
Abstract
Background
Benzimidazoles and triazoles are useful structures for research and development of new pharmaceutical molecules and have received much attention in the last decade because of their highly potent medicinal activities.
Findings
A simple and efficient synthesis of triazole was carried out by treatment of 2-(4-azidophenyl)-1H-benzo[d]imidazole (6) with different types of terminal alkynes in t-BuOH/H2O, sodium ascorbate, and Zn(OTf)2, screened for cytotoxicity assay and achieved good results. A series of new benzimidazole-linked 1,2,3-triazole (8a-i) congeners were synthesized through cyclization of terminal alkynes and azide. These synthesized congeners 8a-i were evaluated for their cytotoxicity against five human cancer cell lines. These benzimidazole-linked 1,2,3-triazole derivatives have shown promising activity with IC50 values ranging from 0.1 to 43 μM. Among them, the compounds (8a, 8b, 8c, and 8e) showed comparable cytotoxicity with adriamycin control drug.
Conclusions
In conclusion, we have developed a simple, convenient, and an efficient convergent approach for the synthesis of benzimidazole-linked 1,2,3-triazole congeners as agents.

Synthesis of 1,2,3-triazole derivatives
Similar content being viewed by others


Findings
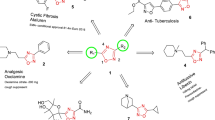
Cancer is one of the terrible diseases which cause uncontrolled growth of group of cells. It remains a mean threat to human beings and cause death [1],[2]. Recently, many researchers developed safe and effective ways of treating this disease and to search for novel chemotherapeutic agents. The benzimidazole nucleus is an important pharmacophore in medicinal chemistry. The synthesis of novel benzimidazole derivatives remains a main focus of modern drug discovery. The versatility of new generation benzimidazole would represent a fruitful pharmacophore for further development of better medicinal agents. Many researchers have been attracted to benzimidazole derivatives because of their wide range of biological activity. Over the past years, there is a considerable interest in the development and pharmacology of benzimidazole. They are of wide interest because of their diverse biological activity and clinical applications [3]. Some of benzimidazole derivatives have also exhibit antimicrobial [4],[5], antitumor [6], anti-inflammatory [7], antihypertensive [8], and antiviral [9] activities. Benzimidazole moiety can also be extracted from naturally occurring compounds such as vitamin B12 and its derivatives, and it is similar to the structure of purins. Pyrrolo[1,2-a]benzimidazoles represent a new class of antitumour agent exhibiting cytotoxic activity against a variety of cancer cell lines [10]. Benzimidazole containing anticancer agent, [Hoechst-33342], 2′-(4-ethoxyphenyl)-5-(4-methyl-1-piperazinyl)-2,5′-bis-1H-benzimidazole (1), has been reported as inhibitor of topoisomerase-I [11],[12]. The other derivative of benzimidazole [Hoechst-33258] (2 Figure 1) [13],[14] shows both in vitro antitumour activity, as inhibitor of DNA topoisomerase-I [15]. Hoechst 33258 a fluorescent reagent and as initially found to be active against L1210 murine leukemia. During phase I trial in humans, some responses were seen in pancreatic cancer. However, a subsequent phase II trial did not show any objective responses.

Biologically active benzimidazole derivatives.
In addition, triazoles also display wide spectrum of biological activities and are widely employed as pharmaceuticals and agrochemicals. Triazoles are reported to possess antibacterial, antifungal, and antihelminthic activities [16]-[21]. They have been regarded as an interesting unit in terms of biological activity [22],[23], and some of them have also shown significant anticancer activity in many of the human cell lines [24].
In view of the biological importance of benzimidazole and 1,2,3-triazoles, to know the combined effect of both benzimidazole and 1,2,3-triazole moieties, it was considered worthwhile to synthesize certain new chemical entities having benzimidazole and 1,2,3-triazole pharmacophores in a single molecular framework, and here we have used Zn(OTf)2 catalyst instead of CuSO4. All of these congeners have been evaluated for their anticancer activity against a panel of five human cancer cell lines (Figure 1).
