
Abstract
Background
Teleost fishes exhibit remarkably diverse and plastic sexual developmental patterns. One of the most astonishing is the rapid socially controlled female-to-male (protogynous) sex change observed in bluehead wrasses (Thalassoma bifasciatum). Such functional sex change is widespread in marine fishes, including species of commercial importance, yet its underlying molecular basis remains poorly explored.
Methods
RNA sequencing was performed to characterize the transcriptomic profiles and identify genes exhibiting sex-biased expression in the brain (forebrain and midbrain) and gonads of bluehead wrasses. Functional annotation and enrichment analysis were carried out for the sex-biased genes in the gonad to detect global differences in gene products and genetic pathways between males and females.
Results
Here we report the first transcriptomic analysis for a protogynous fish. Expression comparison between males and females reveals a large set of genes with sex-biased expression in the gonad, but relatively few such sex-biased genes in the brain. Functional annotation and enrichment analysis suggested that ovaries are mainly enriched for metabolic processes and testes for signal transduction, particularly receptors of neurotransmitters and steroid hormones. When compared to other species, many genes previously implicated in male sex determination and differentiation pathways showed conservation in their gonadal expression patterns in bluehead wrasses. However, some critical female-pathway genes (e.g., rspo1 and wnt4b) exhibited unanticipated expression patterns. In the brain, gene expression patterns suggest that local neurosteroid production and signaling likely contribute to the sex differences observed.
Conclusions
Expression patterns of key sex-related genes suggest that sex-changing fish predominantly use an evolutionarily conserved genetic toolkit, but that subtle variability in the standard sex-determination regulatory network likely contributes to sexual plasticity in these fish. This study not only provides the first molecular data on a system ideally suited to explore the molecular basis of sexual plasticity and tissue re-engineering, but also sheds some light on the evolution of diverse sex determination and differentiation systems.
Similar content being viewed by others


Background
Sexual dimorphism is ubiquitous in nature: males and females differ not only in their gonadal structure and function, but also in many aspects of their morphology, physiology, and behavior [1–3]. While sex-determination mechanisms are relatively conserved in mammals and birds, teleost fishes show remarkably diverse sexual developmental patterns, including both genetic and environmental sex-determination (GSD and ESD) systems [1, 4, 5]. Such diversity probably arises from the extreme sexual plasticity characteristic of teleost fishes. For example, in fishes with GSD systems, sex is determined during early development stages and individuals remain in the same sex for a lifetime (defined as gonochorism) [4]. However, this primary sex differentiation guided by genetic signals can be interrupted or even reversed by temperature or endocrine-disrupting chemicals [6–11]. More extreme cases are found in fishes with ESD systems including sequential hermaphroditism in which some adults in a social group undergo functional sex change in response to environmental stimuli (e.g., temperature or social cues) [12–16]. Revealing the mechanisms underlying such sexual plasticity may help us understand how sex is maintained and gain insights into the origin and evolution of sex-determination systems.
The genetic bases of sexual dimorphism have been intensively studied for decades in mammals and birds, but are less well characterized in teleost fishes [1–4, 17, 18]. So far, genetic studies on sex determination in fishes have examined either sex-specific genetic differences or sex-biased gene expression [19–23]. The search for sex-specific genetic markers has not met with much success because, unlike mammals and birds, fishes have relatively young sex chromosomes that are not usually heteromorphic [24–28] with exceptions including some species of salmonids [29, 30], stickleback fishes [31], glass knife fishes [32], and half-smooth tongue sole, Cynoglossus semilaevis [33]. Even in fishes with heterogenic sex-determination systems, sex differences are usually limited to a few loci or certain linkage groups [34–39]. No conserved sex-specific gene has been found in teleost fishes: six sex-determining genes have been reported to evolve separately in different fish lineages [22, 40]. In contrast, studies examining sex-biased gene expression in fishes have yielded many more genes, including some that play conserved roles in vertebrate sex differentiation. However, for most of these sex-biased genes, their detailed molecular functions in fishes remain to be clarified [23, 41–46]. Studies to date have focused mainly on the expression patterns of a limited number of genes during primary sex differentiation stages in gonochoristic fishes [47–53]. Few studies have yet examined sex-biased gene expression in hermaphroditic fishes, with the exception of genetic studies in protandrous black porgy, Acanthopagrus schlegelii [19, 54], and transcriptomic studies in two other protandrous species: sharpsnout seabream, Diplodus puntazzo, and Asian seabass, Lates calcarifer [20, 21]. However, hermaphroditism is phylogenetically widespread in fishes, with protogyny being the most commonly observed pattern [15, 55, 56], but a large-scale analysis of sex-biased gene expression is currently lacking for protogynous fishes.
The bluehead wrasse, Thalassoma bifasciatum, is a diandric (two male phenotypes) protogynous species belonging to the wrasse family (Labridae) and is abundant on coral reefs throughout the Caribbean [57, 58]. This highly social species exhibits two major color phases: females and smaller sneaker males in initial phase (IP) share the same color pattern, while the large males display a distinct terminal phase (TP) phenotype [57–59]. Natural social groups typically consist of one dominant TP male and numerous females as well as a few IP males. Following the loss of the dominant TP male from a social group, both large females and IP males can transform into TP males through sex or role change, although the latter is rarely reported [55, 59, 60]. In females, functional gonadal sex change takes about a week while behavioral sex change can begin within minutes to hours [60, 61]. Importantly, manipulation of the social environment can induce sex change in females, which makes the bluehead wrasse a useful model for investigating sexual plasticity.
Significant progress has been made in understanding the ecology and the neuroendocrine bases of sex change in this species, but detailed mechanisms still remain elusive, especially at the molecular level [55, 62]. According to the Animal Genome Size Database [63], the haploid DNA contents (C-value) of bluehead wrasse is 0.98 picogram (1 picogram = 978 megabase pair). However, its genome and transcriptome sequences are not available yet. In this study, we took advantage of RNA sequencing technology and captured the transcriptomic profiles in the brain and gonads of TP male, female, and intersex bluehead wrasses. To identify genes exhibiting sex-biased expression in the brain (forebrain and midbrain) and gonads of the bluehead wrasse, we generated a de novo transcriptome assembly for read mapping and compared gene expression patterns at the isoform level between control females and TP males. We also conducted functional annotation and enrichment analysis on the genes showing sex-biased expression in the gonad to detect sex-biased genetic pathways that could contribute to gonadal sex differences in bluehead wrasses.
Methods
Sample collection
Sex change was induced in large females by the removal of dominant TP males from established social groups in the wild [61, 64, 65]. Twenty fishes were captured before or during the daily spawning period around high tide from patch reefs off the coast of Key Largo in late May 2012. All fishes were euthanized with an overdose of MS-222 (Sigma) within 2 min of capture, and the brain and gonads were dissected immediately. These experiments were performed in accordance with guidelines established by the Institutional Animal Care and Use Committee at North Carolina State University (NCSU).
One gonadal lobe and the whole brain were preserved in RNAlater (Life Technologies, Inc.) on ice, followed by storage at −20 °C for less than 1 week and transfer to −80 °C until RNA extraction. The other gonadal lobe was fixed in 4 % paraformaldehyde/1X PBS overnight at 4 °C, followed by storage in 1X PBS before being fixed in paraffin for histological sectioning and HE (hematoxylin and eosin) staining (Histology Laboratory, College of Veterinary Medicine, NCSU) to determine the gonadal status [66]. Before RNA extraction, the hindbrain (corpus cerebelli, pons, and medulla) was removed from each brain. Only the forebrain/midbrain was used for RNA sequencing, because the forebrain and midbrain contain regions belonging to the social behavior network and mesolimbic reward system, two neural circuits that are involved in the regulation of social decision-making [67], and thus may be key integrators and drivers of socially induced sex change.
RNA extraction
The tissues were homogenized using TissueLyser II (QIAGEN®) (Center for Neuroendocrinology, Department of Anatomy, University of Otago). Forebrain/midbrain and gonadal total RNA were extracted with TRI reagent (Invitrogen) using chloroform (forebrain/midbrain) or bromochloropropane (gonads) as the phase separation reagent. Samples were then DNase-treated (TURBO DNA-free Kit, Ambion) and total RNA-cleaned (NucleoSpin RNA XS columns, Macherey-Nagel). RNA integrity was assessed on an Agilent 2100 Bioanalyzer. Sex-changing gonads consistently showed RNA profiles with a strong peak of low molecular weight RNA, which possibly corresponds to massive 5S RNA expression in atretic ovaries and masks the 18S and 28S rRNA peaks used for calculating RNA integrity numbers (RIN). Such patterns were also observed in ovaries and intersex gonads of thicklip gray mullets, Chelon labrosus [68], and ovaries of protandrous sharpsnout seabream, Diplodus puntazzo [20]. Therefore, RIN values could not serve as useful measures of RNA integrity in these sex-changing gonads of bluehead wrasses. For brain RNA, samples with RIN values above 6.0 were used for RNA-seq. Total RNA concentration was measured by Qubit 2.0 Fluorometer (Qubit RNA HS Assay Kit, Life Technologies), and samples were diluted to 10 ng/μL.
RNA sequencing
Total RNA from 12 forebrain/midbrain and 12 gonadal samples (3 control females, 3 TP males, and 6 intersex fish), 500 ng per sample, were sent to the Otago Genomics and Bioinformatics Facility at the University of Otago under contract to New Zealand Genomics Limited for library construction and RNA sequencing. Twenty-four multiplexed libraries were prepared with the Illumina TruSeq Stranded mRNA Sample Prep Kit and 100-bp paired-end reads were generated using 8 flow cell lanes on the HiSeq 2000 platform. The insert size was designed to produce a small overlap between paired reads.
Read pre-processing
Read quality was first assessed with FastQC (v0.10.1) [69]. Quality filtering was performed using Trimmomatic (v0.25) [70]: low quality reads were trimmed if average Phred quality scores were less than 20 within a 3-bp sliding window and discarded if the length was below 40 bp after trimming (Trimmomatic parameters: SLIDINGWINDOW:3:20 MINILEN:40). Read pairs were processed with FLASH (v1.2.4) [71]. Overlapping read pairs were joined and used for assembly along with the non-merged read pairs.
