
Abstract
Purpose
In threatened diversity hotspots, such as mountain cloud forests, microbiome studies have focused essentially on bacteria. Unlike prokaryotic microbiomes, the study of the microeukaryotes has largely been restricted to the visual identification of specific groups. Herein, microeukaryotic edaphic diversity from a pristine mountain cloud forest (MCF) of Mexico was analyzed via the metabarcoding of the ITS1 region of ribosomal DNA.
Materials and methods
An exploratory triangular sampling was conducted in the mountain cloud forest located in El Relámpago Mount, Santiago Comaltepec, Oaxaca, Mexico. Each vertex was located adjacent to a dominant plant species in the ecosystem (Oreomunnea mexicana and Alsophila salvinii). After DNA extraction the ITS1 region (rDNA) was amplified. Microeukaryotic sequences were filtered by computational subtraction against the ITS2 Database. Next, alpha and beta diversity indexes were calculated, and the relationship between abiotic variables and diversity patterns were inferred by means of a Canonical Correspondence Analysis.
Results
Overall, 138 inferred sequence variants were identified, including 87 protists, 35 animals (microfauna), and 16 algae. Within the animals, the nematodes were the dominant group, chlorophytes dominated algae, and in Protista, no dominance patterns were observed given the high diversity and equitability of this group. Soil available carbon, carbon degrading enzymes and the pH play a key role in modeling the community structure. Remarkably, high beta diversity levels were obtained, evidencing a strong spatial heterogeneity at the small scale.
Conclusions
The ITS metabarcoding proved to be a useful tool to conduct multi-taxa diversity assessments for microeukaryotes, allowing the identification of alpha and beta diversity patterns and overcoming limitations of sampling and the direct observation of individuals. The results presented in this work evidenced high microeukaryotic diversity levels in the soil of MCF and encourage future studies aiming to explore the taxonomic diversity of individual taxa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Mountain Cloud Forests (MCF) harbor an immense biodiversity and a high percentage of endemisms, despite its restricted extension (1% of the Mexican territory) and fragmented distribution (restricted to altitudes between 600 and 3,500 masl) (Rzedowski 1996; Bubb et al. 2004). This is due to characteristic environmental conditions such as constant cloud immersion, rugged relief, low luminosity, and the convergence of boreal and neotropical species (Gual-Díaz and Rendón-Correa 2014). Due to the continuous disturbance and destruction of these forests, MCF have been recognized worldwide as a priority for conservation since they play a very important role in hydrological cycles and are biodiversity hotspots (Bubb et al. 2004; FAO 2006; CONABIO 2010).
The inventory of the diversity in MCF has increased for groups of higher plants and animals (i.e. mammals and birds). However, the characterization of the belowground diversity is still in progress (Rzedowski 1996; Bubb et al. 2004; CONABIO 2010). Members of this edaphic community such as algae, protists, nematodes, and microarthropods, hereafter referred to as microeukaryotes, participate as key elements in the degradation of organic matter and nutrient cycling (Hättenschwiler et al. 2005; Jacquiod et al. 2016). For example, nematodes and protists regulate fungal and bacterial populations by preying on them. This causes changes in community composition and activity of these microorganisms, and hence affects decomposition processes, nutrient turnover, and plant-microbiota interactions (Bonkowski 2004; Thakur and Geisen 2019). In addition, algae (i.e. glaucophytes, rhodophytes, chlorophytes and streptophytes) participate in the incorporation of carbon and nitrogen in the early stages of soil formation and serve as food for other microeukaryotes (Thorn and Lynch 2007; Friedl and Rybalka 2012; Jacquiod et al. 2016).
Around 60% of the nutrients ingested by soil microeukaryotes are excreted to the soil, having a direct effect on the nutrient cycles (Bardgett and Griffiths 1997; Hättenschwiler et al. 2005; Bardgett 2005). For example, protists release through their excreta N that is immobilized by bacteria, incrementing available N in soil by 37% (Koller et al. 2013). Particularly, microarthropods (i.e. mites, springtails) consume, fragment, and process the organic matter that enters the system, facilitating its future decomposition. In addition, when moving, they not only modify the physical structure of the microhabitat, altering the microenvironmental conditions and creating niches for microorganisms, but also transport bacteria and fungi, favoring colonization in new sites (Hättenschwiler 2005).
Despite their important role in soil trophic webs, in Mexico, of the approximately 1,400 recorded species of protists, only 36 have been isolated from soils (Gallegos-Neyra et al. 2014; Mayén-Estrada et al. 2014a, b). For algae, of the more than 2,000 reported species, only 4 come from terrestrial habitats (Pedroche et al. 1993; Novelo 2012). For both groups, the records are biased towards freshwater and marine ecosystems. In addition, due to their small size and the scarcity of genetic/molecular work conducted for these groups, records of edaphic species in Mexico are rare (Gallegos-Neyra et al. 2014). Regarding the microfauna of the MCF, most of the studies rely on direct extraction of organisms and their morphological identification, highlighting the presence of Coleoptera, Diptera, Hemiptera, Hymenoptera, Orthoptera; as well as Acari and Collembola (Parada-Barrera 1987; Morón-Ríos and Huerta-Lwanga 2006; Bernal-Rojas et al. 2009; De la Rosa and Negrete-Yankelevich 2012; Beltrán-Villanueva et al. 2016).
