
Abstract
Effective and compound-sparing methods to evaluate promising drug delivery systems are a prerequisite for successful selection of formulations in early development stages. The aim of the study was to develop a small-scale in situ method to determine drug release and supersaturation in highly concentrated suspensions of enabling formulations. Mesoporous magnesium carbonate (MMC), which delivers the drug in an amorphous form, was selected as a drug carrier. Five model compounds were loaded into the MMC at a 1:10 ratio using a solvent evaporation technique. The μDiss Profiler was used to study the drug release from MMC in fasted-state simulated intestinal fluid. To avoid extensive light scattering previously seen in particle-rich suspensions in the μDiss Profiler, an in-house-designed protective nylon filter was placed on the in situ UV probes. Three types of release experiments were conducted for each compound: micronized crystalline drug with MMC present, drug-loaded MMC, and drug-loaded MMC with 0.01% w/w hydroxypropyl methyl cellulose. The nylon filters effectively diminished interference with the UV absorption; however, the release profiles obtained were heavily compound dependent. For one of the compounds, changes in the UV spectra were detected during the release from the MMC, and these were consistent with degradation of the compound. To conclude, the addition of protective nylon filters to the probes of the μDiss Profiler is a useful contribution to the method, making evaluations of particle-rich suspensions feasible. The method is a valuable addition to the current ones, allowing for fast and effective evaluation of advanced drug delivery systems.
Similar content being viewed by others


INTRODUCTION
A major hurdle during drug development is the poor water solubility of discovery compounds; as many as 75% of the drugs under development suffer from low aqueous solubility (1). Methods to circumvent this, and hence increase the bioavailability of poorly water-soluble drugs (PWSDs), include the use of supersaturating drug delivery systems (SDDSs) (2). In these systems, the drug is typically delivered in its amorphous form, a high-energy form resulting in an increased apparent solubility (2,3). The addition of a crystallization inhibitor, e.g., a polymer, can stabilize the supersaturation. Common SDDSs are solubilizing formulations (e.g., self-emulsifying drug delivery systems promoting supersaturation during digestion), amorphous solid dispersions, and inorganic carriers loaded with the amorphous form of the drug (3). In 1992, mesoporous silica was introduced by the Mobil Oil company and since then several types of mesoporous material have been produced (4,5). Large pore volumes and high specific surface areas give these materials a variety of possible applications including improved drug dissolution of PWSDs (4,6). Once loaded into the carrier, the drug is in an amorphous form and recrystallization is inhibited through spatial constraints (7).
In 2013, a template-free mesoporous carrier, mesoporous magnesium carbonate (MMC), was introduced. The synthesis is performed under low temperatures, and no strong organic solvents nor surfactants are required during manufacturing (8). Magnesium carbonate is GRAS-listed by the FDA, and acute toxicity studies of MMC do not show any signs of toxicity in animals (9). MMC has also been shown to be a promising excipient in drug delivery systems by stabilizing the drugs in an amorphous state (10). MMC has proven to be compatible with a variety of drugs, indicating it as a new drug carrier with general applicability. Due to the diffusion-controlled release, the release rate can be tailored by altering the particle size of the MMC (11,12,13). Loading of MMC has so far been conducted by simple solvent evaporation, and studies of release from drug-loaded MMC have previously been performed using the traditional USP 2 dissolution bath (10,12). This does, however, demand significant amounts of material. As the sampling must also be done manually, it also limits the possibility of studying rapid release and time-sensitive events. The μDiss Profiler, a small-scale dissolution apparatus, enables in situ UV measurements of drug dissolution. This apparatus has been successfully used to determine dissolution rates and intrinsic dissolution rates (IDR) from powder, controlled suspensions, and compressed discs (14,15,16). It has also been used to study supersaturation obtained from SDDSs and release from drug-loaded MMC (11,17). However, particle-rich suspensions of MMC produce significant light scattering which interferes with the UV absorption measurements. To overcome this issue, we previously proposed a method to estimate the initial release rate from high concentrations of drug-loaded MMC in which the amount of MMC in the assay is gradually increased. The release rate is linearly dependent on the amount of drug-loaded MMC added, enabling extrapolation to higher concentrations (11). This provides an insight into the initial release rate in high concentrations of loaded carrier, but it does not provide information about the full release event, such as the degree of supersaturation and time to recrystallization. Therefore, we targeted the development of a methodology that would enable studies of both the immediate release and degree of supersaturation obtained from drug-loaded MMC. A small-scale in situ method was developed making use of the μDiss Profiler and protective filters. The method may also be applicable for studies of release from other complex formulations that produce particle-rich suspensions. It should be noted that the pore size of the nylon filter used herein (11 μm) suggests that the method might not be suitable for performance assessment of nanosuspensions.