De novo transcriptome assembly
Filtered short reads with high-quality scores were assembled de novo with Trinity [72] (r2014-03-23, default kmer 25, minimum contig length of 200 bp), an assembler developed for efficient and robust de novo reconstruction of transcriptomes from RNA-seq data [20, 73, 74]. Since our libraries were made using the dUTP method [75], we specified the library type by setting the “strand-specific library type (—SS_lib_type)” as “RF.” We also used the “—jaccard_clip” option to reduce chimeric fusion of transcripts [76].
Quality checking
Assembled contigs were first searched against CEGMA (Core Eukaryotic Genes Mapping Approach, v2.5) KOGs (the eukaryotic orthologous groups) [77]. We then ran “TransDecoder” (v1.0) [76] to check the chimeric rate in our assembly (if two large open reading frames were found in one contig, it would be reported as chimeric). Full-length transcript analysis was carried out using the Trinity function “analyze_blastPlus_topHit_coverage.pl” with BLAST+ (BLASTN, E-value cut-off 10−50) against 17 bluehead wrasse expressed sequence tags (ESTs) and 19,712 Nile tilapia protein sequences (Ensembl release 75) [76, 78, 79]. Finally, we manually checked the sequences of all the candidate genes based on read mapping and visualization in Integrative Genomics Viewer (IGV, v2.3.40) [80].
Annotation
The assembly was searched against the UniProt (Swiss-Prot and TrEMBL) protein database [81] with BLAST+ (BLASTX, E-value cut-off 10−10, keeping the top hit) [78] for taxonomic distribution and bacterial contamination detection. Information on taxa was obtained using an in-house Perl script, and the numbers of each taxon were manually checked.
We then conducted BLASTX searches of the assembled contigs against the Ensembl (release 75) Nile tilapia (Oreochromis niloticus), zebrafish (Danio rerio), and medaka (Oryzias latipes) protein databases (E-value cut-off 10−10, keeping the top hit) [78, 79].
Contigs with no hit in the protein databases were searched against the Ensembl (release 77) zebrafish non-coding RNA (ncRNA) database (BLASTN, E-value cut-off 10−5, keeping the top hit) and mapped to the tilapia genome downloaded from Ensembl (release 79, BLASTN, E-value cut-off 10−10) [78, 79]. Finally, putative open reading frames (ORFs) were searched in both annotated and unannotated contigs using OrfPredictor (v2.3) [82].
Read mapping and differential expression analysis
The de novo transcriptome assembly served as a reference for read mapping. Raw reads were aligned to the assembly with Bowtie (v0.12.9) [83] and transcript abundance estimation was calculated with RNA-seq by expectation maximization (RSEM, v1.2.12) [84] using the align_and_estimate_abundance.pl script from the Trinity package [76]. RSEM expected counts for each contig (representing the isoform) were used for downstream differential expression analysis in R (v3.1.0) [85] using the DESeq package (v1.20.0) [86].
Comparisons between TP males and females were conducted separately for the brain and gonadal samples (3 samples for 2 conditions each) using the DESeq function nbinomTest [86]. Principal component analysis (PCA) [87] and the heatmap.2 function in the gplots package [88] were used to visualize global similarities and differences among either the brain or gonadal samples. Contigs with very low expression in either gonad or brain (average expected counts of mapped reads fewer than 1 per sample) were excluded prior to differential expression analysis to improve the statistical power [89]. All samples (including intersex samples) were used for estimating dispersions. p value adjustment was performed using the false discovery rate controlling procedure [90]. Contigs with an adjusted p value less than 0.05 and a fold change larger than 2 were reported as significantly differentially expressed between sexes in the gonad, while contigs with an adjusted p value less than 0.05 were reported as significantly differentially expressed between sexes in the brain.
Gene ontology and pathway analysis
Contigs showing significantly sex-biased expression in the gonad were searched against the Ensembl zebrafish protein database (BLASTX, E-value cut-off 10−10). Matched zebrafish protein IDs were converted to unique Ensembl zebrafish gene IDs via BioMart [91]. These gene IDs were imported into the Database for Annotation, Visualization, and Integrated Discovery (DAVID, v6.7) [92] for functional annotation and enrichment analysis, using the default zebrafish database in DAVID (v6.7) as the background. Gene ontology (GO) [93] and pathway analysis [94] was carried out only for the gonad because there were not enough differentially expressed contigs detected in the brain. GO terms of level one and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with a fold enrichment above 1.2 and p value below 0.05 are shown in Figs. 4 and 5. GO terms and KEGG pathways with p values below 0.05 after adjustment using the Benjamini and Hochberg (BH) procedure are indicated by stars.
Results and discussion
De novo transcriptome assembly
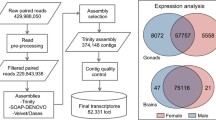
The Illumina HiSeq 2000 sequencing produced more than two billion 100-bp paired-end reads (1,106,170,692 read pairs). The raw sequence data in FASTQ format have been submitted to the National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database and are accessible under accession number SRP06302. After trimming, 1,586,678,582 (71.7 %) high-quality reads were retained for the transcriptome assembly.
The de novo assembled transcriptome using Trinity [72] resulted in 230,626 contigs with a N50 of 1,146 bp and minimum and maximum contig lengths of 201 and 27,427 bp, respectively. There are 77,632 contigs having a length of 500 bp or more. Short contigs (<500 bp) were retained for annotation and mapping because many neuropeptides have a short protein sequence.
Assembly quality was assessed by three means: the representation of core eukaryotic genes, predicted chimeric rate, and full-length recovery of the bluehead wrasse expressed sequence tags (ESTs) and Nile tilapia protein sequences (Ensembl release 75) [79]. All CEGMA KOGs (the eukaryotic orthologous groups) [77] were present in this assembly (98 % complete, 100 % partial). The predicted chimeric rate [76] was 3.2 %. All of the bluehead ESTs were recovered (BLASTN, E-value ≤10−50): 14 ESTs with >90 % recovery and 3 ESTs with 70–80 % recovery. Fifty-eight percent of Nile tilapia protein sequences had a match in the bluehead wrasse transcriptome assembly with alignment coverage above 90 % (BLASTX, E-value ≤10−10).
Transcriptome annotation
The bluehead wrasse transcriptome assembly was searched against the UniProt (Swiss-Prot and TrEMBL) protein database [81] (BLASTX, E-value ≤10−10, keeping the top hit). In total, 41,799 contigs (18 %) had significant hits to 34,275 unique protein sequences. Of these sequences, 94 % came from ray-finned bony fishes while only 63 contigs matched to bacterial sequences (Fig. 1). This indicates negligible contamination of bacteria, which is consistent with expectations that our assembly comprises mainly brain and gonadal coding RNAs.

Taxonomic distribution of top BLASTX hits in UniProt protein database. Numbers of unique hits of protein sequences in each group are shown in the parentheses
Searching against the Ensembl protein databases [79] (BLASTX, E-value ≤10−10, keeping the top hit), we found 16–17 % of the assembled contigs had a significant BLASTX match (E-value ≤10−10, keeping the top hit). Of 26,763 input Nile tilapia protein sequences, 20,182 sequences were found in our assembly (Table 1). Similar results have been reported in two other non-model fish transcriptomes [20, 95].
Surprisingly, a large portion (82 %) of the contigs had no significant BLASTX match to any known protein sequence. These sequences were searched against the Ensembl zebrafish ncRNA database [79] (BLASTN, E-value ≤10−5, 8819 input ncRNA sequences), but only 93 contigs had a match, including processed or antisense transcripts with no protein product, miRNA, miscRNA, snoRNA etc. Putative ORFs were searched in both annotated and unannotated contigs using OrfPredictor [82]. The length distribution of the longest ORFs is shown in Fig. 2. Briefly, over 98 % of the contigs contained ORFs, but most (69 %) were smaller than 300 bp. Almost all of the unannotated contigs (>99 %) had an ORF smaller than 600 bp. These contigs may represent novel protein-coding transcripts, fragmented UTRs, pre-mature mRNA sequences with retained introns, or polyadenylated non-coding RNAs of potential biological importance. At present, however, it is still challenging to provide complete annotations for a de novo assembled transcriptome, especially for a non-model teleost fish for which few genomic resources are available. Multiple BLAST searches provide a powerful means for automated annotations but are limited by available sequences, sequence similarity among homologues, and alignment sensitivity. Future genome sequencing and more information on alternatively spliced isoforms and non-coding RNAs will improve our current annotation. It will be useful to revisit these data as more gene sequences become available.

Length distribution of the longest ORFs in assembled contigs (blue: unannotated contigs, pink: annotated contigs)
Differential gene expression between female and TP male
All of the contigs were kept for read mapping, and the RSEM [84] expected value table of contigs (representing isoforms) was used for expression analysis. This allows detection of isoform-specific expression patterns and clustering of the contigs based on both annotations and expression patterns.
In total, 889,652,430 raw read pairs (from control females and TP males) were mapped back to the reference transcriptome assembly [83]. In this paper, we focused on the sex-biased gene expression; thus, we only report the comparison between females and TP males.
Global gene expression patterns in the brain and gonad
Comparisons between TP males and females were conducted separately for the brain and gonadal samples (3 samples for 2 conditions each) using the DESeq function nbinomTest [86]. p value adjustment was performed using Benjamini and Hochberg (BH) procedure [90]. Principal component analysis (PCA) plots [87] and heatmaps [88] both showed that sex differences in gene expression were much more pronounced between the ovary and testis than those between male and female brains (Fig. 3b, c). Consistently, a large number of contigs showed sex-biased expression in the gonad (fold change ≥2 and BH adjusted p value ≤0.05), while only eight contigs showed sex-biased expression in the brain (BH adjusted p value ≤0.05). Similar global expression patterns were also reported in zebrafish [44, 96] and sharpsnout seabream [20], which may reflect the functional and regulatory differences between the brain and gonads.

