
Abstract
Background
Toxascaris leonina is one of the most common intestinal parasites of canids and felids. In this study, we characterised the entire mitochondrial (mt) genome sequence of T. leonina from the cheetah and compared it with that of T. leonina from the dog.
Results
The entire mt genome sequence of T. leonina from the cheetah is 14,685 bp in size, which is 375 bp longer than that from the dog, and it is 408 bp longer than that from the South China tiger. The overall nucleotide sequence (except for the non-coding region) identity was 92.8% between the two mt genomes of T. leonina from the cheetah and the dog. For the 12 protein-coding genes, sequence difference between T. leonina from the cheetah and the dog was 5.0–9.7% at the nucleotide level and 1.0–7.2% at the amino acid level. Moreover, comparison of mt cox1 sequences among T. leonina isolates (n = 23) from different hosts revealed substantial nucleotide differences (10.6%). Phylogenetic analysis showed the separation of T. leonina from canid and felid hosts into three distinct clades.
Conclusions
Taken together, these mtDNA datasets indicate that T. leonina from canid and felid hosts represents a species complex. Our results have implications for further studies of the molecular epidemiology, systematics and population genetics of this nematode.
Similar content being viewed by others


Background
Nematodes (roundworms) are common parasites which inhabit in the gastrointestinal tract of humans, and domestic and wild animals, and most of them can cause significant economic losses and public health problems [1]. Toxascaris leonina (Nematoda: Ascarididae) is a common nematode of various animals, including dogs, cats, wolves, tigers, lions and foxes. Although it has a diverse range of definitive hosts, to date, it has been the only species described in the genus Toxascaris. Adult T. leonina parasitize in the small intestines of the definitive hosts and can cause serious disease in young animals [2]. Importantly, larvae of T. leonina can also infect humans, posing a potential public health problem [3].
The species identification of parasites is of significance for studying epidemiology, systematics, diagnostics and population genetics of parasites [4]. Usually, nematodes are identified based on morphological features, hosts, and geographical distributions. However, it may not always be possible to identify to the species level nematodes from different hosts and geographical locations based on these criteria. To overcome the limitation of morphological taxonomy, molecular/DNA markers have been extensively used for species identification and differentiation of various nematodes [5,6,7]. Recently, our preliminary comparative study has indicated that the mitochondrial (mt) cox1 gene sequence of T. leonina from cheetahs and dogs differed by 6.8% [8]. In addition, substantial nucleotide difference in part of the mt nad1 (9.0%) and nad4 (10.8%) were also detected between T. leonina from canid and felid hosts [9]. These findings raised the possibility that T. leonina may represent a species complex. However, this hypothesis was proposed based on a small molecular dataset. Therefore, this hypothesis should be tested by using more datasets. Mt genome (containing 36–37 genes) has been considered a suitable marker for the species identification and differentiation of many nematodes [10]. The mt genomes of T. leonina from canids (such as the dog) and felids (such as the South China tiger) have been sequenced and characterised previously [11, 12], but the T. leonina mt genome sequence from the South China tiger has not yet been deposited in the GenBank NCBI database. Moreover, it is yet unknown whether there are significant differences in the entire mt nucleotide and amino acid sequences between T. leonina from the dog and those from the South China tiger [12].
Therefore, in the present study, we (i) characterized the entire mt genome sequence of T. leonina from the cheetah, (ii) compared it with that of T. leonina from the dog, and (iii) then tested the hypothesis that T. leonina represents a species complex.
Methods
Parasites and DNA isolation
All adult worms (n = 14; coded TC1–TC14) of T. leonina were obtained from a cheetah (Acinonyx jubatus) in Changsha Ecological Zoo, China. This cheetah was dewormed at importation to Changsha in 2012. This cheetah died in 2018 and 14 adult nematode specimens were collected by the Zoo veterinarians at necropsy. Adult nematode specimens were washed separately in physiological saline, identified preliminarily to the species level based on morphological characters [13], fixed in 70% (v/v) ethanol and stored at − 20 °C until further study was performed. Some representative examined specimens were deposited in the Specimen Museum and Resource Bank of Parasites, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (accession no. SMRBP-LVRI-2018001-5), for retrieval and comparative study. Total genomic DNA was isolated from individual worms using sodium dodecyl sulphate/proteinase K treatment, followed by spin-column purification (Wizard® SV Genomic DNA Purification System, Promega). Single specimen was further identified as T. leonina by PCR-based sequencing of ITS1 and ITS2 rDNA [14], and both regions had 100% nucleotide homology with those of T. leonina from Lynx lynx deposited in GenBank (GenBank accession no. JF837179). However, these sequences differed by 3% in ITS1 compared with the sequence of T. leonina from a wolf in China (GenBank: JF837174), and by 8% in ITS2 compared with the sequence of T. leonina from a golden jackal in Croatia (GenBank: MF495479).