Metabarcoding (simultaneous identification of multiple taxa from an environmental DNA sample) presents several advantages in the study of edaphic communities since it does not involve the extraction, manipulation and description of organisms; and succeeds in the detection of cryptic, rare and non-cultivable species. This approach allows a quick and precise determination of the relative abundance of organisms and diversity patterns (Hamilton et al. 2009; Drummond et al. 2015; Ruppert et al. 2019; Lin et al. 2020), increasing the knowledge generated from traditional methodologies.
In this work we implemented a metabarcoding approach of the ITS1 region of ribosomal DNA to describe the diversity and structure of soil microeukaryotic communities in relation to key environmental variables in a pristine location of MCF in Mexico. We expect that this proxy will allow the detection of a high microeukariotic diversity at the small spatial scale (within a 10 m triangular transect) in accordance with former reports acknowledging this ecosystem as diversity hotspot, and that large diversity patters will be identified in relation to key edaphic variables paving the way for further comprehensive research on specific taxa. As a result, we report 87 protist, 35 microfaunal and 16 algal ASVs. We detected differences in community structure comparing three sites influenced by different plant species: Oreomunnea mexicana, Alsophila salvinii, and a decaying trunk. These differences were explained by the particular environmental variables of each site, mainly pH, dissolved organic carbon, ammonium, and the activity of enzymes such as polyphenol oxidase and β -glucosidase. In addition to environmental variables, the differences can also be explained by possible interactions between microeukaryotes and other microorganisms such as bacteria and fungi on which they feed.
2 Materials and methods
2.1 Study site and Sampling
Sampling was carried out in the MCF located in El Relámpago Mount (17° 35′ 30.4″ N, 96° 23′ 57.1″ W), Santiago Comaltepec, Oaxaca, Mexico. This forest belongs to the largest and most continuous patch of MCF in Mexico, an area of critical priority for conservation (Toledo-Aceves et al. 2011; del Mar et al. 2015). It is characterized by humid temperate climate with temperatures ranging from 10–26 °C, annual rainfall of 800–4,000 mm (INEGI 2005). Dominant soil group is acidic humic Acrisol (Sundue 2017).
Three sampling sites were established along an equilateral 10 m-triangular transect (spatial sampling method that increases the chance of achieving a high interstrata variance; Bąk 2014) at 2,219 masl. The first vertex was located adjacent to the base of an individual of O. mexicana (Juglandaceae), the second point next to an individual of the fern A. salvinii (Cyatheaceae), and the third point under a decomposing tree trunk. This sampling design was chosen to better represent the heterogeneity of the site. In each plot a 1 m x 1 m x 1 m triangular subplot was delimited, where three soil subsamples were collected on each vertex. Soil cores were collected at a depth of 25 cm (excluding organic horizon) using a 10 cm diameter corer. Subsamples were placed in hermetically sealed plastic bags, kept in the dark at a temperature of 4 °C for analysis in the laboratory.
2.2 Environmental DNA isolation and sequencing
Direct DNA extraction of 0.25 g from each subsample was obtained using the DNeasy PowerSoil extraction kit (Qiagen) following the manufacturer's instructions. The DNA was quantified using the Qubit® 2.0 Fluorometer (Invitrogen by Life Technologies) and sequenced using the Illumina MiSeq (2 × 300) paired-end format at the Genomic Services Laboratory (LANGEBIO, Irapuato, Guanajuato, Mexico). The ITS region (rDNA) was amplified using the primer set ITS1-F and ITS2 (Gardes and Bruns 1993), with a yield of about 25,000 reads per sample.
2.3 Bioinformatics analysis
The ITS reads were processed with the dada2 v1.13.1 (Callahan et al. 2016) package of R v4.0.2 (R Core Team 2020). Firstly, the primers and adapters were eliminated and then the ASVs (amplicon sequence variant) were determined. Sequences were filtered, excluding reads with bases other than G, C, T, and A (maxN = 0) and keeping those that presented < 1 expected errors, considering their quality scores (maxEE = c (1,1), truncQ = 3). For each subsample, the ASV inference was made for the reads in both directions (5′ 3 '-3′ 5'), considering the specific error rates of each run. Subsequently, the paired sequences that presented a minimum of 50 bp of overlap were assembled and those that did not coincide completely in the overlapping region were eliminated. Finally, chimeras were eliminated with the “removeBimeraDenovo” function of dada2 and singletons (unique sequence between readings) were excluded. To filter the microeukaryotic sequences, the ITS2 Database was used via a Basic Local Alignment Search Tool (BLAST) to identify significant similarities. Sequences matching to unidentified organisms, fungal species or higher plants, were discarded from the analysis; and only those that paired with protists, algae, nematodes and microarthropods were retained.
To attain a more precise identification of the organisms (i.e. phylum or order), reference sequences from the GenBank were incorporated in character matrices and aligned using MUSCLE v3.8.31 (Edgar 2004). Phylogenetic trees were built using the principle of parsimony using the MESQUITE version 3.61 program (Madisson and Madisson 2019). We performed heuristic searches by the Subtree pruning and regrafting (SPR) algorithm. Branch support was assessed by bootstrap analysis (100 replicates). We recovered the most parsimonious tree by a majority-rule consensus.