Gene expression patterns in the brain (right) and gonads (left). a Numbers of differentially expressed contigs between TP males (M) and females (F). b PCA plots of brain and gonadal samples (green: female, blue: TP male). c Heatmaps showing the expression of top 100 contigs in brain and gonads (ordered by average normalized read counts across the row; red: lower expression, green: higher expression; M: TP male, F: female)
A large set of genes showed significant sex-biased expression in the gonad
Expression analysis revealed a large set of transcripts differentially expressed between ovary and testis of the bluehead wrasse (fold change ≥2 and BH adjusted p value ≤0.05). Contigs showing male-biased expression were twice as abundant as those showing female-biased expression, although contigs with the highest expression in the gonad were female-biased (Fig. 3a, c). Of the 13,768 male-biased contigs, 6769 (49 %) contigs had a significant BLASTX match in the UniProt protein databases (E-value ≤10−10), including 6279 hits in the Ensembl zebrafish protein database. Of the 6415 female-biased contigs, 4246 (66 %) had a significant BLASTX match in the UniProt protein databases (E-value ≤10−10), including 4074 hits in the Ensembl zebrafish protein database.
Enriched gene ontology terms and pathways in testis and ovary
Contigs showing sex-biased expression in the gonad were mapped to the Ensembl zebrafish protein database and further converted to their equivalent Ensembl zebrafish gene IDs (4824 male-biased, 3373 female-biased) via BioMart [91]. These gene IDs (Additional file 1) were searched against the DAVID (v6.7) [92] zebrafish database to detect which GO terms [93] and KEGG pathways [94] were enriched in the testis and ovary of bluehead wrasses, respectively. As a result, 4080 (male-biased) and 2989 (female-biased) DAVID IDs were reported, of which 30–50 % were assigned with GO terms and about 20 % were mapped to KEGG pathways.
Significantly enriched GO terms (level 1) in the ovary and testis are shown in Fig. 4. In general, the ovary was enriched for metabolic process, while the testis was enriched for signal transduction and receptor activity. Similarly, the pathway enrichment analysis also found that ovaries are enriched for RNA and protein metabolism, while testes are enriched for signal transduction (Fig. 5).

Top GO terms enriched in the ovary (pink) and testis (blue). Enriched GO terms with modified Fisher exact test p value below 0.05 and fold enrichment above 1.2 are shown here. Stars indicate GO terms with BH adjusted p value below 0.05

KEGG pathways enriched in the ovary (pink) and testis (blue). Enriched pathways with modified Fisher exact test p value below 0.05 and fold enrichment above 1.2 are shown here. Stars indicate pathways with BH adjusted p value below 0.05
Interestingly, the top pathway enriched in the testis was “neuroactive ligand-receptor interaction” (Fig. 4), which includes receptors for many neuropeptides (Fig. 6a, b). Within this pathway, receptors of norepinephrine, epinephrine, melatonin, oxytocin/isotocin (IT), and vasopression/vasotocin (AVT) were significantly over-expressed in the testis (Fig. 6a) while receptors of dopamine, serotonin (5-HT), and neuropeptide FF were significantly over-expressed in the ovary (Fig. 6b). Some of these neuropeptides have been suggested to play important roles at the onset of protogynous sex change [55, 56, 62]. Briefly, norepinephrine and vasotocin have a promoting effect on gonadal or behavioral sex change, while dopamine and serotonin have an inhibitory effect [97–100]. The function of isotocin and melatonin in sex-changing fishes are largely unknown due to limited information. Nevertheless, the interesting point here is that these neuropeptides were thought to act on the brain and regulate gonadal sex change indirectly through the hypothalamic-pituitary-gonadal (HPG) axis [55, 101, 102]. However, their receptors are widely expressed in the gonad. Some recent studies suggest these neuropeptides can act directly on the gonad, in addition to their classical actions through the HPG axis. For example, vasotocin and melatonin are reported to regulate oocyte maturation in catfish [103] and carp [104], whereas vasotocin and catecholamines (dopamine, norepinephrine, and epinephrine) are shown to modulate ovarian steroidogenesis in catfish in a biphasic manner [103, 105]. Further studies on expression and function of these neuropeptides in both the brain and gonads of sex-changing fishes are warranted.

The neuroactive ligand-receptor interaction pathway was significantly enriched in the testis (a) but not in the ovary (b). Red stars indicate the DE genes significantly up-regulated (fold change above 2 and BH adjusted p value below 0.05) in the testis (a) or the ovary (b)
Another pathway enriched in the testis is related to steroid hormone biosynthesis (Fig. 4). Steroid hormones are known to play a critical role in sex differentiation across vertebrates [4]. Within this pathway, only three genes (cyp19a1a, hsd11b3, hsd17b1) showed significantly female-biased expression in the gonad while 11 genes showed significantly male-biased expression, including cyp11c1, hsd11b2, hsd17b3, cyp11a1, and cyp17a1 (Figs. 7 and 8a). These results are generally consistent with our current knowledge of sexually dimorphic levels of steroid hormones in fish.


Expression patterns of candidate genes in the gonad (a) and brain (b). a Expression patterns of 56 sex-related genes in the gonad of bluehead wrasses. Genes with BH adjusted p value below 0.05 are shown in solid circles (blue: male-biased, red: female-biased). b Expression patterns of 16 genes of interest in TP male and female forebrain/midbrain of bluehead wrasses. Genes (hsd17b3, ugt8, slc6a20) with BH adjusted p value below 0.05 are shown in solid circles. Genes (it, cyp19a1b, esr1, esr2b, and hsd11b2) with pre-adjusted p values below 0.05 prior to BH correction are shown in open circles with a cross. Genes (avt, hsd11b3, cyp11c1, gnrh1, gnrh2, gnrh3, kiss1, and kiss2) with p values above 0.05 before and after BH correction are shown in open circles. Genes showing male- or female-biased expression are colored in blue or red, respectively
In teleost fishes, 17β-estradiol (E2) and 11-ketotestosterone (11-KT) function as the major estrogen and androgen, respectively [4]. Testosterone (T) serum levels can also be high in males and females, but T can be converted into either E2 by aromatase (cyp19a1a in the gonad) or 11-KT by 11β-hydroxylase (cyp11b or cyp11c1 in zebrafish) and 11β-hydroxysteroid dehydrogenase 2 (hsd11b2) [4, 106, 107]. In protogynous species, steroid hormones play a central role in controlling sex change: high plasma E2 levels prevent females from changing into males, whereas blocking E2 production or injecting 11-KT in females can induce sex change [108]. As expected in our study, cyp19a1a expression was only detected in the ovary, whereas cyp11c1 and hsd11b2 expression were significantly higher in the testis (Fig. 8a, Table 2, and Additional file 2). The brain aromatase gene cyp19a1b was also detected in the gonad of rainbow trout [109], but our data showed almost no expression of cyp19a1b in bluehead wrasse gonads. In addition, the genes that encode androgen receptors (ar1 and ar2) showed no sex-biased expression, while the estrogen receptor genes (esr1, esr2a, and esr2b) had higher expression in the testis, although the male-biased expression of esr2b was not statistically significant (Fig. 8a, Table 2, and Additional file 2). Such male-biased expression of estrogen receptors has been shown during late sexual differentiation for Nile tilapia (70dah) [49] and rainbow trout (60-110dpf) [110]. A recent study on rainbow trout also showed significantly elevated testicular expression of esr1a and esr2a during the final stage of spermiation, while esr1b and esr2b were expressed at early stages of testicular development [111]. In the same study, androgen implants up-regulated testicular esr1a, esr2a, and esr2b expression but down-regulated cyp19a1a expression, whereas estrogens reduced testicular cyp19a1a expression but increased the expression of cyp19a1b and esr1b. These findings all suggest a potential role for estrogens and their receptors in teleost testicular development.
Interestingly, cyp11c1 and hsd11b2 are also involved in cortisol (or glucocorticoid, GC) production: Cyp11c1 converts 11-deoxycortisol to cortisol while Hsd11b2 converts cortisol to its inactive form cortisone, and Hsd11b3 (or Hsd11b1-like) could convert cortisone to cortisol [107, 112]. Cortisol treatment has been reported to cause masculinization of genetic females of medaka [113, 114], Japanese flounder [2, 115], southern flounder [116], and pejerrey [117]. In Japanese flounder, cortisol was suggested to cause female-to-male sex reversal by suppressing cyp19a1a expression [20, 115]. In zebrafish [118] and pejerrey [112], cortisol treatment of larvae elevated hsd11b2 expression, while cortisol also enhanced in vitro 11-KT synthesis in pejerrey testes. Sexually dimorphic expression of cyp11c1, hsd11b2, hsd11b3, and nr3c1 (nuclear receptor subfamily 3, group C, member 1 or glucocorticoid receptor) found in the gonad of bluehead wrasse (Fig. 8a, Table 2, and Additional file 2) suggests that local cortisol production could be important for gonadal sex differences. Moreover, cortisol treatment can induce protogynous sex change in three-spot wrasse [119], but a peak in serum cortisol levels appears to be a key event during gonadal sex change in both protandrous and protogynous species [120, 121]. The specific role of cortisol in regulating gonadal sex change remains to be clarified.
Expression patterns of genes involved in sex determination/differentiation
The processes of sex determination and differentiation can be viewed as a battle for primacy between a male regulatory gene network (e.g., dmrt1, sf-1, amh, sox9) and female genetic pathways involving foxl2 and Rspo1/Wnt/β-catenin signaling [122, 123]. Despite the diverse regulatory mechanisms, expression patterns of these genes are generally consistent across taxa [1, 40, 122, 124]. In our study, male-pathway genes all showed significantly higher expression in the testis (e.g., dmrt1, sf-1, amh, amhr2, sox9a/b, sox8, and gsdf). In contrast, a few genes involved in the female-pathway (e.g., rspo1, wnt4b) showed unexpected expression patterns (Fig. 8a, Table 2, and Additional file 2).
First, two paralogues of forkhead box L2 genes (referred to as foxl2 and foxl3) were detected in the gonad of the bluehead wrasse. Foxl2 and foxl3 probably originated from an ancient genome duplication event; they are present ubiquitously in fish lineages but foxl3 was repeatedly lost in the tetrapods [125]. Foxl2 is critical for maintenance of female differentiation [126, 127] and can up-regulate cyp19a1a expression together with Sf-1 [128]. Foxl3 was proposed to play a role in testicular development, but its exact function remains elusive. In our study, foxl2 was expressed at higher levels in the ovary while foxl3 expression was higher in the testis of bluehead wrasses (Fig. 8a, Table 2, and Additional file 2). Such sexually dimorphic expression was also found in European sea bass, and the gonadal expression of foxl2 and foxl3 varied significantly during the reproductive cycles [125].
