 122:281–301
Guo Q, Brady M, Gunn RN (2009) A biomathematical modeling approach to central nervous system radioligand discovery and development. J Nucl Med 50:1715–1723
Guo N, Guo W, Kralikova M, Jiang M, Schieren I, Narendran R, Slifstein M, Abi-Dargham A, Laruelle M, Javitch JA, Rayport S (2010) Impact of D2 receptor internalization on binding affinity of neuroimaging radiotracers. Neuropsychopharmacology 35:806–817
Guterstam J, Jayaram-Lindstrom N, Cervenka S, Frost JJ, Farde L, Halldin C, Franck J (2013) Effects of amphetamine on the human brain opioid system—a positron emission tomography study. Int J Neuropsychopharmacol 16:763–769
Haahr ME, Fisher PM, Jensen CG, Frokjaer VG, Mahon BM, Madsen K, Baare WF, Lehel S, Norremolle A, Rabiner EA, Knudsen GM (2014) Central 5-HT4 receptor binding as biomarker of serotonergic tonus in humans: a [11C]SB207145 PET study. Mol Psychiatry 19:427–432
Halldin C, Stone-Elander S, Farde L, Ehrin E, Fasth KJ, Langstrom B, Sedvall G (1986) Preparation of 11C-labelled SCH 23390 for the in vivo study of dopamine D-1 receptors using positron emission tomography. Int J Rad Appl Instrum A 37:1039–1043
Halldin C, Farde L, Hogberg T, Mohell N, Hall H, Suhara T, Karlsson P, Nakashima Y, Swahn CG (1995) Carbon-11-FLB 457: a radioligand for extrastriatal D2 dopamine receptors. J Nucl Med 36:1275–1281
Halldin C, Foged C, Chou YH, Karlsson P, Swahn CG, Sandell J, Sedvall G, Farde L (1998) Carbon-11-NNC 112: a radioligand for PET examination of striatal and neocortical D1-dopamine receptors. J Nucl Med 39:2061–2068
Halldin C, Gulyas B, Langer O, Farde L (2001) Brain radioligands—state of the art and new trends. Q J Nucl Med 45:139–152
Halldin C, Lundberg J, Sovago J, Gulyas B, Guilloteau D, Vercouillie J, Emond P, Chalon S, Tarkiainen J, Hiltunen J, Farde L (2005) [11C]MADAM, a new serotonin transporter radioligand characterized in the monkey brain by PET. Synapse 58:173–183
Hartvig P, Valtysson J, Antoni G, Westerberg G, Langstrom B, Ratti Moberg E, Oye I (1994) Brain kinetics of (R)- and (S)-[N-methyl-11C]ketamine in the rhesus monkey studied by positron emission tomography (PET). Nucl Med Biol 21:927–934
Hartvig P, Valtysson J, Lindner KJ, Kristensen J, Karlsten R, Gustafsson LL, Persson J, Svensson JO, Oye I, Antoni G et al (1995) Central nervous system effects of subdissociative doses of (S)-ketamine are related to plasma and brain concentrations measured with positron emission tomography in healthy volunteers. Clin Pharmacol Ther 58:165–173
Hendry N, Christie I, Rabiner EA, Laruelle M, Watson J (2011) In vitro assessment of the agonist properties of the novel 5-HT1A receptor ligand, CUMI-101 (MMP), in rat brain tissue. Nucl Med Biol 38:273–277
Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KK, Bolam JP, Ince E, Yi H, Levey AI (1995) Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci 15:5222–5237
Hervas I, Artigas F (1998) Effect of fluoxetine on extracellular 5-hydroxytryptamine in rat brain. Role of 5-HT autoreceptors. Eur J Pharmacol 358:9–18
Higgitt A, Lader M, Fonagy P (1986) The effects of the benzodiazepine antagonist Ro 15-1788 on psychophysiological performance and subjective measures in normal subjects. Psychopharmacology (Berl) 89:395–403
Hillmer AT, Wooten DW, Farhoud M, Higgins AT, Lao PJ, Barnhart TE, Mukherjee J, Christian BT (2013) PET imaging of acetylcholinesterase inhibitor-induced effects on alpha4beta2 nicotinic acetylcholine receptor binding. Synapse 67:882–886
Hirani E, Sharp T, Sprakes M, Grasby P, Hume S (2003) Fenfluramine evokes 5-HT2A receptor-mediated responses but does not displace [11C]MDL 100907: small animal PET and gene expression studies. Synapse 50:251–260
Horti AG, Gao Y, Kuwabara H, Dannals RF (2010) Development of radioligands with optimized imaging properties for quantification of nicotinic acetylcholine receptors by positron emission tomography. Life Sci 86:575–584
Houle S, Ginovart N, Hussey D, Meyer JH, Wilson AA (2000) Imaging the serotonin transporter with positron emission tomography: initial human studies with [11C]DAPP and [11C]DASB. Eur J Nucl Med 27:1719–1722
Houston GC, Hume SP, Hirani E, Goggi JL, Grasby PM (2004) Temporal characterisation of amphetamine-induced dopamine release assessed with [11C]raclopride in anaesthetised rodents. Synapse 51:206–212
Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, Kapur S (2012) The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry 69:776–786
Hume SP, Hirani E, Opacka-Juffry J, Osman S, Myers R, Gunn RN, McCarron JA, Clark RD, Melichar J, Nutt DJ, Pike VW (2000) Evaluation of [O-methyl-11C]RS-15385-197 as a positron emission tomography radioligand for central alpha2-adrenoceptors. Eur J Nucl Med 27:475–484
Hume S, Hirani E, Opacka-Juffry J, Myers R, Townsend C, Pike V, Grasby P (2001) Effect of 5-HT on binding of [11C]WAY 100635 to 5-HT1A receptors in rat brain, assessed using in vivo microdialysis and PET after fenfluramine. Synapse 41:150–159
Hwang DR, Kegeles LS, Laruelle M (2000) (−)-N-[11C]propyl-norapomorphine: a positron-labeled dopamine agonist for PET imaging of D2 receptors. Nucl Med Biol 27:533–539
Ihalainen JA, Tanila H (2002) In vivo regulation of dopamine and noradrenaline release by alpha2A-adrenoceptors in the mouse prefrontal cortex. Eur J Neurosci 15:1789–1794
Innis RB, Malison RT, al-Tikriti M, Hoffer PB, Sybirska EH, Seibyl JP, Zoghbi SS, Baldwin RM, Laruelle M, Smith EO (1992) Amphetamine-stimulated dopamine release competes in vivo for [123I]IBZM binding to the D2 receptor in nonhuman primates. Synapse 10:177–184
Invernizzi R, Belli S, Samanin R (1992) Citalopram’s ability to increase the extracellular concentrations of serotonin in the dorsal raphe prevents the drug’s effect in the frontal cortex. Brain Res 584:322–324
Jakobsen S, Pedersen K, Smith DF, Jensen SB, Munk OL, Cumming P (2006) Detection of alpha2-adrenergic receptors in brain of living pig with 11C-yohimbine. J Nucl Med 47:2008–2015