2.4 Data analyses
Alpha diversity (α) was calculated using the Shannon-Wiener and Simpson Indexes. The beta diversity (β) was calculated by means of the Jaccard Index and the Bray-Curtis distances. Also, to explore the relationship between the abundance of ASVs and environmental variables (pH, dissolved carbon, dissolved phosphorus, ammonium, and enzymatic activity of polyphenol oxidase, β-glucosidase, and phosphomonoesterase; data synchronously obtained from the exact same sites and retrieved from Velez et al. 2021, significant variables were chosen after a redundancy analysis) a Canonical Correspondence Analysis (CCA) was performed. These analyses were run using the vegan package (Oksanen et al. 2020) in the R v4.0.2 program (R Core Team 2020). Lastly, an Olmsted-Tukey corner test (Sokal and Rohlf 1981) was performed to visualize the community structure recognizing dominant, constant, occasional and rare species (González-Acosta et al. 2005). It was calculated and plotted in Excel using the natural logarithm of the abundances and the frequencies of appearance of each ASV.
3 Results
Through Illumina MiSeq sequencing, 682,361 raw ITS1 reads were obtained. After quality filtering 178,089 reads were removed, resulting in 504,272 retained reads that were further analyzed. Subsequently, 2771 ASVs were inferred, out of which 668 ASVs paired with microeukaryotic sequences. Particularly: 103 ASVs matched with animalia, 38 ASVs with algae (plantae), 330 ASVs with protists and 197 ASVs matched with various microeukaryotic groups. The ASV delimitation was further refined using a phylogenetic approach, leading to the identification of 138 ASVs. Protists represented 63% of the total richness, followed by animals with 25% and plantae representing only 12%.
3.1 Diversity patterns
3.1.1 Animalia
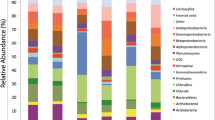
The animal fraction of microeukaryotes was composed of 15 nematodes ASVs, 15 arthropods, 2 mites and 3 platyhelminthes. Samples from site 1 (O. mexicana) and site 3 (trunk) presented the dominance of the ASV Nematoda 1. On the other hand, the structure of the community in site 2 (A. salvinii) presented a greater representation of arthropods, hexapods and different nematode ASVs with relative abundances in similar proportions to each other (Fig. 1). Regarding α diversity patterns, site 1 presented communities with high Simpson dominance, especially in subsample 1. On the contrary, for subsamples 5 and 6 of site 2, Simpson dominance was low, presenting the highest diversity values in the Inverse Simpson and Shannon Index (Table 1).

Diversity of ASVs corresponding to Animalia (microfauna). The x axis shows the three sites with their respective subsamples and the y axis indicates the relative abundance of the ASVs. The red numbers above the bars indicate the total number of ASVs identified in the subsample
When analyzing β diversity, a high heterogeneity was observed on ASV composition between sites and subsamples (Fig. 2A). Nonetheless, regarding the abundances, site 1 and 3 differed from site 2 (Fig. 2B). The structure of the microfauna community was mostly represented by 23 rare ASVs, eight dominant ASVs corresponding to nematodes, hexapods, arthropods and Sarcoptiform mites, two constant ASVs and two occasional ASVs (Fig. 3).

A Jaccard distances and B Bray-Curtis distances between subsamples. The y axis indicates the distance between subsamples. The blue numbers correspond to the subsamples from site 1, the green numbers correspond to site 2, and the red numbers correspond to site 3

Olmstead-Tukey diagram for Animalia. The x axis shows the frequency of appearance of the ASVs and the y axis the logarithm of the total abundance of each ASV plus 1. The quadrants of Occasional, Dominant, Rare and Constant ASVs are shown, delimited by the means of each axis
3.1.2 Protista
The protists' community included 20 ASVs affiliated to the Alveolata superphylum, five for Heterokonta, 17 for Rhizaria and 16 ASVs for Amoebozoa. In total, 29 ASVs could not be assigned to any phylum or superphylum, so they were classified as part of the SAR supergroup or as Protist. Given the high richness of protists, a high heterogeneity was observed in the composition of the communities by subsample. The lowest richness values were presented in the subsamples 8 and 9 from site 3 (trunk), while the highest richness was observed in the subsamples 1 and 4 (Fig. 4). In general, the α diversity of protists in all subsamples was high, particularly in subsamples 5 and 6 from site 2 (A. salvinii), and subsample 1 from site 1 (O. mexicana). Site 3 was the least diverse of the sites, however, the dominance values are very low and the Shannon's index indicated high diversity (Table 2).

Diversity of ASVs corresponding to the kingdom Protista. The x axis shows the three sites with their respective subsamples and the y axis indicates the relative abundance of the ASVs. The red numbers above the bars indicate the total number of ASVs identified in the subsample
The community composition was similar among the subsamples of sites 1 and 2, segregating site 3 (Fig. 5A). On the other hand, based on the abundances per subsample, no particular grouping was observed (Fig. 5B). In general, the community presented a balance between the number of dominant and rare ASVs, 34 and 45 respectively; six occasional and two constant ASVs. Dominant ASVs corresponded to the Rhizaria superphylum, whereas rare ASVs were represented by the Alveolata superphylum (Fig. 6).