Long-PCR amplification and sequencing
The primers (Additional file 1: Table S1) were designed according to well-conserved regions of the mt genome sequences of T. leonina from the dog (GenBank: NC023504) [11]. These primers were then used to amplify the entire mt genome by long-PCR as seven overlapping fragments (2–4 kb each) from the genomic DNA. The PCR reactions contained ~ 20 ng of genomic DNA and were carried out in 50-μl reaction volumes containing 25 μl 2× Phusion Emweald Amp MAX HS PCR Master Mix (TaKaRa, Dalian, China), 2 μl of each primer, 2 μl DNA and 19 μl of ddH2O in a thermocycler (Bio-Rad, California, USA). Cycling amplification was executed under the following conditions: 94 °C for 2 min (initial denaturation), then 94 °C for 30 s (denaturation), 50–55 °C for 45 s (annealing), 66 °C (~ 1 kb region) for 5 min (extension) for 9 cycles, followed by 94 °C for 30 s, 50–55 °C for 45 s, and 66 °C for 5 min for 21 cycles, and a final extension at 68 °C for 10 min. Each PCR reaction yielded a single band detected in a 1.0% (w/v) agarose gel upon ethidium-bromide staining. PCR products were column-purified and then sequenced from both directions.
Sequence analyses
Contiguous sequences were assembled manually and aligned with previously published mt genome sequence of T. leonina from the dog using Clustal X program v.1.83 [15] to identify gene boundaries. The open-reading frames (ORFs) and each protein-coding gene (PCG) were predicted using the program MEGA 5.0 [16], then the translation start and stop codons were identified based on comparison with those reported previously [11]. Putative secondary structures of 22 tRNA genes were identified using tRNAscan-SE [17] with manual adjustment, and two rRNA genes were predicted by comparison with those of T. leonina from the dog [11].
Phylogenetic analyses
The deduced amino acid sequences of 12 PCGs were aligned individually with those of published mt genomes of other selected species of the Ascaridoidea, using Thelazia callipaeda as the outgroup [18]. All amino acid sequences were aligned using MAFFT 7.122 [19] and then concatenated into a single alignment. The regions and sites of ambiguous alignment were eliminated using Gblocks online server [20]. Bayesian inference (BI) was used for phylogenetic analyses as described previously [6, 11]. Phylograms were drawn using the program FigTree v.1.4.
Sequencing of cox1 and analysis
The primers JB3/JB4.5 were used for PCR amplification and then sequencing of a portion of the cox1 gene from multiple T. leonina samples from the cheetah. These newly generated sequences and previously published cox1 sequences of T. leonina were aligned using the software MAFFT 7.122. Phylogenetic analysis was performed using the same methods as described above.
Results and discussion
General features of the mt genome of T. leonina from the cheetah
The entire mt genome of T. leonina from the cheetah is 14,685 bp in size. This mt genome contains 12 PCGs, 22 tRNA genes and two rRNA genes (Additional file 1: Table S2). The nucleotide content of the mt genome sequence of T. leonina from the cheetah is biased toward A+T (71.1%), in accordance with mt genomes of other nematodes sequenced to date (such as Ascaris suum, Ascaris lumbricoides, Toxocara canis, Toxocara cati) [6, 21]. All mt genes of T. leonina from the cheetah are transcribed in the same direction. Gene content, arrangement and composition of the mt genome of T. leonina from the cheetah are the same as those of other nematodes, such as T. canis, T. cati and Toxocara malaysiensis [21]. Additionally, the genes in the mt genome of T. leonina from the cheetah overlap by 1 bp between tRNA-Trp and tRNA-Glu (Additional file 1: Table S2). There are 12 intergenic regions (from 1 to 10 bp in length) in the mt genome of T. leonina from the cheetah (Additional file 1: Table S2). In addition, the mt genome of T. leonina from the cheetah contains two non-coding (AT-loop) regions (1450 bp) (Additional file 1: Table S2).
