
Abstract
Background
The emergence of antibiotic-resistant pathogens has created an urgent need for novel antimicrobial treatments. Advances in next-generation sequencing have opened new frontiers for discovery programmes for natural products allowing the exploitation of a larger fraction of the microbial community. Polyketide (PK) and non-ribosomal pepetide (NRP) natural products have been reported to be related to compounds with antimicrobial and anticancer activities. We report here a new culture-independent approach to explore bacterial biosynthetic diversity and determine bacterial phyla in the microbial community associated with PK and NRP diversity in selected soils.
Results
Through amplicon sequencing, we explored the microbial diversity (16S rRNA gene) of 13 soils from Antarctica, Africa, Europe and a Caribbean island and correlated this with the amplicon diversity of the adenylation (A) and ketosynthase (KS) domains within functional genes coding for non-ribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs), which are involved in the production of NRP and PK, respectively. Mantel and Procrustes correlation analyses with microbial taxonomic data identified not only the well-studied phyla Actinobacteria and Proteobacteria, but also, interestingly, the less biotechnologically exploited phyla Verrucomicrobia and Bacteroidetes, as potential sources harbouring diverse A and KS domains. Some soils, notably that from Antarctica, provided evidence of endemic diversity, whilst others, such as those from Europe, clustered together. In particular, the majority of the domain reads from Antarctica remained unmatched to known sequences suggesting they could encode enzymes for potentially novel PK and NRP.
Conclusions
The approach presented here highlights potential sources of metabolic novelty in the environment which will be a useful precursor to metagenomic biosynthetic gene cluster mining for PKs and NRPs which could provide leads for new antimicrobial metabolites.
Similar content being viewed by others

Background
Environmental bacteria are a source of natural product diversity which formed the basis of early drug development work on antibiotics. Over 80% of all antibiotics deployed in the clinic, as well as ~ 47% of anticancer drugs originate from natural products [1, 2]. Decades of antibiotic misuse, both in the clinic and in agriculture, have led to the worldwide antibiotic crisis with multi-drug-resistant pathogens posing a significant threat to human health [3]. The current dearth of antimicrobial compounds with novel modes of action means that we need to expand our exploration of natural products by investigating the biosynthetic potential of the uncultured soil bacteria through genome mining and metagenomic approaches [4, 5]. Natural products from bacteria have been widely used in human and veterinary medicine and prokaryotic genome analysis has demonstrated that the genes responsible are clustered and many contain non-ribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) enzymes [5]. There is clear evidence that in silico/in vitro combined strategy for identifying NRPS and PKS could provide a rich source of new antimicrobial agents [6].
Genome sequencing has revolutionised natural product discovery with the identification of biosynthetic gene clusters (BGCs) encoding for the production of natural products accounting > 10% of some bacterial genomes such as in the case of Actinomycetes [5, 7]. It has been observed that not all the identified BGC sequences could be linked to an antimicrobial product under laboratory conditions due to difficulties with expressing BGCs in culture to a level which facilitates natural product detection and elucidation [8]. Indeed, the production of biologically active natural products in natural environments is a tightly regulated process controlled by a wide range of environmentally activated responses such as gacA and gacS which control phenazine production in Pseudomonas [9] and signalling factors such as γ-butyrolactone and furans in the Actinomycetales [10]. Little is known about the factors stimulating the production of such signalling molecules and the natural role of antibiotic biosynthesis in soil has long been debated. However, evidence suggests natural products likely have a role in signalling and protection, with recent studies showing antibiotics playing a key role in antagonism within ant communities and as protective agents in wasps’ nests [11]. Recent genomic evidence has also demonstrated that the less well-characterised Bacteroidetes, Verrucomicrobia and Planctomycetes harbour novel BGCs [12,13,14], though the functions of these are widely unknown.
Exploring the uncultured soil bacteria for natural products still remains challenging [15]. Efforts have been made to recover single cells from the natural environment. Single cell isolation has now become a key route to understanding the metabolism of uncultured cells, using SiC-Seq to recover the genomes [16]. Such methods are difficult to apply to bacteria intimately associated with soil particles, but large-scale screening efforts combined with iChip technology have uncovered novel genera with bioactive properties [17]. In addition to single cell isolation techniques, analysis of microbial community DNA has enabled the exploitation of metabolic diversity using metagenomic libraries combined with expression screening [18, 19]. However, this approach is challenging if large BGCs such as in the case of natural products need to be recovered. Furthermore, little information was provided in relation to the taxonomic drivers of this diversity [20, 21]. The application of co-occurrence statistics has enabled the linkage of structure to function in microbial communities. Thus, within microbiomes, the ability to understand the importance of diversity in relation to metabolic function has been improved [22].
