
Abstract
The RYR1 gene, which encodes the sarcoplasmic reticulum calcium release channel or type 1 ryanodine receptor (RyR1) of skeletal muscle, was sequenced in 1988 and RYR1 variations that impair calcium homeostasis and increase susceptibility to malignant hyperthermia were first identified in 1991. Since then, RYR1-related myopathies (RYR1-RM) have been described as rare, histopathologically and clinically heterogeneous, and slowly progressive neuromuscular disorders. RYR1 variants can lead to dysfunctional RyR1-mediated calcium release, malignant hyperthermia susceptibility, elevated oxidative stress, deleterious post-translational modifications, and decreased RyR1 expression. RYR1-RM-affected individuals can present with delayed motor milestones, contractures, scoliosis, ophthalmoplegia, and respiratory insufficiency.
Historically, RYR1-RM-affected individuals were diagnosed based on morphologic features observed in muscle biopsies including central cores, cores and rods, central nuclei, fiber type disproportion, and multi-minicores. However, these histopathologic features are not always specific to RYR1-RM and often change over time. As additional phenotypes were associated with RYR1 variations (including King-Denborough syndrome, exercise-induced rhabdomyolysis, lethal multiple pterygium syndrome, adult-onset distal myopathy, atypical periodic paralysis with or without myalgia, mild calf-predominant myopathy, and dusty core disease) the overlap among diagnostic categories is ever increasing. With the continuing emergence of new clinical subtypes along the RYR1 disease spectrum and reports of adult-onset phenotypes, nuanced nomenclatures have been reported (RYR1- [related, related congenital, congenital] myopathies). In this narrative review, we provide historical highlights of RYR1 research, accounts of the main diagnostic disease subtypes and propose RYR1-related disorders (RYR1-RD) as a unified nomenclature to describe this complex and evolving disease spectrum.
Similar content being viewed by others


Introduction
Congenital myopathies (CM), a term first coined by Victor Dubowitz [1], are a group of inherited, non-dystrophic neuromuscular disorders characterized by specific clinical features and skeletal muscle histopathology [2]. Pathogenic variations in the RYR1 gene, a relatively large gene in the human genome, are the most common cause of CM and contribute to the clinical, histopathological, and genetic heterogeneity of CMs.
The RYR1 gene (19q 13.2) encodes a calcium (Ca2+) release channel located in the terminal cisternae of the sarcoplasmic reticulum (SR) of skeletal muscle that is activated by CaV1.1 voltage sensor proteins in the transverse tubule membrane during excitation-contraction (EC) coupling [3]. The purification of RyR1 Ca2+ release channels in 1988 [4] and the subsequent discovery in 1991 of pathogenic RYR1 variants [5] led to the association of impaired Ca2+ homeostasis with malignant hyperthermia (MH) susceptibility (MIM # 145600) [6]. MH is a potentially fatal disorder of skeletal muscle Ca2+ regulation. MH episodes are triggered by exposure to certain volatile anesthetics (e.g., sevoflurane, desflurane) and depolarizing muscle relaxants (succinylcholine). MH episodes are characterized by uncontrolled muscle hypermetabolism, with a clinical presentation including hypercapnia, sinus tachycardia, masseter muscle rigidity, and hyperthermia [5, 7]. Guidelines from the European Malignant Hyperthermia Group (EMHG) and Malignant Hyperthermia Association of the United States (MHAUS) continue to inform the use of anesthetic drugs in individuals susceptible to MH [8, 9]. In certain cases, extreme heat conditions, fever, and/or exertion can result in symptoms that mimic MH episodes (enhanced heat response or EHS) [10,11,12]. Variations in the RYR1 gene remain the leading cause of MH susceptibility; however, few MH-associated variations in other genes such as CACNA1S and STAC3 have been described [13].
Functional analyses of RyR1 channels resulting from putative RYR1 disease variants revealed multiple causative mechanisms including (1) increased sensitivity of RyR1 channels to activators (e.g., caffeine, halothane, CaV1.1 voltage sensors) as observed in MH resulting in uncontrolled channel opening and Ca2+ release [14], (2) enhanced RyR1 Ca2+ leak [15], (3) reduction in RyR1 Ca2+ permeation leading to reduced Cav1.1-mediated SR Ca2+ release, a process referred to as excitation-contraction uncoupling [16, 17], and (4) dramatic reduction in RyR1 channel expression [6]. In spite of these advances, many putative RYR1 variants have not yet been directly tested/associated with Ca2+ release dysfunction, and thus, are classified as variants of uncertain significance (VUS) [18].
With a likely underestimated disease prevalence of 1:90,000 individuals [19], RYR1-RM is considered the most common form of non-dystrophic muscle disease in humans [20]. Of note, genetic variants resulting in MH susceptibility are more common, affecting approximately 1:3000–1:8500 [13], with a recent estimate from an exome analysis of a cohort of 870 individuals suggesting a prevalence of 1 in 400 [21]. RYR1-RM is inherited in both autosomal dominant and recessive manners and de novo cases have also been described. Clinical features suggestive of RYR1-RM are extensive, with mild to severe symptoms ranging from delayed motor milestones, proximal muscle weakness, hypotonia, and fatigue, to kyphoscoliosis, ophthalmoplegia, and moderate to severe respiratory insufficiency, which is more often apparent in recessive cases [22,23,24].
Historically, RYR1-RM subtypes were diagnosed and named based primarily on muscle biopsy histopathologic features such as central cores, cores with rods, central nuclei, fiber type disproportion, and multi-minicores. A number of earlier cases were described prior to RYR1 gene identification and association with disease and, thus, may have been classified differently today. However, these histopathologic features are not unique to RYR1-RM, can be dynamic over time, may vary based on biopsy site, and may be absent when biopsy is performed at an early age [25, 26] or reflect a consequence of the gene dose (heterozygous = MH susceptibility versus homozygous = clinical myopathy) [27]. There are also several clinical and histopathologic similarities between the main RYR1-RM diagnostic categories of central core disease (CCD; MIM # 117000), multi-mini core disease (MmD; MIM #255320), core-rod myopathy (CRM), centronuclear myopathy (CNM), and congenital fiber-type disproportion (CFTD) [28, 29].
RYR1-related disorders can be viewed as occurring along a spectrum [30]. This spectrum includes RYR1 variant-associated clinical phenotypes including King-Denborough syndrome, congenital neuromuscular disease with uniform type 1 fiber (CNMDU1), dusty core disease, rhabdomyolysis-myalgia syndrome, atypical periodic paralysis, and bleeding abnormalities [31,32,33,34,35,36,37,38]. Additionally, the spectrum of RYR1-related disorders has further expanded following reports of inherited, adult-onset phenotypes [39, 40].
Nuanced terminology is evident within the literature (including RYR1-related, RYR1-associated, RYR1-related congenital, RYR1-congenital) myopathies [28, 41,42,43]. The number of non-dystrophic neuromuscular disorders associated with RYR1 genetic variations reflect the importance of the RyR1 protein in normal muscle function. Numerous informative reviews have been published on the RYR1 disease spectrum [3, 13, 44,45,46,47,48,49]. This narrative review describes key historical milestones that led to the recognition of RYR1-related disorders as an overarching entity occurring along a complex clinical and histopathological spectrum. Here, we also summarize the current state of knowledge on the primary disease subtypes and propose a unified nomenclature that encompasses current and future phenotypes.
Methods
As a preface, this is a historical narrative review. With input from subject-matter experts in RYR1-related disorders (including malignant hyperthermia), and information in landmark publications such as Magee and Shy [50] and Dubowitz and Pearse [51], the following search strategies were used for this narrative review: (1) computer search of databases for articles on congenital myopathies, and specifically, core myopathies; (2) review of congenital myopathy textbooks [52,53,54,55]; and (3) review of key articles of historical significance from reference lists of retrieved publications.
PubMed, ScienceDirect, and Scopus databases were searched without setting date limits or language exclusions. Chapters or sections from specialized neuromuscular textbooks or journals not annotated or available online were requested through the NIH Library. A timeline approach of landmark discoveries, starting from the earliest possible reports of congenital myopathies with related phenotypic features to that of RYR1 subtypes was used as a framework for this historical perspective.
Historical perspective of RYR1-related disorders
As with most historical reports, to our knowledge, none of the reported cases highlighted in this review prior to the advent of genetic sequencing have been resolved or unequivocally attributed to variations in the RYR1 gene. Other genes currently associated with the histopathologic features of the main RYR1-related disorder subtypes are summarized in Table 1 and the historical timeline of their emergence is depicted in Fig. 1.

Timeline of significant discoveries/milestones in the evolution of RYR1-RD. 1900s to present: In the twentieth century, congenital myopathy diagnoses were based primarily on muscle biopsy histopathologic features. Advancements in next-generation sequencing enabled more precise identification of RYR1-related phenotypes. Solving of the RyR1 structure at near-atomic resolution provided valuable insight into RyR1 channel function and disease mechanisms. These advances paved the way for the first RYR1-RM clinical trial completed in 2018 and the Phase 1 Rycal trial which began enrolling participants in 2020
Early twentieth century: amyotonia congenita
Most CMs in the early twentieth century were originally misclassified as amyotonia congenita, a condition initially described by neurologist Herman Oppenheim in 1900 [62] as general or local hypotonia of muscles in early infancy, with hyporeflexia, limb paralysis, absence of muscle wasting and familial involvement, and tendency to improve (Fig. 1).
In the early 1900s, the diagnosis of amyotonia congenita was given in cases involving sparing of the muscles supplied by the cranial nerves with muscular weakness and atony attributed to delayed muscle development. However, there were similarities in the clinical presentation of amyotonia congenita and infantile spinal muscular atrophy described by Werdnig and Hofmann in 1891 [63], a condition of progressive muscular flaccidity and weakness involving axial and proximal muscles due to loss of anterior horn cells in the spinal cord, as post-mortem records of some fatal cases of amyotonia congenita revealed lesions in the ventral horn cells and neuropathic atrophy [64]. These findings and the lack of clearly defined differentiating clinical features at the time resulted in an alternative erroneous hypothesis that the two conditions were variations of the same disease [65, 66].
Early twentieth century: post-operative heat stroke
Cases of hyperpyrexia associated with anesthesia during and immediately following surgical procedures were first reported in 1900 [67,68,69,70,71]. In 1919, written documents regarding the deaths of a mother and son anesthetized with chloroform and ether by G.A. Jones and E. Penny recounted muscle rigidity, violent and persistent respiratory muscle spasms, and rapid pulse [72]. However, the body temperature of the patients was not mentioned in this suggestive hereditary susceptibility to chloroform.