Experimental section
All chemicals and reagents were obtained from Aldrich (Sigma-Aldrich, St. Louis, MO, USA) and Lancaster (Alfa Aesar, Johnson Matthey Company, Ward Hill, MA, USA) and were used without further purification. Reactions were monitored by TLC and performed on silica gel glass plates containing 60 F-254, and visualization on TLC was achieved by UV light or iodine indicator. 1H and 13C NMR spectra were recorded on Gemini Varian-VXR-unity (Palo Alto, California) (300 and 100 MHz) instrument. Chemical shifts (d) are reported in ppm downfield from internal TMS standard. ESI spectra were recorded on Micromass, Quattro LC (McKinley Scientific, Sparta, NJ, USA) using ESI + software with capillary voltage 3.98 kV and ESI mode positive ion trap detector. Melting points were determined with an electrothermal melting point apparatus and are uncorrected.
Chemistry
The synthesis of novel benzimidazole linked triazole (8a-i) derivatives is carried out as shown in Scheme 1. The key intermediate for the preparation of the new analogs is 2-(4-azidophenyl)-1H-benzo[d]imidazole (6). The mixture of O-phenylenediamine (3) and 4-aminobenzoic acid (4) was mixed with a sufficient quantity of polyphosphoric acid. The resulting solution was stirred at 250°C for 4 h, to afford compound 5. Compound 5 was diazotizated followed by azidation to afford compound 6. Compound 6 upon treatment with different types of terminal alkynes in t-BuOH/H2O, sodium ascorbate, and Zn(OTf)2 afforded compounds (8a-i).

Synthesis 1,2,3-triazoles.
4-(1H-Benzo[d]imidazol-2-yl)benzenamine(5)
A mixture of the O-phenylenediamine (3) (500 mg, 3.64 mmol) and the 4-aminobenzoic acid (4) (394 mg, 3.64 mmol) was dissolved in sufficient quantity of polyphosphoric acid (PPA). The mixture was heated slowly to 250°C for 4 h, permitted to cool to room temperature, quenched with excess of 10% Na2CO3 solution, and extracted with ethyl acetate. Then, the mixture was dried over anhydrous Na2SO4, and the crude product was purified by column chromatography with ethyl acetate/hexane (6:4) to afford pure compound 5, 946 mg in 97% yield. Mp: 209°C to 211°C, 1H NMR (300 MHz, DMSO-d6): δ 6.68 (d, 2H, J =7.3 Hz), 7.14 (br s, 2H), 7.50 (br s, 2H), 7.85 (d, 2H, J =7.1 Hz). IR (neat, cm−1): γmax 404.3; 501.4; 537.9; 607.7; 742.3; 833.4; 960.1; 1,009.1; 1,108.6; 1,178.6; 1,225.7; 1,272.2; 1,397.2; 1,444.1; 1,499.2; 1,612.4; 1,701.4; 2,750.1; 2,853.1; 2,921.8; 3,355.4; 3,435.0; MS (ESI): 210 [M + H]+.
2-(4-Azidophenyl)-1H-benzo[d]imidazole(6)
The amine derivative (5) (500 mg, 2.39 mmol) was dissolved in 10% aq HCl at room temperature. This reaction mixture upon cooling to 0°C and addition of a solution of NaNO2 (165 mg, 2.39 mmol) was stirred for 10 min at 0°C to 5°C. Sodium azide (186 mg, 2.87 mmol) was added, and the mixture was stirred at room temperature for 2 h. The reaction was worked up by dilution with ethyl acetate. The organic layer was washed with brine and dried over Na2SO4. After evaporation of the solvent, the crude product was purified by column chromatography with ethyl acetate/hexane (3:7) to afford pure compound 6, 536 mg in 95% yield; Mp: 317°C to 319°C, 1H NMR (300 MHz, DMSO-d6): δ 7.16 to 7.26 (m, 2H), 7.32 (d, 2H, J =9.0 Hz), 7.50 to 7.69 (dd, 2H, J =40.0, 38.5 Hz), 8.22 (d, 2H, J =8.3 Hz), 12.97 (s, 1H). IR (neat, cm−1): γmax 500.6; 541.4; 694.3; 743.3; 838.1; 963.4; 1,011.1; 1,115.8; 1,175.5; 1,283.5; 1,395.2; 1,438.8; 1,485.1; 1,604.6; 1,726.8; 2,120.1; 2,414.9; 2,856.9; 2,918.2; 3,055.5; 3,422.9; MS (ESI): 236 [M + H]+.