MATERIAL AND METHODS
Materials
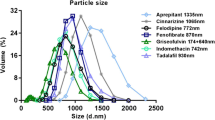
Five PWSDs were selected and MMC was synthesized in-house. MMC has been extensively characterized as a drug carrier in previous work and had a mean pore size of 5 nm, a specific surface area of 650 m2/g, and a total pore volume of 0.81 cm3/g (11,18). Hydroxypropyl methyl cellulose (HPMC) (Methocel E4M Premium CR) was generously donated by Colorcon (Dartford, UK). Fenofibrate, tolfenamic acid, tamoxifen, hydrocortisone, and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St Louis, MO, US). Tamoxifen and ketoconazole were purchased from Toronto Research Chemicals (Toronto, CAN). All drugs were used as received. The nylon net filter, 11-μm pore size, used in the protective filters were purchased from Merck Millipore (Billerica, MA, US). FaSSGF/FaSSIF/FeSSIF powder was purchased from biorelevant.com (Croydon, UK).
Micronization of the Crystalline Material
Each crystalline drug compound (461–500 mg) was micronized in a planetary ball mill (Model PM 100 Retsch, Germany), to reduce particle size and thereby increase the dissolution rate. The drug was added, together with ten small steel beads (Ø 5 mm) to the milling bowl, and milled for 20 min at 600 rpm.
Loading of the Carrier
Loading of the MMC was performed using a modified soaking method previously described by Zhang et al. (10). The drugs were loaded into the MMC at a 1:10 ratio by dissolving 1 mg/mL drug in ethanol, and then adding 10 mg/mL MMC to the solution. The loading degree used was chosen to ensure that the all compounds adsorbed into the MMC were in an amorphous state, and was furthermore intentionally kept low to enable challenging conditions (i.e., need for high amounts of MMC particles) during the release studies. The suspension was stirred on a magnetic stirrer at 500 rpm for 24 h. The ethanol was then evaporated at 70 °C during a time period of 24 h.
Characterization of Loaded Carrier
The thermal behavior of the drugs and drug-loaded MMC was determined with a Q2000 Differential Scanning Calorimeter (TA Instrument Co., USA). The DSC cell was calibrated with indium (melting temperature, Tm = 156.59 °C and heat of fusion, Hf = 28.57 J/g) and then purged with 50 mL/min of nitrogen. For determining the Tm of the crystalline drug, 2.2–4.3 mg was weighed into an aluminum pan that was then sealed with an aluminum lid containing pinholes. The samples were equilibrated at 0 °C and thereafter heated at 10 °C/min to 29–53 °C above the Tm of the drug. To increase the sensitivity, and confirm the absence of crystallinity, modulated DSC (mDSC) was performed on the drug-loaded MMC. In these measurements, 9.2–10.2 mg of drug-loaded MMC was weighed into the aluminum pans and each sample was equilibrated at 0 °C. It was then modulated at ± 0.5 °C every 60 s and heated at 1 °C/min to a temperature 20–53 °C above the Tm of the drug. Onset values of Tm are reported.
Drug Release Studies from Loaded MMC
Preparation of Dissolution Media
Fasted-state simulated intestinal fluid version 1 (FaSSIF) was prepared in accordance with the instructions from the manufacturer. The ready-to-use simulated intestinal fluid powder was dissolved in phosphate buffer (PhB), pH 6.5. To increase the complexity of the system and allow for studies of stabilization of supersaturation, a medium was prepared in which 0.01% w/w HPMC was pre-dissolved in PhB. Two modified versions of FaSSIF, at pH 7.5 and 8.5, were also made by adjusting the pH of the PhB (originally set to pH 6.5).
