
Abstract
Background
Chromosome microdissection is one of the most important techniques in molecular cytogenetic research. Cotton (Gossypium Linnaeus, 1753) is the main natural fiber crop in the world. The resistance gene analog (RGA) cloning after its single chromosome microdissection can greatly promote cotton genome research and breeding.
Results
Using the linker adaptor PCR (LA-PCR) with the primers of rice disease-resistance homologues, three nucleotide sequences PS016 (KU051681), PS054 (KU051682), and PS157 (KU051680) were obtained from the chromosome Ah01 of upland cotton (cv. TM-1). The Blast results showed that the three sequences are the nucleotide binding site-leucine rich repeat (NBS-LRR) type RGAs. Clustering results indicated that they are homologous to these published RGAs. Thus, the three RGAs can definitely be confirmed as NBS-LRR class of RGAs in upland cotton.
Conclusions
Using single chromosome microdissection technique, DNA libraries containing cotton RGAs were obtained. This technique can promote cotton gene cloning, marker development and even the improvement of cotton genome research and breeding.
Similar content being viewed by others


Background
Chromosome microdissection is one of the most important techniques in molecular cytogenetic research. Specific chromosome or chromosomal sections are isolated using a glass needle or laser under a microscope, and then are enzymatically digested and amplified to construct DNA library of a single chromosome or chromosomal section. Research focusing on a single chromosome or a chromosomal subsection can greatly reduces subsequent work, such as identifying, screening and minimizing the whole genome screening. This technique has been widely used in Drosophila, humans and many other animals since its establishment [1,2,3,4,5,6,7,8] Subsequently, the technique has been widely adapted to apply in herbaceous plants including barley, wheat, rice, and tomato [9,10,11,12,13,14,15,16,17,18] and woody plants such as pomelo and poplar [19, 20].
Plants have developed defensive mechanisms to protect themselves from pathogen infection through a number of evolutionary processes. The gene-for-gene hypothesis proposed by Flor is based on the interactions between pathogenic fungi and host plants and constitutes the theoretical basis of cloning avirulence genes from pathogens and resistance genes (R genes) from plants [21]. So far, many R genes have been cloned from different host plants using positional cloning and transposon tagging methods. However, considering the large number of physiological races of pathogens, transposon tagging and positional cloning methods are clearly inefficient. Thus new strategies and methods should be adopted to accelerate the cloning of disease R genes. Due to the conserved domains of R genes, homologous sequence amplification or the homologous sequence-based candidate gene approach would be a good choice; actually, these techniques have been quickly adopted by the scientific community. A great progress has been made in recent years for obtaining disease RGAs from many plant species [22,23,24,25,26,27,28]. Additionally, some of these RGAs were used as probes for linkage analysis and positioning [22,23,24, 27].
As the primary natural fiber crop, cotton (Gossypium hirsutum) plays an important role in the world’s economy. However, cotton cells contain large amounts of secondary metabolites, and their chromosomes are small in size and nearly identical to each other. These prevent in somehow to well prepare the chromosomes from the cells and clearly distinguish them from their karyotypes, and thus cytogenetic research of cotton is still lagging behind other plant species, such as rice and wheat. As a typical tetraploid plant species [29,30,31,32,33,34,35], there are two sub-genome (A1A1D1D1, 2n = 4× = 52) and high number of nucleotide sequence repeats in cotton genome. There are greater uncertainties in interpreting whole genome while assembling or annotating [33, 34]. Microdissection of a single chromosome or its subsections using direct micromanipulation techniques and gene microcloning through molecular biology should be one easy way to slove this problem. However, currently, there is only one report about single chromosome microdissection that was from somatic cells [36], there is no any report on chromosome microdissection from pollen mother cells (PMC) and on gene microcloning from single chromosome.