2-(4-(4-(3,4,5-Trimethoxyphenyl)-1H-1,2,3-triazol-1-yl)phenyl)-1H-benzo[d]imidazole (8a)
A mixture of the corresponding azide 6 (200 mg, 0.85 mmol) and the corresponding alkyne 7a (163 mg, 0.85 mmol) was dissolved in t-BuOH/H2O 1:1 (20 mL). Sodium ascorbate (33 mg, 20 mol%) and Zn(OTf)2 (300 mg, 5 mol%) were added. After stirring for 4 h, water/ice (40 mL) was added. The product was either worked up by filtration, followed by rinsing with aqueous 5% NH3 (×3) and cold ether (×2), or by extraction with dichloromethane (4 × 100 mL). The combined organic layers were washed with aqueous 5% NH3 (3 × 100 mL) and brine (100 mL) and dried over anhydrous MgSO4. The solvent was removed in vacuo, and the crude product was purified by column chromatography with ethyl acetate/hexane (3:7) to afford pure compound 8a, 347 mg in 95% yield. Mp: 270°C to 272°C, 1H NMR (400 MHz, CDCl3): δ 3.71 (s, 3H), 3.89 (s, 6H), 7.24 to 7.28 (m, 4H), 7.63 to 7.66 (m, 2H), 8.17 (d, 2H, J =8.4 Hz), 8.41 (d, 2H, J =8.4 Hz), 9.44 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 57.2, 61.4, 110.2, 114.8, 117.1, 122.1, 123.3, 125.3, 128.4, 129.8, 140.1, 141.8, 143.3, 148.7, 152.4, 154.1; MS (ESI): 428 [M + H]+.
The other derivatives are also prepared according to the same procedure and described in Additional file 1.
Biological evaluation
In vitro cytotoxicity assay
The synthesized compounds 8a-i were evaluated for their anticancer activity in selected human cancer cell lines of A375, B-16, colon-205, MCF-7, and A-549 by using MTT assay. All the compounds (8a-i) exhibited significant anticancer activity with IC50 values ranging from 0.1 to 43 μM, while the positive control, adriamycin, demonstrated the IC50 in the range of 0.03 to 3.5 μM respectively, in the cell lines employed as shown in Table 1.
Procedure for MTT assay
Toxicity of test compound in cells was determined by MTT assay based on mitochondrial reduction of yellow MTT tetrazolium dye to a highly colored blue formazan product. Cells (1 × 104) (counted by Trypan blue exclusion dye method) in 96-well plates were incubated with compounds with series of concentrations tested for 48 h at 37°C in RPMI/DMEM/MEM with 10% FBS medium. Then, the above media was replaced with 90 μl of fresh serum free media and 10 μl of MTT reagent (5 mg/ml), and plates were incubated at 37°C for 4 h, thereafter the above media was replaced with 200 μl of DMSO and incubated at 37°C for 10 min. The absorbance at 570 nm was measured on a spectrophotometer (SpectraMax, Molecular devices, Sunnyvale, CA, USA). IC50 values were determined from plot: percent inhibition (from control) versus concentration.
Additional file
References
El-Azab AS, ElTahir KEH: Design and synthesis of novel 7-aminoquinazoline derivatives: antitumor and anticonvulsant activities Bio Org. Med Chem 2012, 22: 1879–1885.
El Azab AS, Al Omar MA, Abdel Aziz AAM, Abdel Aziz NI, El Sayed MAA, Aleisa AM, Sayed Ahmed MM, Abdel Hamide SG: Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: molecular docking study. Eur J Med Chem 2010, 45: 4188–4198. 10.1016/j.ejmech.2010.06.013
Brunton LL, Lazo JS, Parker KL: The pharmacological basis of therapeutics. 11th edition. Mc Graw-Hill, New York; 2006.