Evaluation of the Nylon Filters
All release experiments were conducted with the μDiss Profiler (pION, MA) in accordance with previously described protocols (19). The solubility of all compounds was determined in FaSSIF pH 6.5. To explore the pH dependency, the solubility of tolfenamic acid in FaSSIF pH 7.5 was also determined, as was the solubility of tamoxifen at pH 7.5 and 8.5. The path lengths (2–20 mm) of the in situ UV probes were selected on the basis of the drug solubility in the selected medium and the expected degree of supersaturation. The medium was preheated to 37 °C prior to the release determinations and each channel was calibrated using a separate calibration curve. The solubility studies were performed by adding an excess of drug to the vials and then 15 mL preheated FaSSIF, upon which the determination started. To evaluate the capacity of the protective filters to diminish interference with the UV absorbance measurements that we have previously observed (see, e.g., Supplementary data, Fig. S1), three setups were used: micronized crystalline drug with MMC, drug-loaded MMC, and drug-loaded MMC in the presence of 0.01% w/w HPMC (Fig. 1). To ensure that the conditions were consistent throughout the study, equivalent amounts of MMC were used in the preparation of the standard curve. A fixed amount of sample (54.5 ± 0.1 mg MMC and 5.5 ± 0.1 mg micronized drug or 60 ± 0.1 mg drug-loaded MMC, i.e., 4 mg/mL material) was weighed into the vials, and the nylon filter was placed on the in situ UV probes to protect them from the accumulation of particles. The protective nylon filters (4 × 8 cm) were folded and the edges were heat-welded together, creating a tube. The determinations started upon addition of 15 mL preheated dissolution medium. For the hydrocortisone-loaded MMC in FaSSIF and FaSSIF containing 0.01% w/w HPMC, 120 ± 0.1 mg (i.e., 8 mg/mL material) drug-loaded MMC was used for the studies of supersaturation. Analysis was performed using a second derivative of the spectra and a single wavelength.

Illustration of the dissolution/release studies performed for each drugs. (a) Micronized crystalline drug in FaSSIF. (b) Micronized crystalline drug and MMC in FaSSIF. (c) Drug-loaded MMC in FaSSIF. (d) Drug-loaded MMC in FaSSIF containing 0.01% w/w HPMC. In studies B–D, protective nylon filters were placed on the in situ UV probes to avoid interference of the UV read
Determination of Degradation Products by UPLC-MS
A shift in the UV spectrum occurred during the release of fenofibrate-loaded MMC, so UPLC-MS was used to determine the origin of the shift. Two samples were weighed into Eppendorf tubes, one with 8 mg fenofibrate-loaded MMC and the other with 0.7 mg crystalline fenofibrate and 7.2 mg MMC. Two milliliters of Milli-Q water was added to the tubes, which were then placed on a plate shaker at 37 °C for 24 h. The supernatants were recovered by centrifugation at 37 °C for 15 min at 23,000×g (Heraeus Megafuge 8R, ThermoScientific) and analyzed by obtaining a full MS spectrum (QTRAP 6500, AB Sciex). A stock solution of fenofibrate dissolved in DMSO was used as reference. Separation was performed on an Agilent 1290 LC-system, with a Waters BEH C18 2.1 × 50 mm (1.7 μm) column, and the run was made in positive mode. The mobile phase consisted of (A) 5% acetonitrile, 0.1% formic acid, and 94.9% Milli-Q water; and (B) 95% acetonitrile, 0.1% formic acid, and 4.9% Milli-Q water. A constant flow rate of 0.5 mL/min was used for the gradient elution as follows: A was constant at 99% for 1.0 min, then decreased linearly to 20% for 7.30 min. A was thereafter decreased to 10% for 0.2 min, kept constant for 0.5 min, and then increased to 99% for 0.5 min.
Statistical Analysis
The mean and standard deviation were determined for all measurements. The area under the curve (AUC) was calculated for the release profiles using GraphPad Prism 7.03, and one-way ANOVA analysis was performed on the AUC values for each compound (GraphPad Software, Inc., San Diego, US). Tukey’s multiple comparison test with a single pooled variance was performed to determine differences between the three setups. Limit for significance was set to p < 0.05.
RESULTS AND DISCUSSION
Characterization of the Loaded Carrier
The PWSDs were selected on the basis of their physicochemical properties. Tm values for all compounds are shown in Table I. No Tm was detected for compounds loaded into the MMC (Supplementary data, Fig.S2). This lack of drug crystallinity when incorporated to the MMC is consistent with previously published results (10,11). For all compounds, a solvent (water) endotherm was observed in the thermograms (Supplementary data, Fig.S2). This indicates that the drug-loaded MMC had absorbed some water during storage. However, the absorbed water did not seem to affect the stability of the system, since no recrystallization was detected.