A Jaccard distances and B Bray-Curtis distances between subsamples. The y axis indicates the distance between subsamples. The blue numbers correspond to the subsamples from site 1, the green numbers correspond to site 2, and the red numbers correspond to site 3

Olmstead-Tukey Diagram for Protista. The x axis shows the frequency of appearance of the ASVs and the y axis the logarithm of the total abundance of each ASV plus 1. The quadrants of Occasional, Dominant, Rare and Constant ASVs are shown, delimited by the means of each axis
3.1.3 Plantae
Within the algae, four ASVs were assigned to Chlorophyta, 11 to Rhodophyta and one to Glaucophyta. In general, the algal communities per subsample were dominated by chlorophytes. Most of the rhodophytes were exclusive ASVs of site 3 (trunk), the one with the highest richness (Fig. 7). Subsample 8 from site 3 was the most diverse, followed by subsample 3 from site 1 (O. mexicana). Although site 3 presented the most diverse subsample, it also included the subsample with the highest dominance (subsample 7). In site 2 (A. salvinii), subsamples 4 and 5 showed high dominance (Table 3).

Diversity of ASVs corresponding to the algae of the kingdom Plantae. The x axis shows the three sites with their respective subsamples and the y axis indicates the relative abundance of the ASVs. The red numbers above the bars indicate the total number of ASVs identified in the subsample
According to Jaccard distances, subsamples in site 3 clustered together. In contrast, subsamples from site 1 presented interspersed clustering with those from site 2 (Fig. 8A). Regarding the Bray-Curtis distances by abundance, no grouping pattern was observed since the abundances were highly variable (Fig. 8B). Within the algal community, 9 ASVs were rare and most of these were identified as rhodophytes and the only reported glaucophyte; 4 ASVs were dominant chlorophytes ASVs, and both the occasional (1 ASV) and constant (2 ASVs) ASVs were also rhodophytes (Fig. 9).

A Jaccard distances and B Bray-Curtis distances between subsamples. The y axis indicates the distance between subsamples. The blue numbers correspond to the subsamples from site 1, the green numbers correspond to site 2, and the red numbers correspond to site 3

Olmstead-Tukey diagram for Plantae. The x axis shows the frequency of appearance of the ASVs and the y axis the logarithm of the total abundance of each ASV plus 1. The quadrants of Occasional, Dominant, Rare and Constant ASVs are shown, delimited by the means of each axis
3.2 Relationship between environmental variables and diversity
3.2.1 Animalia
The CCA revealed differences in richness and diversity between sites that can be related to the local environmental conditions. The community of site 1 (O. mexicana) is related to a high activity of the enzyme β-glucosidase, low concentration of ammonium and dissolved organic phosphorus. Site 2 (A. salvinii) is characterized by the high activity of phosphomonoesterase, polyphenol oxidase, and concentration of dissolved organic carbon. And finally, at site 3 (trunk) there is a high concentration of ammonium and dissolved organic phosphorus, and the highest pH values (Fig. 10).

Canonical Correspondence Analysis. The x axis represents component 1, the y axis represents component 2, the blue arrows correspond to the environmental variables (GluSid: β-glucosidase enzyme, DOP: dissolved organic phosphorus, NH4: ammonium, pH, PMonEst: phosphomonoesterase enzyme, DOC: dissolved organic carbon and Lac: polyphenol oxidase enzyme), the ASVs are shown in red and the points indicate the position of each site. The CCA1 reflects an 84% explained proportion and the CCA2 a 15%
3.2.2 Protista
Sites 2 (A. salvinii) and 3 (trunk) were associated with the concentration of dissolved nutrients (C, P and ammonium) as well as the enzymatic activity of polyphenol oxidase and phosphomonoesterase. On the contrary, the communities of site 1 (O. mexicana) were subjected to an environment with limited resources since it was related negatively with all the characterized variables (Fig. 11).

Canonical Correspondence Analysis. The x axis represents component 1, the y axis represents component 2, the blue arrows correspond to the environmental variables (GluSid: β-glucosidase enzyme, DOP: dissolved organic phosphorus, NH4: ammonium, pH, PMonEst: phosphomonoesterase enzyme, DOC: dissolved organic carbon and Lac: polyphenol oxidase enzyme), the ASVs are shown in red and the points indicate the position of each site. The CCA1 reflects a 57% explained proportion and the CCA2 a 42%
3.2.3 Plantae
Differences were observed in the algae communities associated with the environmental variables. Site 1 (O. mexicana) showed a negative relationship with pH, that is, it was the most acidic site, where Chlorophyta sp. 4 and Rhodophyta sp. 8 occurred as exclusive ASVs. Site 2 (A. salvinii) was linked with polyphenol oxidase and phosphomonoesterase enzymes, as well as dissolved organic carbon concentration. Also, as the CCA showed, site 2 was the only one not having any exclusive ASVs. Finally, site 3 (trunk) was associated with dissolved organic phosphorus and ammonium (Fig. 12).

