
Abstract
Background
Studying mitochondrial (mt) genomics has important implications for various fundamental areas, including mt biochemistry, physiology and molecular biology. In addition, mt genome sequences have provided useful markers for investigating population genetic structures, systematics and phylogenetics of organisms. Toxocara canis, Toxocara cati and Toxocara malaysiensis cause significant health problems in animals and humans. Although they are of importance in human and animal health, no information on the mt genomes for any of Toxocara species is available.
Results
The sizes of the entire mt genome are 14,322 bp for T. canis, 14029 bp for T. cati and 14266 bp for T. malaysiensis, respectively. These circular genomes are amongst the largest reported to date for all secernentean nematodes. Their relatively large sizes relate mainly to an increased length in the AT-rich region. The mt genomes of the three Toxocara species all encode 12 proteins, two ribosomal RNAs and 22 transfer RNA genes, but lack the ATP synthetase subunit 8 gene, which is consistent with all other species of Nematode studied to date, with the exception of Trichinella spiralis. All genes are transcribed in the same direction and have a nucleotide composition high in A and T, but low in G and C. The contents of A+T of the complete genomes are 68.57% for T. canis, 69.95% for T. cati and 68.86% for T. malaysiensis, among which the A+T for T. canis is the lowest among all nematodes studied to date. The AT bias had a significant effect on both the codon usage pattern and amino acid composition of proteins. The mt genome structures for three Toxocara species, including genes and non-coding regions, are in the same order as for Ascaris suum and Anisakis simplex, but differ from Ancylostoma duodenale, Necator americanus and Caenorhabditis elegans only in the location of the AT-rich region, whereas there are substantial differences when compared with Onchocerca volvulus,Dirofiliria immitis and Strongyloides stercoralis. Phylogenetic analyses based on concatenated amino acid sequences of 12 protein-coding genes revealed that the newly described species T. malaysiensis was more closely related to T. cati than to T. canis, consistent with results of a previous study using sequences of nuclear internal transcribed spacers as genetic markers.
Conclusion
The present study determined the complete mt genome sequences for three roundworms of human and animal health significance, which provides mtDNA evidence for the validity of T. malaysiensis and also provides a foundation for studying the systematics, population genetics and ecology of these and other nematodes of socio-economic importance.
Similar content being viewed by others


Background
Mitochondria are sub-cellular organelles involved in oxidative phosphorylation, offering energy to organisms. They play important roles in cellular metabolism, living and apoptosis. Within these organelles, most metazoan species possess a compact, circular mitochondrial (mt) genome, which varies in size from 14 to 20 kb [1]. The metazoan mt genome usually contains a complement of genes encoding 12–13 protein subunits of the enzymes involved in oxidative phosphorylation, 22 transfer RNAs, and two ribosomal RNAs. There are no introns within genes, and no to limted spacer regions between genes [1, 2]. Studying mt genomes has important implications for various fundamental areas, including mt biochemistry, physiology and molecular biology. In addition, mt genome sequences have provided useful markers for investigating population genetic structures, systematics and phylogenetics of organisms due to their maternal inheritance, higher mutation rates than nuclear genes and relatively conserved genome structures [3–5].
Although Nematoda is the second largest animal phylum, to date only over 40 complete mitochondrial DNA sequences of nematode species have been deposited in GenBank™ [6–15]. In the order Ascaridada, the mt genomes of only two species have been sequenced [6, 15]. The lack of knowledge of mt genomics for parasitic nematodes in this order is a major limitation for population genetic and phylogenetic studies of the pathogens in the order Ascaridada including the species in the genus Toxocara.
Toxocara canis, Toxocara cati and Toxocara malaysiensis are the common ascaridoid nematodes of dogs and cats, causing significant health problems. Infection of dogs with T. canis is quite common, with the prevalence ranging from 5.5% to 64.7% [16–18]. Infection of cats with T. cati is also found worldwide, with infection rates of up to 25.2% to 66.2% [19–21]. More importantly, T. canis and T. cati are of public health significance due to their larvae of being able to invade humans and cause diseases such as ocular larva migrans (OLM), visceral larva migrans (VLM), eosinophilic meningoencephalitis (EME) and/or covert toxocariasis (CT) [22–24]. Although they are of importance in human and animal health, there is still no information on the mt genomes available for any of Toxocara species.