In recent years, Dmrt1 (doublesex and mab-3 related transcription factor 1) has received much attention due to its conserved role in vertebrate testicular differentiation and maintenance [127, 129–132]. Dmrt1 expression was significantly higher in the testis than in the ovary of the bluehead wrasse (Fig. 8a, Table 2, and Additional file 2). Dmrt1 and Foxl2 have been proposed to have antagonistic effects on cyp19a1a expression to control gonadal sex fate [124, 127]. This hypothesis has been supported by studies in tilapia where knockout of cyp19a1a or foxl2 expression caused gonadal sex reversal in females while dmrt1 and cyp11b2 (11β-hydroxylase) were co-expressed in follicular cells surrounding the degenerating oocytes [127]. Moreover, foxl2 expression decreased while dmrt1 expression increased during female-to-male sex change in honeycomb grouper [133]. However, such shifts in foxl2 and dmrt1 expression did not occur until the late transitioning stage, which was downstream of declining E2 levels [134]. Foxl2 showed no strong sexually dimorphic expression in the gonad of protogynous three-spotted wrasses, and its expression even increased during aromatase-inhibitor-induced sex change [135]. Thus, the roles of foxl2 and dmrt1 may be species-specific in sex-changing fishes. Further manipulative studies will be especially useful for elucidating the precise functions of these key genes in sex-changing fishes.
Most genes involved in the ovary-specific Rspo1/Wnt/β-catenin signaling pathway showed sexually dimorphic expression in the gonad of bluehead wrasses (Fig. 8a, Table 2, and Additional file 2). However, some of these genes displayed an expression pattern that was opposite to our expectations based on studies from mammalian models [17, 136]. For example, ctnnb1 (β-catenin) was highly expressed in both the ovary and testis of bluehead wrasse, but its expression was significantly female-biased. In contrast, rspo1 (R-spondin-1) and wnt4b (wingless-type MMTV integration site family, member 4b) were expressed at much lower levels in the bluehead wrasse gonads, but they both showed significantly male-biased expression. Similar sexually dimorphic expression patterns of ctnnb1, rspo1, and wnt4b were also reported in east cichlid fishes [137]. Fst (follistatin) is downstream to Wnt4 signaling [17, 136, 138]. We detected a few fst-like genes in the bluehead wrasse gonad: fstl3 and fstl5 both showed male-biased expression, while two long isoforms of fstl4 showed either female- or male-biased expression. It has been well-established in mammals that Rspo1, β-catenin, Wnt4, and Fst are key players in early ovarian differentiation [17, 136, 139], but information is limited regarding their roles in teleost fishes. Research to date, including our current study, reveals complicated expression patterns of these genes in fishes [18, 20, 53, 140–143]. Collectively, these data suggest ctnnb1 likely plays a conserved role in both establishing and maintaining female sex differentiation across vertebrate taxa, while other genes involved in Rspo1/Wnt/β-catenin signaling pathway may participate in both ovarian and testicular development in fishes. More manipulative studies are needed to better characterize the roles of these genes in teleost fishes and to test whether the male-biased expression of rspo1 and wnt4b is involved in protogynous sex change.
The RA (retinoid acid) signaling pathway is important in ovarian differentiation because RA controls the sex-specific timing of meiosis initiation [144–146]. RA level is regulated by Aldh1a (retinal dehydrogenase) and Cyp26 enzymes: Aldh1a2 increases RA level and initiates meiosis, while Cyp26a1 and Cyp26b1 decrease RA level and prevent germ cells from entering into meiosis [145]. Our study revealed higher expression of aldh1a2 and cyp26b1 but lower expression of cyp26a1 and genes encoding RA receptors (raraa, rarab, and rarb) in the testis of bluehead wrasses (Fig. 8a, Table 2, and Additional file 2). These patterns are consistent with findings in Nile tilapia [145] and mice [147]. In addition, Cyp26b1 prevents stra8 (stimulated by retinoic acid gene 8) expression in mouse testes [147, 148]. Stra8 is lost in teleost fishes [149], but we found stra6, the receptor for retinol-binding protein 4 [150], in bluehead wrasse gonads. Its expression is much lower in the testis than in the ovary, which is consistent with high expression of cyp26b1 in the testis (Fig. 8a, Table 2, and Additional file 2). Interestingly, studies in mice suggest that dmrt1 expression is essential to maintain male-sex fate because it can protect the testis from transdifferentiation into ovary by RA signaling [131, 132]. Another study in mice also supports the hypothesis that Sox9 and Sf-1 up-regulate cyp26b1 to maintain the male fate of germ cells in testes, while Foxl2 acts to antagonize cyp26b1 expression in ovaries [151]. Taken together, the RA signaling pathway may play a key role in regulating gonadal sex change in hermaphroditic fishes and warrants further investigation.
Lastly, accumulating evidence suggests that epigenetic modifications also participate in the regulation of sex differentiation and sex change [152–157]. Transcripts of mRNA encoding DNA methyltransferases (Dnmt) and histone deacetylases (Hdac) or acetyltransferases (Hat) were detected in the bluehead wrasse, and most showed sex-biased expression in the gonad (Table 2, and Additional file 2). However, because the epigenetic mechanisms underlying sex differentiation are still poorly understood, we cannot infer any detailed functions of these genes from their expression patterns. Future studies are needed to reveal their molecular functions in sex differentiation and sex change.
Few sex-biased genes detected in the forebrain/midbrain
The brain represents a key site where environment stimuli and internal signals are integrated to regulate vertebrate physiology and behavior. Sex differences in the brain have been a major and growing focus in neuroscience [2, 3]. In mammals, sex differences in the brain are likely established by both organizational effects of sex steroid hormones and cellular autonomous regulation based on sex chromosomes [2]. Teleost brains, however, appear to show less sex bias in brain structure or gene expression [20, 45, 96]. Thus, the sex differences observed in teleost brains may be due primarily to the activational influences of steroid hormones [158], which may also explain the brain sexual lability of teleost fishes.
In our study, expression analysis using the DESeq package revealed seven up-regulated contigs and one down-regulated contig in the TP male bluehead wrasse forebrain/midbrain (Additional file 3). Only four of these contigs had a significant BLASTX match in the UniProt protein databases (E-value ≤10−10): 17β-hydroxysteriod dehydrogenase (hsd17b3), UDP glycosyltransferase 8 (ugt8), solute carrier family 6 (proline IMINO transporter) member 20 (slc6a20; also BLASTs to zebrafish slc6a19b), and a novel gene with unknown function. Larger numbers of sex-biased genes have been reported in the brains of other fishes (e.g., zebrafish [96], seabream [20], and black-faced blenny [159]). We conducted differential expression analysis at the isoform (represented by contigs) level with the most conservative software (DESeq) and stringent cut-offs (BH adjusted p value below 0.05) in order to reduce false positives [160]. We also included six intersex samples from the same experimental group in dispersion estimation and read count normalization (see “Methods” section). Such stringent analyses are likely to detect fewer but more reliable sex-biased contigs.
17β-hydroxysteroid dehydrogenase (Hsd17b3), the enzyme converting androstenedione to testosterone, was significantly up-regulated at the transcriptional level in the forebrain/midbrain of TP males (Fig. 8b and Additional file 3). Analyses in zebrafish have also shown male-biased expression of hsd17b3 at the whole-brain level [96], suggesting conserved sex differences in local testosterone production in the brain. In teleosts, testosterone in the brain can be converted to E2 by the brain isoform of aromatase (cyp19a1b) or to 11-KT by Cyp11b and Hsd11b2 [161]. Although not significantly different after BH correction, cyp19a1b showed a 1.9-fold up-regulation in female brains, while hsd11b2 was 1.7-fold higher in TP male brains (Fig. 8b and Additional file 3), suggesting a potentially higher E2 synthesis in female brains and 11-KT synthesis in TP male brains. Also, not significant after BH correction but likely biologically relevant, estrogen receptor 1 (esr1) and esr2b were up-regulated in TP male brains compared to female brains (Fig. 8b and Additional file 3). These patterns suggest that local neurosteroid production and signaling likely contribute to sex differences in the brain [161, 162] and are consistent with the previously documented influences of estrogen on behavioral sex change in bluehead wrasses [163].
The significance of the sexually dimorphic patterns of expression for other genes uncovered in the brain of the bluehead wrasse is unclear. Ugt8 was significantly up-regulated in the forebrain/midbrain of TP males, while slc6a20 showed an opposite pattern (Fig. 8b and Additional file 3). Wong et al. [96] also found ugt8 to be up-regulated at the whole-brain level in male zebrafish. In mammals, UGT8 synthesizes galactocerebrosides, a major component of the myelin sheath surrounding nerves [164, 165]. Knocking out ugt8 in mice reduces myelin thickness and nerve conduction, resulting in tremor and motor weakness [166, 167]. SLC6A20 transports proline and other imino acids and N-methylated amino acids across cell membranes [168]. Proline has been implicated in neuromodulation [169] and has been shown to modulate glutaminergic neurotransmission in mammals [170, 171]. There is currently no information on distribution or function of UGT8 and SLC6A20 in teleost brains. Sex differences in ugt8 and slc6a20 expression within the forebrain/midbrain of bluehead wrasses may translate into differences in neurotransmission and behavior, but these possibilities require more research to address.
We did not find significant differences in the expression of a number of key neuropeptide genes (Fig. 8b and Additional file 3), including arginine vasotocin (avt), isotocin (it), gonadotropin-releasing hormone (gnrh), and kisspeptin (kiss), that are known to be involved in socio-sexual behavior and/or reproduction in teleost fishes [172–175] and implicated in the regulation of socially induced sex change (reviewed in [55, 62]). Avt and it mRNAs are highly expressed in both TP male and female brains, but only it showed male-biased expression in our dataset, although this sex difference in it expression was not statistically significant after BH correction (Fig. 8b and Additional file 3). Avt mRNA expression was shown to be male-biased in the magnocellular preoptic area of bluehead wrasses [176] and to increase with behavioral sex change [64], but such differences may be masked due to the lower neuroanatomical resolution of whole-forebrain/midbrain sampling. The role of isotocin in sex change is less studied. However, it was shown that the number of isotocin-immunoreactive neurons in the preoptic area of bluebanded gobies (Lythrypnus dalli), a bi-directional sex-changing species, was higher in females than in males [177]. This sex-biased pattern appears to be opposite to our data for bluehead wrasses, although future studies are needed to determine if TP males have significantly higher expression than females and if isotocin plays a major role in regulating sex change.
