
Abstract
Background
Teleosts are characterized by a remarkable breadth of sexual mechanisms including various forms of hermaphroditism. Sparidae is a fish family exhibiting gonochorism or hermaphroditism even in closely related species. The sparid Diplodus puntazzo (sharpsnout seabream), exhibits rudimentary hermaphroditism characterized by intersexual immature gonads but single-sex mature ones. Apart from the intriguing reproductive biology, it is economically important with a continuously growing aquaculture in the Mediterranean Sea, but limited available genetic resources. Our aim was to characterize the expressed transcriptome of gonads and brains through RNA-Sequencing and explore the properties of genes that exhibit sex-biased expression profiles.
Results
Through RNA-Sequencing we obtained an assembled transcriptome of 82,331 loci. The expression analysis uncovered remarkable differences between male and female gonads, while male and female brains were almost identical. Focused search for known targets of sex determination and differentiation in vertebrates built the sex-specific expression profile of sharpsnout seabream. Finally, a thorough genetic marker discovery pipeline led to the retrieval of 85,189 SNPs and 29,076 microsatellites enriching the available genetic markers for this species.
Conclusions
We obtained a nearly complete source of transcriptomic sequence as well as marker information for sharpsnout seabream, laying the ground for understanding the complex process of sex differentiation of this economically valuable species. The genes involved include known candidates from other vertebrate species, suggesting a conservation of the toolkit between gonochorists and hermaphrodites.
Similar content being viewed by others


Background
Teleosts exhibit remarkably diverse patterns of sex modes. The way males and females develop, and the molecular mechanisms underlying those differences, vary dramatically among taxa. In teleosts, the decision on an individual’s sex (‘sex determination’) may be due to genetic and/or environmental factors [1–3] with an evident epigenetic component [4]. Apart from the genes that determine sex, downstream genes and pathways drive the development and maintenance of sex-specific phenotypes. Those processes define sex differentiation. The genes involved in sex determination and differentiation form the necessary toolkit leading to the sex-specific phenotypes. However, the picture becomes more complicated in cases of hermaphroditism, a rather common sexual system among teleosts.
The molecular processes underlying sex have been deeply studied in model vertebrates like human, mouse, chicken and African clawed frog [5–8]. Several studies on fish have unveiled the genes responsible for sex determination in gonochoristic species, such as Dmy in medaka [9, 10], Amhr2 in Tiger pufferfish [11], Sdy in Rainbow trout [12] and Amhy in Patagonian pejerrey [13]. Other studies have revealed loci linked to sex in various species (e.g. [14] for stickleback; [15] for tilapia; [16] for Atlantic halibut; [17] for turbot; [18] for zebrafish), even in the hermaphrodite Gilthead seabream [19, 20]. All these studies illustrate the large diversity in sex determination among teleosts. However, the genes involved in sex differentiation are considered conserved across vertebrates [21, 22], even though alternative scenarios have also been suggested [23]. To get an overview of the genetic toolkit deployed for the development and maintenance of the differences between sexes, whole-transcriptome approaches are required [24]. Several transcriptomic analyses have characterized the expression differences that underlie the two sex phenotypes in fish (e.g. [25–30]) and revealed that the main pathways are present in most species, although the role of each gene might change.
Most studies on understanding sex differentiation have been conducted on gonochoristic taxa. Hermaphroditism, however, is common among teleosts and has evolved repeatedly in different lineages [31]. The two sexual systems are sometimes observed even among phylogenetically-close species, suggesting that the molecular pathways involved might not differ dramatically. Sparidae is a teleost family with a wide variety of sex mechanisms [32–34]. Sparids exhibit either gonochorism or various forms of hermaphroditism, such as simultaneous, sequential or rudimentary hermaphroditism (simultaneous: presence of both male and female gonads; sequential: an individual develops first as a functional male and then changes to female or vice versa; rudimentary: immature individuals carry both male and female immature gonad types and during maturation one of the two types develops fully, determining the sex). A recent study on the ancestral reconstruction of sexual patterns in sparids revealed both gonochorism and hermaphroditism in almost every group of the family [31]. Therefore, there is a great potential for understanding the molecular mechanisms underlying gonochorism and hermaphroditism within Sparidae [31, 35]. To date, considerable effort to unravel the genes involved in sex determination, sex differentiation and sex change have been conducted mainly on the protandrous black porgy, Acanthopagrus schlegelii[35–42]. Further knowledge comes from QTL studies on the protandrous Gilthead seabream, Sparus aurata[19, 20], but current knowledge concerning other Sparidae species is poor.
The Sharpsnout seabream, Diplodus puntazzo, is a sparid of great importance for the industry of fisheries and aquaculture. This species is also particularly interesting from an evolutionary point of view, as it has one of the most spectacular reproductive systems; it is rudimentary hermaphrodite with some instances of protandry [43, 44]. Several studies have investigated various aspects of its biology, including reproduction and development (e.g. [44–46]), but the available information concerning the species’ genetic content is limited. To date, this is the case for most sparids, except for the two protandrous species of Sparus aurata and Acanthopagrus schlegelii, which account for more than 90% of the Sparidae sequences available in GenBank. With modern sequencing technologies, this lack of knowledge can be overcome and ultimately allow the comparison of the genetic networks involved in sex determination and differentiation among closely related species that exhibit different reproductive modes.
Here, we sought to identify and understand the molecular toolkit underlying the sex differences of the expressed transcriptome in the rudimentary hermaphrodite sharpsnout seabream. To that end, we employed an RNA-Sequencing (RNA-Seq) approach [47] aiming at capturing the gene content of sharpsnout seabream that exhibits sex-biased expression pattern. We chose to study both gonad and brain tissues. Gonads were selected to get a comprehensive overview of the genes responsible for the divergence of the primary sex-related structure; brain was included to increase the species genic information and to understand how sex affects brain functions, as shown in gonochoristic fishes (see [48] and references therein). Our results revealed major expression differences between male and female gonads, but only minor differences between male and female brains. Most genes involved in primary pathways of sex determination and differentiation in other vertebrates are present also in the sharpsnout seabream; this implies a conservation of pathways between gonochorists and hermaphrodites. Finally, we constructed a dataset of genes and a resource of genetic markers that will assist future genetic research at the species and family level.
Methods
Sample collection
Animal care was carried out according to the “Guidelines for the treatment of animals in behavioural research and teaching” [49]. Hatchery produced sharpsnout seabream (from eggs spawned in October 2010) were reared in tanks supplied with flow-through seawater under ambient conditions at the Institute of Marine Biology, Biotechnology and Aquaculture. Fish were fed daily to apparent satiation using a commercial extruded feed (SKRETTING, Norway and IRIDA S.A., Greece). During the reproductive period (October-December) of 2012 (when fish were 2+ years old), fish were sampled randomly from the population and were euthanized in a mixture of seawater and ice. Selected individuals were examined for sexual maturation, based on the presence of releasable sperm from the males or the presence of vitellogenic oocytes in the females. At that time, sperm could not be collected from any of the males, but histological evaluation indicated the presence of intratesticular spermatozoa. Females were immature, containing only primary oocytes in their ovaries.
Gonad and brain tissues were sampled from four male and four female individuals (16 samples in total) in a sterile and RNase-free way and fixed immediately in RNAlater (Applied Biosystems, Foster City, CA, USA). Tissues for RNA extraction were stored at 4°C overnight and then transferred to -80°C until further processing.
RNA extraction and sequencing
To avoid biases regarding different expression in different parts of the same organ, we homogenized the whole brain and male gonad samples whereas in the case of female gonads, because of their large size (1.2 g - 4.3 g) we excised three different parts of the organ (the posterior, the anterior and the middle tissue) and homogenized it in QIAzol Lysis buffer (QIAGEN®) following supplier’s recommendations.
The disruption and homogenization occurred using TissueLyser II (QIAGEN®) and steal beads of 5 mm diameter (QIAGEN®). We used Qiagen’s miRNeasy extraction kit, as we targeted on extracting both mRNA and miRNA (only mRNA was analyzed in this study). The quantity of the isolated RNA was measured spectrophotometrically with NanoDrop® ND-1000 (Thermo Scientific), while its quality was tested on an agarose gel (electrophoresis in 1.5% w/v) and further on an Agilent Technologies 2100 Bioanalyzer (Agilent Technologies). The majority of the samples had an RNA Integrity Number (RIN) value higher than 8 (Table 1). This was not the case for any of the female gonad samples. Further effort to improve RNA extraction did not yield any higher RIN value. Although NanoDrop values were of the expected range (260/280 ratio [1.98-2.07]), the agarose gel and Bioanalyzer revealed the presence of an enormous peak at 100 bp (Additional file 1: Figure S1). However, several teleost female gonads show a similar RNA profile as reported earlier [50]. This high peak corresponds to 5S rRNA and possibly hampers the correct estimation of the 18S and 28S rRNA peaks. This was consistent in all female gonad extractions and possibly affected the RIN number as the observed pattern deviated greatly from the expected pattern of RNA content.