The objectives of the present study were to fill some of these knowledge gaps by determining the structure, organization and sequence of the complete mt genomes of T. canis, T. cati and T. malaysiensis of human and animal significance, and to provide mtDNA evidence for the recently described species T. malaysiensis [25, 26]. Also, the features of mt genomes of the three ascaridoid nematodes, such as the gene arrangements, structures, compositions, as well as translation and initiation codons and codon usage patterns were compared with those of other nematodes in the same order Ascaridata, namely Ascaris suum and Anisakis simplex. The phylogenetic relationships among these ascaridoid nematodes were also investigated using the protein-coding amino acid sequences.
Results and Discussion
General features of the mt genome of three Toxocara species
The complete mt genomes of T. canis, T. cati and T. malaysiensis are 14,322 bp, 14,029 bp and 14266 bp in length, respectively. These complete mt genome sequences have been deposited in the GenBank™ under the accession numbers AM411108 (for T. canis), AM411622 (for T. cati) and AM412316 (for T. malaysiensis). All three mt genomes contain 12 protein-coding genes (cox 1-3, nad 1-6, nad 4L, atp 6 and cyt b), 22 transfer RNA genes and two ribosomal RNA genes, but lack an atp 8 gene (Table 1). These circular genomes are typical of other nematode mitochondrial genomes except for Trichinella spiralis in which the atp 8 gene is encoded. All genes are transcribed in the same direction as found in other members of the secernentean nematodes sequenced to date, but in contrast to T. spiralis and Xiphinema americanus [8, 13].
The mt genome arrangement of T. canis, T. cati and T. malaysiensis are the same as those of A. suum and A. simplex and almost identical to the genome structures of strongylida nematodes Ancylostoma duodenale, Necator americanus, Cooperia oncophora and rhabditid nematode Caenorhabditis elegans, with the exception of the relative position of an AT-rich region and the number of non-coding regions (NCR). In the mt genomes of the three Toxocara species, the AT-rich region is located between trn S2 and trn N, whereas it is positioned between trn A and trn P in A. duodenale, N. americanus, Co. oncophora and C. elegans. The only NCR found in Toxocara spp. was between nad 4 and cox 1, as has been identified in the aforementioned species. The second NCR located between nad 3 and nad 5 in the hookworms A. duodenale and N. americanus was not found in Toxocara spp. [9]. The genome structures of these Toxocara species differ significantly from those of Onchocerca volvulus, Strongyloides stercoralis, Dirofiliria immitis, X. americanus and T. spiralis in the location of the AT-rich region, and some tRNA and protein-coding genes. Comparison of the gene arrangements of these three Toxocara species with those of the other nine representatives of the secernentean nematodes suggests that T. canis, T. cati and T. malaysiensis are more closely related to A. suum and A. simplex than to C. elegans, A. duodenale, N. americanus, Co. oncophora, O. volvulus,D. immitis and S. stercoralis.
The nucleotide compositions of the entire mtDNA sequences for T. canis, T. cati and T. malaysiensis are biased toward A and T, with T being the most favored nucleotide and C the least favored, in accordance with mt genomes of other nematodes. The content of A+T is 68.57% for T. canis (21.9% A, 46.7% T, 22.0% G and 9.4% C), which is the lowest of the nematodes studied to date (Table 2). The content of A+T is 69.95% for T. cati (22.3% A, 47.7% T, 20.9% G and 9.1% C) and 68.86% for T. malaysiensis (21.7% A, 47.2% T, 22.0% G and 9.1% C), respectively (Table 2).
Protein-coding genes and codon usage patterns
The boundaries between protein-coding genes of mt genomes of T. canis, T. cati and T. malaysiensis were determined by aligning their sequences and by identifying translation initiation and termination codons with those of A. suum. For each of the three Toxocara species, the lengths of protein-coding genes cox 3, nad 1, nad 2, nad 3 and nad 4 are the same as those of A. suum, whereas the lengths of genes atp 6, nad 4L,nad 5 and nad 6 are reduced, and the lengths of cox 2 and cyt b are increased (Table 1) when compared to those of A. suum. The length of cox 1 in T. canis and T. cati is the same as that of A. suum, but the length of cox 1 in T. malaysiensis is increased (Table 3).