Two known enzymes for the production of natural products in bacteria are NRPS and PKS. These enzymes are coded by core genes within BGCs which contain all of the information for the biosynthesis of a defined bioactive metabolite, including regulatory elements, transporters and resistant genes [23, 24]. These core genes contain domains that are conserved across all BGCs of that type. The nucleotide identities of such domains can be used to determine phylogenetic relationships between related BGCs, in addition to giving an indication of the natural product encoded. This has been validated at a genomic level and underpins widely used bioinformatics tools such as NaPDoS [25] and anti-SMASH [26]. Recently, we have developed a target assay based on the adenylation (A) domain of the NRPS and the ketosynthase (KS) domain of PKS resulting in amplification of non-conserved regions of these genes that can be used for defining novel metabolic capability [27]. Previous studies using similar targets, focused on soil across the USA, provided evidence that NRP and PK diversity varies with soil type and actinomycete richness [4]. A further survey of Australian soils suggested that both pH and latitude drive BGCs diversity [28]. However, on a global scale across multiple habitats, the taxonomic drivers of BGC diversity are still to be determined.
In collaboration with the British Antarctic Survey (BAS), we became interested in microbial communities within permafrost soils and found preliminary evidence for diversity that could facilitate the exploitation of novel metabolism for bioactive natural products. In particular, BGCs with low similarity to already known clusters belonging to Actinobacteria and Proteobacteria have previously been isolated and identified through functional metagenomic library screening [27, 29].
The aim of the current study is to understand how differences in microbial community composition drives PK and NRP diversity in a range of soils including those from more extreme environments such as an Antarctic fellfield and the Sahara Desert.
Our experimental design focused on the identification of community structure-function correlations to elucidate the microbial groups driving the diversity in the A and KS domains of the NRPS and PKS biosynthetic genes, respectively. Results presented here shed new light on approaches to discover the potential of unique habitats for PK and NRP BGC diversity.
Materials and methods
Soil sample collection and processing
Soil samples were collected from 13 sites across different countries (Algerian Sahara Desert, Mars Oasis in Antarctica, Iceland, Sourhope and Warwick in UK, Tuscany and South Tyrol in Italy, Kilkenny in Ireland, Cayo Blanco and Trinidad in Cuba; Additional file 1: Table S1) under DEFRA licence 51993/194938/3 and sampling permission compliant with national biodiversity legislation. Chemical analyses were performed by YARA Analytical Service, LanCrop Laboratories, Grimbsy, UK. Samples were collected with sterile equipment from the top 10 cm of the soil layer, stored at 4 °C for transport and immediately frozen at −30 °C upon arrival. Large soil samples (300–1000 g) were thawed and three individual samples (0.5 g) were taken from each soil for DNA extraction using the FastDNA® SPIN Kit for soil (MP Biomedicals) according to the manufacturer’s instructions. A Nanodrop spectrophotometer (ThermoFisher) was used to quantify DNA and DNA integrity was confirmed with agarose gel electrophoresis.
Amplicon library preparation
A total of 117 MiSeq paired-end libraries (2 × 300 bp) were prepared using the Illumina® Nextera XT Library preparation kit for the V3–V4 region of the 16S rRNA gene [30], the A domain of the NRPS gene and the KS domain of the PKS gene [27] (Additional file 1: Table S2). Amplification of the 16S rRNA gene target was performed according to the manufacturer’s instructions (Illumina). Reactions were optimized for A and KS amplicons, with 40 cycles of amplification being used to improve the yield of amplicon DNA recovered (Additional file 1: Table S2).
Sequence analysis
All amplicon reads are available at the European Nucleotide Archive (study PRJEB11689) and were processed using the EBI Metagenomics analysis pipeline v.3 [31] in order to predict protein coding sequences that could be used for taxonomic analysis (project ERP013097). A custom pipeline composed of PANDAseq [32], USEARCH v.8.1.1861 [33], UPARSE [34], QIIME [35] v.1.9.1 and Kaiju [36] was also used for microbial and functional gene diversity analysis to validate results between different pipelines. In this custom pipeline, paired-end reads were assembled using PANDAseq with a minimum overlap of 10 bp. USEARCH algorithms were then used to de-replicate and sort by size the sequences, discarding singletons. Operational taxonomic units (OTUs) were clustered de novo using the UPARSE algorithm and chimeras were removed based on the prediction of UCHIME. A summary of sequence and observed OTU counts per each dataset is reported in Additional file 1: Tables S3 and S4. A and KS amplicons were initially clustered into OTUs using 95% and 97% similarity but results were comparable (Additional file 1: Table S5) and 97% was selected for all further analyses. The community structure (16S rRNA gene amplicons) was also resolved at 97% OTU similarity. The most abundant sequence per each OTU was selected as the representative sequence. Mitochondrial or chloroplasts sequences were also removed from 16S rRNA gene sequences using the RDP gold database as a reference (in QIIME). Rarefaction was performed using 17,000 sequences for 16S rRNA gene and 8500 or 2600 sequences for A and KS datasets, as cut-offs to obtain OTU tables for further comparative analyses.
Taxonomic assignment of reads
Taxonomy was assigned to 16S rRNA gene OTUs using the RDP classifier and the Greengenes database.
Raw A and KS reads were processed and analysed with the EBI Metagenomics analysis pipeline, which performs protein prediction using FragGeneScan v1.15 [37]. Additional manual taxonomic annotation of the predicted protein coding sequences derived from A and KS reads were performed using the Unipept [38] software with the NCBI taxonomy classification system. Kaiju web software was also used for the taxonomic annotation of A and KS OTUs [36].