Finally, all 16 samples were used for library preparation and sequencing as 100 bp paired reads in 1.5 lanes of a HiSeq2500 following the protocols of Illumina Inc. (San Diego, CA) in the Genomics Resources Core Facility of Weill Cornell Medical College.
Read pre-processing
Read quality was assessed with FastQC [51] and subjected to quality control with FASTX_Toolkit [52]. Adapters were trimmed with fastx_clipper. Quality trimming was conducted with fastq_quality_trimmer (minimum quality 25, minimum read length 50). Reads were further quality controlled by excluding those with more than 5% of low-quality (quality threshold 25) nucleotides using fastq_quality_filter. After filtering, read pairs were reconstructed with a custom Perl script.
Transcriptome assembly and annotation
For the assembly, we pooled the filtered reads of all samples and implemented three different trials using SOAP-DENOVO [53] (kmer = 35, min length 200 nucleotides), Velvet/Oases [54, 55]) (kmer = 35, min length 200 nucleotides) and Trinity [56] (trinityrnaseq_r2013-02-25; default kmer 25, min length 200 nucleotides). The three candidate assemblies were evaluated by BLASTn [57] against Oreochromis niloticus, Oryzias latipes and Gasterosteus aculeatus cDNA dataset downloaded from Ensembl database [58] with an e-value threshold of 10-9. The assembly produced by Trinity had the highest number of significant similarity with unique genes from all three teleost species and was selected.
To assess the assembled transcripts and exclude the spurious ones, we pooled all the reads and mapped them to the selected assembly with Bowtie [59] within RSEM [60] using the script available in trinity utilities run_RSEM_align_n_estimate.pl. Putative transcripts with less than 1% of a locus reads mapped to that particular isoform (IsoPct < 1) were eliminated (as proposed in [61]). The same was done for those with Fragments Per Kilobase of transcript per Million mapped reads (FPKM) values less than 0.3. The choice of FPKM threshold was based on BLASTn similarity searches (e-value threshold 10-10) against O. niloticus cDNA sequences. The unfiltered transcripts dataset had significant similarity with 18,527 O. niloticus genes, while the filtered assembly had significant similarity with 17,346 out of 21,462 genes reported in Ensembl. This dataset constituted the final assembled transcriptome given that the selected threshold value resulted in elimination of 255,891 possibly spurious transcripts, while the hits to unique genes of O. niloticus were only slightly reduced.
To annotate the assembled transcripts, we conducted a BLASTx similarity search against the NCBI protein database nr (e-value threshold 10-9; keeping the top ten hits). BLASTx was done in parallel using NOBlast [62]. The output was used in Blast2GO [63] where gene ontology terms were retrieved and assigned to the transcripts (only the longest transcript was used per locus). The open reading frames (ORF) were extracted per sequence with the EMBOSS program getorf. InterProScan [64] was run on the longest ORF per transcript. The run was done in parallel splitting the query in 100 subqueries and merging the output with custom scripts. GO terms derived from InterProScan were merged with GO terms derived from BLASTx against nr. In cases where accurate orthology inference was of interest (e.g. for identifying the orthologs of the genes associated with sex in other taxa), we implemented a reciprocal BLASTn hit approach of the annotated sequences of medaka, tilapia and other fish from Ensembl against sharpsnout seabream assembled transcripts. When the gene of interest had significant similarity with the assembled transcripts, we used the top hit in a BLASTn search towards the transcriptome of the starting species. If that step returned the initial transcript, we assumed orthology between the two.
Differential expression
The paired reads of each sample were mapped to the assembly with Bowtie and abundance was estimated with RSEM v. 1.2.4, as implemented in the trinity script run_RSEM_align_n_estimate.pl. The estimated expected counts for each sample, at the gene level, were extracted and used for the analysis of differential expression conducted in DESeq [65], a software considered accurate and conservative for differential expression analyses [66, 67]. Samples were grouped according to sex and expression was compared for each tissue separately, following the developers’ manual (FDR threshold of 0.05). Due to a reported difficulty in assessing differential expression when a gene is expressed only in one group [66], we considered as significant those genes only when the expression in the other group was higher than 30 normalized counts in total.
Detection of genetic markers
The assembled sequences were scanned for microsatellites with Phobos [68]. We detected non-exact Short Tandem Repeats (STRs) with 2–10 repeat unit length and a minimum length of 20 nucleotides. A custom Perl script was used to parse the output.
Single Nucleotide Polymorphisms (SNPs) were detected with SAMTools [69] and VCFtools [70]. Alignment (.bam) files produced through mapping for gonads and brain were merged for each individual. Alignment files were further filtered for mapping quality (threshold 75), number of mismatches to the reference (‘ΝΜ’ threshold 10) and for proper pair mapping with BamTools [71]. Then, they were sorted and piled (with SAMTools function ‘mpileup’) to detect candidate SNPs. Custom Perl scripts were used to eliminate SNPs that formed clusters (>1 variant every 4 bases) to exclude hypervariable regions where accurate alignment is difficult. Those with SNP QUAL < 25, total high quality bases coverage below 15 and minor allele read ratio < 0.2 were excluded [72]. In each locus we kept only the SNPs belonging to the transcript with the highest SNP number. Finally, SNPs were categorized to synonymous, non-synonymous and those that belong to the transcripts UTRs. For that, we first retrieved the predicted ORF per transcript with the highest similarity to tilapia proteins (e-value threshold 10-10). Then, we evaluated whether each SNP is located within the coding region defined by the ORF, and for those located within the coding region if it causes a synonymous or a non-synonymous mutation using a Perl script.
To identify SNPs for which genotyping can be conducted robustly we applied extra filtering steps. Those included the genotype quality (‘GQ’) score > =20 as provided by SAMtools and VCFtools pipeline and required minimum coverage of the SNP site per sample > 5 for all samples. Note that the genotype quality is a function of the probability that the genotype call is wrong given that the site is variable. SNPs that passed the extra criteria were used for a PCA and a relatedness analysis with the R package SNPRelate [73]. For the Relatedness analysis, we estimated Identity By Descent with the Maximum Likelihood method and calculated the kinship coefficient. Finally, all scripts used in the study are available upon request.
Results
Sharpsnout seabream assembled transcriptome
Illumina HiSeq2500 sequencing yielded 429,988,050 paired reads (214,994,025 read pairs) (Table 1). The filtering process resulted in 229,843,938 paired and 74,027,237 single or orphan high quality reads used for the transcriptome assembly.
The initial assembly process produced 374,149 putative transcripts (N50: 1,712 nucleotides, mean length: 871 nucleotides). Following further editing and transcript quality assessment (see Methods), we limited our dataset to 118,258 transcripts belonging to 82,331 loci (N50: 2,092; mean length: 1,352 nucleotides) (Figure 1). The unfiltered transcripts dataset had significant similarity to 18,527 O. niloticus genes, while the filtered assembly had significant similarity to 17,346 out of 21,462 genes reported in Ensembl. Given that the filtering resulted in elimination of 255,891 possibly spurious transcripts, while the hits to unique genes of O. niloticus were only slightly reduced, the restricted dataset constituted the final transcriptome. Out of the 82,331 loci, 31,895 had significant BLASTx similarity hits with publicly available protein sequences, 22,574 of the loci were assigned GO terms and 40,823 had an InterProScan protein domain match. The great majority of the top-hits of the assembled transcripts had significant similarity with sequences of Maylandia zebra, Oreochromis niloticus, Takifugu rubripes, Oryzias latipes and Dicentrarchus labrax. Further, 14,041 of the genes without any significant similarity to known proteins had InterProScan protein domains assigned.

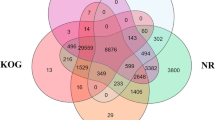
The pipeline followed to build the assembly and the male versus female expression profiles. On the left, flow chart of the steps implemented from raw reads to the selection of the final assembled loci. On the right, Venn diagram showing the loci commonly and differentially expressed in the two sexes for brains and gonads.