Conclusions
The genetic basis of sexual dimorphism in teleost fishes and the molecular mechanisms underlying the protogynous and protandrous sex change common to teleosts remain to be fully elucidated. In this study, we took advantage of high-throughput sequencing technology to generate the first high-quality transcriptome for a protogynous fish, the bluehead wrasse. This resource will make future comparative and experimental analyses of protogynous sex change possible. We also identified a large number of genes that exhibit sexually dimorphic expression in the gonad and several sex-biased genes in the forebrain/midbrain of bluehead wrasses. These genes include most known vertebrate sex-related genes as well as numerous novel genes that currently lack annotation but may well have important biological roles in sex differentiation and/or sex change. In addition, we find that most candidate genes implicated or known to be involved in sex determination and differentiation in other vertebrate systems showed conserved expression patterns in the bluehead wrasse with a few exceptions. This suggests that some subtle variability in the standard sex-determination regulatory network, although having evolved from a conserved toolkit, could be responsible for the sexual plasticity in these fishes. Overall, this study provides not only key data on the molecular basis of sexual dimorphism in the brain and gonad of bluehead wrasse, but also valuable resources for investigating the molecular pathways that underpin this extraordinary example of sexual plasticity in response to environmental influences. Further examination of the gene expression dynamics across the process of protogynous sex change will uncover the genetic cascade that progressively re-engineers a female into a male.
Availability of supporting data
All sequencing data have been uploaded to NCBI Sequence Read Archive under accession number SRP06302.
References
Gamble T, Zarkower D. Sex determination. Curr Biol. 2012;22:R257–62.
Jazin E, Cahill L. Sex differences in molecular neuroscience: from fruit flies to humans. Nat Rev Neurosci. 2010;11:9–17.
McCarthy MM, Arnold AP, Ball GF, Blaustein JD, De Vries GJ. Sex differences in the brain: the not so inconvenient truth. J Neurosci. 2012;32:2241–7.
Devlin RH, Nagahama Y. Sex determination and sex differentiation in fish: an overview of genetic, physiological, and en vironmental influences. Aquaculture. 2002;208:191–364.
Barske LA, Capel B. Blurring the edges in vertebrate sex determination. Curr Opin Genet Dev. 2008;18:499–505.
Paul-Prasanth B, Bhandari RK, Kobayashi T, Horiguchi R, Kobayashi Y, Nakamoto M, et al. Estrogen oversees the maintenance of the female genetic program in terminally differentiated gonochorists. Sci Rep. 2013;3:2862.
Kobayashi H, Iwamatsu T. Sex reversal in the medaka Oryzias latipes by brief exposure of early embryos to estradiol-17beta. Zool Sci. 2005;22:1163–7.
Sato T, Endo T, Yamahira K, Hamaguchi S, Sakaizumi M. Induction of female-to-male sex reversal by high temperature treatment in Medaka, Oryzias latipes. Zool Sci. 2005;22:985–8.
Kobayashi T, Kajiura-Kobayashi H, Nagahama Y. Induction of XY sex reversal by estrogen involves altered gene expression in a teleost, tilapia. Cytogenet Genome Res. 2003;101:289–94.
Kwon JY, McAndrew BJ, Penman DJ. Treatment with an aromatase inhibitor suppresses high-temperature feminization of genetic male (YY) Nile tilapia. J of Fish Bio. 2002;60:625–36.
Bhandari RK, Nakamura M, Kobayashi T, Nagahama Y. Suppression of steroidogenic enzyme expression during androgen-induced sex reversal in Nile tilapia (Oreochromis niloticus). Gen Comp Endocrinol. 2006;145:20–4.
Nakamura M, Kobayashi Y, Miura S, Alam MA, Bhandari RK. Sex change in coral reef fish. Fish Physiol Biochem. 2005;31:117–22.
Godwin J. Socially controlled sex change in fishes. In Encyclopedia of Fish Physiology: From Genome to Environment. Edited by Farrell A. Elsevier Inc; 2011. p. 662–9.
Munday PL, Buston PM, Warner RR. Diversity and flexibility of sex-change strategies in animals. Trends Ecol Evol. 2006;21:89–95.
Avise JC, Mank JE. Evolutionary perspectives on hermaphroditism in fishes. Sex Dev. 2009;3:152–63.
Godwin J. Social determination of sex in reef fishes. Semin Cell Dev Biol. 2009;20:264–70.
Ungewitter EK, Yao HHC. How to make a gonad: cellular mechanisms governing formation of the testes and ovaries. Sex Dev. 2013;7:7–20.
Herpin A, Adolfi MC, Nicol B, Hinzmann M, Schmidt C, Klughammer J, et al. Divergent expression regulation of gonad development genes in medaka shows incomplete conservation of the downstream regulatory network of vertebrate sex determination. Mol Biol Evol. 2013;30:2328–46.
Wu G-C, Tomy S, Lee M-F, Lee Y-H, Yueh W-S, Lin C-J, et al. Sex differentiation and sex change in the protandrous black porgy, Acanthopagrus schlegeli. Gen Comp Endocrinol. 2010;167:417–21.
Manousaki T, Tsakogiannis A, Lagnel J, Sarropoulou E, Xiang JZ, Papandroulakis N, et al. The sex-specific transcriptome of the hermaphrodite sparid sharpsnout seabream (Diplodus puntazzo). BMC Genomics. 2014;15:655.
Ravi P, Jiang J, Liew WC, Orban L. Small-scale transcriptomics reveals differences among gonadal stages in Asian seabass (Lates calcarifer). Reprod Biol Endocrinol. 2014;12:5.
Kikuchi K, Hamaguchi S. Novel sex-determining genes in fish and sex chromosome evolution. Dev Dyn. 2013;242:339–53.
Sun F, Liu S, Gao X, Jiang Y, Perera D, Wang X, et al. Male-biased genes in catfish as revealed by RNA-seq analysis of the testis transcriptome. PLoS ONE. 2013;8:e68452.
Parise-Maltempi PP, da Silva EL, Rens W, Dearden F, O’Brien PCM, Trifonov V, et al. Comparative analysis of sex chromosomes in Leporinus species (Teleostei, Characiformes) using chromosome painting. BMC Genet. 2013;14:60.
Takehana Y et al. Chapter 15—frequent turnover of sex chromosomes in the medaka fishes. In: Naruse K, editor. Medaka : a model for organogenesis, human disease, and evolution. Berlin: Springer; 2011. p. 229–40.
Charlesworth D, Charlesworth B, Marais G. Steps in the evolution of heteromorphic sex chromosomes. Heredity. 2005;95:118–28.
Tanaka K, Takehana Y, Naruse K, Hamaguchi S, Sakaizumi M. Evidence for different origins of sex chromosomes in closely related Oryzias fishes: substitution of the master sex-determining gene. Genetics. 2007;177:2075–81.
Arkhipchuk VV. Role of chromosomal and genome mutations in the evolution of bony fishes. Hydrobiol J. 1995;31:55–65.
Iturra P, Lam N, La Fuente De M, Vergara N, Medrano JF. Characterization of sex chromosomes in rainbow trout and coho salmon using fluorescence in situ hybridization (FISH). Genetica. 2001;111:125–31.
Phillips RB, Park LK, Naish KA. Assignment of Chinook salmon (Oncorhynchus tshawytscha) linkage groups to specific chromosomes reveals a karyotype with multiple rearrangements of the chromosome arms of rainbow trout (Oncorhynchus mykiss). G3 (Bethesda). 2013;3:2289–95.
Ross JA, Urton JR, Boland J, Shapiro MD, Peichel CL. Turnover of sex chromosomes in the stickleback fishes (Gasterosteidae). PLoS Genet. 2009;5:e1000391.
Henning F, Moysés CB, Calcagnotto D, Meyer A, de Almeida-Toledo LF. Independent fusions and recent origins of sex chromosomes in the evolution and diversification of glass knife fishes (Eigenmannia). Heredity. 2011;106:391–400.
Chen S, Zhang G, Shao C, Huang Q, Liu G, Zhang P, et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat Genet. 2014;46:253–60.
Kamiya T, Kai W, Tasumi S, Oka A, Matsunaga T, Mizuno N, et al. A trans-species missense SNP in Amhr2 is associated with sex determination in the tiger pufferfish, Takifugu rubripes (fugu). PLoS Genet. 2012;8:e1002798.
Yano A, Guyomard R, Nicol B, Jouanno E, Quillet E, Klopp C, et al. An immune-related gene evolved into the master sex-determining gene in rainbow trout, Oncorhynchus mykiss. Curr Biol. 2012;22:1423–8.
Liu F, Sun F, Li J, Xia JH, Lin G, Tu RJ, et al. A microsatellite-based linkage map of salt tolerant tilapia (Oreochromis mossambicus x Oreochromis spp.) and mapping of sex-determining loci. BMC Genomics. 2013;14:58.
Bradley KM, Breyer JP, Melville DB, Broman KW, Knapik EW, Smith JR. An SNP-based linkage map for zebrafish reveals sex determination loci. G3 (Bethesda). 2011;1:3–9.
Shirak A, Seroussi E, Cnaani A, Howe AE, Domokhovsky R, Zilberman N, et al. Amh and Dmrta2 genes map to tilapia (Oreochromis spp.) linkage group 23 within quantitative trait locus regions for sex determination. Genetics. 2006;174:1573–81.
Hattori RS, Murai Y, Oura M, Masuda S, Majhi SK, Sakamoto T, et al. A Y-linked anti-Mullerian hormone duplication takes over a critical role in sex determination. Proc Natl Acad Sci U S A. 2012;109:2955–9.
Mei J, Gui J-F. Genetic basis and biotechnological manipulation of sexual dimorphism and sex determination in fish. Sci China Life Sci. 2015;58:124–36.
Penman DJ, Piferrer F. Fish gonadogenesis. Part I: genetic and environmental mechanisms of sex determination. Rew Fish Sci. 2008;16:16–34.