Drug Release Studies from Loaded MMC
Evaluation of Nylon Filters
The protective filters diminished the interference from particle concentrations of 4–8 mg/mL (Fig. 2). Thus, satisfying release profiles could be obtained for all compounds except fenofibrate when the filters were used (i.e., no scattering effects were observed in the absorption spectra). The release from the drug-loaded MMC was rapid for all other compounds (Fig. 2), and the addition of the filters to the UV probes did not seem to cause any significant delay in the determination of concentration. Diffusion barrier effects on drug release have been previously studied in the μDiss Profiler by Ahnfelt et al. (20). They saw a correlation between equilibrium time and pore size, where smaller pore sizes increased the time to reach equilibrium. This stresses the issue of selecting the correct pore size, depending on the objective of the study. The pore size of the nylon filter used was in micrometer range (11 μm), and the method described herein might therefore not be suitable for performance assessment of suspensions containing nanoparticles. Based on our release profiles (Fig. 2), the pore size used was large enough to allow for diffusion of solvent and dissolved drug, but small enough to block most of the MMC particles. The particle size of the MMC/drug-loaded MMC was polydisperse, and smaller particles might have penetrated the filter, but these did not seem to affect the measurements.

Release/dissolution profiles of compounds investigated. Fenofibrate excluded due to changes in UV spectrum of compound. Supersaturation is seen for both ketoconazole and hydrocortisone. No conclusions can be made with regard to supersaturation for tolfenamic and tamoxifen, due to the increase in pH resulting from the dissolution of MMC. The stabilizing effect of the HPMC is clearly seen in case of ketoconazole, whereas only a slight increase in concentration was seen for hydrocortisone and tamoxifen when HPMC was added to the FaSSIF
An important aspect when selecting filter material is the potential adsorption of the drug and/or carrier. This work focused on supersaturated systems, and the degree of adsorption of the drug to the filters was not evaluated. For one of the compounds studied (hydrocortisone), non-saturated conditions were applied. The micronized hydrocortisone with MMC present reaches a plateau equivalent to the amount of drug added, 360 μg/mL (i.e., 5.5 mg drug in 15 mL FaSSIF) (Fig. 2b). This indicates limited adsorption of this lipophilic drug onto the filter. However, compound-specific adsorption to the filter material needs to be evaluated when, e.g., sink conditions are used.
The release profiles from the drug-loaded MMCs were compound dependent. Supersaturation was observed during the release of ketoconazole and hydrocortisone (Fig. 2a, b). The degree of supersaturation was higher for ketoconazole than hydrocortisone and can be explained by the amount that had to be added in relation to the Sapp of the drug. For ketoconazole, amounts equivalent to 12 × Sapp were used, whereas only 1.6 × Sapp was used for hydrocortisone, because of the large difference in solubility between the two drugs. The 15.1-fold higher solubility of hydrocortisone requires large amounts of drug to be added into the vial, which means that the number of MMC particles must also be significantly higher to study supersaturation. The higher supersaturation level of ketoconazole compared to hydrocortisone also explains why ketoconazole crystallizes more rapidly.
The HPMC in the dissolution media significantly stabilized the supersaturation of ketoconazole, which was reflected by an increase in AUC (Fig. 3a). (This is consistent with the results of Yang et al. (11), who studied the stabilization effects of HPMC in concert with MMC). The precipitation kinetics of ketoconazole were slower when HPMC was present, suggesting that the HPMC mainly prevented crystal growth rather than nucleation (Fig. 2a). The same trend was seen for hydrocortisone and tamoxifen, although the correlation was weaker (Figs. 2b, d and 3b, c). To more thoroughly investigate the capacity of HPMC to inhibit crystal growth of hydrocortisone, higher degrees of supersaturation need to be explored. For tolfenamic acid, no significant difference in AUC was seen when HPMC was added to the dissolution media (Fig. 3d).