Comparative expression profiling between males and females
The global gene expression pattern observed in the tissues of the eight individuals used in this study is summarized in a principal component analysis (PCA) (Figure 2). The picture obtained shows a clear separation between female and male gonads with remarkable variation within the two groups. On the contrary, brain expression patterns are similar between males and females in this general overview.

Principal component analysis of the expression profiles. On top, the global expression profiles of gonad and brain samples of male and female individuals analyzed in a PCA. At the bottom, brain and gonad samples are analyzed separately. The brain sample pointed by the arrow is considered an outlier and was excluded from the differential expression analyses.
A separate PCA of the brain global expression values showed a scattered pattern with some overlap between the groups of males and females (Figure 2). Notably, one of the male brain samples had an extreme profile compared to the other male or female samples. To test whether this was due to library construction error, we mapped the reads to Sparus aurata ribosomal RNA (rRNA) to find no relation of rRNA content to the outlier (for the majority of samples 0–0.05% of the reads were mapped on rRNA). Thus, this sample was excluded from the differential expression analysis. For gonads, global expression patterns showed once again a clear separation of males and females (Figure 2). Sex-specific expression patterns were evaluated separately for brain and gonad tissues and are described below.
Male vs. female gonads expression patterns
In gonads, male and female expression patterns differed greatly. Out of the total 82,331 loci, 71,387 had an estimated abundance of at least one pair of reads in gonad samples. The differential expression analysis revealed 5,558 loci up-regulated in female and 8,072 loci up-regulated in male gonads (Figure 1; Additional file 2: Figure S2; Additional file 3: Table S1). Clustering of the samples based on the top 40 differentially expressed genes grouped males and females separately (Figure 3).

Heatmap of the differentially expressed genes for gonad and brain samples. Heatmap of the variance-stabilized transformed count data of gonad and brain samples for the differentially expressed genes between male and female individuals. Note that only the top 40 genes are included in the gonad heatmap; for the brain heatmap all genes are included.
The putative functions of those genes include development, signal transduction and metabolism (Figure 4). To test whether certain functions are more frequently observed in one of the two sexes, we conducted a Fisher’s exact test in Blast2GO comparing the GO terms of the male versus the female over-expressed genes (each gene was represented by the longest transcript; p-value 0.05; Table 2; see Additional file 4: Table S2 for complete list). The main GO terms associated with the genes over-expressed in females are involved in metabolism, while in males in signaling and regulation of transcription.

Gene Ontology profiling of gonad and brain samples. Pie charts representing the GO terms (Biological Process) associated with the genes over-expressed in gonad and brain samples. For gonads only terms with > 700 sequences are shown.
Sex determination and differentiation genes in gonads
The expression profiles of genes known to be involved in sex determination and differentiation (as reviewed in [28]) were closely examined (Table 3). Orthology with other genes was assessed with the reciprocal best hit approach (See Methods). Out of 44 target genes, 42 are found within the assembled transcriptome. The only genes not found were Fgf9 and Dmrt6. Dmr6 is absent from the teleost lineage according to Ensembl orthology inference and Fgf9 might be not expressed, not sequenced or lost from sharpsnout seabream. Sixteen genes are significantly differentially expressed between the two gonad types, while 15 change in the ‘expected’ direction of expression, assuming that the expression direction observed in other vertebrates is conserved in sharpsnout seabream. Only Dhh shows an overexpression in female gonads, opposite to the expected.
Further data mining revealed eight loci containing the GO term “sex differentiation” or “sex determination”, and 34 more related to the terms “female” or “male” (e.g. female/male gonad development, female/male meiosis, female pregnancy, etc.). From those 42 genes in total, 17 were significantly differentially expressed in the gonads (Additional file 5: Table S3) including Dmrt1 and Ctnnb1.
To search for potential genes that are present or absent from the two gonad tissues, we listed those differentially expressed loci that had an estimated abundance of absolutely zero counts in one gonad type, but at least a minimum of expression in the other (mean count > 30). We found 95 loci fulfilling those criteria (Additional file 6: Table S4). The majority (89 out of 95) exhibited a male-biased expression. Those loci were used in a similarity search (BLASTp) against tilapia proteins (e-value threshold 10-10). Our search retrieved 21 tilapia genes as top hits (Additional file 6: Table S4). Through Ensembl BioMART interface, we retrieved the scaffolds those genes are located in tilapia and downloaded the orthologous genes for stickleback and human. This comparison showed that some of the tilapia genes are located in the same scaffold. Search in stickleback showed that some of the genes located in different scaffolds in tilapia are found in the same chromosome in stickleback. Finally, the human ortholog of the genes Fam70A and Xpnpep2 are both located on the X chromosome (Additional file 6: Table S4).
Male vs. female brain expression patterns
In brain, 75,184 loci had an estimated abundance of more than one pair of reads. Male and female overall expression patterns were indistinguishable (Figure 2). However, the comparison of brain expression patterns (after excluding the outlier sample) between males and females revealed 68 genes (Figure 1; Additional file 3: Table S1) with significant expression difference between the two sexes (21 were over-expressed in females and 47 in males). Clustering of the samples based on all differentially expressed genes led to the grouping of one female with the males as observed also in the PCA conducted in total expression profiles (Figure 3; Figure 2).The main GO terms associated with the genes found in brain comparison are related to transposable elements, developmental processes, visual perception and signaling among others (Figure 4). Fisher’s exact test did not reveal any significantly over-represented term in the brain dataset compared to the whole assembly, probably due to the small size of the dataset.
Genetic marker discovery
We searched for two types of markers, SNPs and STRs. For the SNP discovery, we combined the reads obtained from the brain and gonad of each individual to increase the individual coverage and then applied various quality filters (see Methods, Table 4). In total, we found 85,189 SNPs, that pass the quality criteria, located in 30,291 loci (Additional file 7: Table S5). From those loci, 9,997 are significantly similar to known tilapia cDNA sequences and contain at least one predicted ORF. From the total SNPs within those 9,997 loci (30,278 SNPs), ~10% were in the first, ~7% in the second and ~41% in the third codon position. The rest were in the non-coding regions (5′ and 3′ UTRs) (Figure 5).

Genetic markers distribution at the coding loci. SNPs and STRs within coding regions are categorized according to their distribution at the codon position (for SNPs only) or 5′ and 3′UTRs.
From the loci containing SNPs, ~25% were significantly differentiated in males and females (37 in brain and 7,592 in gonads). Especially for the gonads, in which the total number of differentially expressed genes was 13,630, this is way more than expected by chance (two-sample z test on proportions; p-value < 0.05).
The required coverage and quality criteria applied secure a robust genetic marker discovery. In an attempt to genotype all individuals in certain loci, we applied further criteria requiring certain depth and quality per individual SNP calling (see Methods). Those filters reduced dramatically the number of SNPs that are exploitable for genotyping to 1,009 SNPs (Additional file 8: Table S6). The individual genotypes for all these SNPs were used in a PCA (Additional file 9: Figure S3) and a relatedness analysis (Additional file 10: Table S7). The average kinship coefficient (probability that two alleles randomly chosen from two individuals are Identical By Descent) between pairs is ~0.10 (min: 0.04; max: 0.18). No parent-offspring relationship is observed (expected kinship coefficient 0.25). Note that linkage disequilibrium-based SNP pruning was not conducted due to lack of information regarding linkage of SNPs.
Our search for STRs revealed 29,076 candidates located in 16,377 loci (23,759 transcripts) (Additional file 7: Table S5). From the discovered microsatellites, 12,625 STRs (~43%) are found in genes that code for proteins based on similarity searches with tilapia cDNA and 7,804 had a predicted ORF > 60 aa. The latter were used to study the STR distribution in the 5′UTR, 3′UTR and coding regions (Figure 5). The unit-length distribution of the discovered STRs is shown in Figure 6. Finally, STRs were found in 18 genes differentially expressed in brain and 3,713 in gonads.

Unit length distribution of identified STRs. Plot of STRs frequency based on unit length.
Discussion
The analyses presented explore the transcriptomic landscape of female and male gonad and brain tissues providing the first assessment of the molecular toolkit underlying the reproductive biology of a rudimentary hermaphrodite fish. We investigated the expression differences observed between sexes and tracked the expression of known targets of sex determination and differentiation. Finally, we provided a catalogue of SNPs and STRs that will assist following genetic research of the economically important sharpsnout seabream.