Piferrer F, Guiguen Y. Fish gonadogenesis. Part II: molecular biology and genomics of sex differentiation. Rew Fish Sci. 2008;16:35–55.
Siegfried KR. In search of determinants: gene expression during gonadal sex differentiation. J Fish Biol. 2010;76:1879–902.
Small CM, Carney GE, Mo Q, Vannucci M, Jones AG. A microarray analysis of sex- and gonad-biased gene expression in the zebrafish: Evidence for masculinization of the transcriptome. BMC Genomics. 2009;10:579.
Sreenivasan R, Cai M, Bartfai R, Wang X, Christoffels A, Orban L. Transcriptomic analyses reveal novel genes with sexually dimorphic expression in the zebrafish gonad and brain. PLoS ONE. 2008;3:e1791.
Nakamoto M, Matsuda M, Wang DS, Nagahama Y, Shibata N. Molecular cloning and analysis of gonadal expression of Foxl2 in the medaka, Oryzias latipes. Biochem Biophys Res Commun. 2006;344:353–61.
Tao W, Yuan J, Zhou L, Sun L, Sun Y, Yang S, et al. Characterization of gonadal transcriptomes from Nile tilapia (Oreochromis niloticus) reveals differentially expressed genes. PLoS ONE. 2013;8:e63604.
Shibata Y, Paul-Prasanth B, Suzuki A, Usami T, Nakamoto M, Matsuda M, et al. Expression of gonadal soma derived factor (GSDF) is spatially and temporally correlated with early testicular differentiation in medaka. Gene Expr Patterns. 2010;10:283–9.
Ijiri S, Kaneko H, Kobayashi T, Wang D-S, Sakai F, Paul-Prasanth B, et al. Sexual dimorphic expression of genes in gonads during early differentiation of a teleost fish, the Nile tilapia Oreochromis niloticus. Biol Reprod. 2008;78:333–41.
Kobayashi T, Kajiura-Kobayashi H, Guan GJ, Nagahama Y. Sexual dimorphic expression of DMRT1 and Sox9a during gonadal differentiation and hormone-induced sex reversal in the teleost fish Nile tilapia (Oreochromis niloticus). Dev Dyn. 2008;237:297–306.
Vizziano D, Randuineau G, Baron D, Cauty C, Guiguen Y. Characterization of early molecular sex differentiation in rainbow trout, Oncorhynchus mykiss. Dev Dyn. 2007;236:2198–206.
Baron D, Guiguen Y. Gene expression during gonadal sex differentiation in rainbow trout (Oncorhynchus mykiss): from candidate genes studies to high throughout genomic approach. Fish Physiol Biochem. 2003;28:119–23.
Nicol B, Guiguen Y. Expression profiling of Wnt signaling genes during gonadal differentiation and gametogenesis in rainbow trout. Sex Dev. 2011;5:318–29.
Wu G-C, Chang C-F. The switch of secondary sex determination in protandrous black porgy, Acanthopagrus schlegeli. Fish Physiol Biochem. 2013;39:33–8.
Godwin J. Neuroendocrinology of sexual plasticity in teleost fishes. Front Neuroendocrinol. 2010;31:203–16.
Larson ET. Neuroendocrine regulation in sex-changing fishes. In: Norris DO, editor. Hormones and Reproduction of Vertebrates. Waltham: Academic; 2010. p. 149–68.
Feddern HA. The spawning, growth, and general behavior of the bluehead wrasse, Thalassoma bifasciatum (Pisces: Labridae). Bull Mar Sci. 1965;896–941.
Warner RR, Robertson DR. Sexual patterns in the labroid fishes of the western Caribbean I: the wrasses. Smithson Contrib Zool. 1978;254:1–27.
Warner RR. Mating behavior and hermaphroditism in coral reef fishes. Am Sci. 1984;72:128–36.
Warner RR, Swearer SE. Social control of sex change in the bluehead wrasse, Thalassoma bifasciatum (Pisces: Labridae). Biol Bull. 1991;181:199–204.
Godwin J, Crews D, Warner RR. Behavioural sex change in the absence of gonads in a coral reef fish. Proc Biol Sci. 1996;263:1683–8.
Lamm MS, Liu H, Gemmell NJ, Godwin JR. The need for speed: neuroendocrine regulation of socially-controlled sex change. Integr Comp Biol. 2015;55:307–22.
Gregory TR. Animal Genome Size Database. 2015. Available online at: http://www.genomesize.com. Accessed 06 Nov 2015.
Semsar K, Godwin J. Social influences on the arginine vasotocin system are independent of gonads in a sex-changing fish. J Neurosci. 2003;23:4386–93.
Semsar K, Godwin J. Multiple mechanisms of phenotype development in the bluehead wrasse. Horm Behav. 2004;45:345–53.
Nakamura M, Hourigan TF, Yamauchi K, Nagahama Y, Grau EG. Histological and ultrastructural evidence for the role of gonadal-steroid hormones in sex change in the protogynous wrasse Thalassoma-Duperrey. Environ Biol Fish. 1989;24:117–36.
O’Connell LA, Hofmann HA. Genes, hormones, and circuits: an integrative approach to study the evolution of social behavior. Front Neuroendocrinol. 2011;32:320–35.
Diaz de Cerio O, Rojo-Bartolomé I, Bizarro C, Ortiz-Zarragoitia M, Cancio I. 5S rRNA and accompanying proteins in gonads: powerful markers to identify sex and reproductive endocrine disruption in fish. Environ Sci Technol. 2012;46:7763–71.
Andrews S. FastQC: A quality control tool for high throughput sequence data. 2010. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 2011;29:644.
Singhal S. De novo transcriptomic analyses for non-model organisms: an evaluation of methods across a multi-species data set. Mol Ecol Resour. 2013;13:403–16.
Li B, Fillmore N, Bai Y, Collins M, Thomson JA, Stewart R, et al. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 2013;15:553.
Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods. 2010;7:709–15.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512.
Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–7.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC bioinformatics. 2009;10:421.
Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–55.
Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–12.
Min XJ, Butler G, Storms R, Tsang A. OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 2005;33:W677–80.
Langmead B: Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics 2010, Chapter 11:Unit 11.7.doi: 10.1002/0471250953.bi1107s32
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria; 2014.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106.
Jolliffe IT. Principal component analysis. 2nd ed. Berlin: Springer; 2002.
Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, et al. gplots: Various R programming tools for plotting data. R package version. 2009;2:4.
Richard Bourgon RGWH. Independent filtering increases detection power for high-throughput experiments. Proc Natl Acad Sci U S A. 2010;107:9546–51.
Benjamini Y, Hochberg Y. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300.
Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, et al. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015;43:W589–98.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Liu S, Zhang Y, Zhou Z, Waldbieser G, Sun F, Lu J, et al. Efficient assembly and annotation of the transcriptome of catfish by RNA-Seq analysis of a doubled haploid homozygote. BMC Genomics. 2012;13:595.
Wong RY, McLeod MM, Godwin J. Limited sex-biased neural gene expression patterns across strains in Zebrafish (Danio rerio). BMC Genomics. 2014;15:905.
Larson ET, Norris DO, Grau EG, Summers CH. Monoamines stimulate sex reversal in the saddleback wrasse. Gen Comp Endocrinol. 2003;130:289–98.
Semsar K, Kandel F, Godwin J. Manipulations of the AVT system shift social status and related courtship and aggressive behavior in the bluehead wrasse. Horm Behav. 2001;40:21–31.
Semsar K, Perreault HAN, Godwin J. Fluoxetine-treated male wrasses exhibit low AVT expression. Brain Res. 2004;1029:141–7.
Lema SC, Sanders KE, Walti KA. Arginine vasotocin, isotocin and nonapeptide receptor gene expression link to social status and aggression in sex-dependent patterns. J Neuroendocrinol. 2015;27:142–57.
Zohar Y, Muñoz-Cueto JA, Elizur A, Kah O. Neuroendocrinology of reproduction in teleost fish. Gen Comp Endocrinol. 2010;165:438–55.
Maruska KP, Fernald RD. Social regulation of gene expression in the hypothalamic-pituitary-gonadal axis. Physiology (Bethesda). 2011;26:412–23.
Joy KP, Chaube R. Vasotocin—a new player in the control of oocyte maturation and ovulation in fish. Gen Comp Endocrinol. 2015.
Maitra SK, Chattoraj A, Mukherjee S, Moniruzzaman M. Melatonin: a potent candidate in the regulation of fish oocyte growth and maturation. Gen Comp Endocrinol. 2013;181:215–22.
Joy KP, Singh V, Chaube R. An in vitro study on catecholamine modulation of ovarian steroidogenic activity in the catfish Heteropneustes fossilis. Gen Comp Endocrinol. 2014;196:91–9.
Guiguen Y, Fostier A, Piferrer F, Chang C-F. Ovarian aromatase and estrogens: a pivotal role for gonadal sex differentiation and sex change in fish. Gen Comp Endocrinol. 2010;165:352–66.
Tokarz J, Möller G, de Angelis MH, Adamski J. Zebrafish and steroids: what do we know and what do we need to know? J Steroid Biochem Mol Biol. 2013;137:165–73.
Higa M, Ogasawara K, Sakaguchi A, Nagahama Y, Nakamura M. Role of steriod hormones in sex change of protogynous wrasse. Fish Physiol Biochem. 2003;28:149–50.
von Schalburg KR, Yasuike M, Davidson WS, Koop BF. Regulation, expression and characterization of aromatase (cyp19b1) transcripts in ovary and testis of rainbow trout (Oncorhynchus mykiss). Comp Biochem Physiol B Biochem Mol Biol. 2010;155:118–25.
Baron D, Houlgatte R, Fostier A, Guiguen Y. Expression profiling of candidate genes during ovary-to-testis trans-differentiation in rainbow trout masculinized by androgens. Gen Comp Endocrinol. 2008;156:369–78.
Delalande C, Goupil A-S, Lareyre J-J, Le Gac F. Differential expression patterns of three aromatase genes and of four estrogen receptors genes in the testes of trout (Oncorhynchus mykiss). Mol Reprod Dev. 2015;82:694–708.
Fernandino JI, Hattori RS, Kishii A, Strüssmann CA, Somoza GM. The cortisol and androgen pathways cross talk in high temperature-induced masculinization: the 11β-hydroxysteroid dehydrogenase as a key enzyme. Endocrinol. 2012;153:6003–11.