(a–d) Area under the curve (AUC) for dissolution/release studies. Drug+MMC: Micronized drug with MMC present (blue bars). Drug-MMC: Drug-loaded MMC in FaSSIF pH 6.5 (red bars). Drug-MMC with HPMC: Drug-loaded MMC in FaSSIF pH 6.5 with 0.01% w/w HPMC (green bars). For ketoconazole (KETO), hydrocortisone (HYDRO), and tamoxifen (TAM), the AUC increased when HPMC was added to the dissolution media, indicating that the HPMC stabilized the supersaturation for these compounds. No statistical significant difference in AUC was seen for any of the runs with tolfenamic acid (TOL).
The release of one of the drugs, hydrocortisone, was incomplete during the time course of the experiment (Fig. 2b), even though the amount added was lower than the solubility of the compound. About 360 μg/mL of hydrocortisone (9% of 4 mg/mL drug-loaded MMC) was added, but only about 320 μg/mL was released (Supplementary data, Tab. S1). Incomplete release from MMC has also been reported by Zhang et al., who speculate that it can be due to the diffusion time through different particle sizes (12). The MMC in our study was polydisperse in terms of size, and this may have affected the amount released from the MMC.
In the experiments where MMC was present, the pH increased due to partial dissolution of the MMC. This also affected the ionic strength and possibly the vesicular structures within the FaSSIF. The buffering capacity of FaSSIF is ~ 12 mM/dpH (21), and a stronger buffer would have been needed to stabilize the pH in this case. The increase in pH significantly influenced the final solubility and supersaturation of tamoxifen and tolfenamic acid, so their solubility was also determined in FaSSIF at pH 7.5. For tamoxifen, the solubility was additionally measured at pH 8.5. For tolfenamic acid, a weak acid with pKa of 3.7, the solubility clearly increased with increasing pH (Fig. 2c). The pH in the dissolution media after the experiment was 8.3–9.4, increasing the crystalline solubility from 66.7 ± 0.8 μg/mL in FaSSIF pH 6.5 to 317.6 ± 4.3 μg/mL in FaSSIF pH 7.5. Because the Sapp at pH 7.5 is almost as high as the amount of drug added, no conclusions can be made about the degree of supersaturation. Indeed, at the higher pH (8.3–9.4), no supersaturation was present. In contrast, tamoxifen, which is a weak base with pKa of 8.5, decreased in solubility from 152.9 ± 10.8 μg/mL at pH 6.5 to 32.1 ± 0.7 μg/mL at pH 8.5. As seen in Fig. 2d, the concentration in the media during the release from the MMC does not even reach the solubility for pH 6.5, but rapidly precipitates to concentration values lower than the Sapp at pH 8.5. Ketoconazole, which has a relatively low pKa (Table I), decreased only slightly in solubility at the higher pH values. To what extent these pH changes would occur in vivo remains to be shown, but the buffering capacity and the composition of the intestinal fluid clearly plays a role in the potential of the pH to drift. The buffering capacity in the intestinal fluid is lower than that in the PhB commonly used in FaSSIF (22), and an increase in local pH in the intestine might be seen due to the dissolution of MMC. The buffering agent in the intestine, bicarbonate, is also of significance when dissolution of MMC is discussed as this might affect to what extent the carrier will dissolve. To obtain a more accurate in vivo prediction of the release from MMC, a study performed in a bicarbonate buffer would be of relevance (22).
Determination of Degradation Products by UPLC-MS
It was not possible to determine release profiles for fenofibrate-loaded MMC, as the UV spectra shifted over time in the release study of fenofibrate (Fig. 4). Since the spectra of fenofibrate and those of the new species formed overlapped at all wavelengths, the concentration of fenofibrate could not be determined with this method. Interestingly, this pattern was not observed in the dissolution of micronized fenofibrate with MMC present, indicating that this shift was specific for the fenofibrate-loaded MMC. Fenofibrate is a prodrug, with an ester function prone to alkaline hydrolysis, leading to the formation of fenofibric acid in a basic environment (23). Since the UV shift was only observed when fenofibrate was loaded into the MMC, it can be speculated that degradation took place within the pores of the MMC, due to the locally high pH in the pore structure of the MMC. To further evaluate if this UV-shift could have been due to degradation, and not a result of particles inducing interferences with the UV absorption, a simplified release study was performed and the samples were analyzed with UPLC-MS. In a parent scan of fenofibrate in DMSO, the retention time (tR) for fenofibrate was 7.55 min (m/z 361) (Supplementary data, Fig.S3a). In the sample of fenofibrate-loaded MMC, no peaks appeared at this time point; however, a new peak was detected with a tR of 5.80 min (Supplementary data, Fig.S3b). This peak corresponded to a mass of 319 m/z, a value consistent with an alkaline ester hydrolysis of fenofibrate into fenofibric acid (23). No peaks were detected in the sample containing crystalline fenofibrate and pure MMC. This may be a result of two properties: (i) dissolution of fenofibrate is too slow for the time scale of this experiment, and (ii) the pH is higher locally inside in the MMC pores than when the fenofibrate is only in contact with MMC (outer) surface. The protective filters allowed detection of the shift in the UV spectra even when there were high amounts of particles. This makes the method useful for determination of various processes during drug release from particle-rich media.