The gonad and brain transcriptome of sharpsnout seabream
Gonad and brain expression profiling was conducted on males and females, with four individuals/biological replicates in each of the two groups. The reason we chose relatively immature individuals was two-fold. First, we sought to capture the transcriptomic differences between the two sexes in a stage where the two gonad types are clearly formed and sex is easily identifiable in this rudimentary hermaphrodite species. Second, we aimed at genes that contribute to sex differentiation, but upstream players that might play a role in sex determination as well. The stage selection was appropriate for characterizing the sex-related transcriptomic profiles of sharpsnout seabream, since we identified the great majority of previously reported sex differentiation and determination genes of various taxa in our assembled sequences.The general pattern observed in the expression analyses revealed a homogeneous global expression among brain samples for both sexes (Figure 2). On the contrary, gonads diverge greatly among individuals. Although, the two sexes clearly form two groups, there is great within-group variation. This might reflect the actual developmental stage of each individual. All individuals used in the study are of the same stage, but we cannot rule out the possibility that within the observed developmental stages different biological and molecular processes take place through a series of finer phases exhibiting alternative expression profiles.
Male vs. female gonads expression patterns
The most striking aspect in the expression profiles of male and female gonads is the magnitude of the discovered differentially expressed genes (~20% of the total 71,387 loci expressed in gonads). A similar pattern is obtained from the PCA where the distances between the two gonad types are comparable to that between any of the two gonad types and the brain (Figure 2). This probably reflects their functional divergence. It is noteworthy that many more genes are over-expressed in male rather than female gonads (~60% of the gonad DE genes). This male-bias has been observed in other fish species gonad comparisons (zebrafish: [25, 26]; tilapia: [30]) or in whole organism male–female comparison (platyfish: [27]). However, given the lack of knowledge on the genetic and/or environmental mechanisms of sex determination and differentiation in sharpsnout seabream (or any other sparid), we cannot infer whether this is linked to the genetic architecture of the species or reflects other biological phenomena. The presence of genes expressed in the gonads of only one of the two sexes may be linked to the genetic architecture of sharpsnout seabream. The syntenic relationship of several of those genes observed in other species suggests a possible syntenic relationship in sharpsnout seabream and a common regulatory mechanism driven by the physical proximity on the genome. This can be tested in the future, e.g. with the construction of a genetic linkage map.
Multiple previous studies on vertebrates provide us with numerous candidate genes with possible involvement in sex determination and differentiation. A detailed search for those genes in the assembled transcriptome of sharpsnout seabream revealed that almost all are present, and some are differentially expressed in the gonads (Table 3). To start with the male phenotype, Dmrt1 is the gene with the most prominent role in sex determination and spermatogenesis across vertebrates [74, 75]. In teleosts, it is highly linked with gonad development and maintenance (see [76] for a review). In Sparidae, Dmrt1 has been implicated in the fate of the ovotestis in the protandrous black porgy Acanthopagrus schlegelii, and eliminating it results in a change from male to female [35, 77]. In our data, Dmrt1 is up-regulated in male gonads implying a strong role in the development and maintenance of the male phenotype in the hermaphrodite sharpsnout seabream. Amh, and its receptor Amhr2, are both main candidates for triggering and maintaining the male phenotype. In our data, Amh is over-expressed in male gonads suggesting an important role in sharpsnout seabream. On the contrary, Amhr2 exhibits similar expression levels in both gonad types. Both Amh and Amhr2 have been linked to gonadal development in the protandrous black porgy [78]. Focused research on sharpsnout seabream and other Sparidae will reveal their specific role. Amh is tightly linked to Sox9 and Sf1[79]. The male factor known to act on Sertoli cells, Sox9, was not differentially expressed in the sharpsnout seabream gonads. Other studies have showed that it is up-regulated in male Siberian sturgeons with undifferentiating gonads - in contrast to mature ones - [80], and it is over-expressed in the male immature gonads of sablefish [81]. Thus, Sox9 might play a role in gonadal development at stages earlier than those studied here (e.g. see [82]). In contrast to Sox9, Sf1 has significantly higher expression in male gonads. This factor might be tightly linked to the observed up-regulation of Amh. Male phenotype is reinforced by the high expression of androgen receptor, while Dax1- another gene involved in testis development- is similarly expressed between male and female gonads. The same pattern has been observed in tilapia [83], seabass [84] and medaka [85] where Dax1 showed no expression difference suggesting a role in both gonad types. Interestingly, Dhh, one of the genes involved in testis differentiation and development in humans and mice [86, 87], is linked with the function of female rather than male gonads in sharpsnout seabream. However, in mammals it is expressed in both testes and ovaries [88–90]. Finally, multiple other genes strongly related with vertebrate male gonads are found within the sharpsnout seabream transcriptome (Additional file 3: Table S1).
Concerning the genes involved in female gonads, we observed significant over-expression in two key players of ovarian development, the ovarian aromatase (Cyp19a1a) and β-catenin (Ctnnb). Cyp19a1 is a central component of ovarian steroidogenesis (converts androgens to estrogens). It is a conserved protein with a strong role in the ovarian development of vertebrates (see [91]). In fish, it is important not only for gonochoristic [92–95], but also hermaphrodite fish, as shown in [38] and confirmed in our data. The second activated key player, Ctnnb, is a member of the Wnt4/β-catenin pathway. However, Wnt4 is not over-expressed in sharpsnout seabream female gonads. This pathway is well-studied in mammals (e.g. [96, 97]), but not much is known for teleosts; the few studies on teleosts show that Wnt4 does not follow the pattern observed in mammals [23, 40, 98]. Finally, two genes that are known markers of ovarian development in mammals tightly linked to Wnt4/β-catenin pathway, R-spondin (Rspo-1) and Follistatin (Fst), were not differentially expressed in sharpsnout seabream gonads. Rspo-1 is involved in the ovarian differentiation in medaka in early stages of development; in later stages, its expression balances between male and female gonads [99]. Fst has been studied in detail in medaka [23] showing a lack of great differences among gonad types. Given the significant up-regulation of Ctnnb, the Wnt4/β-catenin pathway is involved in female gonads function and its significance and role should be deeper studied. Our search for Foxl2, another marker of ovarian development across vertebrates [100], showed that it is expressed similarly in both male and female gonads. This was also the case for the gonads of a protogynous hermaphrodite wrasse, Halichoeres trimaculatus[101], but not for several other gonochoristic fish [e.g. medaka [102, 103], tilapia [104], Southern catfish [105], catfish [106], Rare minnow [107], etc.], which suggests that Foxl2 might play distinct roles in the gonads of hermaphrodite fishes. Finally, estrogen receptors did not exhibit any differentiation between the two sexes.
Genes known to have a role in sex determination and differentiation in vertebrates are active in the hermaphrodite sharpsnout seabream, as shown by the expression patterns in the current study. However, it is still open whether the molecular pathways are conserved compared to other teleosts. The current knowledge and comparisons among teleosts show that both upstream and downstream genes alter their position and function in space and time [23, 95]. Apart from the known candidates, we observed thousands other genes in our data. Those reflect the complex biological processes taking place in the gonads (e.g. cell proliferation, metabolic processes, regulation of transcription, etc.).
Male vs. female brain expression patterns
Brain has a pronounced sexual dimorphism in function in mammals. However, even in model organisms like human, detailed information is only recently being gathered e.g. [108]. In fish brain, sexual dimorphism is less pronounced in gonochorists compared to mammals, and even less in hermaphrodites [109]. Further, the teleost brain is characterized by remarkable sexual plasticity [110]. Our results showed that in sharpsnout seabream, male and female brains have almost identical expression patterns with few exceptions. The observed divergence was smaller than reported in other fishes (e.g. zebrafish: [25]; rainbow trout: [111]; medaka: [109]). This may be due to reduced sex-specificity in rudimentary hermaphrodites brain.
The discovered genes provide a baseline for understanding the brain sexual divergence in sharpsnout seabream (Figure 4). Like in gonads, more genes are over-expressed in male rather than female brains. These genes are involved in development, metabolism, regulation of transcription, vision, etc. Some are even involved in RNA-dependent DNA replication -probably linked to transposable elements. Notably, none of the known sex determining genes is differentially activated in the brain. The genes over-expressed in male brains include several factors linked to sex in other taxa, such as Tcf12 (involved in estrogen/antiestrogen response in the teleost Fathead minnow [112]), Cbln1 (over-expressed in mouse testis compared to ovaries, as part of the male developmental pathway [113–116]), Rs1 (X-linked gene in human associated to a common macular degeneration in males [117]), Ca12 (involved in the function of uterus in mice [118]), Hyou1 (involved in gonadogenesis in mice [119]) and Aqp1 (possible involvement in the water homeostasis of the male reproductive system [120]). From the genes over-expressed in females, Hamp is regulated by estrogens in mice [113] regulating brain iron metabolism [121]. Other genes that might play significant but still unknown roles in the development of female brain are Alx3, NeoVTX subunit alpha, Thap9 and Itga2. All those are candidates for regulating and maintaining the sex-specific brain phenotypes and require deeper investigation.