The inferred nucleotide and amino acid sequences for each of the 12 mt proteins of T. canis, T. cati and T. malaysiensis were compared with those of A. suum and A. simplex. The identity of the nucleotide and amino acid sequences is 71~90% and 72.2~95.8%, respectively (Table 3). Based on identity, cox 1 is the most conserved protein-coding gene, while the cyt b is the least conserved. For all 12 proteins, the amino acid sequence identities are higher when compared between each of the three Toxocara species than between each of Toxocara specis and A. suum or A. simplex. The identities of the nucleotide and amino acid sequences among five ascaridoid species are higher than those among C. elegans, A. duodenale, N. americanus and Co. oncophora (data not shown). These findings reinforce the conclusion that the three Toxocara species are genetically more closely related to A. suum and A. simplex than to C. elegans, A. duodenale, N. americanus and Co. oncophora.
The predicted initiation and termination codons for the protein-coding genes of the three Toxocara species were compared with those of ascaridoid species (A. suum and A. simplex) and with selected species representing different nematode orders including the human hookworm A. duodenale, filarioid worm O. volvulus, and rhabditid species S. stercoralis. The most common start codon for three Toxocara species is TTG (four of 12 protein-coding genes), followed by ATT (three of 12 protein-coding genes for T. canis and T. malaysiensis, two for T. cati), ATA (two of 12 protein-coding genes for T. canis and T. cati, one for T. malaysiensis), and ATG, GTG, GTT and GTA are used as initiation codons. GTG, which is used in the cyt b of T. canis and T. malaysiensis, and cox 3 of T. malaysiensis, is not used as a start codon in the other nematodes compared. GTA used in the cox 2 of T. malaysiensis is also used as a start codon for nad 4L in D. immitis (data not shown). Seven of the 12 protein-coding genes were predicted to have a TAG or TAA translation termination codon. The remaining protein-coding genes were inferred to end with an abbreviated stop codon, such as TA or T. For the three Toxocara species, the 3'-end of most of these genes is immediately adjacent to a downstream tRNA gene (Table 1), which is consistent with the arrangement in the mt genomes of A. suum and A. simplex [6, 15], but in contrast to that of C. elegans where both the nad 1 and nad 3 genes terminate in T or TA, and are followed by the putative ATT translation initiation codon of their downstream protein-coding genes. The protein-coding gene nad 6 ended with an abbreviated stop codon TA is followed by the putative ATT translation initiation codon of its downstream protein coding gene nad 4L, which is similar to C. elegans.
In general, the nucleotides of metazoan mt genomes are not randomly distributed, and such nucleotide bias is often associated with unequal usage of synonymous codons. The mt genome nucleotide composition of nematodes is biased toward A and T. The A+T content of protein-coding genes ranged from 63.2% to 74.9% for all three Toxocara species (Table 2). This bias in nucleotide composition toward AT (Table 2) affects both the codon usage pattern and amino acid composition of proteins. In these three species examined, all 64 possible codons were used, and the most frequently used codon was TTT (Phe) while the least used codon was CGC (Arg). The preferred nucleotide usage at the third codon position of Toxocara mt protein-coding genes reflects the overall nucleotide composition of the mtDNA. At this position, T is the most frequently used, and C the least frequently used. The codons ending in G have higher frequencies than the codons ending in A, which is similar to A. suum, but opposite to C. elegans, A. duodenale and N. americanus.
The protein-coding genes of the three Toxocara genomes are biased toward using amino acids encoded by T-, A- and G-rich codons (data not shown). The AT-rich codons represent amino acids Phe, Ile, Met, Tyr, Asn or Lys, and the GC-rich codons represent Pro, Ala, Arg or Gly. T-rich codons (with ≥2 Ts in a triplet) comprise Phe (13.4% TTT and 0.6% TTC), Leu (9.0% TTG, 3.8% TTA, and 1.5% CTT), Ile (5.3% ATT), Val (6.2% GTT), Tyr (4.1% TAT), Ser (4.0% TCT), and Cys (1.5% TGT), and account for approximately half (49.6%) of the total amino acid composition. A- and G-rich codons (with ≥2 As and Gs, respectively) represent 9.7% and 13.3% of the total amino acid composition, respectively (data not shown). In contrast, the proportion of C-rich codons (with ≥2 Cs) is much lower (4.0%). This codon bias against C is even more evident when only the third codon positions are considered in both four fold and two fold degenerate codon families. When the frequencies of synonymous codons within the AT-rich group were compared, the frequency was always decreased if the third position was substituted with a C. For instance, the relative frequencies of codons for Phe are 13.4% TTT and 0.6% TTC, respectively. This result suggests that the third codon positions mostly reflect mutational bias against C. A greater translational efficiency has also been considered to be a potential cause underlying observed codon usage bias [27].