Annotation of metabolites to A and KS OTUs
Selected A and KS OTUs from the network analysis were associated to potential metabolite families. Blastx against the MIBiG database (version 1.2) [39] was performed with a cut-off expectation value (e-value) of 10−20 to prevent misclassification.
Talent ratio calculation using Integrated Microbial Genomes/Atlas of Biosynthetic Gene Clusters (IMG/ABC) database investigation
In order to determine the biosynthetic potential of communities discovered in the various soil samples, an analysis of specific phyla was done in silico to highlight already existing information on these groups. The IMG/ABC [40] database was manually checked for the number of BGCs and genomes available for each of the main phyla used in the correlation analysis presented in this study. Statistics available on the database were used to retrieve the number of genomes and BGCs (inclusive of PKS, NRPS, saccharides and terpenes) from all available sequenced single isolate genomes deposited in the database (data available on 13/04/2018). BGC evidence (experimentally characterised or predicted only) was also noted. The genetic potential for natural products biosynthesis, here called talent ratio (TR), was calculated for each phylum as the total number of BGC counts divided by the number of genomes for that specific bacterial group. The TR calculation did not consider the different genome average size for each phylum. This indicator was created to investigate theoretical genetic potential for BGC at the phylum level only and not at lower taxonomic ranks.
Statistical analyses
Statistical analyses were performed using QIIME v. 1.9.1 [35] and R studio v 1.1.456 using the packages Vegan and Phyloseq v 1.16.2 [41]. Alpha diversity was investigated with the Simpson inverse index and part of the beta diversity analysis was based on Bray-Curtis dissimilarity matrices. Differences between rarefied and non-rarefied samples were checked using a paired t test (p < 0.05) on the Simpson inverse index. OTU networks were created through QIIME, visualized and further developed using Cytoscape [42]. Analysis of similarities (ANOSIM) was performed on groups observed in principal coordinate analysis (PCoA) to test statistically significant difference between groups of samples. The correlation between phyla (as 16S rRNA gene) and the diversity of the two functional genes was investigated using a Mantel correlation between the generated Bray-Curtis dissimilarity matrices for each marker gene, and further explored with a Procrustes superimposition of PCoA plots generated from Bray-Curtis dissimilarity matrices. These analyses were performed between each respective functional gene (A and KS domain) and each separate phylum (filtered 16S rRNA gene by phylum). The phyla selected for correlation analysis had > 1% abundance in at least 20% of the samples representing the core community covering on average 96% of the total bacterial community population in each sample. A correlation-like statistic, Procrustes randomization test (PROTEST) using Monte Carlo simulations (999 permutations) tested the significance of the Procrustes superimposition by evaluating the non-randomness between two configurations. The goodness-of-fit (M2) value from the PROTEST represented the concordance between the ordinations used in the superimpositions based on the residual sum of squares. The lower the M2 value, which ranges from 0 to 1, the greater the concordance between the data sets [43]. Procrustes plots were generated with QIIME and visualized using Emperor.
Results
A and KS domain diversity across soils
The annotation of translated sequences for A and KS genes obtained through InterPro in the EBI pipeline showed primer specificity for the desired targets. On average, 98% of A reads per sample were assigned to either the AMP-dependent synthetase/ligase domain (IPR000873), the phosphopantetheine binding ACP domain (IPR00081) or the AMP-binding enzyme (IPR002510), while 70–80% of KS reads were assigned either to the thiolase-like (IPR016039) or beta-ketoacyl synthase domains (IPR013030 and IPR013031).
The rarefaction curves demonstrated that for the A and KS domains, diversity was covered by the sequencing with rarefaction not affecting the diversity (A, paired t test, t(33)=3.67, p > 0.05; KS, paired t test, t(33)=0.997, p > 0.05) (Additional file 1: Figure S1). A total of 5834 and 9625 OTUs were observed for the A and KS domains, respectively. Alpha diversity was measured using the Simpson inverse index and ranged across sites from 4.4 to 128.5 for the A domain and from 2.8 to 144 for the KS domain (Additional file 1: Figure S2). The Tuscan soil was significantly more diverse than any of the other soils for both A and KS domains (Tukey’s test, p < 0.05).
The A and KS domain diversity (based on Simpson inverse index) did not show statistically significant Pearson’s correlation to edaphic features such as pH and salinity as electrical conductivity (EC). Linear regression models with one or multiple predictors were also fitted but no statistically significant results were obtained. Beta diversity analysis through PCoA plots of A and KS domains (based on Bray-Curtis dissimilarity matrices) suggested that composition was dependent on geographic location. In particular, for the A domain, European and Cuban samples clustered together, whilst Antarctica, Sourhope, Trinidad and Algeria each formed separate clusters (ANOSIM, R = 0.65, p = 0.001) (Fig. 1). For the KS domain, Cuban and Algerian soils clustered separately, whilst the European, Antarctic, Icelandic, Trinidad and South Tyrol soils had sequences that showed similarity and grouped together (ANOSIM, R = 0.66, p = 0.001) (Fig. 1). To visualize shared OTUs between samples and OTUs present only in specific locations, OTU networks of both A and KS OTUs were constructed (Fig. 2). Network separation was based on the number of shared OTU nodes between samples and sites distinctly separated according to geographic location, supporting the results obtained through PCoA plots. A further investigation of the shared and location-specific OTUs across samples allowed the identification of potential areas with endemic A and KS diversity, such as Antarctica and Cuba. These soils harboured lower community diversity than other samples, such as Tuscany, but showed potential endemic A and KS OTUs.