In Sparidae, brain expression has been studied in black porgy; the expression of three Gnrh genes in different developmental stages of ovaries and testis has been characterized and they were found linked to sexual development [38]. However, in sharpsnout seabream all three genes (Gnrh-I-III) have only basal or no expression in both male and female brains. Further, in the gonads we found significant divergence in the expression of aromatase α, which had no expression in either male or female brains. On the contrary, we found strong expression of aromatase β in the brain and low expression in the gonads of both sexes. In both tissues, no significant expression difference was observed between the sexes for aromatase β. Those are the expected expression patterns of the two aromatases as observed in other teleosts as well (summarized in [48]). Targeted studies and comparative data from other species will elucidate the role each of those genes play and the mechanisms regulating their expression (e.g. environmental, social, genetic, etc.).
Genetic marker discovery through RNA-sequencing
The possibility to reconstruct the expressed genic sequences at a global scale through RNA-Seq, gives access to a plethora of genetic markers widely distributed in the genome. We identified a considerable dataset of STR and SNP markers that can be used as raw material in future population genetics, broodstock management, QTL mapping and Marker Assisted Selection (MAS) in sharpsnout seabream and other phylogenetically close sparid species. Discovering SNP markers can be a relatively easy task in a dataset like the one produced here, as pooling individuals allows identifying polymorphic sites robustly (although this can be proven only through further targeted experiments). However, further filtering of the SNPs for individual genotyping (see Methods) reduced dramatically the available SNPs. Thus, finding SNP markers that pass certain quality filters for all sequenced individuals is not trivial. To increase the number of informative markers, either deeper sequencing of each individual or use of different genotyping methods would be more appropriate (e.g. RNA-Seq with normalized libraries, Genotyping by Sequencing, SNP-chips scanning, etc.). Apart from SNPs, RNA-Seq leads to the discovery of thousands new STR markers in the transcribed regions of the genome, offering the possibility to select appropriate markers for future analyses.
Conclusions
In this study, we conducted a comprehensive search of the genes expressed in the male and female gonad and brain tissues of sharpsnout seabream. We used the information extracted from sequencing the RNA of the two tissue types i) to obtain a global view of the sex-specific expression patterns in a rudimentary hermaphrodite teleost and ii) to gather transcriptomic information that will make future comparative analyses feasible. The picture we obtained refers to the particular developmental stage, and averages among all sub-tissues within brain or gonads. In our results, we found the same genes that are responsible for sex differentiation in numerous other species. Some of them might play a role in the sex determination procedure that takes place early in the development. The whole-transcriptome approach employed lays the foundation for future studies to identify genes responsible not only for the sex determination, but also the gonad development and maintenance in sharpsnout seabream. Similar analyses on the tissue parts and of different developmental stages will provide the dynamic view necessary for a complete understanding of sex development.
Eventually, comparison of the expression profiles in sharpsnout seabream with closely related species that display alternative reproductive modes (e.g. the protandrous Sparus aurata and other protogynous species), will offer impressive insights into the particular genetic toolkits deployed in each mechanism.
Availability of supporting data
Raw reads are deposited in N.C.B.I. sequence read archive under the BioProject ID PRJNA241484.
References
Bull JJ: Evolution of sex determining mechanisms. 1983, Menlo Park, California: Benjamin/Cummings Pub. Co
Valenzuela N, Adams DC, Janzen FJ: Pattern does not equal process: exactly when is sex environmentally determined?. Am Nat. 2003, 161: 676-683.
Ospina-Alvarez N, Piferrer F: Temperature-dependent sex determination in fish revisited: prevalence, a single sex ratio response pattern, and possible effects of climate change. Plos One. 2008, 3 (7): e2837-
Piferrer F: Epigenetics of sex determination and gonadogenesis. Dev Dynam. 2013, 242 (4): 360-370.
Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R, Goodfellow PN: A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990, 346 (6281): 240-244.
Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R: Male development of chromosomally female mice transgenic for Sry. Nature. 1991, 351 (6322): 117-121.
Smith CA, Roeszler KN, Ohnesorg T, Cummins DM, Farlie PG, Doran TJ, Sinclair AH: The avian Z-linked gene DMRT1 is required for male sex determination in the chicken. Nature. 2009, 461 (7261): 267-271.
Yoshimoto S, Okada E, Umemoto H, Tamura K, Uno Y, Nishida-Umehara C, Matsuda Y, Takamatsu N, Shiba T, Ito M: A W-linked DM-domain gene, DM-W, participates in primary ovary development in Xenopus laevis. Proc Natl Acad Sci U S A. 2008, 105 (7): 2469-2474.
Matsuda M, Nagahama Y, Shinomiya A, Sato T, Matsuda C, Kobayashi T, Morrey CE, Shibata N, Asakawa S, Shimizu N, Hori H, Hamaguchi S, Sakaizumi M: DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature. 2002, 417 (6888): 559-563.
Nanda I, Kondo M, Hornung U, Asakawa S, Winkler C, Shimizu A, Shan ZH, Haaf T, Shimizu N, Shima A, Schmid M, Schartl M: A duplicated copy of DMRT1 in the sex-determining region of the Y chromosome of the medaka, Oryzias latipes. Proc Natl Acad Sci U S A. 2002, 99 (18): 11778-11783.
Kamiya T, Kai W, Tasumi S, Oka A, Matsunaga T, Mizuno N, Fujita M, Suetake H, Suzuki S, Hosoya S, Tohari S, Brenner S, Miyadai T, Venkatesh B, Suzuki Y, Kikuchi K: A trans-species missense SNP in Amhr2 is associated with sex determination in the Tiger pufferfish, Takifugu rubripes (fugu). Plos Genet. 2012, 8 (7): e1002798-
Yano A, Guyomard R, Nicol B, Jouanno E, Quillet E, Klopp C, Cabau C, Bouchez O, Fostier A, Guiguen Y: An immune-related gene evolved into the master sex-determining gene in Rainbow trout, Oncorhynchus mykiss. Curr Biol. 2012, 22 (15): 1423-1428.
Hattori RS, Murai Y, Oura M, Masuda S, Majhi SK, Sakamoto T, Fernandino JI, Somoza GM, Yokota M, Strussmann CA: A Y-linked anti-Mullerian hormone duplication takes over a critical role in sex determination. Proc Natl Acad Sci U S A. 2012, 109 (8): 2955-2959.
Peichel CL, Ross JA, Matson CK, Dickson M, Grimwood J, Schmutz J, Myers RM, Mori S, Schluter D, Kingsley DM: The master sex-determination locus in threespine sticklebacks is on a nascent Y chromosome. Curr Biol. 2004, 14 (16): 1416-1424.
Palaiokostas C, Bekaert M, Khan MGQ, Taggart JB, Gharbi K, McAndrew BJ, Penman DJ: Mapping and validation of the major sex-determining region in Nile tilapia (Oreochromis niloticus L.) using RAD sequencing. Plos One. 2013, 8 (7): e68389-
Palaiokostas C, Bekaert M, Davie A, Cowan ME, Oral M, Taggart JB, Gharbi K, McAndrew BJ, Penman DJ, Migaud H: Mapping the sex determination locus in the Atlantic halibut (Hippoglossus hippoglossus) using RAD sequencing. BMC Genomics. 2013, 14: 566-
Martinez P, Bouza C, Hermida M, Fernandez J, Toro MA, Vera M, Pardo B, Millan A, Fernandez C, Vilas R, Sánchez L, Felip A, Piferrer F, Ferreiro I, Cabaleiro S: Identification of the major sex-determining region of turbot (Scophthalmus maximus). Genetics. 2009, 183 (4): 1443-1452.
Bradley KM, Breyer JP, Melville DB, Broman KW, Knapik EW, Smith JR: An SNP-based linkage map for zebrafish reveals sex determination loci. G3. 2011, 1 (1): 3-9.
Loukovitis D, Sarropoulou E, Batargias C, Apostolidis AP, Kotoulas G, Tsigenopoulos CS, Chatziplis D: Quantitative trait loci for body growth and sex determination in the hermaphrodite teleost fish Sparus aurata L. Anim Genet. 2012, 43 (6): 753-759.