Jasinska AJ, Zorick T, Brody AL, Stein EA (2014) Dual role of nicotine in addiction and cognition: a review of neuroimaging studies in humans. Neuropharmacology 84:111–122
Jones AK, Cunningham VJ, Ha-Kawa S, Fujiwara T, Luthra SK, Silva S, Derbyshire S, Jones T (1994) Changes in central opioid receptor binding in relation to inflammation and pain in patients with rheumatoid arthritis. Br J Rheumatol 33:909–916
Jones T, Rabiner EA, Company PETRA (2012) The development, past achievements, and future directions of brain PET. J Cereb Blood Flow Metab 32:1426–1454
Kawamura K, Akiyama M, Yui J, Yamasaki T, Hatori A, Kumata K, Wakizaka H, Takei M, Nengaki N, Yanamoto K, Fukumura T, Zhang MR (2010) In vivo evaluation of limiting brain penetration of probes for alpha2C-adrenoceptor using small-animal positron emission tomography. ACS Chem Neurosci 1:520–528
Kehler J, Kilburn JP, Estrada S, Christensen SR, Wall A, Thibblin A, Lubberink M, Bundgaard C, Brennum LT, Steiniger-Brach B, Christoffersen CT, Timmermann S, Kreilgaard M, Antoni G, Bang-Andersen B, Nielsen J (2014) Discovery and development of 11C-Lu AE92686 as a radioligand for PET imaging of phosphodiesterase10A in the brain. J Nucl Med 55:1513–1518
Kemp JA, Marshall GR, Wong EH, Woodruff GN (1987) The affinities, potencies and efficacies of some benzodiazepine-receptor agonists, antagonists and inverse-agonists at rat hippocampal GABAA-receptors. Br J Pharmacol 91:601–608
Kew JN, Kemp JA (2005) Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berl) 179:4–29
Kim SY, Lee H, Kim HJ, Bang E, Lee SH, Lee DW, Woo DC, Choi CB, Hong KS, Lee C, Choe BY (2011) In vivo and ex vivo evidence for ketamine-induced hyperglutamatergic activity in the cerebral cortex of the rat: potential relevance to schizophrenia. NMR Biomed 24:1235–1242
Kimura Y, Simeon FG, Zoghbi SS, Zhang Y, Hatazawa J, Pike VW, Innis RB, Fujita M (2012) Quantification of metabotropic glutamate subtype 5 receptors in the brain by an equilibrium method using 18F-SP203. Neuroimage 59:2124–2130
Koepp MJ, Gunn RN, Lawrence AD, Cunningham VJ, Dagher A, Jones T, Brooks DJ, Bench CJ, Grasby PM (1998) Evidence for striatal dopamine release during a video game. Nature 393:266–268
Kumar JS, Prabhakaran J, Majo VJ, Milak MS, Hsiung SC, Tamir H, Simpson NR, Van Heertum RL, Mann JJ, Parsey RV (2007) Synthesis and in vivo evaluation of a novel 5-HT1A receptor agonist radioligand [O-methyl-11C]2-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-4-methyl-1,2,4-triazine-3,5(2H,4H)dione in nonhuman primates. Eur J Nucl Med Mol Imaging 34:1050–1060
Kumlien E, Hartvig P, Valind S, Oye I, Tedroff J, Langstrom B (1999) NMDA-receptor activity visualized with (S)-[N-methyl-11C]ketamine and positron emission tomography in patients with medial temporal lobe epilepsy. Epilepsia 40:30–37
Kung HF, Kasliwal R, Pan SG, Kung MP, Mach RH, Guo YZ (1988) Dopamine D-2 receptor imaging radiopharmaceuticals: synthesis, radiolabeling, and in vitro binding of (R)-(+)- and (S)-(−)-3-iodo-2-hydroxy-6-methoxy-N-[(1-ethyl-2-pyrrolidinyl)methyl]benzamide. J Med Chem 31:1039–1043
Landau AM, Doudet DJ, Jakobsen S (2012) Amphetamine challenge decreases yohimbine binding to alpha2 adrenoceptors in Landrace pig brain. Psychopharmacology (Berl) 222:155–163
Lang L, Jagoda E, Schmall B, Vuong BK, Adams HR, Nelson DL, Carson RE, Eckelman WC (1999) Development of fluorine-18-labeled 5-HT1A antagonists. J Med Chem 42:1576–1586
Langmead CJ, Watson J, Reavill C (2008) Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther 117:232–243
Larisch R, Klimke A, Hamacher K, Henning U, Estalji S, Hohlfeld T, Vosberg H, Tosch M, Gaebel W, Coenen HH, Muller-Gartner HW (2003) Influence of synaptic serotonin level on [18F]altanserin binding to 5HT2 receptors in man. Behav Brain Res 139:21–29
Laruelle M (2000) Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab 20:423–451
Laruelle M, Abi-Dargham A, van Dyck CH, Rosenblatt W, Zea-Ponce Y, Zoghbi SS, Baldwin RM, Charney DS, Hoffer PB, Kung HF et al (1995) SPECT imaging of striatal dopamine release after amphetamine challenge. J Nucl Med 36:1182–1190
Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D’Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB (1996) Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci U S A 93:9235–9240
Laruelle M, Iyer RN, al-Tikriti MS, Zea-Ponce Y, Malison R, Zoghbi SS, Baldwin RM, Kung HF, Charney DS, Hoffer PB, Innis RB, Bradberry CW (1997) Microdialysis and SPECT measurements of amphetamine-induced dopamine release in nonhuman primates. Synapse 25:1–14
Lee CM, Farde L (2006) Using positron emission tomography to facilitate CNS drug development. Trends Pharmacol Sci 27:310–316
Lehto J, Hirvonen MM, Johansson J, Kemppainen J, Luoto P, Naukkarinen T, Oikonen V, Arponen E, Rouru J, Sallinen J, Scheinin H, Vuorilehto L, Finnema SJ, Halldin C, Rinne JO, Scheinin M (2015a) Validation of [11C]ORM-13070 as a PET tracer for alpha2C-adrenoceptors in the human brain. Synapse 69:172–181
Lehto J, Virta JR, Oikonen V, Roivainen A, Luoto P, Arponen E, Helin S, Hietamaki J, Holopainen A, Kailajarvi M, Peltonen JM, Rouru J, Sallinen J, Virtanen K, Volanen I, Scheinin M, Rinne JO (2015b) Test-retest reliability of [11C]ORM-13070 in PET imaging of α2C-adrenoceptors in vivo in the human brain. Eur J Nucl Med Mol Imaging 42:120–127
Lehto J, Johansson J, Vuorilehto L, Luoto P, Arponen E, Sheinin H, Rouru J, Scheinin M (2015c) Sensitivity of [11C]ORM-13070 to increased extracellular noradrenaline in the CNS - a PET study in human subjects. Psychopharmacology
Lemaire C, Cantineau R, Guillaume M, Plenevaux A, Christiaens L (1991) Fluorine-18-altanserin: a radioligand for the study of serotonin receptors with PET: radiolabeling and in vivo biologic behavior in rats. J Nucl Med 32:2266–2272
Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, Price DL, Maggio R, Brann MR, Ciliax BJ (1993) Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc Natl Acad Sci U S A 90:8861–8865