Canonical Correspondence Analysis. The x axis represents component 1, the y axis represents component 2, the blue arrows correspond to the environmental variables (GluSid: β-glucosidase enzyme, DOP: dissolved organic phosphorus, NH4: ammonium, pH, PMonEst: phosphomonoesterase enzyme, DOC: dissolved organic carbon and Lac: polyphenol oxidase enzyme), the ASVs are shown in red and the points indicate the position of each site. The CCA1 reflects a 77% explained proportion and the CCA2 a 22%
4 Discussion
To the extent of our knowledge this is the first description of soil microeukaryote diversity in a MCF of Mexico using high-throughput multi-taxa identification from environmental DNA. Former work was based on the direct extraction of the organisms, their morphological description, and identification. This proxy has led to the description of edaphic microfauna in MCF of Jalisco, Chiapas, Oaxaca, Veracruz and Puebla, describing Coleoptera, Diptera, Hemiptera, Chilopoda, Diplopoda, Hymenoptera, Orthoptera and Collembola, as the main orders (Parada-Barrera 1987; Morón-Ríos and Huerta-Lwanga 2006; Bernal-Rojas et al. 2009; De la Rosa and Negrete-Yankelevich 2012; Beltrán-Villanueva et al. 2016). In contrast, our results showed the high representation of Nematoda, the presence of Acari, platyhelminths and unidentified arthropods and an overall diversity of 35 ASVs at the small spatial scale.
Despite algae and protists are known to occur in MCF (e.g. in association with old tree trunks, increasing water-holding capacity and nutrient availability and as photobionts of lichens, Kosecka et al. 2021) reports of these groups in Mexico come mostly from aquatic environments (i.e. freshwater and marine ecosystems), and little is known about their diversity in soils of MCF. For example, from 146 described species of heterotrophic flagellate protists (Rhizaria, Stramenopiles and Excavata) registered for Mexico, only 4 have been isolated from soils and 3 from litter (Mayén-Estrada et al. 2014b); and from the 959 described species of ciliates only 25 have been found in soils (Mayén-Estrada et al. 2014a). Here, we characterized more than 80 ASVs for protists, revealing that soils are potential habitats for a large diversity of protists that needs to be further explored. Remarkably, this approach allowed the detection of potentially non-cultivable protists species that are left out from direct extraction methods. Whereas for soil algal diversity, registers in Oaxaca were limited to the genera Chlamydomonas, Chlorella, Cladophora and Rhizoclonium (Chlorophyta) isolated from Tehuacán-Cuicatlán Valley (Novelo 2012). By overcoming cultivation limitations, we report the occurrence of Rhodophytes and Glaucophytes, paving the way for the exploration of soil diverse environments in MCF and extending the knowledge of the distribution of these groups.
In general, specific clustering patterns in terms of community composition among the study sites were not observed. This reflects a huge heterogeneity in soil at the small spatial scale (1 m). However, we observed differences in terms of the abiotic setting that could contributed to the modeling of microeukaryotic communities in response to variables such as vegetation, litter, and biological activity (Bardgett 2005; Wehner et al. 2016).
Our results evidenced that sites 1 (O. mexicana) and 3 (trunk) were characterized by the dominance of Nematoda 1. This may be related with the high expression of β-glucosidase (produced by carbon decomposer bacteria; Woodward and Wiseman 1982; Baldrian 2006; Sharma et al. 2007), and ammonium (fixed by nitrogen decomposer bacteria and recourse for nitrifying bacteria; Vitousek et al. 2002). According to former work by Leocadio and collaborators (2023), sites 1 and 3 registered the presence of Actinobacteria, Firmicutes and Proteobacteria, confirming the potential availability of resources for opportunistic bacteriophages nematodes. Various studies conclude that bacteriophage nematodes migrate to recourse pulses, explaining their copious occurrence in these sites (Griffiths 1990; Bonkowski 2004; Van der Wal et al. 2012; Shaw et al. 2019). In contrast, site 2 (A. salvinii) reflected the highest diversity values, being related with the enzyme polyphenol oxidase, mainly produced by fungi that decompose lignin (Woodward and Wiseman 1982; Baldrian 2006; Sharma et al. 2007). In accordance to former work demonstrating that the richness and abundance of fungivore nematodes depends on diverse fungal communities (Freckman and Caswell 1985; Zhang et al. 2020), our results confirmed that the presence of fungal-derived exoenzymes are linked to the dominance of this fraction of the microfaunal community.
For protists, the highest diversity values agreed with the presence of A. salvinii (site 2). This diversity pattern may be related with the dissolved carbon concentration and microbial activity (polyphenol oxidase and β-glucosidase), highlighting the role of the rhizospheric nutrients on protists diversity. As rhizospheric carbon compounds serve as resources for microorganisms such as bacteria and fungi, the main prey of protists (Acosta-Mercado and Lynn 2004; Adl and Coleman 2005; Geisen et al. 2018; Samad et al. 2019). This assumption is supported by the copious occurrence of bacterial (Leocadio et al. 2023) and fungal (Velez et al. 2021) diversity in this site.
In terms of algae, the largest diversity was registered in site 3 (trunk), which was associated with high ammonium and dissolved organic phosphorus values, essential for these autotrophic organisms. Both P and N represent determinant factors that positively affect the abundance, biomass, and richness of algae (Lin and Wu 2014; Glaser et al. 2017; Novakovskaya et al. 2022). Also in this site, rhodophytes showed high richness, in accordance with former works reporting that this type of algae proliferates on moist ammonium-rich soils (Thorn and Lynch 2007). Algae typically proliferate on soils devoid of vegetation (Thorn and Lynch 2007). Both sites 1 (O. mexicana) and 2 (A. salvinii), were located at the base of a plant host showing the lowest diversity values. So, it is feasible that this pattern could be linked to negative biotic interactions with plants, microfauna, and protists. Nonetheless this assumption should be further confirmed.