There are many important genes are closely related to disease resistance, fiber development, fiber quality and yield in the Ah01 chromosome of TM-1 upland cotton [37,38,39]. In this study, the Ah01chromosome was microdissected from the Ah01 monosome materials derived from TM-1 (a genetically standard line of upland cotton) using the laser method. A DNA pool was constructed from the single chromosome by amplifying DNA using linker adaptor polymerase chain reaction (LA-PCR). RGAs from this chromosome were then cloned.
Methods
Plant materials
A accession of Ah01 monosome, developed from the genetically standard line of upland cotton at Texas A&M University (U.S.) [40], was used as the primary plant materials. The accession was grown in the Greenhouses at the Institute of Cotton Research, Chinese Academy of Agricultural Sciences, (ICR-CAAS) (Anyang, Henan, China) and is also maintained in the National Wild Cotton Nursery located at Sanya City, Hainan Island, China.
SSR markers and primers
The chromosome-Ah01-specific BAC clone 52D06 was provided by Professor Tianzhen Zhang of Nanjing Agricultural University. The primers of corresponding simple sequence repeat (SSR) marker BNL3580 (F primer: CTTGTTTACATTCCCTTCTTTATACC; R primer: CAAAGGCGAACTCTTCCAAA), degenerate specific primers P1 (5′-GATCCTGAGCTCGAATTCGACCC-3′) and P2 (5′-GGGTCGAATTCGAGCTCAG-3′) were synthesized by Shanghai Sangon Biotech Inc. [41].
Preparation of mitotic metaphase chromosomes
Mitotic metaphase chromosomes were prepared according to a previous report [36] with a few modifications. The slides prepared were kept at −20 °C for a long-term storage or at 4 °C for a short period storage. Slides were baked at 60 °C overnight immediately before use.
Preparation of film-slides
Sampling, fixing, and enzymatic hydrolysis of flower buds were performed according to the protocol of Peng et al. [36]. Enzymatically digested anthers were smeared on film-slides as previously described [36].
Microdissection of single chromosome and LA-PCR amplification
Single chromosome was microdissected using the CellCutPlus Laser micromanipulation system (MMI Company, Swiss) and LA-PCR amplification was conducted as previously described [36]. Positive (~10 pg of genomic DNA added to the initial template) and negative controls (no genomic DNA added to the initial template) were also set up.
Agarose electrophoresis
Two rounds of LA-PCR products were separated through electrophoresis with 1% agarose at 100 V for 30 min. LA-PCR products were observed and photographed under UV light after 40 min staining with ethidium bromide.
Southern hybridization
Southern hybridization was conducted with PCR products, partially digested genomic DNA, positive control (PCR product from genomic DNA as template) and negative control (no template PCR reaction) [42].
SSR amplification
The amplified pool of Ah01 chromosomes and second LA-PCR products were amplified using the chromosome- Ah01- specific SSR primer respectively. The amplified products were checked by polyacrylamide gel electrophoresis (PAGE).
Fluorescence in situ hybridization
Dual-color FISH (fluorescence in situ hybridization) and the detection of metaphase chromosome specimens were performed according to a previous [36]. The second round of LA-PCR products labeled with DIG (Digoxigenin-11-dUTP, Roche) and specific Biotin (Biotin-16-dUTP) labeled bacterial artificial chromosome (BAC) clones (52D06) were used as probes, which were detected by Anti-Digoxigenin-Rhodamine (red) and FITC-Anti-Biotin (green) (Roche Diagnostics, USA), respectively. Cot-1 DNA was used to pre-hybridize for blocking the repetitive sequences. Chromosomes were counterstained by 4′, 6-diamidino-2-phenylindole (DAPI) in VECTASHIELD anti-fade solution (Vector Laboratories, Burlingame, CA). The hybridization signals were observed using a fluorescence microscope with a change-coupled device (CCD) camera (Zeiss Axiokop2 plus). The images were adjusted using Adobe Photoshop CS3 software.