Goker H, Ku C, Boykin DW, Yildiz S, Altanlar N (2002) Synthesis of some new 2-substituted-phenyl-1H-benzimidazole-5-carbonitriles and their potent activity against Candida species. Bioorg Med Chem 10:2589
Ozden S, Atabey D, Yildiz S, Goker H (2005) Synthesis and potent antimicrobial activity of some novel methyl or ethyl 1H-benzimidazole-5-carboxylates derivatives carrying amide or amidine groups. Bioorg Med Chem 13:1587
Mann J, Baron A, Opoku Boahen Y, Johansoon E, Parkmson G, Kelland LR, Neidle S (2001) A new class of symmetric bisbenzimidazole-based DNA minor groove-binding agents showing antitumor activity. J Med Chem 44:138
Achar KS, Hosamani KM, Seetharam HR (2010) Novel benzimidazole derivatives as expected anticancer agents. Eur J Med Chem 45:2048
Kumar JR, Jawahar JL, Pathak DP (2006) Synthesis and pharmacological evaluation of benzimidazole derivatives. Eur J Chem 3:278
Tewari AK, Mishra A (2006) Synthesis and antiviral activity of N-substituted-2-subastituted benzimidazole derivatives. Ind J Chem Sec B 45:489
Schulz WG, Islam I, Skibo EB (1995) Pyrrolo[1,2-a]benzimidazole-based quinones and iminoquinones. The role of the 3-substituent on cytotoxicity. J Med Chem 38:109
Chen A, Yu C, Gatto B, Liu LF (1993) Poisoning of human DNA topoisomerase I by ecteinascidin 743, an anticancer drug that selectively alkylates DNA in the minor groove. Proc Natl Acad Sci USA 96:908131
Chen AY, Yu C, Bodley AL, Peng LF, Liu LF (1993) Topoisomerase I inhibitors and drug resistance. Cancer Res 53:1332
Kraut E, Fleming T, Segal M, Neidhart J, Behrens BC (1999) Hoechst-IR: an Imaging agent that detects necrotic tissue in vivo by binding extracellular DNA. J Invest New Drugs 9:95
Tolner B, Hartly JA, Hochhauser D (2001) Transcriptional regulation of topoisomerase II alpha at confluence and pharmacological modulation of expression by bis-benzimidazole drugs. Mol Pharamcol 59:699
Beerman TA, McHugh MM, Sigmund R, Lown JW, Rao KE, Bathini Y: Effects of analogs of the DNA. Biochim Biophys Acta 1992, 1131: 53–61. 10.1016/0167-4781(92)90098-K
Hardman J, Limbird L, Gilman A: Goodman and Gilman’s. The pharmacological basis of therapeutics. 9th edition. McGraw-Hill, New York; 1996.
Gennaro A, Remington R (1995) A mechanistic analysis of carrier-mediated oral delivery of protein therapeutics. The science and practice of pharmacy. Mack Easton PA II:1327
Richardson K, Whittle PJ (1984) 17 human secreted proteins. Eur Pat Appl EP 115:416, Richardson, K.; Whittle, P. (1984) J Chem Abstr 101:230544
Ammermann E, Loecher F, Lorenz G, Janseen B, Karbach S, Meyer N (1990) The science and practice of pharmacy. Brighton Crop Prot Conf Pests Dis 2:407
Ammermann E, Loecher F, Lorenz G, Janseen B, Karbach S, Meyer N (1991) The science and practice of pharmacy. Chem Abstr 114:223404h
Heindel ND, Reid JR (1980) 4-Amino-3-mercapto-4H-1,2,4-triazoles and propargyl aldehydes: a new route to 3-R-8-aryl-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazepines. J Heterocycl Chem 17:1087
Dehne H: methoden der organischen chemie (Houben-Weyl). 8th edition. Edited by: Schumann E. Thieme, Stuttgart; 1994:305.
Wamhoff H: In comprehensive heterocyclic chemistry. Edited by: Katritzky AR, Rees CW. Pergamon, Oxford; 1984:669. 10.1016/B978-008096519-2.00079-5
De Las Heras FG, Alonso R, Alonso G (1979) Alkylating nucleosides 1. Synthesis and cytostatic activity of N-glycosyl(halomethyl)-1,2,3-triazoles. A new type of alkylating agent. J Med Chem 22:496
Acknowledgements
We thank the management and principal of the Mahatma Gandhi Institute of Technology and the vice-chancellor and the registrar of JNT University Hyderabad for their encouragement.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Harkala, K.J., Eppakayala, L. & Maringanti, T.C. Synthesis and biological evaluation of benzimidazole-linked 1,2,3-triazole congeners as agents. Org Med Chem Lett 4, 14 (2014). https://doi.org/10.1186/s13588-014-0014-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13588-014-0014-x