Overall, our results on the large magnitude of soil microeukaryotic diversity (including members of the Plantae, Animalia and Protista) and community composition patterns in relation to the abiotic setting, demonstrate the efficiency of ITS high-throughput multi-taxa identification using environmental DNA for soil microeukaryotic communities. Nonetheless, this methodology remains to be refined to obtain more precise results. For example, by improving the resolution of taxonomic assignments with taxa-specific primers to amplify the scope. Also, as many of these soil organisms have been poorly studied before, reference sequences are not well represented in public repositories. Pairing genetic studies with traditional methodologies of direct extraction of the organisms and morphological identification, may complement our understanding of determining species more than just genetic units (Bogale et al. 2020).
5 Conclusions
A remarkably diverse microeukaryotic community (including members from three kingdoms of life) was registered at the small spatial scale for Mexico in contrast with former reports based on the direct observation of these organisms in soil samples. Our results evidenced the large magnitude of unseen belowground biota and warn on their functional relevance, encouraging future studies aiming to explore individual taxa. Herein, we demonstrated that belowground microeukaryotes are closely interlinked at the community level with further elements of the microbial community (e.g. fungi and bacteria), potentially reinforced by the effect of plants and edaphic variables such as availably carbon and pH. Our data showed that these organisms may be powerful mutual drivers, with both positive and negative feedbacks. So, a combined aboveground-belowground approach to community and ecosystem ecology in MCF is strongly recommended to improve our understanding of ecosystem functioning and conservation.
The ITS metabarcoding proved to be a useful tool to conduct multi-taxa diversity assessments, allowing the identification of alpha and beta diversity patterns overcoming traditional sampling cultivation limitations. Likewise, complementing these results with the analysis of relevant environmental variables, depicted gross community structuring forces, revealing their potential role in the functioning of an ecosystem as diverse and threatened as the MCF. Considering that the main limitation of metabarcoding is the availability of reference sequences in the repositories, it is necessary to enrich public databases with curated and properly identified sequences to broaden the scope of this technique. Despite verifying the effectiveness of the ITS metabarcoding to assess edaphic microeukaryote communities, we acknowledge the importance of conducting complementary studies that implement classic techniques of direct collection, morphological description coupled to the molecular identification of the organisms.
References
Acosta-Mercado D, Lynn D (2004) Soil ciliate species richness and abundance associated with the rhizosphere of different subtropical plant species. J Eukaryot Microbiol 51:582–588. https://doi.org/10.1111/j.1550-7408.2004.tb00295.x
Adl S, Coleman D (2005) Dynamics of soil protozoa using a direct count method. Biol Fertil Soils 42:168–171. https://doi.org/10.1007/s00374-005-0009-x
Bąk T (2014) Triangular method of spatial sampling. Statistics in Transition 15:9–22
Baldrian P (2006) Fungal laccases - occurrence and properties. FEMS Microbiol Rev 30:215–242. https://doi.org/10.1111/j.1574-4976.2005.00010.x
Bardgett RD, Griffiths B (1997) Ecology and biology of soil protozoa, nematodes and microarthropods. Modern Soil Microbiology. CRC Press, New York, pp 129–163
Bardgett RD (2005) The diversity of life in soil. The Biology of Soil: A community and Ecosystem Approach. Oxford University Press, Oxford, pp 24–55
Beltrán-Villanueva MA, Tamez-Hernández LR, Cruz-Miranda SG, Páez-Gerardo LE (2016) Listado Taxonómico de algunos Artrópodos Edáficas en dos localidades de Xicotepec de Juárez, Puebla. Bol Soc Mex Entomol 2:39–44
Bernal-Rojas A, Castaño-Meneses G, Palacios-Vargas JG, García-Calderón NE (2009) Oribatid mites and springtails from a coffee plantation in Sierra Sur, Oaxaca. Mexico Pesq Agropec Bras 44:988–995. https://doi.org/10.1590/S0100-204X2009000800030
Bogale M, Baniya A, DiGennaro P (2020) Nematode identification techniques and recent advances. Plants (basel, Switzerland) 9:1260. https://doi.org/10.3390/plants9101260
Bonkowski M (2004) Protozoa and plant growth: the microbial loop in soil revisited. New Phytol 162:617–631. https://doi.org/10.1111/j.1469-8137.2004.01066.x
Bubb P, May I, Miles L, Sayer J (2004) Cloud Forest Agenda. UNEP-WCMC, UK
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
CONABIO (2010) El Bosque Mesófilo de Montaña en México: Amenazas y Oportunidades para su Conservación y Manejo Sostenible. CONABIO, México