UV absorbance spectra of fenofibrate-loaded MMC obtained from the μDiss Profiler after 2 min (blue line) vs 60 min (purple line). A shift in the UV spectrum is seen during the release study
CONCLUSIONS
Small-scale methods to evaluate promising drug delivery systems are a prerequisite for successful selection of formulations in early development stages. In this work, we present a method with the μDiss profiler and protective nylon filters to study release from particle-rich suspensions of a complex carrier-mediated system. The setup allows for determination of immediate release and studies of supersaturation in suspensions of 4–8 mg/mL of drug-loaded carrier. The effect of a stabilizer, HPMC, could also be determined. During the drug release of one compound, fenofibrate, a change in the UV spectra was detected, which also showcases the versatility of the method. In conclusion, the method is applicable to studies of a variety of compounds and formulations, filling an existing gap in the current evaluation of promising drug delivery systems.
References
Williams HD, Trevaskis NL, Charman SA, Shanker RM, Charman WN, Pouton CW, et al. Strategies to address low drug solubility in discovery and development. Pharmacol Rev. 2013;65(1):315–499.
Fong SYK, Bauer-Brandl A, Brandl M. Oral bioavailability enhancement through supersaturation: an update and meta-analysis. Expert Opin Drug Deliv. 2017;14(3):403–26.
Brouwers J, Brewster ME, Augustijns P. Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci. 2009;98(8):2549–72.
Xu W, Riikonen J, Lehto V-P. Mesoporous systems for poorly soluble drugs. Int J Pharm. 2013;453(1):181–97.
Kresge C, Leonowicz M, Roth W, Vartuli J, Beck J. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature. 1992;359(6397):710–2.
Moritz M, Geszke-Moritz M. Mesoporous materials as multifunctional tools in biosciences: principles and applications. Mater Sci Eng C. 2015;49:114–51.
Laitinen R, Löbmann K, Strachan CJ, Grohganz H, Rades T. Emerging trends in the stabilization of amorphous drugs. Int J Pharm. 2013;453(1):65–79.
Forsgren J, Frykstrand S, Grandfield K, Mihranyan A, Strømme M. A template-free, ultra-adsorbing, high surface area carbonate nanostructure. PLoS One. 2013;8(7):e68486.
Frykstrand S, Forsgren J, Zhang P, Strømme M, Ferraz N. Cytotoxicity, in vivo skin irritation and acute systemic toxicity of the mesoporous magnesium carbonate Upsalite®. J Biomater Nanobiotechnol. 2015;6(4):257–66.
Zhang P, Forsgren J, Stromme M. Stabilisation of amorphous ibuprofen in Upsalite, a mesoporous magnesium carbonate, as an approach to increasing the aqueous solubility of poorly soluble drugs. Int J Pharm. 2014;472(1–2):185–91.
Yang J, Alvebratt C, Zhang P, Zardán Gómez de la Torre T, Strømme M, Bergström CAS, Welch K. Enhanced release of poorly water-soluble drugs from synergy between mesoporous magnesium carbonate and polymers. Int J Pharm 2017;525(1):183–190.
Zhang P, Zardán Gómez de la Torre T, Forsgren J, Bergström CAS, Strømme M. Diffusion-controlled drug release from the mesoporous magnesium carbonate Upsalite®. J Pharm Sci 2015;105(2):657–663.
Zhang P, Zardán Gómez de la Torre T, Welch K, Bergström CAS, Strømme M. Supersaturation of poorly soluble drugs induced by mesoporous magnesium carbonate. Eur J Pharm Sci 2016;93:468–474.