1950s to 1960s: muscular degeneration
A disease termed muscular degeneration (MD) in pigs, manifested by discoloration of the skeletal musculature, was first reported in 1954 by J. Ludvigsen [73]. The altered musculature appeared gray or pale in color resembling that of chicken meat. MD was often fatal, especially when animals were exposed to exercise or stress. Prior to death the animals had dyspnea, cyanosis, circulatory insufficiency resulting from musculature vasoconstriction, and hyperthermia. Briskey and colleagues referred to this condition as pale, soft, exudative (PSE) tissue [74], and Kjolberg and colleagues referred to this condition as “white muscle disease” [75]. This alteration lowered the quality of the meat and therefore was a significant monetary loss to the meat processors and retailers. PSE was associated with predisposition to accelerated post-mortem glycolysis, and onset of rigor mortis at pH values below 5.9 and temperatures above 35 °C. PSE muscle ultrastructure showed disruption of sarcoplasmic components and protein filaments; however, the tissue appeared macroscopically normal. The economic implications of PSE meat generated a significant amount of research into porcine stress syndrome (PSS) [73, 76,77,78].
1950s: central core disease
In 1956, Magee and Shy [50] investigated muscle biopsy findings from one large family with five affected members. Symptoms included non-progressive, infantile hypotonia with mostly proximal weakness. All biopsies from affected individuals showed similar appearances of larger than normal muscle fibers (up to 240μm in diameter), amorphous central cores in almost all fibers, and myofibrils that stained blue instead of purple with Gömöri trichrome stain. The myopathy resulting in this curious histopathological feature was later referred to as central core disease (CCD) by Greenfield and colleagues in 1958 [79], reflecting lack of oxidative enzyme activity in the amorphous cores due to mitochondrial depletion [51]. Additional histopathological features of CCD include increased internal and central nuclei, presence of rods, minimal to moderate endomysial fibrosis, increased fatty tissue and connective tissue infiltration, Z-line streaming, sarcomeric disorganization, fiber size variation, and predominance of type 1 fibers [79].
1960s: malignant hyperthermia
In 1960, Michael Denborough and Roger Lovell described a case in which a young man nearly died following general anesthesia procedure with halothane for a compound fracture of the tibia and fibula [80]. There had been ten deaths attributable to general anesthesia, specifically ethyl chloride and ether, in the proband’s family. The pattern of inheritance was similar to an incomplete penetrant dominant gene or genes [81]. Available medical records showed that the course of events in the deceased family members had been similar, with convulsion and hyperthermia noted in two cases. This was the first breakthrough in recognizing MH as a heritable condition.
1960s: core-rod myopathy
Shy and colleagues reported on a child with a non-progressive congenital myopathy and curved, thread-like structures at the periphery and center of affected muscle fibers on histopathological examination, and a clinical picture of a “floppy infant” [82]. Histopathologically, the lesion showed a spectrum of muscle fiber size ranging from 8 to 56 μm, with aggregates of palisading rod-like materials in the sub-sarcolemmal areas external to the myofibrils. The rods stained red on Gömöri trichrome stain, showed no polarization with white light or cross-banding, contained no formed inclusions such as mitochondria, and affected fibers had greater phosphorylase activity than unaffected fibers. These features reflected a new morphological muscle cell abnormality with highly organized protein-containing rod formations without cross-striations. This myopathic presentation was named nemaline myopathy because the structures could represent as rods or coils of thread-like structures.
Blurring the distinction between nemaline myopathy and CCD, a case of congenital myopathy with a combination of cores and rods was reported in one family in 1965 by Afifi et al. [83]. Skeletal muscle biopsies from the mother and daughter with autosomal dominant inheritance, congenital, non-progressive myopathy were examined. The daughter’s biopsy exhibited CCD features, while the mother’s biopsy exhibited characteristics of both CCD and nemaline myopathy. The authors suggested that CCD and nemaline myopathy could be manifestations of one disease process, with various means of expression, because it would be most unusual to find two unrelated rare diseases with similar clinical features and inheritance patterns in the same individual.
1960s: centronuclear myopathy
An undescribed, slowly progressive myopathy characterized by the presence of central nuclei in about 85% of muscle fibers was referred to as myotubular myopathy (MTM) by Spiro and colleagues in 1966 [84] and familial centronuclear myopathy (CNM) by Sher and colleagues in 1967 [85]. CNM subtypes were unified histopathologically by small myofibers containing hyperchromatic central nuclei with wrinkled or serrated borders in > 25% of muscle fibers, and rows or aggregates of up to thirty nuclei on hematoxylin and eosin stain. The central nuclei are usually surrounded by an unstained space.
1960s to 1970s: congenital fiber-type disproportion
Following a series of detailed histological studies of variations in muscle fiber types in neuromuscular diseases [86], Michael Brooke coined the term congenital fiber-type disproportion (CFTD) to describe consistently smaller type 1 muscle fibers than type 2 fibers in fourteen patients, with fiber size disproportion (FSD) greater than 12% in the absence of any other histological abnormality [87]. CFTD was initially considered to be a non-specific feature that preceded the development of more specific histologic features, as other CMs presented with FSD and corresponding clinical manifestations [88].
1970s: multi-minicore disease
Multiple small core-like structures on skeletal muscle biopsy in two siblings with CM, termed multicore disease, were first reported by Engel and colleagues in 1971 [89]. Different from the central cores of CCD, these variable, indistinct multiple mini cores of areas with decreased oxidative activity extend only a short distance along the length of the muscle fiber, with some larger mini cores stretching across the fiber width.
1980s to 1990s: RyR1 localization and malignant hyperthermia linkage
The characterization of RyR1 ryanodine binding [90, 91], physiology of PSS and identification of porcine MH (hal) locus [92,93,94], channel activity [95, 96], biochemical purification [97, 98], cloning [99,100,101], and the initial classification of CCD, MmD, and CFTD as “congenital myopathies of unknown aetiology” [102] all took place in the mid-late 1980s. Linkage analysis found MH sensitivity to co-segregate with chromosomal markers in the RYR1 gene with a lod score of 4.20, consistent with MH being caused by mutations in the RYR1 gene in 1990 [103]. These studies naturally prompted the search for specific RYR1 pathogenic variants and led to identification of the first MH causative variant in 1991 (c.1843C>T; p.Cys615Arg) [5, 104] and then subsequently for CCD in 1993 [105].
2004: imaging of intramuscular fatty infiltration as a diagnostic tool
The wide range of phenotypes associated with RYR1 variants, the large size of RYR1, and complex histopathological overlap across different CMs due to variants in other genes encoding sarcolemmal and sarcotubular proteins [29] present challenges in confirmatory diagnosis. RYR1-RM-affected individuals show a consistent pattern of relative sparing of rectus femoris, adductor longus, hamstring muscles, the medial head of the gastrocnemius (except in calf-predominant myopathy) [106], and muscles of the anterior compartment of leg [107]. In 2004, Jungbluth and colleagues used magnetic resonance imaging (MRI) to classify distinct patterns of selective muscle fatty infiltration using in congenital myopathies associated with RYR1 causative variations [107]. As a result, muscle MRI is now used to supplement clinical assessment and genetic testing for the diagnosis of patients with variable histopathology. The term “RYR1-related congenital myopathies” was used to describe the distinct pattern of muscle involvement reported.
2006: generation of first knock-in mouse models of RYR1-related myopathies (RYR1-RM)
Creation of a knock-in mouse heterozygous for the Y524S variation (equivalent to Y522S in humans) by Chelu et al. [108] was the first to represent a murine model of MH. These mice experienced whole body contractions and elevated core temperatures in response to isoflurane exposure or heat stress without uncompensated SR calcium leak or store depletion. In the same year, Yang and colleagues created another valid MH susceptible RYR1 knock-in mouse heterozygous for R163C [109]. The R163C heterozygous mouse SR membranes have a twofold higher affinity (Kd = 35.4 nm) for [H]ryanodine binding compared with wild type.
2011–2012: a spectrum of RYR1-related myopathies (RYR1-RM)
As histological phenotypes associated with RYR1 variations can continually evolve over time in the same patient or vary in individuals with the same variant, terminologies that include these features might not be appropriate for specific designations [110]. The expanding histopathologic (from normal biopsy findings in MH susceptible individuals, histology typical of CCD, MmD, CNM, and CFTD, to cores in MH susceptible individuals, type 1 fiber predominance, and mixed histopathology of cores and rods) and emerging RYR1-RM phenotypes were the focus of the European Neuromuscular Centre (ENMC) international workshops in 2011 and 2016 [27, 111]. These consensus-building workshops delineated RYR1-RM as a spectrum of clinical and pathologic phenotypes, with histolopathologic features ranging from subtle abnormalities such as increased internalized nuclei to prominent and extensive structural cores. The wide range of RYR1-RM histopathological features, with mild myopathic changes and irregular oxidative staining in the late-onset phenotypes and more frequent detection of cores in the congenital myopathies, is suggestive that both are part of a similar spectrum [30]. With reports of adult-onset phenotypes [39, 40] resulting from increased availability of diagnostic exome and whole gene RYR1 sequencing [112], the full spectrum extends beyond birth and early childhood. A confounding factor in assessment of late-onset cases is advancing age, as aged muscle exhibits varying degrees of increased intramuscular fat content on MRI, muscle fiber atrophy and loss, tubular aggregates, and gradual increases in ragged red fibers and cytochrome c oxidase-negative fibers [113].
2015–2016: advances in RyR1 structure-function
The molecular architecture of RyR1, solved using a combination of cryo-electron microscopy (cryo-EM) and x-ray crystallography of soluble subdomains, reveals a homotetrameric complex with a molecular weight of 2.25 million Da, consisting of four protomers (~ 565 kDa each) that interact with other regulatory proteins and ligands [114, 115]. Advancements in cryo-EM and direct electron detector (DED) technology enabled the most comprehensive 3D reconstruction of this 5038-amino acid structure to date. Indeed, between 2015 and 2016, a series of studies reported RyR1 structure at near-atomic resolution, ranging from 3.6 to 4.8 Å [114, 116,117,118]. The cryo-EM reconstructions revealed binding sites for channel agonists and antagonists, as well as the structural basis of channel gating and ligand-dependent activation. Cytoplasmic interacting proteins (e.g., FKBP12 or calstabin1 and calmodulin or CaM) and SR proteins (e.g., triadin and junctin) bind and regulate RyR1 channel activity [119]. In the absence of channel activators (Ca2+, ATP, caffeine), the RyR1 core is rigid and remains in a closed state. In contrast, channel activator binding at different sites on the RyR1 C-terminal domain (CTD) serves to increase transition to and stability of the channel open state [118].