De la Rosa IN, Negrete-Yankelevich S (2012) Distribución espacial de la macrofauna edáfica en bosque mesófilo, bosque secundario y pastizal en la reserva La Cortadura, Coatepec, Veracruz, México. Revista Mexicana De Biodiversidad 83:201–215
del Mar D-S, Escalante R, Basurto S (2015) Is the community-based management of natural resources inherently linked to resilience? An analysis of the Santiago Comaltepec community (Mexico). Revista De Estudios Sobre Despoblación y Desarrollo Rural 18:91–114
Drummond AJ, Newcomb RD, Buckley TR, Xie D, Dopheide A, Potter BC, Heled J, Ross HA, Tooman L, Grosser S, Park D, Demetras NJ, Stevens MI, Russell JC, Anderson SH, Carter A, Nelson N (2015) Evaluating a multigene environmental DNA approach for biodiversity assessment. GigaScience 4:s13742-015-0086-1. https://doi.org/10.1186/s13742-015-0086-1
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 19:113. https://doi.org/10.1186/1471-2105-5-113
FAO (2006) Evaluación de los Recursos Forestales Mundiales. Organización de las Naciones Unidas para la Agricultura y la Alimentación, Rome
Freckman D, Caswell E (1985) The ecology of nematodes in agroecosystems. Ann Rev Phytopathol 23:275–296. https://doi.org/10.1146/annurev.py.23.090185.001423
Friedl T, Rybalka N (2012) Systematics of Green Algae: A Brief Introduction to the Current Status. Progress in Botany, vol 73. Springer, Berlín, pp 259–280
Gallegos-Neyra EM, Lugo-Vázquez A, Calderón-Vega A, Sánchez-Rodríguez MR, Mayén-Estrada R (2014) Biodiversidad de protistas amébidos de vida libre en México. Rev Mex Biodivers 85:10–25
Gardes M, Bruns D (1993) ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118. https://doi.org/10.1111/j.1365-294X.1993.tb00005.x
Geisen S, Mitchell E, Adl S, Bonkowski M, Dunthorn M, Ekelund F, Lara E et al (2018) Soil protists: a fertile frontier in soil biology research. FEMS Microbiol Rev 42:293–323. https://doi.org/10.1093/femsre/fuy006
Glaser K, Baumann K, Leinweber P, Mikhailyuk T, Karsten U (2017) Algal diversity of temperate biological soil crusts depends on land use intensity and affects phosphorus biogeochemical cycling. Biogeosciences Discuss. https://doi.org/10.5194/bg-2017-365
González-Acosta AF, la Cruz-Agüero D, Ruiz-Campos G (2005) Seasonal pattern of the fish assemblage of El Conchalito mangrove swamp, La Paz bay, Baja California Sur. Mexico Hidrobiológica 15:205–214
Griffiths B (1990) A comparison of microbial-feeding nematodes and protozoa in the rhizosphere of different plants. Biol Fertil Soils 9:83–88. https://doi.org/10.1007/BF00335867
Gual-Díaz M, Rendón-Correa A (2014) Bosques Mesófilos de Montaña de México: diversidad, ecología y manejo. CONABIO, México
Hamilton HC, Strickland MS, Wickings K, Bradford MA, Fierer N (2009) Surveying soil faunal communities using a direct molecular approach. Soil Biol Biochem 41:1311–1314. https://doi.org/10.1016/j.soilbio.2009.03.021
Hättenschwiler S, Tiunov AV, Scheu S (2005) Biodiversity and Litter Decomposition in Terrestrial Ecosystems. Annu Rev Ecol Evol Syst 36:191–218. https://doi.org/10.1146/annurev.ecolsys.36.112904.151932
INEGI (2005) Prontuario de información geográfica municipal de los Estados Unidos Mexicanos. Santiago Comaltepec, Oaxaca
Jacquiod S, Stenbæk J, Santos S, Winding A, Sørensen S, Priemé A (2016) Metagenomes provide valuable comparative information on soil microeukaryotes. Res Microbiol 167:436–450. https://doi.org/10.1016/j.resmic.2016.03.003
Kosecka M, Guzow-Krzemińska B, Černajová I, Škaloud P, Jabłońska A, Kukwa M (2021) New lineages of photobionts in Bolivian lichens expand our knowledge on habitat preferences and distribution of Asterochloris algae. Nature 11:8701. https://doi.org/10.1038/s41598-021-88110-0
Koller R, Scheu S, Bonkowski M, Robin C (2013) Protozoa stimulate N uptake and growth of arbuscular mycorrhizal plants. Soil Biol Biochem 65:204–210. https://doi.org/10.1016/j.soilbio.2013.05.020
Leocadio C, López NÁ, Barrios A, Guerra A, Torres YT, Velez P (2023) Diversidad microbiana cultivable del suelo en un bosque mesófilo de montaña prístino en Oaxaca. México Revista Mexicana De Biodiversidad 94:e944980
Lin C, Wu J (2014) Environmental factors affecting the diversity and abundance of soil photomicrobes in Arid Lands of Subtropical Taiwan. Geomicrobiol J 31:350–359. https://doi.org/10.1080/01490451.2013.828135
Lin M, Simons AL, Curd EE, Harrigan RJ, Schneider FD, Ruiz-Ramos DV, Gold Z, Osborne MG, Shirazi S, Schweizer TM, Moore TN et al (2020) A biodiversity composition map of california derived from environmental DNA metabarcoding and earth observation. Ecol Appl. https://doi.org/10.1101/2020.06.19.160374
Maddison WP, Maddison DR (2019) Mesquite: A Modular System for Evolutionary Analysis. Version 3.61. http://mesquiteproject.org
Mayén-Estrada R, Reyes-Santos M, Aguilar-Aguilar R (2014a) Biodiversidad de Ciliophora en México. Rev Mex Biodivers 85:34–43