Transfer RNA genes
Twenty-two tRNA sequences (ranging from 52 to 63 bases in size; see Table 1) were identified in the mt genomes of the three Toxocara species. Their putative secondary structures (not shown) are similar to those of other nematode mtDNAs [6, 7, 9–11, 15], with the exception of T. spiralis [8], and differ from the conventional cloverleaf-like structures found in other metazoan mtDNA molecules. Common features of the predicted secondary structures (not shown) of 22 tRNA genes in T. canis, T. cati and T. malaysiensis mtDNA include a 7 bp amino-acyl stem, a 4 bp DHU stem with a 4–8 bp loop, a 5 bp anticodon stem with a loop of 7 bp (a T always preceding an anticodon as well as an A or a G always following an anticodon), and a TV replacement loop of 6–12 bp with some exceptions, in accordance with other nematodes. The exception is trn S1 (AGN) in which the DHU-arm is lacking.
Ribosomal RNA genes
The rrn S and rrn L genes of the three roundworm species were identified by sequence comparison with those of A. suum. The rrn S is located between trn E and trn S (UCN), and rrn L is located between trn H and nad 3. The two genes are separated from one another by the protein-coding genes nad 3, nad 5, nad 6 and nad 4L. The size of the rrn S gene is 697 bp for T. canis, 696 bp for T. cati and 696 bp for T. malaysiensis. The size of the rrn L gene is 958 bp for T. canis, 955 bp for T. cati and 955 bp for T. malaysiensis. The sizes of the two rRNA genes for the three Toxocara species are similar to those of other nematodes (Table 1). The A+T contents of the rrn S for T. canis, T. cati and T. malaysiensis are 66.9%, 68.8% and 66.4%, respectively, whereas those of the rrn L are higher (72.0%, 73.5% and 70.1%, respectively), and A+T contents of the two genes are the lowest among the nematodes studied to date (Table 2). Sequence identity in the rrn S and rrn L genes is 87.5% and 83.6% between T. canis and T. cati, 87.9% and 82.4% between T. canis and T. malaysiensis, and 91.4% and 85.2% between T. cati and T. malaysiensis, respectively.
Non-coding regions
Like A. suum and A. simplex, the longest non-coding region (AT-region) in the three Toxocara mt genomes is located between the trn S2 and trn N. Their sizes are 985 bp (T. canis), 711 bp (T. cati) and 936 bp (T. malaysiensis), and A+T contents are 79.4% (T. canis), 81.3% (T. cati) and 78.4% (T. malaysiensis), respectively, which are significantly lower than the comparable NCRs of nematodes studied to date (Table 2). Repeated sequence motifs (CR1-CR6) present in the C. elegans AT-rich region [6] are not found in Toxocara spp. However, there are a lot of AT dinucleotide repeats in the AT-region of Toxocara mt genomes of which the longest consists of repeat units (34 base pairs). Similar AT dinucleotide repeats has been found in A. suum [6]. The function or role of these AT repeats is currently unknown [6, 7]. Although nothing is yet known about the replication process(es) in the mtDNA of parasitic nematodes, the high A+T content and the predicted structure of the AT-rich non-coding region suggests an involvement in the initiation of replication [28].
For the three roundworm species, the second longest non-coding region is located between genes cox 1 and nad 4, as in the mt genomes of A. suum [6]. Its length is 111 bp (T. canis), 116 bp (T. cati) and 112 bp (T. malaysiensis), with an A+T content of 86.2%, 74.1% and 74.1%, respectively, and is shorter than that of A. suum (117 bp). The non-coding region for the three Toxocara species could form a hairpin loop structure (AATTTTTAAAAATT).
Phylogenetic analyses
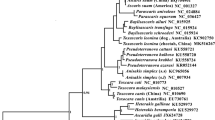
The final alignment of the amino acid sequences of 12 proteins for the six taxa (T. canis, T. cati, T. malaysiensis, A. suum,A. simplex and O. volvulus) yield 3516 characters (2079 variable, 339 parsimony-informative). In all three phylogenetic analyses, three Toxocara species were clustered together (Fig. 1). T. malaysiensis, the recently described Toxocara species from cat [26], was inferred to be the sister species of T. cati with high bootstrap values. This result was consistent with that of a previous study [25] which used sequences of internal transcribed spacers of nuclear ribosomal DNA, thus providing mt DNA evidence for the validity of T. malaysiensis as an ascaridoid of cats. T. malaysiensis is more closely related to T. cati, the common ascaridoid of cats, than to T. canis, the common ascaridoid of canids.