Leysen JE, Gommeren W, Laduron PM (1978) Spiperone: a ligand of choice for neuroleptic receptors. 1. Kinetics and characteristics of in vitro binding. Biochem Pharmacol 27:307–316
Logan J, Dewey SL, Wolf AP, Fowler JS, Brodie JD, Angrist B, Volkow ND, Gatley SJ (1991) Effects of endogenous dopamine on measures of [18F]N-methylspiroperidol binding in the basal ganglia: comparison of simulations and experimental results from PET studies in baboons. Synapse 9:195–207
Lundkvist C, Halldin C, Ginovart N, Nyberg S, Swahn CG, Carr AA, Brunner F, Farde L (1996) [11C]MDL 100907, a radioligland for selective imaging of 5-HT2A receptors with positron emission tomography. Life Sci 58:PL 187–192
Lundquist P, Roman M, Syvanen S, Hartvig P, Blomquist G, Hammarlund-Udenaes M, Langstrom B (2007) Effect on [11C]DASB binding after tranylcypromine-induced increase in serotonin concentration: positron emission tomography studies in monkeys and rats. Synapse 61:440–449
Luoto P, Suilamo S, Oikonen V, Arponen E, Helin S, Herttuainen J, Hietamaki J, Holopainen A, Kailajarvi M, Peltonen JM, Rouru J, Sallinen J, Scheinin M, Virta J, Virtanen K, Volanen I, Roivainen A, Rinne JO (2014) 11C-ORM-13070, a novel PET ligand for brain alpha2C-adrenoceptors: radiometabolism, plasma pharmacokinetics, whole-body distribution and radiation dosimetry in healthy men. Eur J Nucl Med Mol Imaging 41:1947–1956
Lyon RA, Titeler M, Frost JJ, Whitehouse PJ, Wong DF, Wagner HN Jr, Dannals RF, Links JM, Kuhar MJ (1986) 3H-3-N-methylspiperone labels D2 dopamine receptors in basal ganglia and S2 serotonin receptors in cerebral cortex. J Neurosci 6:2941–2949
Ma B, Sherman PS, Moskwa JE, Koeppe RA, Kilbourn MR (2004) Sensitivity of [11C]N-methylpyrrolidinyl benzilate ([11C]NMPYB) to endogenous acetylcholine: PET imaging vs tissue sampling methods. Nucl Med Biol 31:393–397
Maeda J, Suhara T, Ogawa M, Okauchi T, Kawabe K, Zhang MR, Semba J, Suzuki K (2001) In vivo binding properties of [carbonyl-11C]WAY-100635: effect of endogenous serotonin. Synapse 40:122–129
Maier DL, Sobotka-Briner C, Ding M, Powell ME, Jiang Q, Hill G, Heys JR, Elmore CS, Pierson ME, Mrzljak L (2009) [N-methyl-3H3]AZ10419369 binding to the 5-HT1B receptor: in vitro characterization and in vivo receptor occupancy. J Pharmacol Exp Ther 330:342–351
Marcellino D, Kehr J, Agnati LF, Fuxe K (2012) Increased affinity of dopamine for D2-like versus D1-like receptors. relevance for volume transmission in interpreting PET findings. Synapse 66:196–203
Marner L, Gillings N, Madsen K, Erritzoe D, Baare WF, Svarer C, Hasselbalch SG, Knudsen GM (2010) Brain imaging of serotonin 4 receptors in humans with [11C]SB207145-PET. Neuroimage 50:855–861
Marthi K, Bender D, Gjedde A, Smith DF (2002) [11C]Mirtazapine for PET neuroimaging: radiosynthesis and initial evaluation in the living porcine brain. Eur Neuropsychopharmacol 12:427–432
Matusch A, Hurlemann R, Rota Kops E, Winz OH, Elmenhorst D, Herzog H, Zilles K, Bauer A (2007) Acute S-ketamine application does not alter cerebral [18F]altanserin binding: a pilot PET study in humans. J Neural Transm 114:1433–1442
McKernan RM, Whiting PJ (1996) Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci 19:139–143
Meyer JH, Cho R, Kennedy S, Kapur S (1999) The effects of single dose nefazodone and paroxetine upon 5-HT2A binding potential in humans using [18F]-setoperone PET. Psychopharmacology (Berl) 144:279–281
Mick I, Myers J, Stokes PR, Erritzoe D, Colasanti A, Bowden-Jones H, Clark L, Gunn RN, Rabiner EA, Searle GE, Waldman AD, Parkin MC, Brailsford AD, Nutt DJ, Lingford-Hughes AR (2014) Amphetamine induced endogenous opioid release in the human brain detected with [11C]carfentanil PET: replication in an independent cohort. Int J Neuropsychopharmacol 17:2069–2074
Milak MS, Ogden RT, Vinocur DN, Van Heertum RL, Cooper TB, Mann JJ, Parsey RV (2005) Effects of tryptophan depletion on the binding of [11C]-DASB to the serotonin transporter in baboons: response to acute serotonin deficiency. Biol Psychiatry 57:102–106
Milak MS, Severance AJ, Prabhakaran J, Kumar JS, Majo VJ, Ogden RT, Mann JJ, Parsey RV (2011) In vivo serotonin-sensitive binding of [11C]CUMI-101: a serotonin 1A receptor agonist positron emission tomography radiotracer. J Cereb Blood Flow Metab 31:243–249
Millan MJ, Newman-Tancredi A, Lochon S, Touzard M, Aubry S, Audinot V (2002) Specific labelling of serotonin 5-HT1B receptors in rat frontal cortex with the novel, phenylpiperazine derivative, [3H]GR125,743. A pharmacological characterization. Pharmacol Biochem Behav 71:589–598
Millan MJ, Marin P, Bockaert J, Mannoury la Cour C (2008) Signaling at G-protein-coupled serotonin receptors: recent advances and future research directions. Trends Pharmacol Sci 29:454–464
Miller LG, Greenblatt DJ, Barnhill JG, Summer WR, Shader RI (1988) ‘GABA shift’ in vivo: enhancement of benzodiazepine binding in vivo by modulation of endogenous GABA. Eur J Pharmacol 148:123–130
Mirfeizi L, Rybczynska AA, van Waarde A, Campbell-Verduyn L, Feringa BL, Dierckx RA, Elsinga PH (2014) [18F]-(fluoromethoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)propan-2-ol ([18F FPTC) a novel PET-ligand for cerebral beta-adrenoceptors. Nucl Med Biol 41:203–209
Miyake N, Skinbjerg M, Easwaramoorthy B, Kumar D, Girgis RR, Xu X, Slifstein M, Abi-Dargham A (2011) Imaging changes in glutamate transmission in vivo with the metabotropic glutamate receptor 5 tracer [11C]ABP688 and N-acetylcysteine challenge. Biol Psychiatry 69:822–824
Mo YX, Yin YF, Li YM (2014) Neural nAChRs PET imaging probes. Nucl Med Commun 35:135–143
Montgomery AJ, Asselin MC, Farde L, Grasby PM (2007) Measurement of methylphenidate-induced change in extrastriatal dopamine concentration using [11C]FLB 457 PET. J Cereb Blood Flow Metab 27:369–377
Morgan P, Van Der Graaf PH, Arrowsmith J, Feltner DE, Drummond KS, Wegner CD, Street SD (2012) Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving phase II survival. Drug Discov Today 17:419–424