Loukovitis D, Sarropoulou E, Tsigenopoulos CS, Batargias C, Magoulas A, Apostolidis AP, Chatziplis D, Kotoulas G: Quantitative trait loci involved in sex determination and body growth in the Gilthead sea bream (Sparus aurata L.) through targeted genome scan. Plos One. 2011, 6 (1): e16599-
Marı́n I, Baker BS: The evolutionary dynamics of sex determination. Science. 1998, 281 (5385): 1990-1994.
Graham P, Penn JK, Schedl P: Masters change, slaves remain. BioEssays. 2003, 25 (1): 1-4.
Herpin A, Adolfi MC, Nicol B, Hinzmann M, Schmidt C, Klughammer J, Engel M, Tanaka M, Guiguen Y, Schartl M: Divergent expression regulation of gonad development genes in medaka shows incomplete conservation of the downstream regulatory network of vertebrate sex determination. Mol Biol Evol. 2013, 30 (10): 2328-2346.
Piferrer F, Ribas L, Diaz N: Genomic approaches to study genetic and environmental influences on fish sex determination and differentiation. Mar Biotechnol. 2012, 14 (5): 591-604.
Sreenivasan R, Cai MN, Bartfai R, Wang XG, Christoffels A, Orban L: Transcriptomic analyses reveal novel genes with sexually dimorphic expression in the zebrafish gonad and brain. Plos One. 2008, 3 (3): e1791-
Small CM, Carney GE, Mo QX, Vannucci M, Jones AG: A microarray analysis of sex- and gonad-biased gene expression in the zebrafish: evidence for masculinization of the transcriptome. BMC Genomics. 2009, 10: 579-
Zhang ZP, Wang YL, Wang SH, Liu JT, Warren W, Mitreva M, Walter RB: Transcriptome analysis of female and male Xiphophorus maculatus Jp 163 A. Plos One. 2011, 6 (4): e18379-
Forconi M, Canapa A, Barucca M, Biscotti MA, Capriglione T, Buonocore F, Fausto AM, Makapedua DM, Pallavicini A, Gerdol M, De Moro G, Scapigliati G, Olmo E, Schartl M: Characterization of sex determination and sex differentiation genes in Latimeria. Plos One. 2013, 8 (4): e56006-
Sun FY, Liu SK, Gao XY, Jiang YL, Perera D, Wang XL, Li C, Sun LY, Zhang JR, Kaltenboeck L, Dunham R, Liu Z: Male-biased genes in catfish as revealed by RNA-Seq analysis of the testis transcriptome. Plos One. 2013, 8 (7): e68452-
Tao WJ, Yuan J, Zhou LY, Sun LN, Sun YL, Yang SJ, Li MH, Zeng S, Huang BF, Wang DH: Characterization of gonadal transcriptomes from Nile tilapia (Oreochromis niloticus) reveals differentially expressed genes. Plos One. 2013, 8 (5): e63604-
Erisman BE, Petersen CW, Hastings PA, Warner RR: Phylogenetic perspectives on the evolution of functional hermaphroditism in teleost fishes. Integr Comp Biol. 2013, 53 (4): 736-754.
Buxton CD, Garratt PA: Alternative reproductive styles in seabreams (Pisces, Sparidae). Environ Biol Fish. 1990, 28 (1–4): 113-124.
Atz JW: Intersexuality in Fishes. Intersexuality in Vertebrates Including Man. Edited by: Marshall AJ, Armstrong CN. 1964, New York: Academic Press, 145-232.
Mylonas CC, Zohar Y, Pankhurst N, Kagawa H: Reproduction and broodstock management. Sparidae. Edited by: Pavlidis MA, Mylonas CC. 2011, Oxford, UK: Wiley-Blackwell, 95-131.
Wu GC, Chang CF: The switch of secondary sex determination in protandrous black porgy, Acanthopagrus schlegeli. Fish Physiol Biochem. 2013, 39 (1): 33-38.
He CL, Du JL, Lee YH, Sun LT, Chang CF: Differential Dmrt1 transcripts in gonads of the protandrous black porgy, Acanthopagrus schlegeli. Cytogenet Genome Res. 2003, 101: 309-313.
Wu GC, Tomy S, Chang CF: The expression of nr0b1 and nr5a4 during gonad development and sex change in protandrous black porgy fish, Acanthopagrus schlegeli. Biol Reprod. 2008, 78 (2): 200-210.
Wu GC, Tomy S, Lee MF, Lee YH, Yueh WS, Lin CJ, Lau EL, Chang CF: Sex differentiation and sex change in the protandrous black porgy, Acanthopagrus schlegeli. Gen Comp Endocrinol. 2010, 167 (3): 417-421.
Wu GC, Tomy S, Nakamura M, Chang CF: Dual Roles of cyp19a1a in gonadal sex differentiation and development in the protandrous black porgy, Acanthopagrus schlegeli. Biol Reprod. 2008, 79 (6): 1111-1120.
Wu GC, Chang CF: wnt4 as associated with the development of ovarian tissue in the protandrous black porgy, Acanthopagrus schlegeli. Biol Reprod. 2009, 81 (6): 1073-1082.
Tomy S, Wu GC, Huang HR, Chang CF: Age-dependent differential expression of genes involved in steroid signalling pathway in the brain of protandrous black porgy, Acanthopagrus schlegeli. Dev Neurobiol. 2009, 69 (5): 299-313.
Tomy S, Wu GC, Huang HR, Dufour S, Chang CF: Developmental expression of key steroidogenic enzymes in the brain of protandrous black porgy fish, Acanthopagrus schlegeli. J Neuroendocrinol. 2007, 19 (8): 643-655.
Micale V, Perdichizzi F, Basciano G: Aspects of the reproductive biology of the sharpsnout seabream Diplodus puntazzo (Cetti, 1777).1. Gametogenesis and gonadal cycle in captivity during the third year of life. Aquaculture. 1996, 140 (3): 281-291.
Pajuelo JG, Lorenzo JM, Dominguez-Seoane R: Gonadal development and spawning cycle in the digynic hermaphrodite sharpsnout seabream Diplodus puntazzo (Sparidae) off the Canary Islands, northwest of Africa. J Appl Ichthyol. 2008, 24 (1): 68-76.
Klimogianni A, Kalanji M, Pyrenis G, Zoulioti A, Trakos G: Ontogeny of embryonic and yolk-sac larval stage of the sparid sharpsnout sea bream (Diplodus puntazzo Cetti, 1777). J Fish Aquat Sci. 2011, 6: 62-73.
Papadaki M, Papadopoulou M, Siggelaki I, Mylonas CC: Egg and sperm production and quality of sharpsnout sea bream (Diplodus puntazzo) in captivity. Aquaculture. 2008, 276 (1–4): 187-197.
Wang Z, Gerstein M, Snyder M: RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009, 10 (1): 57-63.
Le Page Y, Diotel N, Vaillant C, Pellegrini E, Anglade I, Merot Y, Kah O: Aromatase, brain sexualization and plasticity: the fish paradigm. Eur J Neurosci. 2010, 32 (12): 2105-2115.
Guidelines for the treatment of animals in behavioural research and teaching. Anim Behav. 2001, 61 (1): 271-275. http://www.ncbi.nlm.nih.gov/pubmed/11170716,
de Diaz Cerio O, Rojo-Bartolome I, Bizarro C, Ortiz-Zarragoitia M, Cancio I: 5S rRNA and accompanying proteins in gonads: powerful markers to identify sex and reproductive endocrine disruption in fish. Environ Sci Tech. 2012, 46 (14): 7763-7771.
Babraham Bioinformatics: FastQC A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/,
FASTX-Toolkit software package: http://hannonlab.cshl.edu/fastx_toolkit/,
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW, Yiu SM, Peng S, Xiaoqian Z, Liu G, Liao X, Li Y, Yang H, Wang J, Lam TW, Wang J: SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012, 1 (1): 18-
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18 (5): 821-829.
Schulz MH, Zerbino DR, Vingron M, Birney E: Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012, 28 (8): 1086-1092.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng QD, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A: Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011, 29 (7): 644-U130.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215 (3): 403-410.
Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, García-Girón C, Gordon L, Hourlier T, Hunt S, Juettemann T, Kähäri AK, Keenan S, Komorowska M, Kulesha E, Longden I, Maurel T, McLaren WM, Muffato M, Nag R, Overduin B, Pignatelli M, Pritchard B, Pritchard E, et al: Ensembl 2013. Nucleic Acids Res. 2013, 41 (D1): D48-D55.