Hayashi Y, Kobira H, Yamaguchi T, Shiraishi E, Yazawa T, Hirai T, et al. High temperature causes masculinization of genetically female medaka by elevation of cortisol. Mol Reprod Dev. 2010;77:679–86.
Kitano T, Hayashi Y, Shiraishi E, Kamei Y. Estrogen rescues masculinization of genetically female medaka by exposure to cortisol or high temperature. Mol Reprod Dev. 2012;79:719–26.
Yamaguchi T, Yoshinaga N, Yazawa T, Gen K, Kitano T. Cortisol is involved in temperature-dependent sex determination in the Japanese flounder. Endocrinol. 2010;151:3900–8.
Mankiewicz JL, Godwin J, Holler BL, Turner PM, Murashige R, Shamey R, et al. Masculinizing effect of background color and cortisol in a flatfish with environmental sex-determination. Integr Comp Biol. 2013;53:755–65.
Yamamoto Y, Hattori RS, Kitahara A, Kimura H, Yamashita M, Strussmann CA. Thermal and endocrine regulation of gonadal apoptosis during sex differentiation in pejerrey Odontesthes bonariensis. Sex Dev. 2013;7:316–24.
Tokarz J, Norton W, Möller G, de Angelis MH, Adamski J. Zebrafish 20β-hydroxysteroid dehydrogenase type 2 is important for glucocorticoid catabolism in stress response. PLoS ONE. 2013;8:e54851.
Nozu R, Nakamura M. Cortisol administration induces sex change from ovary to testis in the protogynous wrasse, Halichoeres trimaculatus. Sex Dev. 2015;9:118–24.
Solomon-Lane TK, Crespi EJ, Grober MS. Stress and serial adult metamorphosis: multiple roles for the stress axis in socially regulated sex change. Front Neurosci. 2013;7:210.
Godwin JR, Thomas P. Sex change and steroid profiles in the protandrous anemonefish Amphiprion melanopus (Pomacentridae, Teleostei). Gen Comp Endocrinol. 1993;91:144–57.
Herpin A, Schartl M. Sex determination: switch and suppress. Curr Biol. 2011;21:R656–9.
Wilhelm D, Palmer S, Koopman P. Sex determination and gonadal development in mammals. Physiol Rev. 2007;87:1–28.
Kobayashi Y, Nagahama Y, Nakamura M. Diversity and plasticity of sex determination and differentiation in fishes. Sex Dev. 2013;7:115–25.
Crespo B, Lan-Chow-Wing O, Rocha A, Zanuy S, Gómez A. foxl2 and foxl3 are two ancient paralogs that remain fully functional in teleosts. Gen Comp Endocrinol. 2013;194:81–93.
Uhlenhaut NH, Jakob S, Anlag K, Eisenberger T, Sekido R, Kress J, et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell. 2009;139:1130–42.
Li M-H, Yang H-H, Li M-R, Sun Y-L, Jiang X-L, Xie Q-P, et al. Antagonistic roles of Dmrt1 and Foxl2 in sex differentiation via estrogen production in tilapia as demonstrated by TALENs. Endocrinol. 2013;154:4814–25.
Wang DS, Kobayashi T, Zhou LY, Paul-Prasanth B, Ijiri S, Sakai F, et al. Foxl2 up-regulates aromatase gene transcription in a female-specific manner by binding to the promoter as well as interacting with Ad4 binding protein/steroidogenic factor 1. Mol Endocrinol. 2006;21:712–25.
Herpin A, Schartl M. Dmrt1 genes at the crossroads: a widespread and central class of sexual development factors in fish. FEBS J. 2011;278:1010–9.
Matson CK, Zarkower D. Sex and the singular DM domain: insights into sexual regulation, evolution and plasticity. Nat Rev Genet. 2012;13:163–74.
Matson CK, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature. 2011;476:101–4.
Minkina A, Matson CK, Lindeman RE, Ghyselinck NB, Bardwell VJ, Zarkower D. DMRT1 protects male gonadal cells from retinoid-dependent sexual transdifferentiation. Dev Cell. 2014;29:511–20.
Alam MA, Kobayashi Y, Horiguchi R, Hirai T, Nakamura M. Molecular cloning and quantitative expression of sexually dimorphic markers Dmrt1 and Foxl2 during female-to-male sex change in Epinephelus merra. Gen Comp Endocrinol. 2008;157:75–85.
Bhandari RK, Komuro H, Nakamura S, Higa M, Nakamura M. Gonadal restructuring and correlative steroid hormone profiles during natural sex change in protogynous honeycomb grouper (Epinephelus merra). Zool Sci. 2003;20:1399–404.
Kobayashi Y, Horiguchi R, Nozu R, Nakamura M. Expression and localization of forkhead transcriptional factor 2 (Foxl2) in the gonads of protogynous wrasse, Halichoeres trimaculatus. Biol Sex Differ. 2010;1:3.
Tevosian SG. Genetic control of ovarian development. Sex Dev. 2013;7:33–45.
Böhne A, Heule C, Boileau N, Salzburger W. Expression and sequence evolution of aromatase cyp19a1 and other sexual development genes in East African cichlid fishes. Mol Biol Evol. 2013;30:2268–85.
Yao HHC, Matzuk MM, Jorgez CJ, Menke DB, Page DC, Swain A, et al. Follistatin operates downstream of Wnt4 in mammalian ovary organogenesis. Dev Dyn. 2004;230:210–5.
Chassot A-A, Gregoire EP, Lavery R, Taketo MM, de Rooij DG, Adams IR, et al. RSPO1/β-catenin signaling pathway regulates oogonia differentiation and entry into meiosis in the mouse fetal ovary. PLoS ONE. 2011;6:e25641.
Wu GC, Chang CF. wnt4 is associated with the development of ovarian tissue in the protandrous black porgy, Acanthopagrus schlegeli. Biol Reprod. 2009;81:1073–82.
Nicol B, Guerin A, Fostier A, Guiguen Y. Ovary-predominant wnt4 expression during gonadal differentiation is not conserved in the rainbow trout (Oncorhynchus mykiss). Mol Reprod Dev. 2011;79:51–63.
Zhou L, Charkraborty T, Yu X, Wu L, Liu G, Mohapatra S, et al. R-spondins are involved in the ovarian differentiation in a teleost, medaka (Oryzias latipes). BMC Dev Biol. 2011;12:36.
Vizziano-Cantonnet D, Baron D, Mahè S, Cauty C, Fostier A, Guiguen Y. Estrogen treatment up-regulates female genes but does not suppress all early testicular markers during rainbow trout male-to-female gonadal transdifferentiation. J Mol Endocrinol. 2008;41:277–88.
Lau EL, Lee MF, Chang CF. Conserved sex-specific timing of meiotic initiation during sex differentiation in the protandrous black porgy Acanthopagrus schlegelii. Biol Reprod. 2013;88:150–0.
Feng R, Fang L, Cheng Y, He X, Jiang W, Dong R, et al. Retinoic acid homeostasis through aldh1a2 and cyp26a1 mediates meiotic entry in Nile tilapia (Oreochromis niloticus). Sci Rep. 2015;5:10131.
Bowles J, Knight D, Smith C, Wilhelm D, Richman J, Mamiya S, et al. Retinoid signaling determines germ cell fate in mice. Science. 2006;312:596–600.
MacLean G, Li H, Metzger D, Chambon P, Petkovich M. Apoptotic extinction of germ cells in testes of Cyp26b1 knockout mice. Endocrinol. 2007;148:4560–7.
Li H, MacLean G, Cameron D, Clagett-Dame M, Petkovich M. Cyp26b1 expression in murine Sertoli cells is required to maintain male germ cells in an undifferentiated state during embryogenesis. PLoS ONE. 2008;4, e7501.
Venkatesh B, Kirkness EF, Loh Y-H, Halpern AL, Lee AP, Johnson J, et al. Survey sequencing and comparative analysis of the elephant shark (Callorhinchus milii) genome. PLoS Biol. 2007;5:e101.
Muenzner M, Tuvia N, Deutschmann C, Witte N, Tolkachov A, Valai A, et al. Retinol-binding protein 4 and its membrane receptor STRA6 control adipogenesis by regulating cellular retinoid homeostasis and retinoic acid receptor α activity. Mol Cell Biol. 2013;33:4068–82.
Kashimada K, Svingen T, Feng C-W, Pelosi E, Bagheri-Fam S, Harley VR, et al. Antagonistic regulation of Cyp26b1 by transcription factors SOX9/SF1 and FOXL2 during gonadal development in mice. FASEB J. 2011;25:3561–9.
Zhang Y, Zhang S, Liu Z, Zhang L, Zhang W. Epigenetic modifications during sex change repress gonadotropin stimulation of Cyp19a1a in a teleost ricefield eel (Monopterus albus). Endocrinol. 2013;154:2881–90.
Navarro-Martín L, Viñas J, Ribas L, Díaz N, Gutiérrez A, Di Croce L, et al. DNA methylation of the gonadal aromatase (cyp19a) promoter is involved in temperature-dependent sex ratio shifts in the European sea bass. PLoS Genet. 2011;7:e1002447.
Stromqvist M, Tooke N, Brunstrom B. DNA methylation levels in the 5’ flanking region of the vitellogenin I gene in liver and brain of adult zebrafish (Danio rerio)—sex and tissue differences and effects of 17 alpha-ethinylestradiol exposure. Aquat Toxicol. 2010;98:275–81.
Zhong H, Xiao J, Chen W, Zhou Y, Tang Z, Guo Z, et al. DNA methylation of pituitary growth hormone is involved in male growth superiority of Nile tilapia (Oreochromis niloticus). Comp Biochem Physiol B Biochem Mol Biol. 2014;171:42–8.
Piferrer F. Epigenetics of sex determination and gonadogenesis. Dev Dyn. 2013;242:360–70.
Wen AY, You F, Sun P, Li J, Xu DD, Wu ZH, et al. CpG methylation of dmrt1and cyp19apromoters in relation to their sexual dimorphic expression in the Japanese flounder Paralichthys olivaceus. J Fish Biol. 2013;84:193–205.