Cloning and analysis of RGAs
RGA sequences were obtained by PCR with the Ah01 chromosome second round LA-PCR product as template and P1 and P2 as degenerate specific primers. Positive control (about 10 pg of genomic DNA was added to the initial substrate) and negative control (no template) reactions were also performed. The PCR products were examined by agarose gel electrophoresis and Southern hybridization. The target bands of the PCR products were recovered. Positive clones were obtained, and sequenced, and the sequences were used as a probe BLAST search of homologues in NCBI Genbank database.. Screened homologous RGA clones were sequenced by Shanghai Sangon Biotech Inc. Introns were annotated with ORFinder. The sequences were queried against the tetraploid Gossypium hirsutum genome sequcence [33, 34]. The final obtained sequences were submitted to Genbank. BlastN search was performed in GeneBank using these sequences, and a sequence cluster was created by Phylip.
Results
Chromosomes preparation and microdissection
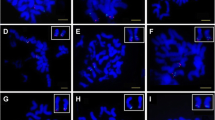
PMCs moderately digested in an enzymatic mixture were stained with carbolfuchsin. The PMCs at metaphase I were used for chromosome preparation (Fig. 1a). The target chromosome Ah01was initially found under low magnification, and then captured under high magnification for collection in a tube containing10 μL proteinase K (50 ng·μL−1) solution. The protocols for cutting and collecting chromosomes are shown in the Figure 1. For comparison, other chromosomes in metaphase I were simultaneously collected in different tubes for SSR-amplified proof after second LA-PCR amplification.

Microdissection and collection of single mono-chromosomes by CellCut Plus laser manipulation. a Film-slide preparations of meiotic metaphase I chromosomes with one monomer chromosome (Ah01). b Film-slide preparations of meiotic metaphase I chromosomes with one microdissected chromosome. c The microdissected chromosome on the cap of a collection tube. Arrow indicates the Ah01 chromosome. Bar: 5 μm
LA-PCR amplification of chromosomal DNA
Two rounds of LA-PCR were conducted to amplify the Ah01 chromosomal DNA. Electrophoresis results (Fig. 2) revealed that a weak DNA smear with sizes ranging from 200 to 1000 bp after the initial LA-PCR (Fig. 2, lane 3), and a strong DNA smearing pattern with sizes ranging from 300 to 2500 bp were generated after the second LA-PCR (Fig. 2, lane 5, 6), because of more products. For the negative controls, there were no bands (Fig. 2, lane 1, 2). The positive control produced a weak initial band (Figure 2, lane 4) and an obvious smearing pattern after the second LA-PCR (lane 7 in Figure 2). These results indicated that the Ah01 chromosome was amplified successfully.

Agarose gel electrophoresis of LA-PCR products. 1, 2: Negative controls 3 Product from the first round LA-PCR. 4, 7: Positive controls. 5, 6: Products from the second round LA-PCR. M: DNA marker
Southern blot analysis
Enzyme-labeled upland cotton genome was used as a probe, and the second LA-PCR products were verified by Southern hybridization with negative and positive controls (Fig. 3). Results of Southern blot showed that the second products of LA-PCR (Fig. 3, lane 4–6) and positive control (Fig. 3, lane 2, 3) had obvious bands, indicating that the amplification products from G. hirsutum genome were ranging from 300 to 2000 bp; that was consistent with the results from agarose gel electrophoresis. There were no bands in the negative control PCR (Figure 3, lane 1).

Southern blotting of products from the second round LA-PCR. 1: The negative control. 2, 3: Positive controls. 4, 5, 6: The second round LA-PCR products. 7: EcoRI digested genomic DNA. M: DNA marker
Verification of SSR amplification
Specific SSR primer from Ah01 chromosome was selected to amplify the second LA-PCR products of chromosome Ah01 and some other chromosomes. Results were checked by PAGE, and it was observed that the Ah01chromosome could amplify a specific band (240 bp), as shown in Fig. 4 (Fig. 4, lane 10). The similar band was obtained using the genome DNA as positive control (Lane 12), no band in negative control (Fig. 4, lane 11) and partly others chromosomes (Fig. 4, lane 1–9).