Annotation
The mt genome of T. leonina from the cheetah encodes 12 PCGs. The most common start codon for the mt genome of T. leonina from the cheetah is TTG (7 of 12 PCGs), followed by ATT (3 of 12 PCGs), GTG (1 of 12 PCGs) and GTT (1 of 12 PCGs) (Additional file 1: Table S2). In this mt genome, the PCGs use TAG, T and TA as a stop codon (Additional file 1: Table S2), consistent with previous studies [11, 12]. Twenty-two tRNA genes in this mt genome range from 52 bp to 62 bp in length. The rrnL and rrnS genes of the mt genome of T. leonina from the cheetah were 959 bp and 700 bp in size, respectively (Additional file 1: Table S2), and the long and short non-coding regions were located between tRNA-SerUCN and tRNA-Asn, and nad4 and cox1, respectively (Additional file 1: Table S2).
Comparative analyses between T. leonina from the cheetah and the dog
The entire mt genome of T. leonina from the cheetah is 375 bp and 408 bp longer than that of T. leonina from the dog and the South China tiger [11, 12], respectively. Nucleotide sequence difference across the entire mt genome (except for the non-coding region sequences) between T. leonina from the cheetah and the dog was 7.2%. This level of mt genome divergence is similar to those between different nematode species. For example, the nucleotide sequence difference was 3.8–9.4% among members of the Pseudoterranova decipiens species complex [22], 11.1% between Ancylostoma duodenale and Ancylostoma caninum [23, 24], 12.5% between T. malaysiensis and T. cati [21], and 6.2% between Baylisascaris schroederi and Baylisascaris ailuri [25]. For the 12 PCGs, the sequence difference between T. leonina from the cheetah and the dog, was 5.0–9.7% (nucleotide sequences) and 1.0–7.2% (aa sequences); these divergence levels are similar to those recorded between other congeneric nematode species. For example, the amino acid sequence divergence between Oesophagostomum dentatum and O. quadrispinulatum was 0.4–8.3% [26], and it was 0.4–7.1% between Angiostrongylus mackerrasae and A. cantonensis [27]. For the 22 tRNAs, sequence difference was 6.0% between T. leonina from the cheetah and the dog. For the rrnL and rrnS genes, sequence difference was both 6.1% between T. leonina from the cheetah and the dog (Table 1).
Nucleotide sequence difference in the mt cox1 gene was also assessed among T. leonina isolates (n = 23; coded TC1–TC23) from canid and felid hosts. Nucleotide differences in cox1 sequences among the T. leonina isolates from felids (n = 17; coded TC1–TC17) occurred at 10 sites (2.7%). Nucleotide differences among the T. leonina isolates from canids (n = 6; coded TC18-TC23) occurred at 21 sites (6.2%). However, comparison of the mt cox1 sequences among all the T. leonina isolates (n = 23) revealed 36 variable sites (10.6%). Our results revealed substantial sequence differences in the cox1 gene among different hosts, consistent with that of previous studies [8, 9].
Taken together, these results support our proposal that T. leonina from different hosts may present a species complex because a previous study has shown that sequence difference in mt genes between closely-related nematode species were 10–20% [28].
Phylogenetic analysis
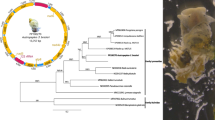
Phylogenetic analyses based on amino acid sequences of 12 PCGs showed clear genetic distinctiveness between T. leonina from the cheetah and the dog, and they were a sister taxon to a clade containing Ascaris spp., Parascaris spp. and Baylisascaris spp. of the family Ascarididae, with strong support (Bayesian posterior probabilities = 1). Basal to these taxa were the families Anisakidae, Toxocaridae, Heterakidae and Ascaridiidae (Fig. 1). The genetic distance between the two T. leonina isolates is approximately the same (looking at tree topologies and branch lengths) as that among the Baylisascaris spp. and the Pseudoterranova spp. (Fig. 1). In addition, phylogenetic analyses based on the mt cox1 sequences also showed support for the separation of T. leonina from different hosts into three distinct clades (Fig. 2). Our results showed that T. leonina isolates from felid hosts were in the same clade; however, T. leonina isolates from canid hosts formed two clades (Bayesian posterior probabilities > 0.71). Toxascaris leonina from red foxes was sister to the two remaining clades (Fig. 2). These results are consistent with a recent study [29].