2018: completion of the first RYR1-RM natural history study and clinical trial
There is no approved treatment for RYR1-RM. Symptom management is primarily supportive, with precautions taken when MH risk is either known or not ascertained. There have been anecdotal reports of positive responses to pyridostigmine, an acetylcholinesterase inhibitor, improving fatigue and energy level [43], and salbutamol, a beta agonist that improves muscle strength and motor function through mechanisms that are not completely understood [120]. Development of patient registries for clinical trial recruitment and funding support from patient advocacy groups such as the Muscular Dystrophy Association (MDA), the RYR-1 Foundation, CureCMD, and congenital muscle disease international registry (CMDIR) have been instrumental in supporting therapeutic development for neuromuscular diseases. The first clinical trial for RYR1-RM was conducted in ambulatory individuals and included a 6-month lead-in natural history phase followed by 6-month intervention with the antioxidant N-acetylcysteine (NAC) [121] (NCT02362425). The rationale for this clinical trial was evidence that NAC rescued elevated oxidative stress and decreased myopathy in murine [122] and zebrafish model systems [123, 124]. All participants exhibited elevated oxidative stress as determined by urine 15-F2t-isoprostane concentration and decreased physical endurance. However, oral treatment with NAC did not impact either outcome [121]. Other therapeutic research opportunities investigating the efficacy of modulators of calcium release from the SR are underway globally. For RYR1-RM-affected individuals, a phase 1 clinical trial testing safety of the RyR stabilizing Rycal molecule S48168 (ARM210) is underway (NCT04141670). Moreover, several drugs already approved for other indications are in the clinical trial pipeline for potential re-purposing, and novel compounds, identified by high-throughput screening, are being tested in pre-clinical studies [125,126,127]. The recent generation of murine model systems that more closely depict RYR1-RM clinical phenotypes [128, 129] and greater understanding of RyR1 structure-function [130] will play a crucial role in identifying RYR1-RM-affected individuals who could benefit most from specific therapeutics. However, the heterogenous nature of the disease suggests that a single treatment is unlikely to be universally efficacious.
Current state of knowledge and differential diagnoses
Central core disease (CCD)
Cores associated with RYR1 variations may be structured or unstructured based on ATPase activity levels (positive, structured; absent, unstructured) and myofibrillar disruption. These cores are typically seen in type I fibers with significant fibro-adipose infiltration [131]. RYR1-associated CCD is predominantly an autosomal dominant condition. In autosomal dominant and de novo cases of CCD, RYR1 variations predominantly affect the RyR1 C-terminal region [56]. RYR1 variations resulting in CCD and MH susceptibility were initially reported to primarily localize in three “hot spot” regions (or domains): domain 1 (N-terminal residues 1–614), domain 2 (central residues 2163–2458), and domain 3 (C-terminal pore/transmembrane residues 4136–4973) [3, 56]. However, more recent information indicates that dominant RYR1 variations can span the entire length of the gene [111].
The clinical features of CCD are variable, and approximately one third of individuals with central cores do not exhibit an overt clinical phenotype [132]. Clinical characteristics of autosomal dominant CCD include hypotonia, developmental motor delay, proximal weakness, myalgia, and orthopedic complications such as scoliosis and hip girdle dislocation. Cardiac, bulbar, and moderate to severe respiratory involvement are rare [6]. Infantile and early childhood presentation with proximal weakness in the hip girdle is typical [55] and, although typically stable over time, slow disease progression has been reported later in life [133]. Recessive cases of CCD are rare but can present with more severe features including, arthrogryposis, respiratory distress and fetal akinesia [29, 134]. CCD is considered allelic to MH susceptibility, which is also predominantly associated with a dominant mode of inheritance [53]. Given this genetic connection, a subset of individuals with CCD diagnoses are MH susceptible, and a subset of MH susceptible individuals present with cores on their muscle biopsies [13]. RyR1 channels with MH- or CCD-associated variants show higher activity and sensitivity to activation than wildtype channels, which ultimately leads to increases in resting Ca2+ concentration [135, 136]. Clinical and histopathologic findings in RYR1-RM-affected individuals are widely variable and often also present in other congenital myopathies. Greater than 90% of cases with typical CCD clinical manifestations and histopathology result from RYR1 variations [56]. However, structures similar to cores are also observed in ACTA1-associated myopathy [137]. Target fibers may be confused with central cores as they are characterized by absence of oxidative enzyme activity, paucity of mitochondria, and disorganized myofibrils in the center, surrounded by a rim of more intense than normal activity on immunohistochemical staining of muscle biopsy [138, 139].
Core-rod myopathy
Although histologically distinct, the presence of cores and rods in the same muscle biopsy examination has been described in other cases with RYR1-RM [110, 140]. RYR1 variations are the most common cause of core-rod myopathy and both dominant and recessive forms have been described [110, 141]. Variations in the NEB [142] and KBTBD13 [143] genes have also been implicated in core-rod myopathy, with KBTBD13-related forms associated with slow muscle movement and proximal weakness [116, 144]. Other genes associated with nemaline myopathy include ACTA1, TPM3, TPM2, TNNT1, and CFL2. Kondo and colleagues reported on a patient diagnosed with severe congenital nemaline myopathy and compound heterozygous variations in RYR1 [145]. Clinical manifestations of this patient included fetal akinesia, severe generalized hypotonia, narrow face with facial muscle weakness, persistent ophthalmoplegia, frog-leg posture, poor-anti-gravity limb movements, respiratory insufficiency, and an improving clinical course [145]. Histologically, nemaline bodies (observed as numerous small rods) were observed in the cytoplasm but not in the nuclei, and small type 1 fibers without central nuclei, fiber degeneration, or cellular infiltration were noted. No central cores or minicores were observed in this patient. The absence of cores and the presence of nemaline rods may differentiate this case from previous reports of recessive RYR1 cases but could also be reflective of delay in core presentation on muscle histology in young patients [146, 147]. Identification of cores or minicores on later biopsies would therefore classify this case as a recessive core-rod myopathy, which is most commonly associated with RYR1 variations [140].
Centronuclear myopathy (CNM)
Mitochondrial oxidative enzyme activity is either concentrated or absent in the centrally nucleated fibers (core-like areas) [148]. The case reported by Spiro and colleagues could be attributable to variation(s) in one of the genes associated with CNM. Of the nine genes currently associated with CNM (RYR1, MTM1, DNM2, BIN1, TTN, MTMR14, SPEG, CCDC78, and CACNA1S), RYR1 variants are the most common cause of autosomal recessive CNM [28]. CNM symptoms predominantly affect skeletal muscles. Clinical features of RYR1-related CNM include extremity muscle weakness (typically severe), foot abnormalities, scoliosis, ophthalmoparesis, and mild to severe respiratory involvement [146]. In individuals with CNM, disease severity is extremely variable with the majority of cases exhibiting compound heterozygous changes. Variations in the MTM1 gene should be investigated first in severely affected males, with analysis of cDNA from muscle tissue recommended if a variation is not identified on genomic DNA [57]. Females with MTM1-related CNM may present with necklace fibers as a histologic marker [149]. DNM2 variations should be investigated first if there is a clear autosomal dominant family history or de novo variant [2, 150].
Congenital fiber-type disproportion (CFTD)
As Brooke’s definition of > 12% FSD rendered CFTD a non-specific diagnosis, FSD greater than 35–55% with clinical features consistent with CM is currently used as a diagnostic criterion [151]. Both dominant and recessive cases of RYR1-related CFTD have been reported [59]. Clinical features reported in CFTD patients include hypotonia, respiratory failure, non-progressive muscle weakness, joint contractures, myopathic facies, ophthalmoparesis, feeding difficulties, and skeletal deformities [59, 152]. It is important to note that many patients initially diagnosed with CFTD develop rods, cores, and central nuclei over time, leading to specific diagnoses that supersede the initial CFTD diagnosis [2, 151]. With an autosomal dominant mode of inheritance, TPM3 is the most common genetic cause of CFTD (25–50% of cases) [58]. RYR1-related CFTD is an autosomal recessive disease accounting for about 20% of CFTD cases [59]. ACTA1 and TPM2 are also uncommon causes of CFTD [153, 154].
Multi-minicore disease (MmD)
The variability associated with the appearance of cores on muscle biopsy led to subsequent reports of multicore disease with various nomenclatures (minicore myopathy, multicore myopathy, multi-minicore myopathy) until a working group of experts agreed on a designation of multi-minicore disease (MmD) [61]. Multi-minicores may affect both type 1 and type 2 muscle fibers and show depletion or absence of mitochondria on electron microscopy, with variable degrees of myofibrillar disorganization, abnormal Z-band material, and regions of sarcomeric disruption. Increased internal nuclei, fibers with slow myosin (type 1 predominance on histochemical stains), prominent connective tissue and adipose tissue in the absence of numerous fibers with developmental myosin, necrosis or endomysial fibrosis are also noted in affected muscles [141, 155]. RYR1-related MmD is inherited in an autosomal recessive pattern with highly variable clinical features [156]. Typical features include hypotonia in infancy, axial muscle weakness, hip girdle weakness, ophthalmoplegia, distal joint laxity, progressive scoliosis, and moderate respiratory/bulbar involvement. Four homogeneous MmD groups have been identified: (1) the classic form, marked by predominantly axial muscle weakness, especially neck flexors, scoliosis, respiratory insufficiency, and limb joint hyperlaxity; (2) the ophthalmoplegia form, with generalized muscle involvement and severe facial weakness; (3) an early-onset form with arthrogryposis; and (4) a slowly progressive form with hand amyotrophy [157]. Other genes associated with MmD include SEPN1 [60], MYH7 [158], and TTN [159]. Pathogenic variants in SEPN1 and RYR1 are responsible for approximately 50% of all cases and SEPN1 variations comprising ~ 75% of the classic form of MmD [60]. RYR1 variations are mostly associated with both the moderate form of MmD with hand involvement and the ophthalmoplegic form [156, 160], both of which typically present as milder than classic SEPN1-associated MmD. Multi-minicores associated with SEPN1 variations are typically smaller in size than those observed in recessive RYR1-related cases [131] and are accompanied by non-specific myopathic changes such as CFTD and Mallory body-like inclusions [161, 162]. Malignant hyperthermia and ophthalmoplegia are not usually noted in SEPN1-related myopathies [163]. On MRI, there is prominent involvement of the sartorius muscle in the thigh, and in more severe cases, the pattern of thigh involvement show similarity with that seen in RYR1-RM [164]. MYH7-, DES-, LMNA- and TTN-related core myopathies are recessive, progressive, and often present with severe cardiomyopathy that can be independent of respiratory insufficiency [158, 159, 165, 166]. Joint hypermobility, although more often a sign of connective tissue disorders, can be a prominent clinical feature of RYR1-related core myopathies [167]. Additionally, the presence of a large number of muscle fibers (up to 50%) with internal or central nuclei has been reported as part of the RYR1-related core myopathy spectrum [168]. Minicores are also non-specific features of congenital muscular dystrophies, dystrophinopathies, neuropathies, short-chain acyl-COA dehydrogenase deficiency, Marfan syndrome, and cardiomyopathies [2, 160]. Nemaline bodies with core-like areas have been described in dominant ACTA1-related myopathy [169] and in a family with recessive CFL2 variations [170]. Multiple core-like areas per fiber were reported in type-1 fibers in DOK7-associated congenital myasthenic syndromes [171] and RYR1-associated atypical periodic paralysis [35]. “Moth-eaten” fibers described in certain muscular dystrophies can also be considered core-like lesions [172]. However, these small core-like lesions are often not confirmed with electron microscopy compared with multiple minicores. Variations in MYH7 and TTN should be considered if there is an associated cardiomyopathy.