PAGE of SSR primer amplification product from single chromosome pool. 1–9: SSR primer amplification products from partial other chromosomes pool with Ah01 chromosome specific primer. 10: SSR primer amplification products from single chromosome pool with chromosome Ah01 special primer (arrow indicated). 11: The negative control. 12: The positive control. M: DNA marker
Fluorescence in situ hybridization
Dual-color FISH was performed using DIG-labeled products of LA-PCR II and specific Biotin-labeled Ah01 chromosome BAC clone (52D06) to probe the metaphase chromosome slide. As shown in Fig. 5, the target chromosomes were accurately identified by means of the specific BAC clone as well as products of LA-PCR II. Meanwhile, partial other chromosomes had weak signal (red light), indicating homologous sequence on these chromosomes.

FISH signals of products from the second round LA-PCR. a Chromosomes stained with DAPI. b Signals fromproducts of LA-PCR II (red). c Signals fromchromosome Ah01 specific BAC (green, arrow indicated). d Signals from dual-FISH
Isolation of RGAs
RGAs were isolated using PCR with P1 and P2 as primers, and TM-1 upland cotton genomic DNA and the second round LA-PCR Ah01 chromosome pool as templates, respectively. Products were detected by gel electrophoresis and Southern hybridization. Using genomic DNA as positive control, a slightly wider DNA smear with sizes ranging from 400 to 1000 bp and a major band of 550 ~ 700 bp was generated (Fig. 6a, Lane3). A narrow DNA smear with sizes ranging from 550 to 800 bp and a main band of 650 bp was generated using the second LA-PCR products as template (Fig. 6a, Lane 2). Southern blot results demonstrated that the products come from the genome of upland cotton (Fig. 6b).

Agarose gel electrophoresis (a) and Southern blotting (b) of P1/P2 primer PCR products. A-1, B-1: Negative controls. A-2, B-2: Single Ah01 chromosome as DNA template. A-3, B-3: Positive controls using 10 pg G. hirsutum genomic DNA as template. B-4: EcoRI digested genomic DNA of G. hirsutum. M: DNA marker
Cloning and validation of RGAs
Main bands of the PCR products were recovered and cloned. Two hundred positive clones (PS001 ~ PS200) were obtained and sequenced, followed by BLAST analysis. Three sequences [(Additional file 1) named PS016 (Genbank ID: KU051681), PS054 (Genbank ID: KU051682) and PS157 (Genbank ID: KU051680)] contained a conserved domain common to the NBS-LRR R genes in plant. Clustering results showed that they were homologous to these published RGAs (Fig. 7). Alignment was made with others RGAs from NCBI (Additional file 2), the results also definitely confirmed that the three RGAs were the NBS-LRR class of RGAs in cotton.

Cluster analysis of single chromosome RGA nucleotide sequenceswith those from other species
Discussion
Identification and microdissection of a single chromosome
Accurate identification of the target chromosome is a key step in chromosome microdissection and cloning. Identification of the target chromosome has mainly relied on the morphological features such as monosome, trisome, nullisome and shape-specific chromosomes [9,10,11, 43,44,45,46,47]. Chromosome banding technique has also been reported as a method to identify chromosomes [48], but this method has not widely used in plants. In this study, monosome chromosome in meiotic metaphase I were easy to identify and isolate from other chromosomes.