Tsinman K, Avdeef A, Tsinman O, Voloboy D. Powder dissolution method for estimating rotating disk intrinsic dissolution rates of low solubility drugs. Pharm Res. 2009;26(9):2093–100.
Avdeef A, Tsinman O. Miniaturized rotating disk intrinsic dissolution rate measurement: effects of buffer capacity in comparisons to traditional Wood’s apparatus. Pharm Res. 2008;25(11):2613–27.
Andersson SBE, Alvebratt C, Bergström CAS. Controlled suspensions enable rapid determinations of intrinsic dissolution rate and apparent solubility of poorly water-soluble compounds. Pharm Res. 2017:1–12.
Edueng K, Mahlin D, Larsson P, Bergström CAS. Mechanism-based selection of stabilization strategy for amorphous formulations: insights into crystallization pathways. J Control Release. 2017;256:193–202.
Cheung O, Zhang P, Frykstrand S, Zheng H, Yang T, Sommariva M, et al. Nanostructure and pore size control of template-free synthesised mesoporous magnesium carbonate. RSC Adv. 2016;6(78):74241–9.
Andersson SBE, Alvebratt C, Bevernage J, Bonneau D, da Costa Mathews C, Dattani R, et al. Interlaboratory validation of small-scale solubility and dissolution measurements of poorly water-soluble drugs. J Pharm Sci. 2016;105(9):2864–72.
Ahnfelt E, Sjögren E, Axén N, Lennernäs H. A miniaturized in vitro release method for investigating drug-release mechanisms. Int J Pharm. 2015;486(1):339–49.
Klein S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010;12(3):397–406.
Krieg BJ, Taghavi SM, Amidon GL, Amidon GE. In vivo predictive dissolution: comparing the effect of bicarbonate and phosphate buffer on the dissolution of weak acids and weak bases. J Pharm Sci.104(9):2894–904.
Rath NP, Haq W, Balendiran GK. Fenofibric acid. Acta Crystallogr C. 2005;61(2):o81–o4.
Acknowledgments
The authors thank Dr. André Mateus for his assistance with the UPLC-MS measurements and Erik Petersson for his help with the initial experimental work. Colorcon (Dartford, UK) is acknowledged for their generous donation of the Methocel E4M Premium CR used in the study.
Funding
This study was financially supported by the Swedish Research Council (grant no. 2014-3929).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclaimer
Maria Strømme declares she is the inventor of MMC and holds shares in the company Disruptive Materials.
Additional information
Guest Editor: Sandra Klein
Electronic supplementary material
Table S1
(DOCX 16 kb)
Figure S1
Dissolution of crystalline tolfenamic acid with 2 mg/mL MMC present. The dissolution study was performed in a phosphate buffer pH 6.8 with 0.001% w/w HPMC pre-dissolved in the media. No protective filters were placed on the in situ UV-probes, and significant interference with the UV read can be seen. (PNG 7 kb)
Figure S2
Thermograms from mDSC for drug-loaded MMC. (a) Ketoconazole-loaded MMC, (b) Hydrocortisone-loaded MMC, (c) Tamoxifen-loaded MMC, (d) Tolfenamic acid-loaded MMC, (e) Fenofibrate-loaded MMC. No crystallinity was detected for any of the drug-loaded MMC at this loading degree. A water endotherm can be seen in all thermograms. (PNG 25 kb)
Figure S3
Mass spectra obtained from release studies of fenofibrate using UPLC-MS in (a) reference sample (crystalline fenofibrate in DMSO) and (b) fenofibrate-loaded-MMC. A peak was detected at retention time 7.55 min in the reference sample consistent with fenofibrate (m/z 361 Da). This was not seen after release from the fenofibrate-loaded MMC, however a new peak appeared at retention time 5.80 min with a m/z consistent with fenofibric acid (m/z 319 Da). (PNG 307 kb)
ESM 1
(PNG 303 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Alvebratt, C., Cheung, O., Strømme, M. et al. A Modified In Situ Method to Determine Release from a Complex Drug Carrier in Particle-Rich Suspensions. AAPS PharmSciTech 19, 2859–2865 (2018). https://doi.org/10.1208/s12249-018-1024-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-018-1024-1