Proposal for a unified nomenclature
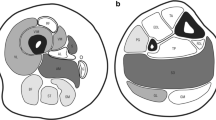
The similarity in muscle histopathology and clinical symptomatology across RYR1-RM subtypes, currently viewed as nosocologically distinct entities, reflects a wide range of manifestations resulting from defects in the same gene. Specifically, patient severity occurs on a spectrum and is often evaluated in the context of mode of inheritance, histopathology, and clinical phenotype (Fig. 2). A combination of factors has made understanding and describing muscle disorders associated with RYR1 variants confusing for health care providers, researchers, and patients/families. Moving forward, we therefore propose the term “RYR1-related disorders (or RYR1-RD)” as a single nomenclature to unify this complex myopathic/non-myopathic and congenital/non-congenital spectrum with the goal of minimizing the following known issues.

Overview of the RYR1 disease spectrum. At time of presentation, clinical severity can vary according to mode of inheritance (dominant, de novo, recessive), histopathologic features, and phenotypes ranging from severe neonatal onset to mild non-progressive muscle weakness. Recessive cases are typically more severe than dominant cases. The majority of histopathological features are associated with more severe clinical phenotypes, though this may not hold true for the core myopathies. Emerging clinical phenotypes associated with RYR1 variations also vary in severity
Dynamic and overlapping histopathology
Historical classification and diagnosis of newly emerging RYR1-RD based on clinico-histopathologic features are complicated because findings often overlap with previously identified phenotypes. The histopathologic features used to diagnose RYR1-RD are variable over time, and therefore, subject to inconclusive findings. The degree of pathological changes may vary by biopsy site or age of the individual when the biopsy was obtained. Very young patients may not present definitively with cores until later in life [173] and some CCD cases with MH susceptibility may not present with cores [131]. Clinical features of dominant CCD typically present on the milder end of the phenotypic spectrum including hypotonia, muscle weakness, and skeletal abnormalities in the absence of cardiac involvement [131]. However, the rare cases of recessive CCD that often exhibit a more severe clinical presentation [174, 175] do not always correlate with histopathologic findings [42].
Delayed diagnosis
Diagnosis is delayed and difficult when nosocologically defining histopathologic features are absent, and in cases with dual morphologic presentations such as core-rod myopathy [131]. Also, the absence of diagnostic morphologic features on muscle biopsy does not exclude a likely pathogenic variation in RYR1 [61]. With nearly 700 RYR1 variations identified to date [176], availability and access to exome sequencing and genetic testing of the entire RYR1 gene can be credited for early and faster diagnosis [177], and expansion of the RYR1 disease spectrum [178]. A recent RYR1-RM natural history study [179] revealed that affected individuals born before the advent of next-generation sequencing (2004) were typically diagnosed as adults, while those born after 2004 were generally diagnosed in early childhood. Additionally, homozygous or compound heterozygous probands often exhibit a profound myopathic phenotype, while heterozygous probands might only experience a triggered phenotype. A genetics-first diagnostic approach is rapidly becoming the standard for confirmation of disorders with known genetic etiology [112, 180]. Nevertheless, the limited sensitivity of next-generation sequencing, requirement for multiple testing in some cases, and interpretation of large numbers of identified VUS in a relatively large gene such as RYR1 still requires a robust knowledge of the suggestive features associated with each variant. Although complete reliance on histopathologically defined entities such as central cores, multi-minicores and central nuclei is arguably outdated, these muscle biopsy findings remain valuable as diagnostic indicators of disease subclassification that are not otherwise feasible from genetic findings alone. RYR1-RD affected individuals exhibit specific and valuable initial patterns of clinical and histopathologic presentations that can facilitate early diagnosis. As the possibility of an inherited process is not always apparent [181], clinical, histopathologic, and genetic information still need to be evaluated together in order to reduce the diagnostic odyssey of patients and their families [182].
Adult-onset phenotypes
RYR1-RD are typically considered primarily early childhood-onset conditions with proximal or generalized muscle weakness. Recent identification of adult-onset subtypes reflects a departure from the long-held definition of this group as strictly “congenital myopathies” [39, 40]. Considered a late neuromuscular manifestation of MH-related RYR1 variations, late-onset axial myopathy, exertional rhabdomyolysis, and periodic paralysis present throughout the lifespan. Late-onset RYR1-RD present significant diagnostic and genetic counseling challenges, as well as implications for proper anesthetic management of patients and their family members. True adult-onset cases are difficult to establish as the affected individual could have either tolerated or been in denial of mild symptoms experienced during childhood. RYR1-RD phenotypes are still predominantly early childhood onset, and excluding the “congenital” descriptor does not detract from their classification as CM. However, awareness of these adult-onset subtypes allows: (a) clinicians to consider RYR1-RD in the differential diagnostic process across all ages even in neuromuscular disease cases without prior muscle biopsy findings [39], (b) inclusion of older adults in clinical trials testing novel therapies, and (c) collection of more robust natural history data on RYR1-RD across the lifespan [37].
Shared calcium dysregulation
RYR1-RD result from varying disease pathomechanisms that collectively share alterations in a common pathway—intracellular calcium dysregulation resulting from primary RyR1 dysfunction (e.g., reduced RyR1 expression, leaky RyR1 channels, impaired RyR1 interdomain interactions, enhanced sensitivity to modulators, impaired excitation-contraction coupling) [6, 22]. Alterations in calcium homeostasis can also lead to secondary cellular dysfunction including increased oxidative/nitrosative stress, altered post-translational modifications, mitochondrial damage, and disrupted protein-protein/ligand interactions [3, 122, 183]. These downstream effects further drive myopathy and enhance heat responsiveness due to a feed-forward loop [11]. However, RyR1- or cellular/mitochondrial-based calcium dysregulation such as increased mitochondrial calcium uptake, production of damaging reactive oxygen species [3, 122, 183] and upregulation of endoplasmic reticulum stress/unfolded protein response [184], may be RYR1 variant dependent.
Discussion
The complex nature of the RyR1 protein, coupled with the expanding and overlapping disease spectrum of RYR1-RD, presents a timely opportunity to consider a unified nomenclature and classification system for this heterogeneous group of disorders. Here, we propose the use of “RYR1-related disorders (RYR1-RD)” as a single nomenclature to unify this complex myopathic/non-myopathic and congenital/non-congenital spectrum.
Any acceptable change in nomenclature will require a careful, widely discussed and expansive evaluation by experts in the field of neuromuscular disorders. A comprehensive classification system would both incorporate the different disease pathomechanisms associated with RYR1-RD and identify potential consequences that may not be immediately apparent. A unified nomenclature is needed for multiple reasons, but most importantly to facilitate unambiguous communication about related conditions among clinicians, researchers, patients, and the lay public. A relevant example in the neuromuscular disease field is the consensus naming and classification of multi-minicore disease (MmD) by a panel of experts [157]. Following the first reports of MmD [89], numerous cases with variable clinical expression and morphological lesions were reported. This led to multiple histologic descriptors including multicore disease, focal loss of cross-striations, minicore myopathy, myopathy with multiple minicore, or pleocore disease to describe the same disease subtype [60]. In comparison to relatively more homogeneous disorders such as SEPN1-RM [60] and COL6-RM [185] where gene-based nomenclatures were introduced, there is an inherent possibility that a single nomenclature for such a clinically and histopathologically heterogeneous group of disorders may be seen or interpreted as having the same pathomechanisms and genotype-phenotype correlations.
RYR1-RD subtypes also present clear clinicopathological and mode of inheritance differences. For example, there are no reported pedigrees with unequivocal evidence for multigenerational dominant inheritance of RYR1-related CFTD and CNM, both overwhelmingly associated with recessive RYR1 variations. Individuals with core-rod myopathy present with excess ryanodine receptor levels in the cores [140], while MmD and recessive forms of CCD exhibit a marked reduction in RyR1 protein expression [186]. Although type 1 fiber predominance and hypotrophy are observed in most cases, type 1 fiber uniformity without structural changes (cores and rods) is found in over 99% of the type 1 muscle fibers in certain cases with C-terminal RYR1 variants [33]. From a clinical perspective, extraocular muscle involvement is almost exclusively associated with recessive forms of RYR1-RD, whereas enhanced malignant hyperthermia susceptibility is more commonly seen with dominant rather than recessive RYR1 variations [187].
Additionally, some degree of dysfunction in calcium homeostasis and/or E-C coupling is a shared pathomechanism among RYR1-RD. The effects of specific RYR1 variants on these mechanisms depend on mode of inheritance and location on the gene [179]. Therapeutic development for RYR1-RD aiming to prevent RyR1 calcium leak may not be beneficial and could potentially be detrimental, in the context of dominant variants that reduce RyR1 calcium conductance or recessive variants that lead to a dramatic reduction in RyR1 expression. The different pathomechanisms caused by RYR1 variations (e.g., hypersensitivity, enhanced calcium leak, E-C uncoupling, decreased RyR1 channel expression) [6] and the prospect of distinct therapeutic approaches needed to combat these different underlying disease mechanisms are seemingly at odds with the concept of a unified nomenclature. Finally, unified nomenclature may have unintended negative implications for research funding opportunities from patient advocacy organizations focused on specific congenital myopathies and may present challenges in formulation of treatment, biomarkers, and/or clinical outcome measures across subtypes with pathomechanisms that affect varying stages of cellular functions. This could slow the pace of drug development and treatment approval by regulatory agencies.