Morris ED, Yoder KK (2007) Positron emission tomography displacement sensitivity: predicting binding potential change for positron emission tomography tracers based on their kinetic characteristics. J Cereb Blood Flow Metab 27:606–617
Morris ED, Yoder KK, Wang C, Normandin MD, Zheng QH, Mock B, Muzic RF Jr, Froehlich JC (2005) ntPET: a new application of PET imaging for characterizing the kinetics of endogenous neurotransmitter release. Mol Imaging 4:473–489
Moulin-Sallanon M, Charnay Y, Ginovart N, Perret P, Lanfumey L, Hamon M, Hen R, Fagret D, Ibanez V, Millet P (2009) Acute and chronic effects of citalopram on 5-HT1A receptor-labeling by [18F]MPPF and -coupling to receptors-G proteins. Synapse 63:106–116
Mukherjee J, Yang ZY, Das MK, Brown T (1995) Fluorinated benzamide neuroleptics—III. Development of (S)-N-[(1-allyl-2-pyrrolidinyl)methyl]-5-(3-[18F]fluoropropyl)-2, 3-dimethoxybenzamide as an improved dopamine D-2 receptor tracer. Nucl Med Biol 22:283–296
Mukherjee J, Shi B, Christian BT, Chattopadhyay S, Narayanan TK (2004) 11C-Fallypride: radiosynthesis and preliminary evaluation of a novel dopamine D2/D3 receptor PET radiotracer in non-human primate brain. Bioorg Med Chem 12:95–102
Mukherjee J, Christian BT, Narayanan TK, Shi B, Collins D (2005) Measurement of d-amphetamine-induced effects on the binding of dopamine D-2/D-3 receptor radioligand, 18F-fallypride in extrastriatal brain regions in non-human primates using PET. Brain Res 1032:77–84
Munk OL, Smith DF (2011) PET kinetics of radiolabeled antidepressant, [N-methyl-11C]mirtazapine, in the human brain. EJNMMI Res 1:36
Nagy K, Toth M, Major P, Patay G, Egri G, Haggkvist J, Varrone A, Farde L, Halldin C, Gulyas B (2013) Performance evaluation of the small-animal nanoScan PET/MRI system. J Nucl Med 54:1825–1832
Nahimi A, Jakobsen S, Munk OL, Vang K, Phan JA, Rodell A, Gjedde A (2015) Mapping alpha2 adrenoceptors of the human brain with 11C-yohimbine. J Nucl Med 56:392–398
Narendran R, Hwang DR, Slifstein M, Talbot PS, Erritzoe D, Huang Y, Cooper TB, Martinez D, Kegeles LS, Abi-Dargham A, Laruelle M (2004) In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (−)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist radiotracer [11C]-raclopride. Synapse 52:188–208
Narendran R, Slifstein M, Hwang DR, Hwang Y, Scher E, Reeder S, Martinez D, Laruelle M (2007) Amphetamine-induced dopamine release: duration of action as assessed with the D2/3 receptor agonist radiotracer (−)-N-[11C]propyl-norapomorphine ([11C]NPA) in an anesthetized nonhuman primate. Synapse 61:106–109
Narendran R, Frankle WG, Mason NS, Rabiner EA, Gunn RN, Searle GE, Vora S, Litschge M, Kendro S, Cooper TB, Mathis CA, Laruelle M (2009) Positron emission tomography imaging of amphetamine-induced dopamine release in the human cortex: a comparative evaluation of the high affinity dopamine D2/3 radiotracers [11C]FLB 457 and [11C]fallypride. Synapse 63:447–461
Narendran R, Mason NS, Laymon CM, Lopresti BJ, Velasquez ND, May MA, Kendro S, Martinez D, Mathis CA, Frankle WG (2010) A comparative evaluation of the dopamine D2/3 agonist radiotracer [11C] (−)-N-propyl-norapomorphine and antagonist [11C]raclopride to measure amphetamine-induced dopamine release in the human striatum. J Pharmacol Exp Ther 333:533–539
Narendran R, Himes M, Mason NS (2013) Reproducibility of post-amphetamine [11C]FLB 457 binding to cortical D2/3 receptors. PLoS One 8:e76905
Narendran R, Jedema HP, Lopresti BJ, Mason NS, Gurnsey K, Ruszkiewicz J, Chen CM, Deuitch L, Frankle WG, Bradberry CW (2014) Imaging dopamine transmission in the frontal cortex: a simultaneous microdialysis and [11C]FLB 457 PET study. Mol Psychiatry 19:302–310
Nishiyama S, Tsukada H, Sato K, Kakiuchi T, Ohba H, Harada N, Takahashi K (2001) Evaluation of PET ligands (+)-N-[11C]ethyl-3-piperidyl benzilate and (+)-N-[11C]propyl-3-piperidyl benzilate for muscarinic cholinergic receptors: a PET study with microdialysis in comparison with (+)-N-[11C]methyl-3-piperidyl benzilate in the conscious monkey brain. Synapse 40:159–169
Nord M, Finnema SJ, Halldin C, Farde L (2013) Effect of a single dose of escitalopram on serotonin concentration in the non-human and human primate brain. Int J Neuropsychopharmacol 16:1577–1586
Nord M, Finnema SJ, Schain M, Halldin C, Farde L (2014) Test-retest reliability of [11C]AZ10419369 binding to 5-HT1B receptors in human brain. Eur J Nucl Med Mol Imaging 41:301–307
Normandin MD, Schiffer WK, Morris ED (2012) A linear model for estimation of neurotransmitter response profiles from dynamic PET data. Neuroimage 59:2689–2699
Nyback H, Halldin C, Ahlin A, Curvall M, Eriksson L (1994) PET studies of the uptake of (S)- and (R)-[11C]nicotine in the human brain: difficulties in visualizing specific receptor binding in vivo. Psychopharmacology (Berl) 115:31–36
O’Donnell P (2013) How can animal models be better utilized? In: Silverstein SM, Moghaddam B, Wykes T (eds) Schizophrenia: evolution and synthesis. MIT Press, Cambridge, pp 205–216
Okauchi T, Suhara T, Maeda J, Kawabe K, Obayashi S, Suzuki K (2001) Effect of endogenous dopamine on extrastriatal [11C]FLB 457 binding measured by PET. Synapse 41:87–95
Palner M, McCormick P, Parkes J, Knudsen GM, Wilson AA (2010) Systemic catechol-O-methyl transferase inhibition enables the D1 agonist radiotracer R-[11C]SKF 82957. Nucl Med Biol 37:837–843
Paterson LM, Tyacke RJ, Nutt DJ, Knudsen GM (2010) Measuring endogenous 5-HT release by emission tomography: promises and pitfalls. J Cereb Blood Flow Metab 30:1682–1706
Paterson LM, Kornum BR, Nutt DJ, Pike VW, Knudsen GM (2013) 5-HT radioligands for human brain imaging with PET and SPECT. Med Res Rev 33:54–111
Pazos A, Probst A, Palacios JM (1987a) Serotonin receptors in the human brain—III. Autoradiographic mapping of serotonin-1 receptors. Neuroscience 21:97–122
Pazos A, Probst A, Palacios JM (1987b) Serotonin receptors in the human brain—IV. Autoradiographic mapping of serotonin-2 receptors. Neuroscience 21:123–139
Phan JA, Landau AM, Wong DF, Jakobsen S, Nahimi A, Doudet DJ, Gjedde A (2015) Quantification of [11C]yohimbine binding to alpha2 adrenoceptors in rat brain in vivo. J Cereb Blood Flow Metab 35:501–511