Grouping of soil based on the principal components of diversity in two-dimensions (PCoA). Analysis of A domain, KS domain and 16S rRNA genes based on Bray-Curtis dissimilarity matrices and coloured by location. Alg_KII, Algerian KII; Alg_KI, Algerian KI; Alg_B3, Algerian B3; Cuba-Fir, Cayo Blanco (Fir-Shrub); Cuba-Sand, Cayo Blanco (Shrub)

Representation of A and KS domain diversity between samples using OTU networks. Main nodes (black dots) represent soil samples, nodes at the end of edges (grey dots) represent single OTUs. The distance between main nodes is equal to the similarity between samples. Edges connect OTU nodes to sample nodes. A OTU network was constructed for each dataset (A and KS domain separately), and both networks were investigated and represented using a different node degree to represent a all OTUs, b only location endemic OTUs belonging to each soil sample and c only OTUs shared between different locations (not between the same location) (node degree ≥ 4)
Microbial community differences across soils
Rarefaction indicated that the bacterial populations (16S rRNA gene) were sampled to sufficient depth (paired t test, t(38) = 1.128, p > 0.05) (Additional file 1: Figure S1).
Alpha diversity with the Simpson inverse index ranged from 22.2 to 404.9, showing significantly lower 16S rRNA gene diversity for the Algerian B3, Cuban and Antarctic samples compared to the other soils (Tukey’s HSD, p < 0.05) (Additional file 1: Figure S2). Pearson’s correlation analysis did not show statistically significant correlations between 16S rRNA gene diversity (Simpson inverse index) and the edaphic features pH and salinity (as EC). The analysis of beta diversity through PCoA based on Bray-Curtis dissimilarity matrices identified six distinct groups (ANOSIM, R = 0.96, p = 0.001): Antarctica, Iceland, Sourhope, Cuba, Algeria and the remaining European soils (Fig. 1). These results suggested differences in the microbial communities according to their geographic locations which were reflected in the microbial community structure at the phylum level (Additional file 1: Figure S3). The mean overall Bray-Curtis similarity value at the genus level between biological replicates was 90.3% suggesting low variability between replicates from the same geographic location.
Evidence for microbial groups driving A and KS domain diversity
The taxonomic identity of all A and KS OTUs across samples through Kaiju [36] assigned the majority of the OTUs to Bacteria. In particular, for the bacterial A and KS domain dataset, the majority of reads were assigned to Actinobacteria (average 39% and 26% of the A and KS sequences, respectively) and Proteobacteria (average 40% and 24% of the A and KS OTUs) in accordance with the prevalence of these two groups (average 16S rRNA gene relative abundance of 24% and 25%, respectively).
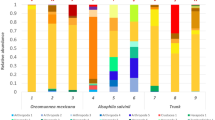
Identity assignment of A and KS domain amino acid sequences of individual soil samples through EBI/Unipept showed similar results to the Kaiju analysis with an average of 95% and 99% of read of A and KS domain, respectively, assigned to Bacteria and the remaining reads were assigned to Archaea and Eukaryota (Additional file 1: Figure S4). However, this classification highlighted some distinct profiles such as the Antarctic soil which had an average of 10.7% and 16.1% of A and KS reads assigned to Verrucomicrobia and Bacteroidetes phyla, respectively. Whereas Sourhope showed an average of 53.9% A reads assigned to Firmicutes (Fig. 3). The highest abundance of KS reads assigned to the phylum Acidobacteria was revealed in the Icelandic soil (average of 6.2%).

Representation at a phylum level of the taxonomic providence of A and KS domain sequences assigned using the EBI/Unipept pipeline. Phyla with a percentage below 1% in at least 20% of the samples were not individually represented. Bar stats represent mean values of triplicate samples for each site
According to Mantel correlation analysis, there was a positive correlation between the main phyla of the microbial communities and the biosynthetic diversity (Table 1). The Mantel correlation values for both domains ranged from 0.81 for the well-known producers Actinobacteria to the lower score of 0.64 for Nitrospirae. In addition, the less well-characterised Bacteroidetes phylum had a correlation coefficient of 0.82 and 0.83 for A and KS domains, respectively, whilst the coefficient for the Verrucomicrobia were 0.82 for A and 0.79 for KS domain (Table 1).