In our opinion, a unified nomenclature should not only encompass the complex clinical and pathological features associated with RYR1 variations (including conditions that do not exhibit an overt myopathy such as MH susceptibility and exertional rhabdomyolysis), but also both accommodate future RYR1-related phenotypes and navigate the field away from utilizing non-specific histopathologic eponyms (Fig. 3).

Distinct and overlapping features of the RYR1-RD spectrum. Individuals with RYR1-RD present with both myopathic and non-myopathic features. Histopathology can overlap among the different subtypes. Additionally, some phenotypes manifest following exposure to pharmacologic, physiologic, or environmental triggers. A classification system based on three distinct and overlapping categories (myopathic, non-myopathic, and triggered) accommodates current and most future subtypes of RYR1-RD
Conclusion
Historically, RYR1-RD have been named and diagnosed based largely on histopathologic findings on muscle biopsy. However, the emergence of new subtypes along the RYR1 disease spectrum complicates diagnoses. As a “genetics-first” approach to inherited disease diagnosis is becoming widely accepted, neuromuscular disorders such as RYR1-RD need established guidelines and consensus principles for classification and naming of emerging phenotypes. The proposal for RYR1-RD as the unifying nomenclature is a first step and could be superceded by a better encompassing terminology. We believe that such a discourse is timely and needed for this widely heterogeneous group of muscle disorders.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed.
Abbreviations
- CM:
-
Congenital myopathies
- Ca2+ :
-
Calcium
- CCD:
-
Central core disease
- CFTD:
-
Congenital fiber-type disproportion
- CNM:
-
Centronuclear myopathy
- CNMDU1:
-
Congenital neuromuscular disease with uniform type 1 fiber
- EC:
-
Excitation-contraction
- MH:
-
Malignant hyperthermia
- MmD:
-
Multi-minicore disease
- RYR1:
-
Ryanodine receptor type 1
- RYR1-RM:
-
Ryanodine receptor type 1-related myopathies
- SR:
-
Sarcoplasmic reticulum
- VUS:
-
Variant of unknown significance
References
Dubowitz V. The “new” myopathies. Neuropediatrics. 1969;1(02):137–48.
North KN, Wang CH, Clarke N, Jungbluth H, Vainzof M, Dowling JJ, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord. 2014;24(2):97–116.
Witherspoon JW, Meilleur KG. Review of RyR1 pathway and associated pathomechanisms. Acta Neuropathol Commun. 2016;4(1):121.
Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature. 1988;331(6154):315–9.
Fujii J, Otsu K, Zorzato F, de Leon S, Khanna VK, Weiler JE, et al. Identification of a mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science (New York, NY). 1991;253(5018):448–51.
Treves S, Jungbluth H, Muntoni F, Zorzato F. Congenital muscle disorders with cores: the ryanodine receptor calcium channel paradigm. Curr Opin Pharmacol. 2008;8(3):319–26.
Kim DC. Malignant hyperthermia. Korean J Anesthesiol. 2012;63(5):391–401.
Hopkins PM, Ruffert H, Snoeck MM, Girard T, Glahn KP, Ellis FR, et al. European malignant hyperthermia group guidelines for investigation of malignant hyperthermia susceptibility. Br J Anaesth. 2015;115(4):531–9.
Ellinas H, Albrecht MA. Malignant hyperthermia update. Anesthesiol Clin. 2020;38(1):165–81.
Groom L, Muldoon SM, Tang ZZ, Brandom BW, Bayarsaikhan M, Bina S, et al. Identical de novo mutation in the type 1 ryanodine receptor gene associated with fatal, stress-induced malignant hyperthermia in two unrelated families. Anesthesiology. 2011;115(5):938–45.
Lanner JT, Georgiou DK, Dagnino-Acosta A, Ainbinder A, Cheng Q, Joshi AD, et al. AICAR prevents heat-induced sudden death in RyR1 mutant mice independent of AMPK activation. Nat Med. 2012;18(2):244–51.
Capacchione JF, Muldoon SM. The relationship between exertional heat illness, exertional rhabdomyolysis, and malignant hyperthermia. Anesth Analg. 2009;109(4):1065–9.
Rosenberg H, Pollock N, Schiemann A, Bulger T, Stowell K. Malignant hyperthermia: a review. Orphanet J Rare Dis. 2015;10:93.
Tong J, Oyamada H, Demaurex N, Grinstein S, McCarthy TV, MacLennan DH. Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J Biol Chem. 1997;272(42):26332–9.
Dirksen RT, Avila G. Altered ryanodine receptor function in central core disease: leaky or uncoupled ca(2+) release channels? Trends Cardiovasc Med. 2002;12(5):189–97.
Loy RE, Orynbayev M, Xu L, Andronache Z, Apostol S, Zvaritch E, et al. Muscle weakness in Ryr1I4895T/WT knock-in mice as a result of reduced ryanodine receptor Ca2+ ion permeation and release from the sarcoplasmic reticulum. J Gen Physiol. 2011;137(1):43–57.
Avila G, O'Brien JJ, Dirksen RT. Excitation--contraction uncoupling by a human central core disease mutation in the ryanodine receptor. Proc Natl Acad Sci U S A. 2001;98(7):4215–20.
Dowling JJ, Lawlor MW, Dirksen RT. Triadopathies: an emerging class of skeletal muscle diseases. Neurotherapeutics. 2014;11(4):773–85.
Amburgey K, McNamara N, Bennett LR, McCormick ME, Acsadi G, Dowling JJ. Prevalence of congenital myopathies in a representative pediatric United States population. Ann Neurol. 2011;70(4):662–5.
Dowling JJ. H DG, Cohn RD, Campbell C. treating pediatric neuromuscular disorders: the future is now. Am J Med Genet A. 2018;176(4):804–41.
Gonsalves SG, Ng D, Johnston JJ, Teer JK, Stenson PD, Cooper DN, et al. Using exome data to identify malignant hyperthermia susceptibility mutations. Anesthesiology. 2013;119(5):1043–53.
Amburgey K, Bailey A, Hwang JH, Tarnopolsky MA, Bonnemann CG, Medne L, et al. Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J Rare Dis. 2013;8:117.
D'Amico A, Bertini E. Congenital myopathies. Curr Neurol Neurosci Rep. 2008;8(1):73–9.
Todd JJ, Razaqyar MS, Witherspoon JW, Lawal TA, Mankodi A, Chrismer IC, et al. Novel variants in individuals with RYR1-related congenital myopathies: genetic, laboratory, and clinical findings. Front Neurol. 2018;9:118.
Rocha J, Taipa R, Melo Pires M, Oliveira J, Santos R, Santos M. Ryanodine myopathies without central cores--clinical, histopathologic, and genetic description of three cases. Pediatr Neurol. 2014;51(2):275–8.
Morgan-Hughes JA, Brett EM, Lake BD, Tome FM. Central core disease or not? Observations on a family with a non-progressive myopathy. Brain. 1973;96(3):527–36.
Jungbluth H, Dowling JJ, Ferreiro A, Muntoni F. 217th ENMC international workshop: RYR1-related myopathies, Naarden, the Netherlands, 29-31 January 2016. Neuromuscul Disord. 2016;26(9):624–33.
Gonorazky HD, Bonnemann CG, Dowling JJ. The genetics of congenital myopathies. Handb Clin Neurol. 2018;148:549–64.
Jungbluth H, Sewry CA, Muntoni F. What's new in neuromuscular disorders? The congenital myopathies. Eur J Paediatr Neurol. 2003;7(1):23–30.
Snoeck M, van Engelen BG, Kusters B, Lammens M, Meijer R, Molenaar JP, et al. RYR1-related myopathies: a wide spectrum of phenotypes throughout life. Eur J Neurol. 2015;22(7):1094–112.
Dowling JJ, Lillis S, Amburgey K, Zhou H, Al-Sarraj S, Buk SJ, et al. King-Denborough syndrome with and without mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord. 2011;21(6):420–7.
King JO, Denborough MA. Anesthetic-induced malignant hyperpyrexia in children. J Pediatr. 1973;83(1):37–40.
Sato I, Wu S, Ibarra MC, Hayashi YK, Fujita H, Tojo M, et al. Congenital neuromuscular disease with uniform type 1 fiber and RYR1 mutation. Neurology. 2008;70(2):114–22.
Witting N, Laforet P, Voermans NC, Roux-Buisson N, Bompaire F, Rendu J, et al. Phenotype and genotype of muscle ryanodine receptor rhabdomyolysis-myalgia syndrome. Acta Neurol Scand. 2018;137(5):452–61.
Matthews E, Neuwirth C, Jaffer F, Scalco RS, Fialho D, Parton M, et al. Atypical periodic paralysis and myalgia: a novel RYR1 phenotype. Neurology. 2018;90(5):e412–e8.
Lopez RJ, Byrne S, Vukcevic M, Sekulic-Jablanovic M, Xu L, Brink M, et al. An RYR1 mutation associated with malignant hyperthermia is also associated with bleeding abnormalities. Sci Signal. 2016;9(435):ra68.
Jungbluth H, Voermans NC. Congenital myopathies: not only a paediatric topic. Curr Opin Neurol. 2016;29(5):642–50.
Garibaldi M, Rendu J, Brocard J, Lacene E, Faure J, Brochier G, et al. 'Dusty core disease' (DuCD): expanding morphological spectrum of RYR1 recessive myopathies. Acta Neuropathol Commun. 2019;7(1):3.
Loseth S, Voermans NC, Torbergsen T, Lillis S, Jonsrud C, Lindal S, et al. A novel late-onset axial myopathy associated with mutations in the skeletal muscle ryanodine receptor (RYR1) gene. J Neurol. 2013;260(6):1504–10.
Jungbluth H, Lillis S, Zhou H, Abbs S, Sewry C, Swash M, et al. Late-onset axial myopathy with cores due to a novel heterozygous dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord. 2009;19(5):344–7.
Jeong HN, Park HJ, Lee JH, Shin HY, Kim SH, Kim SM, et al. Clinical and pathologic findings of Korean patients with RYR1-related congenital myopathy. J Clin Neurol (Seoul, Korea). 2018;14(1):58–65.
Bharucha-Goebel DX, Santi M, Medne L, Zukosky K, Dastgir J, Shieh PB, et al. Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum. Neurology. 2013;80(17):1584–9.