Relationships of Toxascaris leonina with other selected ascaridoidea nematodes based on mitochondrial sequence data. The concatenated amino acid sequences of 12 protein-coding genes were subjected to a Bayesian inference analysis using Thelazia callipaeda as the outgroup. Bayesian posterior probabilities values are indicated

Inferred phylogenetic relationships among representative Toxascaris leonina samples by Bayesian inference based on mitochondrial cox1 nucleotide sequence data, using Ascaris suum as the outgroup. Bayesian posterior probabilities values are indicated
Conclusions
In summary, the present study determined the entire mt genome sequence of T. leonina from the cheetah. Comparative analyses of the mtDNA datasets provided genetic evidence that T. leonina from canid and felid hosts represents a species complex. To further test this hypothesis, additional studies are warranted (i) to explore, in detail, nucleotide variation in rDNA and mtDNA within and among T. leonina populations from different hosts and geographical locations; (ii) to determine the complete mt genomes of T. leonina from other hosts; and (iii) to undertake detailed morphological studies of T. leonina from a range of hosts.
Abbreviations
- mt:
-
mitochondrial
- mtDNA:
-
mitochondrial DNA
- rDNA:
-
ribosomal DNA
- BI:
-
Bayesian inference
- nad2:
-
NADH dehydrogenase subunit 2
- cox1:
-
cytochrome c oxidase subunit 1
- cox2:
-
cytochrome c oxidase subunit 2
- ATP6:
-
ATP synthase F0 subunit 6
- cox3:
-
cytochrome c oxidase subunit 3
- nad3:
-
NADH dehydrogenase subunit 3
- nad5:
-
NADH dehydrogenase subunit 5
- nad4:
-
NADH dehydrogenase subunit 4
- nad4L:
-
NADH dehydrogenase subunit 4L
- nad6:
-
NADH dehydrogenase subunit 6
- cytb:
-
cytochrome b
- nad1:
-
NADH dehydrogenase subunit 3
- tRNA:
-
transfer RNA
- rrnL:
-
large subunit of rRNA
- rrnS:
-
small subunit of rRNA
- ITS:
-
internal transcribed spacers
- BI:
-
Bayesian inference
References
Anderson RC. Nematode parasites of vertebrates, their development and transmission. 2nd ed. Wallingford: CAB International; 2000.
Okulewicz A, Lonc E, Borgsteede FH. Ascarid nematodes in domestic and wild terrestrial mammals. Pol J Vet Sci. 2002;5:5277–81.
Okulewicz A, Perec-Matysiak A, Buńkowska K, Hildebrand J. Toxocara canis, Toxocara cati and Toxascaris leonina in wild and domestic carnivores. Helminthologia. 2012;49:3–10.
Hu M, Chilton NB, Gasser RB. The mitochondrial genomics of parasitic nematodes of socio-economic importance: recent progress, and implications for population genetics and systematics. Adv Parasitol. 2004;56:133–212.
Liu GH, Zhao L, Song HQ, Zhao GH, Cai JZ, Zhao Q, et al. Chabertia erschowi (Nematoda) is a distinct species based on nuclear ribosomal DNA sequences and mitochondrial DNA sequences. Parasit Vectors. 2014;7:44.
Liu GH, Wu CY, Song HQ, Wei SJ, Xu MJ, Lin RQ, et al. Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene. 2012;492:110–6.
Liu GH, Gasser RB, Su A, Nejsum P, Peng L, Lin RQ, et al. Clear genetic distinctiveness between human- and pig-derived Trichuris based on analyses of mitochondrial datasets. PLoS Negl Trop Dis. 2012;6:1539.
Li XY, Jin YC, Nie Y, Su YG, Tang CH, Li F. Sequence comparison and phylogenetic analysis of mitochondrial cox1 gene of Toxascaris leonina in Cheetah. China Anim Health Insp. 2018;35:97–100 (In Chinese).
Song MR, Li KX, Shi X, Hu W, Tan LP, Luo Q, et al. Phylogenetic analysis of three mitochondrial genes of Toxascaris leonina from South China tiger. Chin J Anim Infect Dis. 2015;23:68–75 (In Chinese).
Hu M, Gasser RB. Mitochondrial genomes of parasitic nematodes—progress and perspectives. Trends Parasitol. 2006;22:78–84.
Liu GH, Zhou DH, Zhao L, Xiong RC, Liang JY, Zhu XQ. The complete mitochondrial genome of Toxascaris leonina: comparison with other closely related species and phylogenetic implications. Infect Genet Evol. 2014;21:329–33.
Li K, Yang F, Abdullahi AY, Song M, Shi X, Wang M, et al. Sequence analysis of mitochondrial genome of Toxascaris leonina from a South China Tiger. Korean J Parasitol. 2016;54:803–7.