Piccini P, Pavese N, Brooks DJ (2003) Endogenous dopamine release after pharmacological challenges in Parkinson’s disease. Ann Neurol 53:647–653
Pierson ME, Andersson J, Nyberg S, McCarthy DJ, Finnema SJ, Varnäs K, Takano A, Karlsson P, Gulyas B, Medd AM, Lee CM, Powell ME, Heys JR, Potts W, Seneca N, Mrzljak L, Farde L, Halldin C (2008) [11C]AZ10419369: a selective 5-HT1B receptor radioligand suitable for positron emission tomography (PET). Characterization in the primate brain. Neuroimage 41:1075–1085
Pike VW (2009) PET radiotracers: crossing the blood–brain barrier and surviving metabolism. Trends Pharmacol Sci 30:431–440
Pike VW, McCarron JA, Lammerstma AA, Hume SP, Poole K, Grasby PM, Malizia A, Cliffe IA, Fletcher A, Bench CJ (1995) First delineation of 5-HT1A receptors in human brain with PET and [11C]WAY-100635. Eur J Pharmacol 283:R1–R3
Pike VW, McCarron JA, Lammertsma AA, Osman S, Hume SP, Sargent PA, Bench CJ, Cliffe IA, Fletcher A, Grasby PM (1996) Exquisite delineation of 5-HT1A receptors in human brain with PET and [carbonyl-11C]WAY-100635. Eur J Pharmacol 301:R5–R7
Pinborg LH, Adams KH, Yndgaard S, Hasselbalch SG, Holm S, Kristiansen H, Paulson OB, Knudsen GM (2004) [18F]altanserin binding to human 5-HT2A receptors is unaltered after citalopram and pindolol challenge. J Cereb Blood Flow Metab 24:1037–1045
Pinborg LH, Feng L, Haahr ME, Gillings N, Dyssegaard A, Madsen J, Svarer C, Yndgaard S, Kjaer TW, Parsey RV, Hansen HD, Ettrup A, Paulson OB, Knudsen GM (2012) No change in [11C]CUMI-101 binding to 5-HT1A receptors after intravenous citalopram in human. Synapse 66:880–884
Praschak-Rieder N, Hussey D, Wilson AA, Carella A, Lee M, Dunn E, Willeit M, Bagby RM, Houle S, Meyer JH (2004) Tryptophan depletion and serotonin loss in selective serotonin reuptake inhibitor-treated depression: an [18F]MPPF positron emission tomography study. Biol Psychiatry 56:587–591
Praschak-Rieder N, Wilson AA, Hussey D, Carella A, Wei C, Ginovart N, Schwarz MJ, Zach J, Houle S, Meyer JH (2005) Effects of tryptophan depletion on the serotonin transporter in healthy humans. Biol Psychiatry 58:825–830
Quednow BB, Treyer V, Hasler F, Dorig N, Wyss MT, Burger C, Rentsch KM, Westera G, Schubiger PA, Buck A, Vollenweider FX (2012) Assessment of serotonin release capacity in the human brain using dexfenfluramine challenge and [18F]altanserin positron emission tomography. Neuroimage 59:3922–3932
Quelch DR, Parker CA, Nutt DJ, Tyacke RJ, Erritzoe D (2012) Influence of different cellular environments on [3H]DASB radioligand binding. Synapse 66:1035–1039
Quelch DR, Katsouri L, Nutt DJ, Parker CA, Tyacke RJ (2014a) Imaging endogenous opioid peptide release with [11C]carfentanil and [3H]diprenorphine: influence of agonist-induced internalization. J Cereb Blood Flow Metab 34:1604–1612
Quelch DR, Withey SL, Nutt DJ, Tyacke RJ, Parker CA (2014b) The influence of different cellular environments on PET radioligand binding: an application to D2/3-dopamine receptor imaging. Neuropharmacology 85:305–313
Quelch D, De Santis V, Strege A, Myers J, Wells L, Nutt D, Lingford-Hughes A, Parker C, Tyacke R (2015) Influence of agonist induced internalization on [3H]Ro15-4513 binding—an application to imaging fluctuations in endogenous GABA with positron emission tomography. Synapse 69:60–65
Rabiner EA, Messa C, Sargent PA, Husted-Kjaer K, Montgomery A, Lawrence AD, Bench CJ, Gunn RN, Cowen P, Grasby PM (2002) A database of [11C]WAY-100635 binding to 5-HT1A receptors in normal male volunteers: normative data and relationship to methodological, demographic, physiological, and behavioral variables. Neuroimage 15:620–632
Riad M, Zimmer L, Rbah L, Watkins KC, Hamon M, Descarries L (2004) Acute treatment with the antidepressant fluoxetine internalizes 5-HT1A autoreceptors and reduces the in vivo binding of the PET radioligand [18F]MPPF in the nucleus raphe dorsalis of rat. J Neurosci 24:5420–5426
Riccardi P, Li R, Ansari MS, Zald D, Park S, Dawant B, Anderson S, Doop M, Woodward N, Schoenberg E, Schmidt D, Baldwin R, Kessler R (2006) Amphetamine-induced displacement of [18F]fallypride in striatum and extrastriatal regions in humans. Neuropsychopharmacology 31:1016–1026
Riccardi P, Baldwin R, Salomon R, Anderson S, Ansari MS, Li R, Dawant B, Bauernfeind A, Schmidt D, Kessler R (2008) Estimation of baseline dopamine D2 receptor occupancy in striatum and extrastriatal regions in humans with positron emission tomography with [18F]fallypride. Biol Psychiatry 63:241–244
Richfield EK, Penney JB, Young AB (1989) Anatomical and affinity state comparisons between dopamine D1 and D2 receptors in the rat central nervous system. Neuroscience 30:767–777
Ridler K, Plisson C, Rabiner EA, Gunn RN, Easwaramoorthy B, Abi-Dargham A, Laruelle M, Slifstein M (2011) Characterization of in vivo pharmacological properties and sensitivity to endogenous serotonin of [11C]P943: a positron emission tomography study in Papio anubis. Synapse 65:1119–1127
Rizzo SJ, Edgerton JR, Hughes ZA, Brandon NJ (2013) Future viable models of psychiatry drug discovery in pharma. J Biomol Screen 18:509–521
Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ, Roth BL (2000) Evidence for possible involvement of 5-HT2B receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 102:2836–2841
Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, Barrow R, Yeo R, Lauriello J, Brooks WM (2005) Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry 162:394–396
Ruuskanen JO, Peitsaro N, Kaslin JV, Panula P, Scheinin M (2005) Expression and function of alpha-adrenoceptors in zebrafish: drug effects, mRNA and receptor distributions. J Neurochem 94:1559–1569
Sallinen J, Link RE, Haapalinna A, Viitamaa T, Kulatunga M, Sjoholm B, Macdonald E, Pelto-Huikko M, Leino T, Barsh GS, Kobilka BK, Scheinin M (1997) Genetic alteration of alpha2C-adrenoceptor expression in mice: influence on locomotor, hypothermic, and neurochemical effects of dexmedetomidine, a subtype-nonselective alpha 2-adrenoceptor agonist. Mol Pharmacol 51:36–46