Mayén-Estrada R, Reyes-Santos M, Vicencio-Aguilar ME (2014b) Biodiversidad de protistas (flagelados heterótrofos) en México. Rev Mex Biodivers 85:26–33
Morón-Ríos A, Huerta-Lwanga E (2006) Soil Macrofauna of two successional Evergreen Cloud Forest stages from The Cerro Huitepec Nature Reserve, San Cristóbal de las Casas, Chiapas. México Interciencia 31:611–615
Novakovskaya IV, Patova EN, Dubrovskiy YA, Novakovskiy AB, Kulyugina EE (2022) Distribution of algae and cyanobacteria of biological soil crusts along the elevation gradient in mountain plant communities at the Northern Urals (Russian European Northeast). J Mt Sci 19:637–646. https://doi.org/10.1007/s11629-021-6952-7
Novelo E (2012) Chlorophyta. Flora del Valle de Tehuacán-Cuicatlán. Instituto de Biología, México, pp 1–86
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2020) vegan: Community Ecology Package. R package version 2.5–7. https://CRAN.R-project.org/package=vegan
Parada-Barrera G (1987) Contribución al conocimiento sobre la entomofauna del suelo y la hojarasca, en un Bosque Mesófilo de Montaña en la Sierra de Manantlán, Jalisco. Universidad de Guadalajara, México, México
Pedroche FF, Dreckmann KM, Sentíes A, Margain-Hernández R (1993) Diversidad Algal en México. Rev Soc Mex Hist Nat 44:69–92
R Core Team (2020) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna
Ruppert KM, Kline RJ, Rahman MS (2019) Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA. Glob Ecol Conserv. https://doi.org/10.1016/j.gecco.2019.e00547
Rzedowski J (1996) Análisis preliminar de la flora vascular de los bosques mesófilos de montaña de México. Acta Bot Mex 35:25–44. https://doi.org/10.21829/abm35.1996.955
Samad A, Brader G, Pfaffenbichler N, Sessitsc A (2019) Plant-Associated Bacteria and the Rhizosphere. Modern Soil Microbiology. CRC Press, Nueva York, pp 163–175
Sharma P, Goel R, Capalash N (2007) Bacterial laccases. World J Microbiol Biotechnol 23:823–832. https://doi.org/10.1007/s11274-006-9305-3
Shaw EA, Boot CM, Moore JC, Wall DH, Baron JS (2019) Long-term nitrogen addition shifts the soil nematode community to bacterivore-dominated and reduces its ecological maturity in a subalpine forest. Soil Biol Biochem 130:177–184. https://doi.org/10.1016/j.soilbio.2018.12.007
Sokal RR, Rohlf FJ (1981) Biometría: Principios y métodosestadísticos en la investigación biológica. Blume, Madrid
Sundue MA (2017) Three new species of grammitid ferns (Polypodiaceae) from the fern hunter’s paradise: Sierra Juárez, Oaxaca. Mexico Systematic Botany 42:160–168. https://doi.org/10.1600/036364417X694962
Thakur MP, Geisen S (2019) Trophic regulations of the soil microbiome. Trends Microbiol 27:771–780. https://doi.org/10.1016/j.tim.2019.04.008
Thorn R, Lynch M (2007) Fungi and Eukaryotic Algae. Soil Microbiology and Biochemistry. Elsevier, USA, pp 145–158
Toledo-Aceves T, Meave JA, González-Espinosa M, Ramírez-Marcial N (2011) Tropical montane cloud forests: Current threats and opportunities for their conservation and sustainable management in Mexico. J Environ Manage 92:974–981. https://doi.org/10.1016/j.jenvman.2010.11.007
Van der Wal A, Geydan TD, Kuyper TW, de Boer W (2012) A thready affair: linking fungal diversity and community dynamics to terrestrial decomposition processes. FEMS Microbiol Rev. https://doi.org/10.1111/1574-6976.12001
Velez P, Tapia-Torres Y, García-Oliva F, Gasca-Pineda J (2021) Small-scale variation in a pristine montane cloud forest: evidence on high soil fungal diversity and biogeochemical heterogeneity. PeerJ. https://doi.org/10.7717/peerj.11956
Vitousek PM, Cassman KEN, Cleveland C, Crews T, Field CB, Grimm NB, Sprent JI et al (2002) Towards an ecological understanding of biological nitrogen fixation. Biogeochemistry 57:1–45. https://doi.org/10.1023/A:1015798428743
Wehner K, Norton RA, Blüthgen N, Heethoff M (2016) Specialization of oribatid mites to forest microhabitats-the enigmatic role of litter. Ecosphere 7:e01336. https://doi.org/10.1002/ecs2.1336
Woodward J, Wiseman A (1982) Fungal and other β-d-glucosidases — Their properties and applications. Enzyme Microb Technol 4:73–79. https://doi.org/10.1016/0141-0229(82)90084-9
Zhang Y, Li S, Li H, Wang R, Zhang K-Q, Xu J (2020) Fungi-nematode interactions: Diversity, ecology, and biocontrol prospects in agriculture. J Fungi 6:206. https://doi.org/10.3390/jof6040206
Funding
This research was financially supported by DGAPA-PAPIIT-UNAM IN200921.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Responsible editor: Dulce Flores-Rentería
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rivera-Zizumbo, A.A., Velez, P., Ojeda, M. et al. Implementing ITS1 metabarcoding for the analysis of soil microeukariotic diversity in the Mountain Cloud Forest. J Soils Sediments (2024). https://doi.org/10.1007/s11368-024-03839-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11368-024-03839-1