There are three approaches reported for chromosome isolation. One is flow cytometry, which has facilitated the dissection of large genome into smaller and defined segments for the purpose of gene discovery and genome sequencing in plants [13]. Nevertheless, this method not only requires expensive instrumentation, but also fails to distinguish chromosomes with similar morphological characteristics from one to another, which limit its application in plants to some extent. The second approach is the glass needle method, which involves in dissection of the target chromosome under an optical microscope by a glass needle. The approach is easily operate and independent from high-end instrumentation, which has resulted in effective and widespread application in plants [16, 20]. However, the approach requires the operator to be trained well enough, or there should be much deviation operated by different persons or even in different personal statuses by an operator. The third method is laser cutting [44, 49], in which chromosome specimens are dispersed onto a special carrier covered with a membrane for dissection and collection. In most cases, dissection is much easier than collection. In this study, the CellCutPlus Laser microdissection system was applied to isolate the target chromosome. Initially, cotton chromosomes were spread on the microscope slide coated with film, and then a single chromosome was dissected and automatically collected in a microcentrifuge tube with a sticky cap. This method has a high efficiency and a low risk of contamination.
Confirmation of the chromosomal DNA
LA-PCR is a powerful tool for the amplification of long DNA segments, and it has been widely used in molecular biology [18, 50, 51]. In this research, Ah01 chromosomal DNA was acquired by LA-PCR after microdissection of the target chromosome. Prior to subsequent steps, the PCR products were examined by agarose electrophoresis, Southern blot analysis, SSR primer confirmation and confirmed by FISH. Combining several confirmation methods could achieve multiple analyses, and ensure that amplification products were from the target chromosome.
Significance of generating RGAs from specific single chromosome of cotton
R genes have been isolated from the whole genome or its cDNA in woody plants [7, 19, 20, 39]. In this study, RGAs were isolated from a single chromosome dissected from upland cotton, clarifying the source and location. Efficiency of downstream work was greatly improved due to the isolation of a single chromosome from the entire genome. It has been reported that the R genes family frequently clusters on a certain chromosomal segment [52]. Acquired RGAs could be transformed to molecular marks, and serve to construct a genetic map due to the clear linkage relationship from one chromosome. In addition, it will contribute to the development of map-based cloning, thus, generating RGAs from a specific chromosome has many advantages.
Conclusions
Although cotton is one major crop in the world like rice, wheat and maize, its cytogenetic studies falls much behind others due to the smaller and identical chromosomes in morphology as well as large amounts of secondary metabolites within its cells. All these factors make the cotton chromosome preparation difficult. Here, we successfully developed a technique to separate a single chromosome from upland cotton PMC (monosome cells) with the laser cutting. Using this technology, we also microcloned three RGAs from the DNA pool constructed with the single chromosomes (Ah01). The three RGAs belong to the nucleotide binding site-leucine rich repeat (NBS-LRR) gene family. The techniques will promote the cloning of cotton R genes and marker assistant improvement of cotton genetics and breeding.
References
Scalenghe F, Turco E, Edström JE, Pirrotta V, Melli M. Microdissection and cloning of DNA from a specific region of Drosophila melanogaster polytene chromosomes. Chromosoma. 1981;82(2):205–16.
Lüdecke HJ, Senger G, Clasussen U, Horsthemke B. Cloning defined regions of the human genome by microdissection of banded chromosomes and enzymatic amplification. Nature. 1989;338(6213):348–50.
Senger G, Lüdecke HJ, Horsthemke B, Claussen U. Microdissection of banded human chromosomes. Hum Genet. 1990;84(6):507–11.
Engelen JJM, Albrechts JCM, Hamers GJH, Geraedts JPM. Asimple and efficient method for microdissection and microfish. J Med Genet. 1998;35(4):265–8.
Hori T, Suzuki Y, Solovei I, Saitoh Y, Hutchison N, Ikeda JE, Macgregor H, Mizuno S. Characterization of DNA sequences constituting the terminal heterochromatin of the chicken Z chromosome. Chromosom Res. 1996;4:411–26.
Sarker N, Hawken RJ, Takahashi S, Alexander LJ, Awata T, Schook LB, Yasue H. Directed isolation and mapping of microsatellites from swine chromosome 1q telomeric region through microdissection and RH mapping. Mamm Genome. 2001;12:524–7.