Sanchez C, Bergqvist PB, Brennum LT, Gupta S, Hogg S, Larsen A, Wiborg O (2003) Escitalopram, the S-(+)-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with potent effects in animal models predictive of antidepressant and anxiolytic activities. Psychopharmacology (Berl) 167:353–362
Sander CY, Hooker JM, Catana C, Normandin MD, Alpert NM, Knudsen GM, Vanduffel W, Rosen BR, Mandeville JB (2013) Neurovascular coupling to D2/D3 dopamine receptor occupancy using simultaneous PET/functional MRI. Proc Natl Acad Sci U S A 110:11169–11174
Sandiego CM, Nabulsi N, Lin SF, Labaree D, Najafzadeh S, Huang Y, Cosgrove K, Carson RE (2013) Studies of the metabotropic glutamate receptor 5 radioligand [11C]ABP688 with N-acetylcysteine challenge in rhesus monkeys. Synapse 67:489–501
Sandiego CM, Gallezot JD, Lim K, Ropchan J, Lin SF, Gao H, Morris ED, Cosgrove KP (2015) Reference region modeling approaches for amphetamine challenge studies with [11C]FLB 457 and PET. J Cereb Blood Flow Metab 35:623–629. doi:10.1038/jcbfm.2014.237
Scheinin M, Sallinen J, Haapalinna A (2001) Evaluation of the alpha2C-adrenoceptor as a neuropsychiatric drug target studies in transgenic mouse models. Life Sci 68:2277–2285
Schiffer WK, Mirrione MM, Biegon A, Alexoff DL, Patel V, Dewey SL (2006) Serial microPET measures of the metabolic reaction to a microdialysis probe implant. J Neurosci Methods 155:272–284
Scott DJ, Stohler CS, Koeppe RA, Zubieta JK (2007) Time-course of change in [11C]carfentanil and [11C]raclopride binding potential after a nonpharmacological challenge. Synapse 61:707–714
Scott DJ, Stohler CS, Egnatuk CM, Wang H, Koeppe RA, Zubieta JK (2008) Placebo and nocebo effects are defined by opposite opioid and dopaminergic responses. Arch Gen Psychiatry 65:220–231
Selvaraj S, Turkheimer F, Rosso L, Faulkner P, Mouchlianitis E, Roiser JP, McGuire P, Cowen PJ, Howes O (2012) Measuring endogenous changes in serotonergic neurotransmission in humans: a [11C]CUMI-101 PET challenge study. Mol Psychiatry 17:1254–1260
Seneca N, Finnema SJ, Farde L, Gulyas B, Wikstrom HV, Halldin C, Innis RB (2006) Effect of amphetamine on dopamine D2 receptor binding in nonhuman primate brain: a comparison of the agonist radioligand [11C]MNPA and antagonist [11C]raclopride. Synapse 59:260–269
Shiue CY, Shiue GG, Mozley PD, Kung MP, Zhuang ZP, Kim HJ, Kung HF (1997) P-[18F]-MPPF: a potential radioligand for PET studies of 5-HT1A receptors in humans. Synapse 25:147–154
Shiue C, Pleus RC, Shiue GG, Rysavy JA, Sunderland JJ, Cornish KG, Young SD, Bylund DB (1998) Synthesis and biological evaluation of [11C]MK-912 as an alpha2-adrenergic receptor radioligand for PET studies. Nucl Med Biol 25:127–133
Shotbolt P, Tziortzi AC, Searle GE, Colasanti A, van der Aart J, Abanades S, Plisson C, Miller SR, Huiban M, Beaver JD, Gunn RN, Laruelle M, Rabiner EA (2012) Within-subject comparison of [11C]-(+)-PHNO and [11C]raclopride sensitivity to acute amphetamine challenge in healthy humans. J Cereb Blood Flow Metab 32:127–136
Shrestha SS, Liow JS, Lu S, Jenko K, Gladding RL, Svenningsson P, Morse CL, Zoghbi SS, Pike VW, Innis RB (2014) 11C-CUMI-101, a PET radioligand, behaves as a serotonin 1A receptor antagonist and also binds to alpha1 adrenoceptors in brain. J Nucl Med 55:141–146
Sibley DR, De Lean A, Creese I (1982) Anterior pituitary dopamine receptors. Demonstration of interconvertible high and low affinity states of the D-2 dopamine receptor. J Biol Chem 257:6351–6361
Sibon I, Benkelfat C, Gravel P, Aznavour N, Costes N, Mzengeza S, Booij L, Baker G, Soucy JP, Zimmer L, Descarries L (2008) Decreased [18F]MPPF binding potential in the dorsal raphe nucleus after a single oral dose of fluoxetine: a positron-emission tomography study in healthy volunteers. Biol Psychiatry 63:1135–1140
Skinbjerg M, Ariano MA, Thorsell A, Heilig M, Halldin C, Innis RB, Sibley DR (2009) Arrestin3 mediates D2 dopamine receptor internalization. Synapse 63:621–624
Skinbjerg M, Liow JS, Seneca N, Hong J, Lu S, Thorsell A, Heilig M, Pike VW, Halldin C, Sibley DR, Innis RB (2010) D2 dopamine receptor internalization prolongs the decrease of radioligand binding after amphetamine: a PET study in a receptor internalization-deficient mouse model. Neuroimage 50:1402–1407
Skinbjerg M, Sibley DR, Javitch JA, Abi-Dargham A (2012) Imaging the high-affinity state of the dopamine D2 receptor in vivo: fact or fiction? Biochem Pharmacol 83:193–198
Slifstein M, Narendran R, Hwang DR, Sudo Y, Talbot PS, Huang Y, Laruelle M (2004) Effect of amphetamine on [18F]fallypride in vivo binding to D2 receptors in striatal and extrastriatal regions of the primate brain: single bolus and bolus plus constant infusion studies. Synapse 54:46–63
Slifstein M, Kegeles LS, Xu X, Thompson JL, Urban N, Castrillon J, Hackett E, Bae SA, Laruelle M, Abi-Dargham A (2010) Striatal and extrastriatal dopamine release measured with PET and [18F] fallypride. Synapse 64:350–362
Slifstein M, van de Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, Hackett E, Girgis R, Ojeil N, Moore H, D’Souza D, Malison RT, Huang Y, Lim K, Nabulsi N, Carson RE, Lieberman JA, Abi-Dargham A (2015) Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry 72(4):316–324. doi:10.1001/jamapsychiatry.2014.2414
Smith DF, Stork BS, Wegener G, Ashkanian M, Jakobsen S, Bender D, Audrain H, Vase KH, Hansen SB, Videbech P, Rosenberg R (2009) [11C]Mirtazapine binding in depressed antidepressant nonresponders studied by PET neuroimaging. Psychopharmacology (Berl) 206:133–140
Sobrio F (2013) Radiosynthesis of carbon-11 and fluorine-18 labelled radiotracers to image the ionotropic and metabotropic glutamate receptors. J Labelled Comp Radiopharm 56:180–186
Staley JK, Van Dyck CH, Tan PZ, Al Tikriti M, Ramsby Q, Klump H, Ng C, Garg P, Soufer R, Baldwin RM, Innis RB (2001) Comparison of [18F]altanserin and [18F]deuteroaltanserin for PET imaging of serotonin2A receptors in baboon brain: pharmacological studies. Nucl Med Biol 28:271–279