Illingworth MA, Main M, Pitt M, Feng L, Sewry CA, Gunny R, et al. RYR1-related congenital myopathy with fatigable weakness, responding to pyridostigimine. Neuromuscul Disord. 2014;24(8):707–12.
Fusto A, Moyle LA, Gilbert PM, Pegoraro E. Cored in the act: the use of models to understand core myopathies. Dis Model Mech. 2019;12(12):dmm041368.
Jungbluth H, Treves S, Zorzato F, Sarkozy A, Ochala J, Sewry C, et al. Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nat Rev Neurol. 2018;14(3):151–67.
Lawal TA, Todd JJ, Meilleur KG. Ryanodine receptor 1-related myopathies: diagnostic and therapeutic approaches. Neurotherapeutics. 2018;15(4):885–99.
Mickelson JR, Knudson CM, Kennedy CF, Yang DI, Litterer LA, Rempel WE, et al. Structural and functional correlates of a mutation in the malignant hyperthermia-susceptible pig ryanodine receptor. FEBS Lett. 1992;301(1):49–52.
Lawal TA, Wires ES, Terry NL, Dowling JJ, Todd JJ. Preclinical model systems of ryanodine receptor 1-related myopathies and malignant hyperthermia: a comprehensive scoping review of works published 1990-2019. Orphanet J Rare Dis. 2020;15(1):113.
Mickelson JR, Louis CF. Malignant hyperthermia: excitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol Rev. 1996;76(2):537–92.
Magee KR, Shy GM. A new congenital non-progressive myopathy. Brain : a journal of neurology. 1956;79(4):610–21.
Dubowitz V, Pearse AG. Oxidative enzymes and phosphorylase in central-core disease of muscle. Lancet (London, England). 1960;2(7140):23–4.
Carpenter SKG. Pathology of skeletal muscle. 2nd ed. New York: Oxford University Press; 2001.
CLN W-P. The congenital myopathies. In: GHJD K, Bushby K, Griggs R, editors. Disorders of voluntary muscle. 8th ed. New York: Cambridge University Press; 2010. p. 282–98.
Sewry CAJ-MC, Brown SC, Muntoni F. Disease of muscle. In: SLD L, Ellison D, editors. Greenfield’s neuropathology. 8th ed. London: Hodder Arnold; 2008. p. 1786–91.
Jungbluth HSC, Muntoni F. The congenital myopathies. In: Rosenberg RNPJ, editor. Rosenberg’s molecular and genetic basis of neurological and psychiatric disease. 5th ed. Massachusetts: Academic Press; 2015. p. 1121–8.
Wu S, Ibarra MC, Malicdan MC, Murayama K, Ichihara Y, Kikuchi H, et al. Central core disease is due to RYR1 mutations in more than 90% of patients. Brain. 2006;129(Pt 6):1470–80.
Tosch V, Vasli N, Kretz C, Nicot AS, Gasnier C, Dondaine N, et al. Novel molecular diagnostic approaches for X-linked centronuclear (myotubular) myopathy reveal intronic mutations. Neuromuscul Disord. 2010;20(6):375–81.
Lawlor MW, Dechene ET, Roumm E, Geggel AS, Moghadaszadeh B, Beggs AH. Mutations of tropomyosin 3 (TPM3) are common and associated with type 1 myofiber hypotrophy in congenital fiber type disproportion. Hum Mutat. 2010;31(2):176–83.
Clarke NF, Waddell LB, Cooper ST, Perry M, Smith RLL, Kornberg AJ, et al. Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum Mutat. 2010;31(7):E1544–E50.
Ferreiro A, Quijano-Roy S, Pichereau C, Moghadaszadeh B, Goemans N, Bönnemann C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet. 2002;71(4):739–49.
Jungbluth H, Beggs A, Bonnemann C, Bushby K, Ceuterick-de Groote C, Estournet-Mathiaud B, et al. 111th ENMC international workshop on multi-minicore disease. 2nd international MmD workshop, 9-11 November 2002, Naarden, the Netherlands. Neuromuscul Disord. 2004;14(11):754–66.
Oppenheim H. Ueber allgemeine und localisierte Atonie der Muskulatur (Myatonie) im frühen Kindesalter. Eur Neurol. 1900;8(3):232–3.
Werdnig G. Zwei frühinfantile hereditäre Fälle von progressiver Muskelatrophie unter dem Bilde der Dystrophie, aber anf neurotischer Grundlage. Archiv für Psychiatrie und Nervenkrankheiten. 1891;22(2):437–80.
Greenfield JG, Stern RO. The anatomical identity of the Werdnig-Hoffmann and Oppenheim forms of infantile muscular atrophy. Brain. 1927;50(3-4):652–86.
Tuthill CR, Levy MG. Amyotonia: report of a case with histologic observations. Am J Dis Child. 1931;41(3):591–602.
Macleod JG, Macdonald RM. Infantile muscular atrophy of spinal origin: a report of two cases. Arch Dis Child. 1942;17(89):30–40.
Johnson AB. Heat-stroke as a post operative complication. JAMA. 1900;35:1685.
Brewer GE. Heat-stroke as a post operative complication, abstract of new York academy of medicine-surgical section (Nov. 12, 1990). JAMA. 1900;35:1685.
Gibson C. Heat-stroke as a post operative complication. JAMA. 1900;35:1685.
Tuttle JPT. Heat-stroke as a post operative complication. JAMA. 1900;35:1685.
Moschcowitz AV. Heat-stroke as a post operative complication. JAMA. 1900;35:1685.
Harrison GG, Isaacs H. Malignant hyperthermia. An historical vignette. Anaesthesia. 1992;47(1):54–6.
Ludvigsen J. UndersGgelser over den sikaldte “muskeldegeneration” 110 s svin I. 272. beretning jra jorspigslaboratoriet. Statens husdyrbrugsudvalg, K@benhav. 1954.
Briskey EJ. Pre-slaughter treatments as related to post-mortem changes in pork muscle. In: 5th European Conj Meat Research Paper 16; 1959.
Kjolberg O, Manners DJ, Lawrie RA. Alpha-(1-4)-Glucosans. 16. The molecular structure of some pig-muscle glycogens. Biochem J. 1963;87(2):351–3.
Ludvigsen J. On the hormonal regulation of vasomotor reactions during exercise with special reference to the action of adrenal cortical steroids. Acta Endocrinol. 1957;26(4):406–16.
Briskey EJ. Etiological status and associated studies of pale, soft, exudative porcine musculature. Adv Food Res. 1964;13:89–178.
Forrest JC, Will JA, Schmidt GR, Judge MD, Briskey EJ. Homeostasisin animals (Sus domesticus) during exposure to a warm environment. J Appl Physiol. 1968;24(1):33–9.
Greenfield JGCT, Shy GM. The prognostic value of the muscle biopsy in the floppy infant. Brain. 1958;81(4):461–84.
Denborough MA, Lovell R. Anesthetic deaths in a family. Lancet (London, England). 1960;2(45).
Denborough MA, Forster JF, Lovell RR, Maplestone PA, Villiers JD. Anaesthetic deaths in a family. Br J Anaesth. 1962;34:395–6.
Shy GM, Engel WK, Somers JE, Wanko T. Nemaline myopathy. A new congenital myopathy. Brain. 1963;86:793–810.
Afifi AK, Smith JW, Zellweger H. Congenital nonprogressive myopathy. Central core disease and Nemaline myopathy in one family. Neurology. 1965;15:371–81.
Spiro AJ, Shy GM, Gonatas NK. Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Arch Neurol. 1966;14(1):1–14.
Sher JH, Rimalovski AB, Athanassiades TJ, Aronson SM. Familial centronuclear myopathy: a clinical and pathological study. Neurology. 1967;17(8 Pt 1):727–42.
Brooke MH, Engel WK. The histographic analysis of human muscle biopsies with regard to fiber types. 4. Children's biopsies. Neurology. 1969;19(6):591–605.
Brooke M. Clinical studies in myology. Proceedings of the 2nd International Congress on Muscle Diseases. Congenit Fiber-Type Disproportion. 1973:147–59.
Martin JJ, Clara R, Ceuterick C. Joris C. Is congenital fibre type disproportion a true myopathy? Acta Neurol Belg. 1976;76(5-6):335–44.
Engel AG, Gomez MR, Groover RV. Multicore disease. A recently recognized congenital myopathy associated with multifocal degeneration of muscle fibers. Mayo Clin Proc. 1971;46(10):666–81.
Pessah IN, Waterhouse AL, Casida JE. The calcium-ryanodine receptor complex of skeletal and cardiac muscle. Biochem Biophys Res Commun. 1985;128(1):449–56.
Fleischer S, Ogunbunmi EM, Dixon MC, Fleer EA. Localization of Ca2+ release channels with ryanodine in junctional terminal cisternae of sarcoplasmic reticulum of fast skeletal muscle. Proc Natl Acad Sci U S A. 1985;82(21):7256–9.
O'Brien PJ. Etiopathogenetic defect of malignant hyperthermia: hypersensitive calcium-release channel of skeletal muscle sarcoplasmic reticulum. Vet Res Commun. 1987;11(6):527–59.
Davies W, Harbitz I, Fries R, Stranzinger G, Hauge JG. Porcine malignant hyperthermia carrier detection and chromosomal assignment using a linked probe. Anim Genet. 1988;19(3):203–12.
Lister D. The physiology and biochemistry of the porcine stress syndrome. In: Tarrant PV, Eikelenboom G, Monin G, editors. Evaluation and control of meat quality in pigs: a seminar in the CEC agricultural research Programme, held in Dublin, Ireland, 21-22 November 1985. Dordrecht: Springer Netherlands; 1987. p. 3–16.
Smith JS, Coronado R, Meissner G. Sarcoplasmic reticulum contains adenine nucleotide-activated calcium channels. Nature. 1985;316(6027):446–9.
Meissner G. Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J Biol Chem. 1986;261(14):6300–6.
Imagawa T, Smith JS, Coronado R, Campbell KP. Purified ryanodine receptor from skeletal muscle sarcoplasmic reticulum is the Ca2 + -permeable pore of the calcium release channel. J Biol Chem. 1987;262(34):16636–43.
Inui M, Saito A, Fleischer S. Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J Biol Chem. 1987;262(4):1740–7.
Zorzato F, Fujii J, Otsu K, Phillips M, Green NM, Lai FA, et al. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1990;265(4):2244–56.
Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339(6224):439–45.
Marks AR, Tempst P, Hwang KS, Taubman MB, Inui M, Chadwick C, et al. Molecular cloning and characterization of the ryanodine receptor/junctional channel complex cDNA from skeletal muscle sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1989;86(22):8683–7.