Schmutz SM, Berryere TG, Moker JS, Thue TD, Winkelman DC. Gene mapping from a bovine 1;29 DNA library prepared with chromosome microdissection. Mamm Genome. 1994;5(3):138–41.
Taguchi T, Akimaru K, Hirai H, Hirai Y, Mwenda JM, Yuri K. A probe generated by chromosome microdissection, useful for analyzing Y chromosome evolution in old world monkeys. Chromosom Res. 2003;11:147–52.
Sandery MJ, Forster JW, Macadam SR, Blunden R, Jones RN, Brown SDM. Isolation of sequence common to A- and B-chromosomes of rye (Secaleceresle) by microcloning. Plant Mol Biol Report. 1991;9(1):21–30.
Jung C, Claussen U, Horsthemke B, Fischer F, Herrmann RG. A DNA library from an individual Beta patellaris chromosome conferring nematode resistance obtained by microdissection of meiotic metaphase chromosomes. Plant Mol Biol. 1992;20(3):503–11.
Schondelmaier J, Martin R, Jahoor A, Houben A, Graner A, Koop HU, et al. Microdissection and microcloning of the barley (Hordeumvulgare L.) chromosome 1 HS. Theor Appl Genet. 1993;86(5):629–36.
Albani D, Côté MJ, Armstrong KC, Chen QF, Segal A, Robert LS. PCR amplification of microdissected wheat chromosome arms in a simple “single tube” reaction. Plant J. 1993;4(5):899–903.
Arumuganathan K, Martin GB, Telenius H, Tanksley SD, Earle ED. Chromosome 2-specific DNA clones from flow-sorted chromosomes of tomato. Mol Gen Genet. 1994;242(5):511–58.
Liu B, Segal G, Vega JM, Feldman M, Abbo S. Isolation and characterization of chromosome-specific DNA sequences from a chromosome arm genomic library of common wheat. Plant J. 1997;11(5):959–65.
Song WQ, Li XL, Xu WS, Chen RY. Microdissection and PCR amplification of rye chromosome. Acta Bot Sin. 1998;40(2):158–62.
Zhou YH, Hu ZM, Dang BY, Wang H, Deng XD, Wang LL, et al. Microdissection and microcloning of rye (Secale cereal L.) chromosome 1R. Chromosoma. 1999;108:250–5.
Scutt CP, Kamisugi Y, Gilmartin PM, Sakai F. Laser isolation of plant sex chromosomes: studies on the DNA composition of the X and Y sex chromosomes of Silenelatifolia. Genome. 1997;40(5):705–15.
Dang BY, Hu ZM, Zhou YH, Cui LH, Wang LL, Chen ZH. Construction of single-chromosome DNA library from Lilium regale Wilson. Chin Sci Bull. 1998;43(5):434–9.
Huang D, Wu W, Zhou Y, Hu Z, Lu L. Microdissection and molecular manipulation of single chromosomes in woody fruit trees with small chromosomes using pomelo (Citrus grandis) as a model: I. Construction of single chromosomal DNA libraries. Theor Appl Genet. 2004;108(7):1366–70.
Zhang Y, Zhang SG, Qi LW, Liu B, Gao JM, Chen CB, et al. Construction of poplar (Populustremula) chromosome 1-specific DNA library by using a microdissection technique. Plant Mol Biol Report. 2005;23(2):129–38.
Flor HH. Current status of the gene-for-gene concept. Annu Rev Phytopathol. 1971;9:275–96.
Kanazin V, Marek LF, Shoemaker RC. Resistance gene analogs are conserved and clustered in soybean. Proc Natl Acad Sci U S A. 1996;93(21):11746–50.
Yu YG, Buss GR, Maroof MA. Isolation of a superfamily of candidate disease-resistance genes in soybean based on a conserved nucleotide-binding site. Proc Natl Acad Sci U S A. 1996;93(21):11751–6.