Stepanov V, Zonker B, Gobbi L, Finnema SJ, Honer M, Borroni E, Halldin C (2013) Carbon-11 radiolabelling of RO6899880—a novel GABAA agonist. J Label Compd Radiopharm 56:S311
Stokes PR, Myers JF, Kalk NJ, Watson BJ, Erritzoe D, Wilson SJ, Cunningham VJ, Riano Barros D, Hammers A, Turkheimer FE, Nutt DJ, Lingford-Hughes AR (2014) Acute increases in synaptic GABA detectable in the living human brain: a [11C]Ro15-4513 PET study. Neuroimage 99:158–165
Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, Krystal JH, Nutt D, Barker GJ (2012) Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry 17:664–665
Sun W, Ginovart N, Ko F, Seeman P, Kapur S (2003) In vivo evidence for dopamine-mediated internalization of D2-receptors after amphetamine: differential findings with [3H]raclopride versus [3H]spiperone. Mol Pharmacol 63:456–462
Talbot PS, Frankle WG, Hwang DR, Huang Y, Suckow RF, Slifstein M, Abi-Dargham A, Laruelle M (2005) Effects of reduced endogenous 5-HT on the in vivo binding of the serotonin transporter radioligand 11C-DASB in healthy humans. Synapse 55:164–175
Tsukada H, Nishiyama S, Kakiuchi T, Ohba H, Sato K, Harada N (1999a) Is synaptic dopamine concentration the exclusive factor which alters the in vivo binding of [11C]raclopride?: PET studies combined with microdialysis in conscious monkeys. Brain Res 841:160–169
Tsukada H, Nishiyama S, Kakiuchi T, Ohba H, Sato K, Harada N, Nakanishi S (1999b) Isoflurane anesthesia enhances the inhibitory effects of cocaine and GBR12909 on dopamine transporter: PET studies in combination with microdialysis in the monkey brain. Brain Res 849:85–96
Tsukada H, Harada N, Ohba H, Nishiyama S, Kakiuchi T (2001) Facilitation of dopaminergic neural transmission does not affect [11C]SCH23390 binding to the striatal D1 dopamine receptors, but the facilitation enhances phosphodiesterase type-IV activity through D1 receptors: PET studies in the conscious monkey brain. Synapse 42:258–265
Tsukada H, Miyasato K, Kakiuchi T, Nishiyama S, Harada N, Domino EF (2002) Comparative effects of methamphetamine and nicotine on the striatal [11C]raclopride binding in unanesthetized monkeys. Synapse 45:207–212
Tsukada H, Nishiyama S, Fukumoto D, Ohba H, Sato K, Kakiuchi T (2004) Effects of acute acetylcholinesterase inhibition on the cerebral cholinergic neuronal system and cognitive function: functional imaging of the conscious monkey brain using animal PET in combination with microdialysis. Synapse 52:1–10
Tsukada H, Miyasato K, Nishiyama S, Fukumoto D, Kakiuchi T, Domino EF (2005a) Nicotine normalizes increased prefrontal cortical dopamine D1 receptor binding and decreased working memory performance produced by repeated pretreatment with MK-801: a PET study in conscious monkeys. Neuropsychopharmacology 30:2144–2153
Tsukada H, Nishiyama S, Fukumoto D, Sato K, Kakiuchi T, Domino EF (2005b) Chronic NMDA antagonism impairs working memory, decreases extracellular dopamine, and increases D1 receptor binding in prefrontal cortex of conscious monkeys. Neuropsychopharmacology 30:1861–1869
Udo de Haes JI, Bosker FJ, Van Waarde A, Pruim J, Willemsen AT, Vaalburg W, Den Boer JA (2002) 5-HT1A receptor imaging in the human brain: effect of tryptophan depletion and infusion on [18F]MPPF binding. Synapse 46:108–115
Udo de Haes JI, Harada N, Elsinga PH, Maguire RP, Tsukada H (2006) Effect of fenfluramine-induced increases in serotonin release on [18F]MPPF binding: a continuous infusion PET study in conscious monkeys. Synapse 59:18–26
Valette H, Bottlaender M, Dolle F, Coulon C, Ottaviani M, Syrota A (2005) Acute effects of physostigmine and galantamine on the binding of [18F]fluoro-A-85380: a PET study in monkeys. Synapse 56:217–221
Vallone D, Picetti R, Borrelli E (2000) Structure and function of dopamine receptors. Neurosci Biobehav Rev 24:125–132
Van der Graaf PH, Danhof M (1997) Analysis of drug-receptor interactions in vivo: a new approach in pharmacokinetic-pharmacodynamic modelling. Int J Clin Pharmacol Ther 35:442–446
Van der Mey M, Windhorst AD, Klok RP, Herscheid JD, Kennis LE, Bischoff F, Bakker M, Langlois X, Heylen L, Jurzak M, Leysen JE (2006) Synthesis and biodistribution of [11C]R107474, a new radiolabeled alpha2-adrenoceptor antagonist. Bioorg Med Chem 14:4526–4534
Van Laere KJ, Sanabria-Bohorquez SM, Mozley DP, Burns DH, Hamill TG, Van Hecken A, De Lepeleire I, Koole M, Bormans G, de Hoon J, Depre M, Cerchio K, Plalcza J, Han L, Renger J, Hargreaves RJ, Iannone R (2014) 11C-MK-8278 PET as a tool for pharmacodynamic brain occupancy of histamine 3 receptor inverse agonists. J Nucl Med 55:65–72
Varnäs K, Nyberg S, Halldin C, Varrone A, Takano A, Karlsson P, Andersson J, McCarthy D, Smith M, Pierson ME, Soderstrom J, Farde L (2011) Quantitative analysis of [11C]AZ10419369 binding to 5-HT1B receptors in human brain. J Cereb Blood Flow Metab 31:113–123
Varrone A, Sjoholm N, Eriksson L, Gulyas B, Halldin C, Farde L (2009) Advancement in PET quantification using 3D-OP-OSEM point spread function reconstruction with the HRRT. Eur J Nucl Med Mol Imaging 36:1639–1650
Vernaleken I, Klomp M, Moeller O, Raptis M, Nagels A, Rosch F, Schaefer WM, Cumming P, Grunder G (2013) Vulnerability to psychotogenic effects of ketamine is associated with elevated D2/3-receptor availability. Int J Neuropsychopharmacol 16:745–754
Villemagne VL, Wong DF, Yokoi F, Stephane M, Rice KC, Matecka D, Clough DJ, Dannals RF, Rothman RB (1999) GBR12909 attenuates amphetamine-induced striatal dopamine release as measured by [11C]raclopride continuous infusion PET scans. Synapse 33:268–273
Volkow ND, Wang GJ, Fowler JS, Logan J, Schlyer D, Hitzemann R, Lieberman J, Angrist B, Pappas N, MacGregor R et al (1994) Imaging endogenous dopamine competition with [11C]raclopride in the human brain. Synapse 16:255–262
Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Hitzemann R, Chen AD, Dewey SL, Pappas N (1997) Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature 386:830–833