A number of Procrustes transformation superimpositions were performed on all data and are presented (Additional file 1: Figure S5 and Figure S6) with 16S rRNA gene against A and KS domain data. The correlation with A domains was stronger than for KS, with M2 of 0.36 (protest, p < 0.001) compared to 0.52 (protest, p < 0.001). Separate superimposition of each phylum demonstrated the strong relationship between Actinobacteria and A domain diversity (M2 = 0.36, p < 0.001). Similarly, for the KS domain, Bacteroidetes (M2 = 0.22, p < 0.001) and Proteobacteria (M2 = 0.25, p < 0.001) showed a significant goodness of fit with the KS domain superimposition. In particular, this analysis illustrated A and KS domain diversity potentially related to Actinobacteria, Bacteroidetes and Verrucomicrobia in Antarctic soil showing a closer co-location of the datasets on the superimposed PCoA plot.
Antarctica: a case study for selected functional diversity
As demonstrated by beta diversity and network analysis, the Antarctic soil harboured an endemic community of bacteria and biosynthetic A and KS domains. Taxonomic analysis of shared and location-specific A OTUs recovered from the network analysis (Fig. 2) illustrated that OTUs shared across all sites belonged to Actinobacteria, Proteobacteria and Cyanobacteria species, whilst those endemic to Antarctica also belonged to the Bacteroidetes phylum. The same analysis was performed on KS OTUs, whereby only OTUs belonging to the Actinobacteria were shared across all sites, whilst many of the endemic OTUs belonged to the PVC (Planctomycetes-Verrucomicrobia-Chlamydiae) group. This suggests that Actinobacteria-derived A and KS domains are more widely distributed than those belonging to other phyla.
The assignment of previously characterised compounds (available from the MiBIG database) to Antarctic soil A and KS OTUs indicated that biosynthetic capability for a variety of possible metabolites was present (Fig. 4). Potential metabolites with antimicrobial activity such as teixobactin [17] or antitumor activity as demonstrated for the polyether salinomycin [44] were matched in the database. A large proportion of the total Antarctic endemic OTUs (11% and 59% of A and KS OTUs, respectively) failed to match any of the compounds available in the database suggesting potential for novel endemic metabolites from BGCs.

Matched compounds of A and KS location-specific and shared Antarctic OTUs to the MIBiG database. All OTUs from the three Antarctic samples were subject to Blastx analysis for the A and KS domains separately
Determining the talent ratio for different phyla
The top most abundant phyla across all the soil samples contained some groups poorly represented in genome databases, for example Verrucomicrobia. An audit of the IMG/ABC database illustrated the Actinobacteria and Proteobacteria were by far the most talented bacteria present in the database (talent ratio of 35.83 and 13.02, respectively). A total of 150 genomes were available for Verrucomicrobia with 920 predicted BGCs associated with this group. This represents a small fraction in comparison to the most abundant phyla represented in the database such as the Proteobacteria with 27,431 genomes available and 357,065 BGCs counted (Table 2). However, the potential for natural product BGCs, expressed as the talent ratio (TR), showed that Verrucomicrobia are comparable to more represented groups such as Cyanobacteria, which are better known and characterised for natural product biosynthesis. Similarly, Bacteroidetes which only counted 2162 sequenced genomes and 22,885 BGCs have a TR of 10.58 which is similar to that of Firmicutes (Table 2).
Discussion
The current study provides an insight into the main phyla involved in driving the A and KS domain diversity in a range of soils, focusing on the inter-relationship between structure and function of the microbial community. The geographic segregation of different microbial communities revealed in this study highlights the potential for novel PK and NRP discovery in soils exposed to extreme conditions, such as those of Antarctica, the Algerian Saharan Desert or the pristine Cuban Cayo-Blanco regions. The Antarctic has been the subject of intense study, providing evidence of endemicity amongst the terrestrial metazoans, with endemics also having been identified in cultured and uncultured Cyanobacteria and green algae [45]. We posit that the global ubiquity hypothesis [46] is challenged by our observation that metabolic endemism occur in Antarctica but also in Cuba and Algeria. A recent study on an extreme oligotrophic oasis provided strong evidence of high diversity and endemicity for culturable Actinobacteria [47]. The phylogenetic uniqueness of Streptomyces species recovered from these desert soils provided additional support for migration limitation emphasising the Baas Becking hypothesis of environmental selection. We believe that the Antarctic site at Mars Oasis provides similar endemism for PK and NRP metabolite, further implying dispersal limitation.
Correlation analysis between the main phyla and the A and KS domain diversity showed an association between NRPS or PKS genes and less characterised phyla such as Bacteroidetes and Verrucomicrobia, especially in the Antarctic soils. These two phyla are additional microbial players in metabolite diversity together with the well-known producers Actinobacteria [48], Proteobacteria [49], Firmicutes [50] and Cyanobacteria [51]. BGCs from human-associated Bacteroidetes were recently identified [52] and a limited number of Verrucomicrobia genomes were analysed, identifying possible novel NRPS and PKS genes [12]. The majority of the BGCs reported for both groups have only been predicted and not yet experimentally characterised, but genome mining approaches as well as the results from our study suggest that these groups represent potential reservoirs of novel BGCs. The current study emphasises the potential for less well-characterised phyla being involved in NRP and PK diversity of soil, even though only a small fraction of OTUs were taxonomically matched to these groups. The low number of (annotated) sequences for these phyla in databases may have resulted in fewer matches. Continued sequence analysis combined with experimental characterisation of the predicted clusters will provide an understanding of the biosynthetic potential of these untapped bacterial phyla and the roles in soil ecology of the metabolites that they synthesize.