Speulman E, Bouchez D, Holub EB, Beynon JL. Disease resistance gene homologs correlate with disease resistance loci of Arabidopsis thaliana. Plant J. 1998;14(4):467–74.
Collins NC, Webb CA, Seah S, Ellis JG, Hulbert SH, Pryor A. The isolation and mapping of disease resistance gene analogs in maize. Mol Plant-Microbe Interact. 1998;11(10):968–78.
Grube RC, Radwanski ER, Jahn M. Comparative genetics of disease resistance within the solanaceae. Genetics. 2000;155(2):873–87.
Pan QL, Liu YS, Budai-Hadrian O, Sela M, Carmel-Goren L, Zamir D, et al. Comparative genetics of nucleotide binding site-leucine rich repeat resistance gene homologues in the genomes of two dicotyledons: tomato and Arabidopsis. Genetics. 2001;155(1):309–22.
Deng Z, Huang S, Ling P, Chen C, Yu C, Weber CA, et al. Cloning and characterization of NBS-LRR class resistance-gene candidate sequences in citrus. Theor Appl Genet. 2000;101(5–6):814–22.
Rong J, Bowers JE, Schulze SR, Waghmare VN, Rogers CJ, Pierce GJ, et al. Comparative genomics of Gossypiumand Arabidopsis: unraveling the consequences of both ancient and recent polyploidy. Genome Res. 2005;15(9):1198–210.
Chen ZJ, Scheffler BE, Dennis E, Triplett BA, Zhang TZ, Guo WZ, et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007;145:1304–10.
Wang KB, Wang ZW, Li FG, Ye WW, Wang JY, Song GL, et al. The draft genome of a diploid cotton Gossypiumraimondii. Nat Genet. 2012;44(10):1098–103.
Li FG, Fan GY, Wang KB, Sun FM, Yuan YL, Song GL, et al. Genomesequence of the cultivatedcotton Gossypiumarboretum. Nat Genet. 2014;46(6):567–72.
Li FG, Fan GY, Lu CR, Xiao GH, Zou CS, Kohel RJ, et al. Genomesequence of cuotivateduplandcotton (Gossypiumhirsutum TM-1) providesinsightsintogenomeevolution. Nat Biotechnol. 2015;33(5):524–30.
Zhang TZ, Hu Y, Jiang WK, Fang L, Guan XY, Chen JD, et al. Sequencing of allotetraploidcotton (Gossypiumhirsutum l. acc. tM-1) provides a resource for fiberimprovement. Nat Biotechnol. 2015;33(5):531–7.
Liu X, Zhao B, Zheng HJ, Hu Y, Lu G, Yang CQ, et al. Gossypiumbarbadensegenomesequenceprovidesinsightinto the evolution of extra-long staplefiber and sprcializedmetabolites. Sci Report. 2015;5:14139. doi:10.1038/srep14139.
Peng RH, Liu F, Hu X, Wang CY, Li SH, Zhang XD, et al. Microdissection and microcloning of chromosome 5 in Gossypium arboretum. Plant Mol Biol Report. 2012;30(5):1218–28.
Yu Y, Yuan DJ, Liang SG, Li XM, Wang XQ, Lin ZX, et al. Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypiumhirsutum and G. barbadense. BMC Genomics. 2011;12:15.
Meng XP, Li FG, Liu CL, Zhang CJ, Wu ZX, Chen YJ. Isolation and characterizationof an ERF transcriptionfactor gene from cotton (Gossypiumbarbadense L.). Plant Mol Biol Report. 2010;28(1):176–83.
Zhang F, Liu X, Zuo KJ, Zhang JQ, Sun XF, Tang KX. Molecular cloning and characterization of a novel Gossypium barbadense L. RAD-like gene. Plant Mol Biol Report. 2011;29(2):324–33.
Zhang XD, Wang KB, Li MX, Li SH, Wang CY, Song GL, et al. Observing behavior of monosome and telosome chromosomes of Gossypium hirsutum and their identification approach. Acta Gossypii Sinica. 2000;12(6):302–5.