Wagner HN Jr, Burns HD, Dannals RF, Wong DF, Langstrom B, Duelfer T, Frost JJ, Ravert HT, Links JM, Rosenbloom SB, Lukas SE, Kramer AV, Kuhar MJ (1983) Imaging dopamine receptors in the human brain by positron tomography. Science 221:1264–1266
Watabe H, Endres CJ, Breier A, Schmall B, Eckelman WC, Carson RE (2000) Measurement of dopamine release with continuous infusion of [11C]raclopride: optimization and signal-to-noise considerations. J Nucl Med 41:522–530
Watkins JC, Jane DE (2006) The glutamate story. Br J Pharmacol 147 Suppl:S100–8
Wilson AA, McCormick P, Kapur S, Willeit M, Garcia A, Hussey D, Houle S, Seeman P, Ginovart N (2005) Radiosynthesis and evaluation of [11C]-(+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b] [1,4]oxazin-9-ol as a potential radiotracer for in vivo imaging of the dopamine D2 high-affinity state with positron emission tomography. J Med Chem 48:4153–4160
Wing VC, Payer DE, Houle S, George TP, Boileau I (2014) Measuring cigarette smoking-induced cortical dopamine release: a [11C]FLB-457 PET study. Neuropsychopharmacology 40(6):1417–1427
Wong DF, Waterhouse R, Kuwabara H, Kim J, Brasic JR, Chamroonrat W, Stabins M, Holt DP, Dannals RF, Hamill TG, Mozley PD (2013) 18F-FPEB, a PET radiopharmaceutical for quantifying metabotropic glutamate 5 receptors: a first-in-human study of radiochemical safety, biokinetics, and radiation dosimetry. J Nucl Med 54:388–396
Woodward ND, Cowan RL, Park S, Ansari MS, Baldwin RM, Li R, Doop M, Kessler RM, Zald DH (2011) Correlation of individual differences in schizotypal personality traits with amphetamine-induced dopamine release in striatal and extrastriatal brain regions. Am J Psychiatry 168:418–426
Wu J, Ishikawa M, Zhang J, Hashimoto K (2010) Brain imaging of nicotinic receptors in Alzheimer’s disease. Int J Alzheimers Dis 2010:548913
Wyckhuys T, Verhaeghe J, Wyffels L, Langlois X, Schmidt M, Stroobants S, Staelens S (2013) N-acetylcysteine- and MK-801-induced changes in glutamate levels do not affect in vivo binding of metabotropic glutamate 5 receptor radioligand 11C-ABP688 in rat brain. J Nucl Med 54:1954–1961
Yamamoto S, Onoe H, Tsukada H, Watanabe Y (2007) Effects of increased endogenous serotonin on the in vivo binding of [11C]DASB to serotonin transporters in conscious monkey brain. Synapse 61:724–731
Yamamoto S, Nishiyama S, Kawamata M, Ohba H, Wakuda T, Takei N, Tsukada H, Domino EF (2011) Muscarinic receptor occupancy and cognitive impairment: a PET study with [11C](+)3-MPB and scopolamine in conscious monkeys. Neuropsychopharmacology 36:1455–1465
Yamanaka H, Yokoyama C, Mizuma H, Kurai S, Finnema SJ, Halldin C, Doi H, Onoe H (2014) A possible mechanism of the nucleus accumbens and ventral pallidum 5-HT1B receptors underlying the antidepressant action of ketamine: a PET study with macaques. Transl Psychiat 4:e342
Yatham LN, Liddle PF, Shiah IS, Lam RW, Adam MJ, Zis AP, Ruth TJ (2001) Effects of rapid tryptophan depletion on brain 5-HT2 receptors: a PET study. Br J Psychiatry 178:448–453
Zhang L, Villalobos A, Beck EM, Bocan T, Chappie TA, Chen L, Grimwood S, Heck SD, Helal CJ, Hou X, Humphrey JM, Lu J, Skaddan MB, McCarthy TJ, Verhoest PR, Wager TT, Zasadny K (2013) Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J Med Chem 56:4568–4579
Zhang L, Drummond E, Brodney MA, Cianfrogna J, Drozda SE, Grimwood S, Vanase-Frawley MA, Villalobos A (2014) Design, synthesis and evaluation of [3H]PF-7191, a highly specific nociceptin opioid peptide (NOP) receptor radiotracer for in vivo receptor occupancy (RO) studies. Bioorg Med Chem Lett 24:5219–5223
Zimmer L, Luxen A (2012) PET radiotracers for molecular imaging in the brain: past, present and future. Neuroimage 61:363–370
Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, Meyer CR, Koeppe RA, Stohler CS (2001) Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science 293:311–315
Zubieta JK, Ketter TA, Bueller JA, Xu Y, Kilbourn MR, Young EA, Koeppe RA (2003) Regulation of human affective responses by anterior cingulate and limbic mu-opioid neurotransmission. Arch Gen Psychiatry 60:1145–1153
Zubieta JK, Bueller JA, Jackson LR, Scott DJ, Xu Y, Koeppe RA, Nichols TE, Stohler CS (2005) Placebo effects mediated by endogenous opioid activity on mu-opioid receptors. J Neurosci 25:7754–7762
Acknowledgments
The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115008 of which resources are composed of EFPIA in-kind contribution and financial contribution from the European Union’s Seventh Framework Programme (FP7/2007–2013).
Sjoerd Finnema has received compensation as a member of a scientific advisory board of F. Hoffmann-La Roche. Mika Scheinin has contract research relationships with Orion Corporation, AstraZeneca, Hoffmann-La Roche, Lundbeck, and Pfizer, and has received speaker’s fees and research support from Orion Corporation. Jukka Sallinen and Mika Scheinin are listed as inventors on U.S. Patent no. 5,902,807: “Method for the treatment of mental illness in mammals and a composition therefore” (1999). Jukka Sallinen and Mo Shahid are employed by Orion Corporation. Lars Farde and Erik Wong either are, or have been, employed by AstraZeneca Pharmaceuticals, respectively. Sarah Grimwood is employed by Pfizer. Edilio Borroni is employed by Hoffmann-La Roche. Benny Bang-Andersen is employed by Lundbeck. The other authors have no conflicts of interest to declare.
We would like to acknowledge Adam Schwartz (Lilly), Svante Nyberg (Astra Zeneca), Jacob Nielsen (Lundbeck), Zoë Hughes (Pfizer), Eugenii A. (Ilan) Rabiner (Imanova and King’s College, London), and Paul Maguire (Novartis) for their contributions to NEWMEDS work-package 5.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Finnema, S.J., Scheinin, M., Shahid, M. et al. Application of cross-species PET imaging to assess neurotransmitter release in brain. Psychopharmacology 232, 4129–4157 (2015). https://doi.org/10.1007/s00213-015-3938-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-015-3938-6