Hiraki T, Takeuchi A, Tsumaki T, Zempo B, Kanda S, Oka Y, et al. Female-specific target sites for both oestrogen and androgen in the teleost brain. Proc R Soc Lond B Biol Sci. 2012;279:5014–23.
Schunter C, Vollmer SV, Macpherson E, Pascual M. Transcriptome analyses and differential gene expression in a non-model fish species with alternative mating tactics. BMC Genomics. 2013;15:167.
Soneson C, Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC bioinformatics. 2013;14:91.
Diotel N, Do Rego J-L, Anglade I, Vaillant C, Pellegrini E, Vaudry H, et al. The brain of teleost fish, a source, and a target of sexual steroids. Front Neurosci. 2011;5:137.
Le Page Y, Diotel N, Vaillant C, Pellegrini E, Anglade I, Mérot Y, et al. Aromatase, brain sexualization and plasticity: the fish paradigm. Eur J Neurosci. 2010;32:2105–15.
Marsh-Hunkin KE, Heinz HM, Hawkins MB, Godwin J. Estrogenic control of behavioral sex change in the bluehead wrasse, Thalassoma bifasciatum. Integr Comp Biol. 2013;53:951–9.
Schulte S, Stoffel W. Ceramide UDPgalactosyltransferase from myelinating rat brain: purification, cloning, and expression. Proc Natl Acad Sci U S A. 1993;90:10265–9.
Bosio A, Binczek E, Le Beau MM, Fernald AA, Stoffel W. The human gene CGT encoding the UDP-galactose ceramide galactosyl transferase (cerebroside synthase): cloning, characterization, and assignment to human chromosome 4, band q26. Genomics. 1996;34:69–75.
Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, et al. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–19.
Zöller I, Büssow H, Gieselmann V, Eckhardt M. Oligodendrocyte-specific ceramide galactosyltransferase (CGT) expression phenotypically rescues CGT-deficient mice and demonstrates that CGT activity does not limit brain galactosylceramide level. Glia. 2005;52:190–8.
Takanaga H, Mackenzie B, Suzuki Y, Hediger MA. Identification of mammalian proline transporter SIT1 (SLC6A20) with characteristics of classical system imino. J Biol Chem. 2005;280:8974–84.
Kowalczuk S, Bröer A, Munzinger M, Tietze N, Klingel K, Bröer S. Molecular cloning of the mouse IMINO system: an Na+− and Cl—dependent proline transporter. Biochem J. 2005;386:417–22.
Bröer S. The SLC6 orphans are forming a family of amino acid transporters. Neurochem Int. 2006;48:559–67.
Wyse ATS, Netto CA. Behavioral and neurochemical effects of proline. Metab Brain Dis. 2011;26:159–72.
Goodson JL, Bass AH. Social behavior functions and related anatomical characteristics of vasotocin/vasopressin systems in vertebrates. Brain Res Rev. 2001;35:246–65.
Oka Y. Three types of gonadotrophin-releasing hormone neurones and steroid-sensitive sexually dimorphic kisspeptin neurones in teleosts. J Neuroendocrinol. 2009;21:334–8.
Godwin J, Thompson R. Nonapeptides and social behavior in fishes. Horm Behav. 2012;61:230–8.
Mechaly AS, Viñas J, Piferrer F. The kisspeptin system genes in teleost fish, their structure and regulation, with particular attention to the situation in Pleuronectiformes. Gen Comp Endocrinol. 2013;188:258–68.
Godwin J, Sawby R, Warner RR, Crews D, Grober MS. Hypothalamic arginine vasotocin mRNA abundance variation across sexes and with sex change in a coral reef fish. Brain Behav Evol. 2000;55:77–84.
Black MP, Reavis RH, Grober MS. Socially induced sex change regulates forebrain isotocin in Lythrypnus dalli. Neuroreport. 2004;15:185–9.
Albalat R, Brunet F, Laudet V, Schubert M. Evolution of retinoid and steroid signaling: vertebrate diversification from an amphioxus perspective. Genome Biol Evol. 2011;3:985–1005.
Nagahama Y. 17α,20β-Dihydroxy-4-pregnen-3-one, a maturation-inducing hormone in fish oocytes: mechanisms of synthesis and action. Steroids. 1997;62:190–6.
Huang W, Zhou L, Li Z, Gui J-F. Expression pattern, cellular localization and promoter activity analysis of ovarian aromatase (Cyp19a1a) in protogynous hermaphrodite red-spotted grouper. Mol Cell Endocrinol. 2009;307:224–36.
Zhang Y, Zhang W, Zhang L, Zhu T, Tian J, Li X, et al. Two distinct cytochrome P450 aromatases in the orange-spotted grouper (Epinephelus coioides): cDNA cloning and differential mRNA expression. J Steroid Biochem Mol Biol. 2004;92:39–50.
Baron D, Houlgatte R, Fostier A, Guiguen Y. Large-scale temporal gene expression profiling during gonadal differentiation and early gametogenesis in rainbow trout. Biol Reprod. 2005;73:959–66.
Alam MA, Kobayashi Y, Hirai T, Nakamura M. Isolation, characterization and expression analyses of FSH receptor in protogynous grouper. Comp Biochem Physiol A Physiol. 2010;156:364–71.
Miyake Y, Sakai Y, Kuniyoshi H. Molecular cloning and expression profile of sex-specific genes, Figla and Dmrt1, in the protogynous hermaphroditic fish, Halichoeres poecilopterus. Zool Sci. 2012;29:690–710.
Shen X, Cui J, Yang G, Gong Q, Gu Q. Expression detection of DMRTs and two sox9 genes in Takifugu rubripes (Tetraodontidae, Vertebrata). J Ocean Univ China. 2007;6:182–6.
Shin HS, An KW, Park MS, Jeong MH, Choi CY. Quantitative mRNA expression of sox3 and DMRT1 during sex reversal, and expression profiles after GnRHa administration in black porgy, Acanthopagrus schlegeli. Comp Biochem Physiol B Biochem Mol Biol. 2009;154:150–6.
Xia W, Zhou L, Yao B, Li C-J, Gui J-F. Differential and spermatogenic cell-specific expression of DMRT1 during sex reversal in protogynous hermaphroditic groupers. Mol Cell Endocrinol. 2007;263:156–72.
Wang DS, Zhou LY, Kobayashi T, Matsuda M, Shibata Y, Sakai F, et al. Doublesex- and Mab-3-related transcription factor-1 repression of aromatase transcription, a possible mechanism favoring the male pathway in tilapia. Endocrinol. 2010;151:1331–40.
Klüver N, Pfennig F, Pala I, Storch K, Schlieder M, Froschauer A, et al. Differential expression of anti-Müllerian hormone (amh) and anti-Müllerian hormone receptor type II (amhrII) in the teleost medaka. Dev Dyn. 2007;236:271–81.
Horiguchi R, Nozu R, Hirai T, Kobayashi Y, Nagahama Y, Nakamura M. Characterization of gonadal soma-derived factor expression during sex change in the protogynous wrasse, Halichoeres trimaculatus. Dev Dyn. 2013;242:388–99.
Degani G. Expression of SOX3 and SOX9 genes in gonads of blue gourami. Adv Biol Chem. 2014;4:322–30.
Takehana Y, Matsuda M, Myosho T, Suster ML, Kawakami K, Shin TI, et al. Co-option of Sox3 as the male-determining factor on the Y chromosome in the fish Oryzias dancena. Nat Commun. 2014;5:4157.
Liu Q, Lu H, Zhang L, Xie J, Shen W, Zhang W. Homologues of sox8 and sox10 in the orange-spotted grouper Epinephelus coioides: sequences, expression patterns, and their effects on cyp19a1a promoter activities in vitro. Comp Biochem Physiol B Biochem Mol Biol. 2012;163:86–95.
Chiang EF, Pai CI, Wyatt M, Yan YL, Postlethwait J, Chung B. Two sox9 genes on duplicated zebrafish chromosomes: expression of similar transcription activators in distinct sites. Dev Biol. 2001;231:149–63.
Yokoi H, Kobayashi T, Tanaka M, Nagahama Y, Wakamatsu Y, Takeda H, et al. Sox9 in a teleost fish, medaka (Oryzias latipes): evidence for diversified function of Sox9 in gonad differentiation. Mol Reprod Dev. 2002;63:5–16.
Du Q-Y, Wang F-Y, Hua H-Y, Chang Z-J. Cloning and study of adult-tissue-specific expression of Sox9 in Cyprinus carpio. J Genet. 2007;86:85–91.
Nakamoto M, Wang D-S, Suzuki A, Matsuda M, Nagahama Y, Shibata N. Dax1 suppresses P450arom expression in medaka ovarian follicles. Mol Reprod Dev. 2007;74:1239–46.
Acknowledgements
We would like to thank Brian Haas for his help with the de novo transcriptome assembly and Alice Dannis, Thomas Buckley, Margaret Ryan, and Lei Ma for their helpful input on the RNA-seq data processing and analysis. We are grateful to Erica Todd for her valuable comments on drafts of the manuscript.
Financial support for this study has been provided by the Marsden Fund [UOO1308], a University of Otago Research Grant, and funds from the Department of Anatomy at the University of Otago, W.M. Keck Center for Behavioral Biology at North Carolina State University, and Department of Biological Sciences at North Carolina State University. HL is supported by the University of Otago PhD scholarship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NG and JG conceived and designed the experiments. ML and JG collected the samples. ML and HL conducted the laboratory work. HL and KR conducted the in silico work with the help of MB. HL analysed the results with the help of all the other authors. HL drafted the manuscript with the help of ML. NG, JG, MB and KR edited the manuscript. All authors have read and approved the final manuscript.
Additional files
Additional file 1: Table S1.
Ensembl zebrafish gene IDs of sex-biased contigs in the gonad for GO and pathway enrichment analysis in DAVID.
Additional file 2: Table S2.
Results of differential expression analysis for the gonad (blue: male-biased contigs, red: female-biased contigs).
Additional file 3: Table S3.
Results of differential expression analysis for the brain (blue: male-biased contigs, red: female-biased contigs).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, H., Lamm, M.S., Rutherford, K. et al. Large-scale transcriptome sequencing reveals novel expression patterns for key sex-related genes in a sex-changing fish. Biol Sex Differ 6, 26 (2015). https://doi.org/10.1186/s13293-015-0044-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13293-015-0044-8