Walton J, Thomas PK. Classification of neuromuscular diseases. J Neurol Sci. 1988;86(2):333–60.
MacLennan DH, Duff C, Zorzato F, Fujii J, Phillips M, Korneluk RG, et al. Ryanodine receptor gene is a candidate for predisposition to malignant hyperthermia. Nature. 1990;343(6258):559–61.
Gillard EF, Otsu K, Fujii J, Duff C, de Leon S, Khanna VK, et al. Polymorphisms and deduced amino acid substitutions in the coding sequence of the ryanodine receptor (RYR1) gene in individuals with malignant hyperthermia. Genomics. 1992;13(4):1247–54.
Quane KA, Healy JM, Keating KE, Manning BM, Couch FJ, Palmucci LM, et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat Genet. 1993;5(1):51–5.
Jokela M, Tasca G, Vihola A, Mercuri E, Jonson PH, Lehtinen S, et al. An unusual ryanodine receptor 1 (RYR1) phenotype: mild calf-predominant myopathy. Neurology. 2019;92(14):e1600–e9.
Jungbluth H, Davis MR, Muller C, Counsell S, Allsop J, Chattopadhyay A, et al. Magnetic resonance imaging of muscle in congenital myopathies associated with RYR1 mutations. Neuromuscul Disord. 2004;14(12):785–90.
Chelu MG, Goonasekera SA, Durham WJ, Tang W, Lueck JD, Riehl J, et al. Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J. 2006;20(2):329–30.
Yang T, Riehl J, Esteve E, Matthaei KI, Goth S, Allen PD, et al. Pharmacologic and functional characterization of malignant hyperthermia in the R163C RyR1 knock-in mouse. Anesthesiology. 2006;105(6):1164–75.
Monnier N, Romero NB, Lerale J, Nivoche Y, Qi D, MacLennan DH, et al. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum Mol Genet. 2000;9(18):2599–608.
Jungbluth H, Dowling JJ, Ferreiro A, Muntoni F. 182nd ENMC international workshop: RYR1-related myopathies, 15-17th April 2011, Naarden, the Netherlands. Neuromuscul Disord. 2012;22(5):453–62.
Westra D, Schouten MI, Stunnenberg BC, Kusters B, Saris CGJ, Erasmus CE, et al. Panel-based exome sequencing for neuromuscular disorders as a diagnostic service. J Neuromusc Dis. 2019;6(2):241–58.
Palmio J, Udd B. Borderlines between sarcopenia and mild late-onset muscle disease. Front Aging Neurosci. 2014;6:267.
Zalk R, Clarke OB, Des Georges A, Grassucci RA, Reiken S, Mancia F, et al. Structure of a mammalian ryanodine receptor. Nature. 2015;517(7532):44–9.
Lobo PA, Van Petegem F. Crystal structures of the N-terminal domains of cardiac and skeletal muscle ryanodine receptors: insights into disease mutations. Structure (London, England : 1993). 2009;17(11):1505–14.
Yan Z, Bai X, Yan C, Wu J, Li Z, Xie T, et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. 2015;517(7532):50–5.
Efremov RG, Leitner A, Aebersold R, Raunser S. Architecture and conformational switch mechanism of the ryanodine receptor. Nature. 2015;517(7532):39–43.
Desgeorges A, Clarke OB, Zalk R, Yuan Q, Condon KJ, Grassucci RA, et al. Structural basis for gating and activation of RyR1. Cell. 2016;167(1):145–57.e17.
Samso M. A guide to the 3D structure of the ryanodine receptor type 1 by cryoEM. Protein Sci. 2017;26(1):52–68.
Messina S, Hartley L, Main M, Kinali M, Jungbluth H, Muntoni F, et al. Pilot trial of salbutamol in central core and multi-minicore diseases. Neuropediatrics. 2004;35(5):262–6.
Todd JJ, Lawal TA, Witherspoon JW, Chrismer IC, Razaqyar MS, Punjabi M, et al. Randomized controlled trial of N-acetylcysteine therapy for RYR1-related myopathies. Neurology. 2020;94(13):e1434–e44.
Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera SA, Boncompagni S, et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133(1):53–65.
Hirata H, Watanabe T, Hatakeyama J, Sprague SM, Saint-Amant L, Nagashima A, et al. Zebrafish relatively relaxed mutants have a ryanodine receptor defect, show slow swimming and provide a model of multi-minicore disease. Development (Cambridge, England). 2007;134(15):2771–81.
Dowling JJ, Arbogast S, Hur J, Nelson DD, McEvoy A, Waugh T, et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain. 2012;135(Pt 4):1115–27.
Volpatti JR, Endo Y, Knox J, Groom L, Brennan S, Noche R, et al. Identification of drug modifiers for RYR1-related myopathy using a multi-species discovery pipeline. eLife. 2020;9.
Murayama T, Kurebayashi N, Ishigami-Yuasa M, Mori S, Suzuki Y, Akima R, et al. Efficient high-throughput screening by endoplasmic reticulum ca(2+) measurement to identify inhibitors of ryanodine receptor ca(2+)-release channels. Mol Pharmacol. 2018;94(1):722–30.
Rebbeck RT, Singh DP, Janicek KA, Bers DM, Thomas DD, Launikonis BS, et al. RyR1-targeted drug discovery pipeline integrating FRET-based high-throughput screening and human myofiber dynamic ca(2+) assays. Sci Rep. 2020;10(1):1791.
Elbaz M, Ruiz A, Bachmann C, Eckhardt J, Pelczar P, Venturi E, et al. Quantitative RyR1 reduction and loss of calcium sensitivity of RyR1Q1970fsX16 + A4329D cause cores and loss of muscle strength. Hum Mol Genet. 2019.
Brennan S, Garcia-Castaneda M, Michelucci A, Sabha N, Malik S, Groom L, et al. Mouse model of severe recessive RYR1-related myopathy. Hum Mol Genet. 2019.
Pancaroglu R, Van Petegem F. Calcium channelopathies: structural insights into disorders of the muscle excitation-contraction complex. Annu Rev Genet. 2018;52:373–96.
Sewry CA, Muller C, Davis M, Dwyer JS, Dove J, Evans G, et al. The spectrum of pathology in central core disease. Neuromuscul Disord. 2002;12(10):930–8.
Quinlivan RM, Muller CR, Davis M, Laing NG, Evans GA, Dwyer J, et al. Central core disease: clinical, pathological, and genetic features. Arch Dis Child. 2003;88(12):1051–5.
Lamont PJ, Dubowitz V, Landon DN, Davis M, Morgan-Hughes JA. Fifty year follow-up of a patient with central core disease shows slow but definite progression. Neuromuscul Disord. 1998;8(6):385–91.
Romero NB, Monnier N, Viollet L, Cortey A, Chevallay M, Leroy JP, et al. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain. 2003;126(Pt 11):2341–9.
Dirksen RT, Avila G. Distinct effects on Ca2+ handling caused by malignant hyperthermia and central core disease mutations in RyR1. Biophys J. 2004;87(5):3193–204.
Tong J, McCarthy TV, MacLennan DH. Measurement of resting cytosolic Ca2+ concentrations and Ca2+ store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2+ release channels. J Biol Chem. 1999;274(2):693–702.
Kaindl AM, Rüschendorf F, Krause S, Goebel H-H, Koehler K, Becker C, et al. Missense mutations of ACTA1 cause dominant congenital myopathy with cores. J Med Genet. 2004;41(11):842–8.
Joyce NC, Oskarsson B, Jin LW. Muscle biopsy evaluation in neuromuscular disorders. Phys Med Rehabil Clin N Am. 2012;23(3):609–31.
Kovarsky J, Schochet SS Jr, McCormick WF. The significance of target fibers: a clinicopathologic review of 100 patients with neurogenic atrophy. Am J Clin Pathol. 1973;59(6):790–7.
Scacheri PC, Hoffman EP, Fratkin JD, Semino-Mora C, Senchak A, Davis MR, et al. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology. 2000;55(11):1689–96.
Ferreiro A, Monnier N, Romero NB, Leroy JP, Bonnemann C, Haenggeli CA, et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol. 2002;51(6):750–9.
Scoto M, Cullup T, Cirak S, Yau S, Manzur AY, Feng L, et al. Nebulin (NEB) mutations in a childhood onset distal myopathy with rods and cores uncovered by next generation sequencing. Eur J Hum Genet. 2013;21(11):1249–52.
Gommans IM, Davis M, Saar K, Lammens M, Mastaglia F, Lamont P, et al. A locus on chromosome 15q for a dominantly inherited nemaline myopathy with core-like lesions. Brain. 2003;126(Pt 7):1545–51.
Tosch V, Rohde HM, Tronchere H, Zanoteli E, Monroy N, Kretz C, et al. A novel PtdIns3P and PtdIns(3,5)P2 phosphatase with an inactivating variant in centronuclear myopathy. Hum Mol Genet. 2006;15(21):3098–106.
Kondo E, Nishimura T, Kosho T, Inaba Y, Mitsuhashi S, Ishida T, et al. Recessive RYR1 mutations in a patient with severe congenital nemaline myopathy with ophthalomoplegia identified through massively parallel sequencing. Am J Med Genet A. 2012;158a(4):772–8.
Wilmshurst JM, Lillis S, Zhou H, Pillay K, Henderson H, Kress W, et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol. 2010;68(5):717–26.
Jungbluth H, Muller CR, Halliger-Keller B, Brockington M, Brown SC, Feng L, et al. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology. 2002;59(2):284–7.
Romero NB. Centronuclear myopathies: a widening concept. Neuromuscul Disord. 2010;20(4):223–8.
Bevilacqua JA, Bitoun M, Biancalana V, Oldfors A, Stoltenburg G, Claeys KG, et al. "necklace" fibers, a new histological marker of late-onset MTM1-related centronuclear myopathy. Acta Neuropathol. 2009;117(3):283–91.
Bitoun M, Maugenre S, Jeannet PY, Lacene E, Ferrer X, Laforet P, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 2005;37(11):1207–9.
Clarke NF. Congenital fiber-type disproportion. Semin Pediatr Neurol. 2011;18(4):264–71.
Tubridy N, Fontaine B, Eymard B. Congenital myopathies and congenital muscular dystrophies. Curr Opin Neurol. 2001;14(5):575–82.
Laing NG, Clarke NF, Dye DE, Liyanage K, Walker KR, Kobayashi Y, et al. Actin mutations are one cause of congenital fibre type disproportion. Ann Neurol. 2004;56(5):689–94.