Antarctic soils harboured A and KS domain diversity unique to this habitat in addition to other reads showed similarities to derivatives of potentially useful antibacterial and anticancer drugs such as teixobactin [17], gramicidin [53], salinomycin [44] and actinomycin [54]. The application of this structure-function relationship analysis has demonstrated the importance of continuing the long-term study of this unique Antarctic habitat.
The identification of variables that affect the microbial community is challenging due to the intrinsic heterogeneity of environmental samples, such as the soil matrix, where different abiotic and biotic variables interact at the micro-scale [55]. However, it has been shown for both microorganisms and macroorganisms that environmental variables and geographic location affect biogeographic patterns of diversity [56,57,58,59,60]. The presence of potentially novel chemical structures in natural products might represent an advantage to microorganisms selecting taxa with different functional potentials to adapt to diverse conditions. Network analysis of A and KS OTUs showed segregation of metabolites forming diversity hotspots, sharing a limited number of OTUs with the other environments. Biogeographical studies have shown that limitation to dispersal, such as physical barriers, do not affect microorganisms but that environmental variables act selectively on the metabolic traits present in the microbial community [61].
Conclusions
The current study identified unexploited and unexplored taxa, such as Verrucomicrobia and Bacteroidetes, as potential sources of novel NRP and PK in soils with the implication that geographic isolation was one of the main drivers for microbial community functional diversity. The application of the approach presented in this study will assist in the identification of environments and relevant bacterial groups rich in potentially novel BGCs allowing a more focused targeted approach for drug discovery programmes. The experimental validation of the BGC potential of these less characterised phyla will also promote a better understanding of the role of natural products in the environment.
References
Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70(3):461–77.
Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75(3):311–35.
Amos GC, Gozzard E, Carter CE, Mead A, Bowes MJ, Hawkey PM, Zhang L, Singer AC, Gaze WH, Wellington EM. Validated predictive modelling of the environmental resistome. ISME J. 2015.
Charlop-Powers Z, Owen JG, Reddy BV, Ternei MA, Brady SF. Chemical-biogeographic survey of secondary metabolism in soil. Proc Natl Acad Sci U S A. 2014;111(10):3757–62.
Cimermancic P, Medema Marnix H, Claesen J, Kurita K, Wieland Brown Laura C, Mavrommatis K, Pati A, Godfrey Paul A, Koehrsen M, Clardy J, et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell. 2014;158(2):412–21.
Fritz S, Rajaonison A, Chabrol O, Raoult D, Rolain J-M, Merhej V. Full-length title: NRPPUR database search and in vitro analysis identify an NRPS-PKS biosynthetic gene cluster with a potential antibiotic effect. BMC bioinformatics. 2018;19(1):463.
Letzel AC, Li J, Amos GCA, Millan-Aguinaga N, Ginigini J, Abdelmohsen UR, Gaudencio SP, Ziemert N, Moore BS, Jensen PR. Genomic insights into specialized metabolism in the marine actinomycete Salinispora. Environ Microbiol. 2017;19(9):3660–73.
Amos GCA, Awakawa T, Tuttle RN, Letzel AC, Kim MC, Kudo Y, Fenical W, Moore BS, Jensen PR. Comparative transcriptomics as a guide to natural product discovery and biosynthetic gene cluster functionality. Proc Natl Acad Sci U S A. 2017;114(52):E11121–e11130.
Wang D, Lee SH, Seeve C, Yu JM, Pierson LS 3rd, Pierson EA. Roles of the Gac-Rsm pathway in the regulation of phenazine biosynthesis in Pseudomonas chlororaphis 30-84. Microbiologyopen. 2013;2(3):505–24.
Sidda JD, Song L, Poon V, Al-Bassam M, Lazos O, Buttner MJ, Challis GL, Corre C. Discovery of a family of [gamma]-aminobutyrate ureas via rational derepression of a silent bacterial gene cluster. Chem Sci. 2014;5(1):86–9.
van der Meij A, Worsley SF, Hutchings MI, van Wezel GP. Chemical ecology of antibiotic production by actinomycetes. FEMS Microbiol Reviews. 2017;41(3):392–416.
Letzel AC, Pidot SJ, Hertweck C. A genomic approach to the cryptic secondary metabolome of the anaerobic world. Nat Prod Rep. 2013;30(3):392–428.
Esmaeel Q, Pupin M, Kieu NP, Chataigné G, Béchet M, Deravel J, Krier F, Höfte M, Jacques P, Leclère V. Burkholderia genome mining for nonribosomal peptide synthetases reveals a great potential for novel siderophores and lipopeptides synthesis. MicrobiologyOpen. 2016;5(3):512–26.
Graça AP, Calisto R, Lage OM. Planctomycetes as novel source of bioactive molecules. Frontiers Microbiol. 2016;7:1241.
Borsetto C, Wellington EMH. Bioprospecting soil metagenomes for antibiotics. In: Paterson R, Lima N, editors. Bioprospecting: Success, Potential and Constraints. Cham: Springer International Publishing; 2017. p. 113–36.