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10 (3): R25-
Li B, Dewey CN: RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011, 12: 323-
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, Macmanes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A: De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc. 2013, 8 (8): 1494-1512.
Lagnel J, Tsigenopoulos CS, Iliopoulos I: NOBLAST and JAMBLAST: new options for BLAST and a java application manager for BLAST results. Bioinformatics. 2009, 25 (6): 824-826.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M: Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005, 21 (18): 3674-3676.
Zdobnov EM, Apweiler R: InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001, 17 (9): 847-848.
Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol. 2010, 11 (10): R106-
Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, Mason CE, Socci ND, Betel D: Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14 (9): R95-
Soneson C, Delorenzi M: A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics. 2013, 14: 91-
Mayer C: Phobos: a tandem repeat search tool. http://www.ruhr-uni-bochum.de/ecoevo/cm/cm_phobos.htm,
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S: The sequence alignment/map format and SAMtools. Bioinformatics. 2009, 25 (16): 2078-2079.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R: The variant call format and VCFtools. Bioinformatics. 2011, 27 (15): 2156-2158.
Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, Marth GT: BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011, 27 (12): 1691-1692.
Quinn EM, Cormican P, Kenny EM, Hill M, Anney R, Gill M, Corvin AP, Morris DW: Development of strategies for SNP detection in RNA-seq data: application to lymphoblastoid cell lines and evaluation using 1000 Genomes data. Plos One. 2013, 8 (3): e58815-
Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS: A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012, 28 (24): 3326-3328.
Zarkower D: DMRT Genes in vertebrate gametogenesis. Curr Top Dev Biol. 2013, 102: 327-356.
Matson CK, Zarkower D: Sex and the singular DM domain: insights into sexual regulation, evolution and plasticity. Nat Rev Genet. 2012, 13 (3): 163-174.
Herpin A, Schartl M: Dmrt1 genes at the crossroads: a widespread and central class of sexual development factors in fish. Febs J. 2011, 278 (7): 1010-1019.
Wu GC, Chiu PC, Lin CJ, Lyu YS, Lan DS, Chang CF: Testicular dmrt1 Is involved in the sexual fate of the ovotestis in the protandrous black porgy. Biol Reprod. 2012, 86 (2): 41-
Wu GC, Chiu PC, Lyu YS, Chang CF: The expression of amh and amhr2 is associated with the development of gonadal tissue and sex change in the protandrous black porgy, Acanthopagrus schlegeli. Biol Reprod. 2010, 83 (3): 443-453.
Sekido R, Lovell-Badge R: Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature. 2008, 453 (7197): 930-934.
Berbejillo J, Martinez-Bengochea A, Bedo G, Brunet F, Volff JN, Vizziano-Cantonnet D: Expression and phylogeny of candidate genes for sex differentiation in a primitive fish species, the Siberian sturgeon, Acipenser baerii. Mol Reprod Dev. 2012, 79 (8): 504-516.
Smith EK, Guzman JM, Luckenbach JA: Molecular cloning, characterization, and sexually dimorphic expression of five major sex differentiation-related genes in a Scorpaeniform fish, sablefish (Anoplopoma fimbria). Comp Biochem Phys B. 2013, 165 (2): 125-137.
Kong Y, Grimaldi M, Curtin E, Dougherty M, Kaufman C, White RM, Zon LI, Liao EC: Neural crest development and craniofacial morphogenesis is coordinated by nitric oxide and histone acetylation. Chem Biol. 2014, 21 (4): 488-501.
Wang DS, Kobayashi T, Senthilkumaran B, Sakai F, Sudhakumari CC, Suzuki T, Yoshikuni M, Matsuda M, Morohashi K, Nagahama Y: Molecular cloning of DAX1 and SHP cDNAs and their expression patterns in the Nile tilapia, Oreochromis niloticus. Biochem Bioph Res Co. 2002, 297 (3): 632-640.
Martins RS, Deloffre LA, Mylonas CC, Power DM, Canario AV: Developmental expression of DAX1 in the European sea bass, Dicentrarchus labrax: lack of evidence for sexual dimorphism during sex differentiation. Reprod Biol Endocrinol. 2007, 5: 19-
Nakamoto M, Wang DS, Suzuki A, Matsuda M, Nagahama Y, Shibata N: Dax1 suppresses P450arom expression in medaka ovarian follicles. Mol Reprod Dev. 2007, 74 (10): 1239-1246.
Bitgood MJ, Shen LY, McMahon AP: Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr Biol. 1996, 6 (3): 298-304.
Clark AM, Garland KK, Russell LD: Desert hedgehog (Dhh) gene is required in the mouse testis for formation of adult-type Leydig cells and normal development of peritubular cells and seminiferous tubules. Biol Reprod. 2000, 63 (6): 1825-1838.
O’Hara WA, Azar WJ, Behringer RR, Renfree MB, Pask AJ: Desert hedgehog is a mammal-specific gene expressed during testicular and ovarian development in a marsupial. BMC Dev Biol. 2011, 11: 72-
Spicer LJ, Sudo S, Aad PY, Wang LS, Chun SY, Ben-Shlomo I, Klein C, Hsueh AJW: The hedgehog-patched signaling pathway and function in the mammalian ovary: a novel role for hedgehog proteins in stimulating proliferation and steroidogenesis of theca cells. Reproduction. 2009, 138 (2): 329-339.
Wijgerde M, Ooms M, Hoogerbrugge JW, Grootegoed JA: Hedgehog signaling in mouse ovary: Indian hedgehog and desert hedgehog from granulosa cells induce target gene expression in developing theca cells. Endocrinology. 2005, 146 (8): 3558-3566.
Guiguen Y, Fostier A, Piferrer F, Chang CF: Ovarian aromatase and estrogens: a pivotal role for gonadal sex differentiation and sex change in fish. Gen Comp Endocrinol. 2010, 165 (3): 352-366.
Kitano T, Takamune K, Kobayashi T, Nagahama Y, Abe SI: Suppression of P450 aromatase gene expression in sex-reversed males produced by rearing genetically female larvae at a high water temperature during a period of sex differentiation in the Japanese flounder (Paralichthys olivaceus). J Mol Endocrinol. 1999, 23 (2): 167-176.
Uchida D, Yamashita M, Kitano T, Iguchi T: An aromatase inhibitor or high water temperature induce oocyte apoptosis and depletion of P450 aromatase activity in the gonads of genetic female zebrafish during sex-reversal. Comp Biochem Physiol Mol Integr Physiol. 2004, 137 (1): 11-20.
Karube M, Fernandino JI, Strobl-Mazzulla P, Strussmann CA, Yoshizaki G, Somoza GM, Patino R: Characterization and expression profile of the ovarian cytochrome p-450 aromatase (cyp19A1) gene during thermolabile sex determination in pejerrey, Odontesthes bonariensis. J Exp Zool Part A. 2007, 307A (11): 625-636.
Bohne A, Heule C, Boileau N, Salzburger W: Expression and sequence evolution of aromatase cyp19a1 and other sexual development genes in East African cichlid fishes. Mol Biol Evol. 2013, 30 (10): 2268-2285.
Chassot AA, Bradford ST, Auguste A, Gregoire EP, Pailhoux E, de Rooij DG, Schedl A, Chaboissier MC: WNT4 and RSPO1 together are required for cell proliferation in the early mouse gonad. Development. 2012, 139 (23): 4461-4472.
Li L, Ji SY, Yang JL, Li XX, Zhang J, Zhang Y, Hu ZY, Liu YX: Wnt/beta-catenin signaling regulates follicular development by modulating the expression of Foxo3a signaling components. Mol Cell Endocrinol. 2013, 382 (2): 915-925.
Nicol B, Guerin A, Fostier A, Guiguen Y: Ovary-predominant wnt4 expression during gonadal differentiation is not conserved in the rainbow trout (Oncorhynchus mykiss). Mol Reprod Dev. 2012, 79 (1): 51-63.
Zhou LY, Charkraborty T, Yu XG, Wu LM, Liu G, Mohapatra S, Wang DS, Nagahama Y: R-spondins are involved in the ovarian differentiation in a teleost, medaka (Oryzias latipes). BMC Dev Biol. 2012, 12: 36-
Benayoun BA, Dipietromaria A, Bazin C, Veitia RA: FOXL2: At the crossroads of female sex determination and ovarian function. Adv Exp Med Biol. 2009, 665: 207-226.