Zheng XW, Zhai WX, Li XB, Wang WJ, Xu JC, Liu GQ, et al. NBS-LRR homologous sequence of R gene in rice. Sci China Ser C. 2001;31(1):43–51.
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 1989. p. 1–1060.
Pich U, Houben A, Fuchs J, Meister A, Schubert I. Utility of DNA amplified by degenerate oligonucleotide-primed PCR (DOP-PCR) from the total genome and defined chromosomal regions of field bean. Mol Gen Genet. 1994;243(2):173–7.
Fukui K, Minezawa M, Kamisugi Y, Ishikawa M, Ohmido N, Yanagisawa T, et al. Microdissection of plant chromosomes by argon-ion laser beam. Theor Appl Genet. 1992;84(7–8):787–91.
Houben A, Kynast RG, Heim U, Hermann H, Jones RN, Forster JW. Molecular cytogenetics characterization of the terminal heterochromatic segment of the B-chromosome of rye (Secalecereale). Chromosoma. 1996;105(2):97–103.
Vega M, Abbo S, Feldman M, Levy AA. Chromosome painting in plants: in situ hybridization with a DNA probe from a specific microdissected chromosome arm of common wheat. Proc Natl Acad Sci U S A. 1994;91(25):12041–5.
Saunders RDC, Glover DM, Ashburner M, Siden-Kiamos I, Louis C, Monastirioti M, et al. PCR amplification of DNA microdissected from a single polytene chromosome band: a comparison with conventional microcloning. Nucleic Acids Res. 1989;17(22):9027–37.
Kamisugi Y, Sakai F, Minezawa M. Fujishita M Fukui K recovery of dissected C-band regions in Crepis chromosomes. Theor Appl Genet. 1993;85(6–7):825–8.
Cocca E, Petraccioli A, Morescalchi MA, Odierna G, Capriglione T. Laser microdissection- based analysis of the Y sex chromosome of the Antarctic fish Chionodracohamatus (Notothenioidei, Channichthyidae). Comp Cytogent. 2015;9(1):1–15.
Chen Q, Armstrong K. Characterization of a library from a single microdissected oat (Avena sativa L.) chromosome. Genome. 1995;38(4):706–14.
Ponelies N, Stein N, Weber G. Microamplification of specific chromosome sequences; an improved method for genome analysis. Nucleic Acids Res. 1997;25(17):3555–7.
Hulbert SH, Webb CA, Smith SM, Sun Q. Resistance genecomplexes: evolutionandutilization. Annu Rev Phytopathol. 2001;39:285–312.
Acknowledgements
We deeply thank Prof. Tianzhen Zhang (Nanjing Agricultural University, China) for kindly providing the chromosome-specific BAC clone.
Funding
The research was supported by grants from the National Natural Science Foundation of China (No. 31471548), State Key Laboratory of Cotton Biology Open Fund (No. CB2014A07), the Program for Science & Technology Innovation Talents in Universities of Henan Province (13HASTIT026).
Authors’ contributions
RP and KW designed the study; XC, YL, ZL, FL, YW, ZZ, XC, XW, CW, YW and ZL performed the experiments; YL wrote the manuscript, RP and KW proofread the manuscript. All authors read and approved of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Accession numbers
The sequence data of three newly identified RGAs are deposited in NCBI GeneBank with the accession ID of PS016 (KU051681), PS054 (KU051682), andPS157 (KU051680).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1:
The nucleotide sequences of three newly identified RGA genes. (TXT 1 kb)
Additional file 2:
Alignment of the three RGAs. (PDF 143 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cao, X., Liu, Y., Liu, Z. et al. Microdissection of the Ah01 chromosome in upland cotton and microcloning of resistance gene anologs from the single chromosome. Hereditas 154, 13 (2017). https://doi.org/10.1186/s41065-017-0035-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-017-0035-3