Skrjabin KI, Shikhobalova NP, Mozgovoi AA. Oxyurata and ascaridata. In: Skrjabin KI, editor. Keys to parasitic nematodes, vol. II. New Delhi: Amerind Publishing Co Pvt Ltd; 1991.
Zhu X, Chilton NB, Jacobs DE, Boes J, Gasser RB. Characterisation of Ascaris from human and pig hosts by nuclear ribosomal DNA sequences. Int J Parasitol. 1999;29:469–78.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–82.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Liu GH, Gasser RB, Otranto D, Xu MJ, Shen JL, Mohandas N, et al. Mitochondrial genome of the eyeworm, Thelazia callipaeda (Nematoda: Spirurida), as the first representative from the family Thelaziidae. PLoS Negl Trop Dis. 2013;7:2029.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77.
Li MW, Lin RQ, Song HQ, Wu XY, Zhu XQ. The complete mitochondrial genomes for three Toxocara species of human and animal health significance. BMC Genomics. 2008;9:224.
Liu GH, Nadler SA, Liu SS, Podolska M, D’Amelio S, Shao R, et al. Mitochondrial phylogenomics yields strongly supported hypotheses for ascaridomorph nematodes. Sci Rep. 2016;6:39248.
Hu M, Chilton NB, Gasser RB. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int J Parasitol. 2002;32:145–58.
Jex AR, Waeschenbach A, Hu M, van Wyk JA, Beveridge I, Littlewood DT, et al. The mitochondrial genomes of Ancylostoma caninum and Bunostomum phlebotomum—two hookworms of animal health and zoonotic importance. BMC Genomics. 2009;10:79.
Xie Y, Zhang Z, Wang C, Lan J, Li Y, Chen Z, et al. Complete mitochondrial genomes of Baylisascaris schroederi, Baylisascaris ailuri and Baylisascaris transfuga from giant panda, red panda and polar bear. Gene. 2011;482:59–67.
Lin RQ, Liu GH, Hu M, Song HQ, Wu XY, Li MW, et al. Oesophagostomum dentatum and Oesophagostomum quadrispinulatum: Characterization of the complete mitochondrial genome sequences of the two pig nodule worms. Exp Parasitol. 2012;131:1–7.
Aghazadeh M, Traub RJ, Mohandas N, Aland KV, Reid SA, McCarthy JS, et al. The mitochondrial genome of Angiostrongylus mackerrasae as a basis for molecular, epidemiological and population genetic studies. Parasit Vectors. 2015;8:473.
Blouin MS. Molecular prospecting for cryptic species of nematodes: mitochondrial DNA versus internal transcribed spacer. Int J Parasitol. 2002;32:527–31.
Fogt-Wyrwas R, Dabert M, Jarosz W, Rząd I, Pilarczyk B, Mizgajska-Wiktor H. Molecular data reveal cryptic speciation and host specificity in Toxascaris leonina (Nematoda: Ascarididae). Vet Parasitol. 2019;266:80–3.
Acknowledgements
Not applicable.
Funding
This study was supported by the Planned Programme of Hunan Province Science and Technology Innovation (Grant no. 2018RS3085) and the Training Program for Excellent Young Innovators of Changsha (Grant No. KQ1802035).
Availability of data and materials
The mitochondrial genome and gene sequences of Toxascaris leonina from the cheetah have been deposited in the GenBank database under the accession numbers MK516267; MK522168-MK522181. Five adult individuals of T. leonina collected from the cheetah from Changsha were deposited in the Specimen Museum and Resource Bank of Parasites, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (accession no. SMRBP-LVRI-2018001-5).
Authors’ contributions
GHL and YCJ designed this study and critically revised the manuscript. GHL, YCJ, XYL and JHL performed the experiments, analysed data and drafted the manuscript. GHL and XQZ participated in sample collection and manuscript revision. All the authors read and approved the final manuscript.
Ethics approval and consent to participate
All procedures involving animals in the present study were approved, and this study was approved by the Animal Ethics Committee of Hunan Agricultural University (No. 43321503).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
Sequences of primers used to amplify Long-PCR fragments from Toxascaris leonina. Table S2. Mitochondrial genome organization of Toxascaris leonina from cheetah (Tc) and Toxascaris leonina from dog (Td).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jin, YC., Li, XY., Liu, JH. et al. Comparative analysis of mitochondrial DNA datasets indicates that Toxascaris leonina represents a species complex. Parasites Vectors 12, 194 (2019). https://doi.org/10.1186/s13071-019-3447-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3447-2