Lan F, Demaree B, Ahmed N, Abate A. SiC-Seq: single-cell genome sequencing at ultra high-throughput with microfluidic droplet barcoding. Nature biotechnology. 2017;35(7):640–6.
Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517(7535):455–9.
Brady SF. Construction of soil environmental DNA cosmid libraries and screening for clones that produce biologically active small molecules. Nat Protoc. 2007;2(5):1297–305.
Craig JW, Chang FY, Kim JH, Obiajulu SC, Brady SF. Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl Environ Microbiol. 2010;76(5):1633–41.
Rondon MR, August PR, Bettermann AD, Brady SF, Grossman TH, Liles MR, Loiacono KA, Lynch BA, MacNeil IA, Minor C, et al. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol. 2000;66(6):2541–7.
Reddy BV, Kallifidas D, Kim JH, Charlop-Powers Z, Feng Z, Brady SF. Natural product biosynthetic gene diversity in geographically distinct soil microbiomes. Appl Environ Microbiol. 2012;78(10):3744–52.
Cai Y, Gu H, Kenney T. Learning microbial community structures with supervised and unsupervised non-negative matrix factorization. Microbiome. 2017;5(1):110.
Strieker M, Tanović A, Marahiel MA. Nonribosomal peptide synthetases: structures and dynamics. Curr Opin Struct Biol. 2010;20(2):234–40.
Shen B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr Opin Chem Biol. 2003;7(2):285–95.
Ziemert N, Podell S, Penn K, Badger JH, Allen E, Jensen PR. The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS One. 2012;7(3):e34064.
Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, et al. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Research. 2015;43(W1):W237–43.
Amos GCA, Borsetto C, Laskaris P, Krsek M, Berry AE, Newsham KK, Calvo-Bado L, Pearce DA, Vallin C, Wellington EMH. Designing and implementing an assay for the detection of rare and divergent NRPS and PKS clones in European, Antarctic and Cuban soils. PLoS ONE. 2015;10(9):e0138327.
Lemetre C, Maniko J, Charlop-Powers Z, Sparrow B, Lowe AJ, Brady SF. Bacterial natural product biosynthetic domain composition in soil correlates with changes in latitude on a continent-wide scale. Proc Natl Acad Sci U S A. 2017;114(44):11615–20.
Pearce DA, Newsham KK, Thorne MA, Calvo-Bado L, Krsek M, Laskaris P, Hodson A, Wellington EM. Metagenomic analysis of a southern maritime antarctic soil. Front Microbiol. 2012;3:403.
Herlemann DP, Labrenz M, Jurgens K, Bertilsson S, Waniek JJ, Andersson AF. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011;5(10):1571–9.
Mitchell A, Bucchini F, Cochrane G, Denise H, Hoopen P, Fraser M, Pesseat S, Potter S, Scheremetjew M, Sterk P, et al. EBI metagenomics in 2016 - an expanding and evolving resource for the analysis and archiving of metagenomic data. Nucleic Acids Research. 2016;44(D1):D595–603.
Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC bioinformatics. 2012;13:31.
Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics. 2015;31(21):3476–82.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–8.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6.
Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nature communications. 2016;7:11257.
Rho M, Tang H, Ye Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 2010;38(20):e191.
Mesuere B, Debyser G, Aerts M, Devreese B, Vandamme P, Dawyndt P. The Unipept metaproteomics analysis pipeline. Proteomics. 2015;15(8):1437–42.
Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, de Bruijn I, Chooi YH, Claesen J, Coates RC, et al. Minimum information about a biosynthetic gene cluster. Nat Chem Biol. 2015;11(9):625–31.
Hadjithomas M, Chen IM, Chu K, Ratner A, Palaniappan K, Szeto E, Huang J, Reddy TB, Cimermancic P, Fischbach MA, et al. IMG-ABC: A knowledge base to fuel discovery of biosynthetic gene clusters and novel secondary metabolites. MBio. 2015;6(4):e00932.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13(11):2498–504.
Peres-Neto PR, Jackson DA. How well do multivariate data sets match? The advantages of a Procrustean superimposition approach over the Mantel test. Oecologia. 2001;129(2):169–78.
Zhou S, Wang F, Wong ET, Fonkem E, Hsieh T-C, Wu JM, Wu E. Salinomycin: a novel anti-cancer agent with known anti-coccidial activities. Current medicinal chemistry. 2013;20(33):4095–101.
Vyverman W, Verleyen E, Wilmotte A, Hodgson DA, Willems A, Peeters K, Van de Vijver B, De Wever A, Leliaert F, Sabbe K. Evidence for widespread endemism among Antarctic micro-organisms. Polar Science. 2010;4(2):103–13.
Baas Becking LGM. Geobiologie of inleiding tot de milieukunde. Den Haag: W.P. Van Stockum & Zoon; 1934.
Arocha-Garza HF, Canales-Del Castillo R, Eguiarte LE, Souza V, De la Torre-Zavala S. High diversity and suggested endemicity of culturable Actinobacteria in an extremely oligotrophic desert oasis. PeerJ. 2017;5:e3247.
Bentley SD, Chater KF, Cerdeño-Tárraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature. 2002;417(6885):141–7.