Brandis A, Aronica E, Goebel HH. TPM2 mutation. Neuromusc Disorders. 2008;18(12):1005.
Mathews KD, Moore SA. Multiminicore myopathy, central core disease, malignant hyperthermia susceptibility, and RYR1 mutations: one disease with many faces? Arch Neurol. 2004;61(1):27–9.
Jungbluth H, Zhou H, Hartley L, Halliger-Keller B, Messina S, Longman C, et al. Minicore myopathy with ophthalmoplegia caused by mutations in the ryanodine receptor type 1 gene. Neurology. 2005;65(12):1930–5.
Ferreiro A, Fardeau M. 80th ENMC International Workshop on Multi-Minicore Disease: 1st International MmD Workshop 12–13th May, 2000, Soestduinen, The Netherlands. Neuromuscul Disord. 2002;12(1):60–8.
Cullup T, Lamont PJ, Cirak S, Damian MS, Wallefeld W, Gooding R, et al. Mutations in MYH7 cause multi-minicore disease (MmD) with variable cardiac involvement. Neuromuscul Disord. 2012;22(12):1096–104.
Carmignac V, Salih MA, Quijano-Roy S, Marchand S, Al Rayess MM, Mukhtar MM, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol. 2007;61(4):340–51.
Jungbluth H. Multi-minicore disease. Orphanet J Rare Dis. 2007;2:31.
Ferreiro A, Ceuterick-de Groote C, Marks JJ, Goemans N, Schreiber G, Hanefeld F, et al. Desmin-related myopathy with Mallory body-like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol. 2004;55(5):676–86.
Nance JR, Dowling JJ, Gibbs EM, Bonnemann CG. Congenital myopathies: an update. Curr Neurol Neurosci Rep. 2012;12(2):165–74.
Scoto M, Cirak S, Mein R, Feng L, Manzur AY, Robb S, et al. SEPN1-related myopathies: clinical course in a large cohort of patients. Neurology. 2011;76(24):2073–8.
Mercuri E, Clements E, Offiah A, Pichiecchio A, Vasco G, Bianco F, et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol. 2010;67(2):201–8.
Bertini E, Bosman C, Bevilacqua M, Ricci E, Gagliardi GM, Parisi F, et al. Cardiomyopathy and multicore myopathy with accumulation of intermediate filaments. Eur J Pediatr. 1990;149(12):856–8.
Maidment SL, Ellis JA. Muscular dystrophies, dilated cardiomyopathy, lipodystrophy and neuropathy: the nuclear connection. Expert Rev Mol Med. 2002;4(17):1–21.
Voermans NC, Bonnemann CG, Hamel BCJ, Jungbluth H, van Engelen BG. Joint hypermobility as a distinctive feature in the differential diagnosis of myopathies. J Neurol. 2009;256(1):13–27.
Jungbluth H, Zhou H, Sewry CA, Robb S, Treves S, Bitoun M, et al. Centronuclear myopathy due to a de novo dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromuscul Disord. 2007;17(4):338–45.
Jungbluth H, Sewry CA, Brown SC, Nowak KJ, Laing NG, Wallgren-Pettersson C, et al. Mild phenotype of nemaline myopathy with sleep hypoventilation due to a mutation in the skeletal muscle alpha-actin (ACTA1) gene. Neuromuscul Disord. 2001;11(1):35–40.
Agrawal PB, Greenleaf RS, Tomczak KK, Lehtokari V-L, Wallgren-Pettersson C, Wallefeld W, et al. Nemaline myopathy with minicores caused by mutation of the CFL2 gene encoding the skeletal muscle actin-binding protein, cofilin-2. Am J Hum Genet. 2007;80(1):162–7.
Kinali MBD, Pitt MC, Jungbluth H, Simonds AK, Aloysius A, et al. Congenital myasthenic syndromes in childhood: diagnostic and management challenges. J Neuroimmunol. 2008:6–12.
Pant I, Chaturvedi S, Bala K, Kushwaha S. Muscle histopathology in today's era of molecular genetics: role and limitations. Ann Indian Acad Neurol. 2015;18(4):398–402.
Jungbluth H. Central core disease. Orphanet J Rare Dis. 2007;2:25.
Kossugue PM, Paim JF, Navarro MM, Silva HC, Pavanello RC, Gurgel-Giannetti J, et al. Central core disease due to recessive mutations in RYR1 gene: is it more common than described? Muscle Nerve. 2007;35(5):670–4.
Duarte ST, Oliveira J, Santos R, Pereira P, Barroso C, Conceicao I, et al. Dominant and recessive RYR1 mutations in adults with core lesions and mild muscle symptoms. Muscle Nerve. 2011;44(1):102–8.
Litman RS, Griggs SM, Dowling JJ, Riazi S. Malignant hyperthermia susceptibility and related diseases. Anesthesiology. 2018;128(1):159–67.
Fiszer D, Fisher NA, Carr IM, Shaw M-A, Hopkins PM. Advantage from massive parallel sequencing of RYR1 and CACNA1S in diagnostics of malignant hyperthermia susceptibility. BMC Anesthesiol. 2014;14(1):A13.
Cassandrini D, Trovato R, Rubegni A, Lenzi S, Fiorillo C, Baldacci J, et al. Congenital myopathies: clinical phenotypes and new diagnostic tools. Ital J Pediatr. 2017;43(1):101.
Todd JJ, Sagar V, Lawal TA, Allen C, Razaqyar MS, Shelton MS, et al. Correlation of phenotype with genotype and protein structure in RYR1-related disorders. J Neurol. 2018.
Oliveira J, Goncalves A, Taipa R, Melo-Pires M, Oliveira ME, Costa JL, et al. New massive parallel sequencing approach improves the genetic characterization of congenital myopathies. J Hum Genet. 2016;61(6):497–505.
Arnold WD. Flanigan KM. A practical approach to molecular diagnostic testing in neuromuscular diseases. Phys Med Rehabil Clin N Am. 2012;23(3):589–608.
McDonald CM. Clinical approach to the diagnostic evaluation of hereditary and acquired neuromuscular diseases. Phys Med Rehabil Clin N Am. 2012;23(3):495–563.
Eltit JM, Feng W, Lopez JR, Padilla IT, Pessah IN, Molinski TF, et al. Ablation of skeletal muscle triadin impairs FKBP12/RyR1 channel interactions essential for maintaining resting cytoplasmic Ca2+. J Biol Chem. 2010;285(49):38453–62.
Lee CS, Hanna AD, Wang H, Dagnino-Acosta A, Joshi AD, Knoblauch M, et al. A chemical chaperone improves muscle function in mice with a RyR1 mutation. Nat Commun. 2017;8:14659.
Bonnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol. 2011;7(7):379–90.
Zhou H, Jungbluth H, Sewry CA, Feng L, Bertini E, Bushby K, et al. Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain. 2007;130(Pt 8):2024–36.
Yang T, Ta TA, Pessah IN, Allen PD. Functional defects in six ryanodine receptor isoform-1 (RyR1) mutations associated with malignant hyperthermia and their impact on skeletal excitation-contraction coupling. J Biol Chem. 2003;278(28):25722–30.
Acknowledgements
The authors would like to acknowledge Drs. Joan Austin (Indiana University School of Nursing) and Heinz Jungbluth (Department of Basic and Clinical Neuroscience, King’s College, London) for their constructive feedback on this manuscript. We would also like to recognize the RYR-1 Foundation for their support and advocacy on behalf of individuals affected with RYR1-related disorders, and the European Neuromuscular Centre (ENMC) for facilitating the multiple RYR1-related myopathies workshops where the concept of a historical perspective and a unified nomenclature was borne. This work was supported by the National Institutes of Health, including the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR053349 to SLH and RTD) and the National Institute of Nursing Research Division of Intramural Research.
Funding
National Institute of Nursing Research, Division of Intramural Research, National Institutes of Health, USA and National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, USA (AR053349 to SLH and RTD)
Author information
Authors and Affiliations
Contributions
Tokunbor A. Lawal, Ph.D., NIH, Bethesda, MD, USA, Author, Drafted and revised the manuscript. Joshua J. Todd, Ph.D. , NIH, Bethesda, MD, USA, Author, Reviewed the manuscript and revised for intellectual content. Designed figures and drafted the manuscript for intellectual content. Jessica W. Witherspoon, Ph.D., NIH, Bethesda, MD, USA, Author, Reviewed the manuscript and revised for intellectual content. Carsten G. Bönnemann, M.D., NIH, Bethesda, MD, USA, Author, Revised the manuscript for intellectual content. James J. Dowling MD, Ph.D., University of Toronto, Toronto, Canada, Author, Reviewed the manuscript and revised for intellectual content. Susan L. Hamilton, Ph.D., Baylor College of Medicine, TX, USA, Author, Conceptualized the study and reviewed the manuscript. Katherine G. Meilleur, Ph.D., NIH, Bethesda, MD, USA, Author, Reviewed the manuscript and revised for intellectual content. Robert T. Dirksen, Ph.D., University of Rochester, Rochester, NY, USA, Author, Reviewed the manuscript and revised for intellectual content. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
Dr. Lawal has received support from the RYR-1 Foundation.
Dr. Todd has received support from the RYR-1 Foundation.
Dr. Witherspoon has received support from the RYR-1 Foundation.
Dr. Dirksen is a member of scientific advisory board of the RYR-1 Foundation. Dr. Dirksen has received support from the RYR-1 Foundation, Muscular Dystrophy Association, and the NIH (AR053349).
Dr. Bönnemann is a member of scientific advisory board of the RYR-1 Foundation. Dr. Bönnemann has received funding from Cure CMD.
Dr. Dowling is a member of scientific advisory board of the RYR-1 Foundation and Denature, and a member of the scientific council of the Muscular Dystrophy Association. Dr. Dowling has received support from the RYR-1 Foundation and Muscular Dystrophy Association.
Dr. Hamilton is a member of scientific advisory board of the RYR-1 Foundation. Dr. Hamilton has received the following support from the NIH: R01AR072602, R01AR072475, and R01AR053349. Dr. Hamilton has also received a grant from the Muscular Dystrophy Association and a gift from the McNair Foundation.
Dr. Meilleur has received support from the RYR-1 Foundation.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lawal, T.A., Todd, J.J., Witherspoon, J.W. et al. Ryanodine receptor 1-related disorders: an historical perspective and proposal for a unified nomenclature. Skeletal Muscle 10, 32 (2020). https://doi.org/10.1186/s13395-020-00243-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13395-020-00243-4