Kobayashi Y, Horiguchi R, Nozu R, Nakamura M: Expression and localization of forkhead transcriptional factor 2 (Foxl2) in the gonads of protogynous wrasse, Halichoeres trimaculatus. Biol Sex Differ. 2010, 1 (1): 3-
Nakamoto M, Matsuda M, Wang DS, Nagahama Y, Shibata N: Molecular cloning and analysis of gonadal expression of Foxl2 in the medaka, Oryzias latipes. Biochem Bioph Res Co. 2006, 344 (1): 353-361.
Nakamoto M, Muramatsu S, Yoshida S, Matsuda M, Nagahama Y, Shibatal N: Gonadal sex differentiation and expression of Sox9a2, Dmrt1, and Foxl2 in Oryzias luzonensis. Genesis. 2009, 47 (5): 289-299.
Wang DS, Kobayashi T, Zhou LY, Nagahama Y: Molecular cloning and gene expression of Foxl2 in the Nile tilapia, Oreochromis niloticus. Biochem Bioph Res Co. 2004, 320 (1): 83-89.
Liu Z, Wu F, Jiao B, Zhang X, Hu C, Huang B, Zhou L, Huang X, Wang Z, Zhang Y, Nagahama Y, Cheng CH, Wang D: Molecular cloning of doublesex and mab-3-related transcription factor 1, forkhead transcription factor gene 2, and two types of cytochrome P450 aromatase in Southern catfish and their possible roles in sex differentiation. J Endocrinol. 2007, 194 (1): 223-241.
Sridevi P, Senthilkumaran B: Cloning and differential expression of FOXL2 during ovarian development and recrudescence of the catfish, Clarias gariepinus. Gen Comp Endocrinol. 2011, 174 (3): 259-268.
Jiang W, Yang Y, Zhao D, Liu X, Duan J, Xie S, Zhao H: Effects of sexual steroids on the expression of foxl2 in Gobiocypris rarus. Comp Biochem Physiol B: Biochem Mol Biol. 2011, 160 (4): 187-193.
Trabzuni D, Ramasamy A, Imran S, Walker R, Smith C, Weale ME, Hardy J, Ryten M, Expression NAB: Widespread sex differences in gene expression and splicing in the adult human brain. Nat Commun. 2013, 4: 2771-
Hiraki T, Takeuchi A, Tsumaki T, Zempo B, Kanda S, Oka Y, Nagahama Y, Okubo K: Female-specific target sites for both oestrogen and androgen in the teleost brain. Proc Biol Sci. 2012, 279 (1749): 5014-5023.
Godwin J: Neuroendocrinology of sexual plasticity in teleost fishes. Front Neuroendocrinol. 2010, 31 (2): 203-216.
Vizziano-Cantonnet D, Anglade I, Pellegrini E, Gueguen MM, Fostier A, Guiguen Y, Kah O: Sexual dimorphism in the brain aromatase expression and activity, and in the central expression of other steroidogenic enzymes during the period of sex differentiation in monosex rainbow trout populations. Gen Comp Endocrinol. 2011, 170 (2): 346-355.
Garcia-Reyero N, Kroll KJ, Liu L, Orlando EF, Watanabe KH, Sepulveda MS, Villeneuve DL, Perkins EJ, Ankley GT, Denslow ND: Gene expression responses in male fathead minnows exposed to binary mixtures of an estrogen and antiestrogen. BMC Genomics. 2009, 10: 308-
Ikeda Y, Tajima S, Izawa-Ishizawa Y, Kihira Y, Ishizawa K, Tomita S, Tsuchiya K, Tamaki T: Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. Plos One. 2012, 7 (7): 115f-
Nef S, Schaad O, Stallings NR, Cederroth CR, Pitetti JL, Schaer G, Malki S, Dubois-Dauphin M, Boizet-Bonhoure B, Descombes P, Parker KL, Vassalli JD: Gene expression during sex determination reveals a robust female genetic program at the onset of ovarian development. Dev Biol. 2005, 287 (2): 361-377.
Coveney D, Ross AJ, Slone JD, Capel B: A microarray analysis of the XX Wnt4 mutant gonad targeted at the identification of genes involved in testis vascular differentiation. Gene Expr Patterns. 2007, 7 (1–2): 82-92.
Menke DB, Page DC: Sexually dimorphic gene expression in the developing mouse gonad. Gene Expr Patterns. 2002, 2 (3–4): 359-367.
Friedrich U, Stohr H, Hilfinger D, Loenhardt T, Schachner M, Langmann T, Weber BH: The Na/K-ATPase is obligatory for membrane anchorage of retinoschisin, the protein involved in the pathogenesis of X-linked juvenile retinoschisis. Hum Mol Genet. 2011, 20 (6): 1132-1142.
Gholami K, Muniandy S, Salleh N: In-vivo functional study on the involvement of CFTR, SLC26A6, NHE-1 and CA isoenzymes II and XII in uterine fluid pH, volume and electrolyte regulation in rats under different sex-steroid influence. Int J Med Sci. 2013, 10 (9): 1121-1134.
Ewen K, Baker M, Wilhelm D, Aitken RJ, Koopman P: Global survey of protein expression during gonadal sex determination in mice. Mol Cell Proteomics. 2009, 8 (12): 2624-2641.
Skowronski MT, Leska A, Robak A, Nielsen S: Immunolocalization of aquaporin-1, -5, and -7 in the avian testis and vas deferens. J Histochem Cytochem. 2009, 57 (10): 915-922.
Wang SM, Fu LJ, Duan XL, Crooks DR, Yu P, Qian ZM, Di XJ, Li J, Rouault TA, Chang YZ: Role of hepcidin in murine brain iron metabolism. Cell Mol Life Sci. 2010, 67 (1): 123-133.
Acknowledgements
Financial support for this study has been provided by the Ministry of Education and Religious Affairs, under the Call “ARISTEIA I” of the National Strategic Reference Framework 2007–2013 (SPARCOMP, #36), co-funded by the EU and the Hellenic Republic through the European Social Fund. We would like to thank V. Terzoglou and E. Kaitetzidou for help in RNA extractions, Irini Sigelaki for help in sampling and Dr. Hooman Moghadam, Dr. Gianpaolo Zampicinini, Dr. Jon B. Kristoffersen and Ji Hyoun Kang for valuable discussions. Finally, we would like to thank three anonymous reviewers for their valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
TM, AT, ES, CCM, CST designed experiments. CST, CCM, NP conducted sampling. AT performed laboratory experiments. TM, JL analyzed data. JZX performed library construction and sequencing. TM wrote the manuscript with help from other authors. CST conceived and managed the project. All authors read and approved the final version of the manuscript.
Electronic supplementary material
12864_2014_6346_MOESM1_ESM.pdf
Additional file 1: Figure S1: Female gonad tissue RNA profile. Total RNA profile of a female gonad as retrieved from an agarose gel and the Agilent Bioanalyzer. (PDF 565 KB)
12864_2014_6346_MOESM2_ESM.pdf
Additional file 2: Figure S2: MA plot for gonad and brain samples. Axes represent log2 fold change versus the mean normalized base counts; significantly differentially expressed loci are shown in red. Genes exceeding the axes range are not shown. (PDF 11 MB)
12864_2014_6346_MOESM3_ESM.xlsx
Additional file 3: Table S1: The list of differentially expressed genes in gonad and brain tissues (FDR < 0.05). (XLSX 2 MB)
12864_2014_6346_MOESM4_ESM.xlsx
Additional file 4: Table S2: GO terms over-represented in female or male up-regulated genes in sharpsnout seabream gonads. Fisher’s exact test (adjusted p-value < 005) on the GO terms of male versus female gonads over-expressed loci. (XLSX 40 KB)
12864_2014_6346_MOESM6_ESM.xlsx
Additional file 6: Table S4: Comparative analysis of genes expressed only in the gonads of one sex in sharpsnout seabream. (XLSX 55 KB)
12864_2014_6346_MOESM9_ESM.pdf
Additional file 9: Figure S3: PCA conducted on SNP genotypes. The genotypes of each individual from the restricted set of 1009 SNPs are analyzed in a PCA. (PDF 87 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Manousaki, T., Tsakogiannis, A., Lagnel, J. et al. The sex-specific transcriptome of the hermaphrodite sparid sharpsnout seabream (Diplodus puntazzo). BMC Genomics 15, 655 (2014). https://doi.org/10.1186/1471-2164-15-655
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-655