Liu X, Cheng Y-Q. Genome-guided discovery of diverse natural products from Burkholderia sp. J Ind Microbiol Biotechnol. 2014;41(2):275–84.
Aleti G, Sessitsch A, Brader G. Genome mining: prediction of lipopeptides and polyketides from Bacillus and related Firmicutes. Comput Struct Biotechnol J. 2015;13.
Micallef ML, D’Agostino PM, Sharma D, Viswanathan R, Moffitt MC. Genome mining for natural product biosynthetic gene clusters in the Subsection V cyanobacteria. BMC Genomics. 2015;16(1):669.
Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell. 2014;158(6):1402–14.
Swierstra J, Kapoerchan V, Knijnenburg A, van Belkum A, Overhand M. Structure, toxicity and antibiotic activity of gramicidin S and derivatives. Eur J Clin Microbiol Infect Dis. 2016;35(5):763–9.
Merkel O, Wacht N, Sifft E, Melchardt T, Hamacher F, Kocher T, Denk U, Hofbauer JP, Egle A, Scheideler M, et al. Actinomycin D induces p53-independent cell death and prolongs survival in high-risk chronic lymphocytic leukemia. Leukemia. 2012;26(12):2508–16.
Vos M, Wolf AB, Jennings SJ, Kowalchuk GA. Micro-scale determinants of bacterial diversity in soil. FEMS Microbiol Rev. 2013;37(6):936–54.
Horner-Devine MC, Carney KM, Bohannan BJM. An ecological perspective on bacterial biodiversity. Proceedings of the Royal Society B: Biological Sciences. 2004;271(1535):113–22.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL, Horner-Devine MC, Kane M, Krumins JA, Kuske CR, et al. Microbial biogeography: putting microorganisms on the map. Nat Rev Micro. 2006;4(2):102–12.
Martiny JBH, Eisen JA, Penn K, Allison SD, Horner-Devine MC. Drivers of bacterial β-diversity depend on spatial scale. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(19):7850–4.
Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Micro. 2012;10(7):497–506.
Griffiths RI, Thomson BC, James P, Bell T, Bailey M, Whiteley AS. The bacterial biogeography of British soils. Environ Microbiol. 2011;13(6):1642–54.
Ramette A, Tiedje JM. Biogeography: an emerging cornerstone for understanding prokaryotic diversity, ecology, and evolution. Microbial Ecology. 2007;53(2):197–207.
Acknowledgments
Not applicable
Funding
We gratefully acknowledge the support of European Union Seventh Framework Programme grant agreement No. 289285 and Marie Curie Fellowship for CB, NERC Innovation Fund A My3820 award joint with Warwick Ventures to EMHW and Natural Environment Research Council funding through the British Antarctic Survey’s Long Term Monitoring and Survey Programme to DAP. EMHW acknowledges the support of Biotechnology and Biological Sciences Research Council network grant ‘The exploitation of metagenomics and meta-omics approaches in life science research ComMet’.
Availability of data and materials
The datasets generated and analysed during the current study are available in the European Nucleotide Archive repository under the BioProject PRJEB11689 (https://www.ebi.ac.uk/ena/data/view/PRJEB11689) and in the MGnify website under the study MGYS00000573 (https://www.ebi.ac.uk/metagenomics/studies/MGYS00000573).
Author information
Authors and Affiliations
Contributions
CB, GCAA and EMHW conceived the study. CB, EMHW, RFL, CV, DAP and KKN collected the samples. CB and GCAA conducted the lab work and generated the sequencing data. CB processed and analysed all datasets. ALM processed the EBI pipeline. GCAA, UNR and RDF advised on data analysis pipeline and troubleshooting. CB wrote the manuscript. GCAA, UNR, ALM, DAP, KKN and EMHW revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Characteristics and GPS coordinates of soils used in this study. Table S2. Primers and conditions used to prepare the amplicon libraries. Table S3. Summary of sequence counts per samples and total observed OTU processed with the custom pipeline. Table S4. Sequence counts for each sample for all targeted amplicon processed with the custom pipeline. Table S5. Correlation between phyla (16S rRNA gene diversity) and either A or KS domain diversity in all samples. Figure S1. Rarefaction curves for 16S rRNA gene, A and KS domains diversity. Figure S2. Alpha diversity indices for 16S rRNA gene and A and KS domains for each soil sample. Table S6. Correlation between Bray Curtis dissimilarity matrixes of each taxonomic level and the original OTU table. Figure S3. Community composition at phylum level of each soil sample. Figure S4. Representation at the Superkingdom level of the taxonomic providence of A and KS domain sequences. Figure S5. Procrustes transformation superimposition of 16S rRNA gene (all phyla or separate) against A domain diversity. Figure S6. Procrustes transformation superimposition of 16S rRNA gene (all phyla or separate) against KS domain diversity. (PDF 3191 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Borsetto, C., Amos, G.C.A., da Rocha, U.N. et al. Microbial community drivers of PK/NRP gene diversity in selected global soils. Microbiome 7, 78 (2019). https://doi.org/10.1186/s40168-019-0692-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-019-0692-8