Inferred phylogenetic relationship among the five ascaridoid species (Toxocara canis, T. cati, T. malaysiensis, Ascaris suum, Anisakis simplex) derived from neighbour joining (NJ), maximum parsimony (MP) and maximum likelihood (ML) analyses of amino acid sequences of 12 protein-coding genes from their mitochondrial genomes, using one filarioid species (Onchocerca volvulus) as outgroup. The numbers along branches indicate bootstrap values resulting from different analyses in the order: NJ/MP/ML. Values lower than 50 are given as "-".
Toxocara species was resolved being more closely related to A. suum than to A. simplex with moderate support in the phylogenetic analyses, which was consistent with results of previous morphological and molecular studies [29, 30]. But relationship between A. suum and A. simplex was poorly inferred in the MP and ML analyses (Fig. 1).
Conclusion
Toxocara species are the important socio-economic parasites because they have significant impact on human health. The determined mt genomes of the three roundworms, T. canis, T. cati and T. malaysiensis, add the mtDNA data to the order Ascaridida, which includes a broad range of parasites of major socio-economic importance. Determination of the complete mt genome sequences for three Toxocara species of human and animal health significance provides a foundation for studying the systematics, population genetics and ecology of these and other nematodes of socio-economic importance.
Methods
Parasites and DNA extraction
The ascaridoid nematodes used in the present study were from Zhanjiang city for T. canis, Changsha city for T. cati and Guangzhou city for T. malaysiensis, China, respectively. Adult nematodes of three Toxocara species were obtained from the intestines of dogs and/or cats, washed in physiological saline, identified primarily based on morphological characters to species, fixed in 70% (v/v) ethanol and stored at -20°C until use. Total DNA was isolated from individual nematodes using sodium dodecyl-sulphate/proteinase K treatment, followed by spin-column purification (Wizard Clean-Up, Promega). The specific identity of each individual nematode was verified by species-specific PCR amplification using the sequences of the first and/or second internal transcribed spacers (ITS-1 and/or ITS-2) of ribosomal DNA (rDNA) as the species-specific genetic markers [31, 32].
Long-PCR amplification and sequencing
Using primer set 5F/40R (~9 kb region) and 39F/42R (~6 kb region) [33], the entire mt genome of each Toxocara species was amplified in two overlapping fragments by long-PCR from approximately 20 ng of total genomic DNA purified from an individual nematode, respectively. The primers were designed to mt sequences found to be relatively conserved among A. suum and C. elegans. Primer 5F (forward, 5'-TATGAGCGTCATTTATTGGG-3') and its complementary primer 42R (reverse, 5'-CCCAATAAATGACGCTCATA-3') were designed to the nad 1 gene, while primers 39F (forward, 5'-TAAATGGCAGTCATTAGCGTGA-3') and 40R (reverse, 5'-GAATTAAACTAATATCACGT-3') were designed to the rrn L gene [33]. Long-PCR cycling conditions used were 92°C for 2 min (initial denaturation), then 92°C for 10 s (denaturation), 50°C for 30 s (annealing), and 60°C (for 6 kb) or 68°C (for 9 kb) for 10 min (extension) for 10 cycles, followed by 92°C for 10 s, 50°C for 30 s, and 60°C (for 6 kb) or 68°C (for 9 kb) for 10 min for 20 cycles, with a cycle elongation of 10 s for each cycle and a final extension at 60 or 68°C for 7 min. Each PCR reaction yielded a single band detected in a 0.8% (w/v) agarose gel upon ethidium-bromide staining (not shown). PCR products were sent to TaKaRa Company (Dalian, China) for sequencing using a primer walking strategy, and the following primers were used for sequencing the complete genomes of three Toxocara species: MW1F (5'-ATATTAGATTGTAAATCTAAAG-3'), MW5-1R (5'-ACACGAAAAACCCAAAAATT-3'), MW5-1F (5'-ATTATGTCGTCTTGATAAGGC-3'), MWTCT62F (5'-GCTATTATGATGGAGTGTTTTGTG-3'), MW62-1F (5'-GTTACCTTTTTTACCTGTGCTTGT-3'), MW40R (5'-CTGGTGAACAGATATCAAAGGACATCATC-3'), MW5-2F (5'-TAGATTGGTATATTTTGATTTTAACTTGTCT-3'), MW79F (5'-GGGTTTTAATTTATTTCGTTT-3'), MW78R (5'-GTAGTAGGTGCTTCTACATG-3'), MW62-3F (5'-TCTACTGAGTCTTTTAATTGAG-3'), MWXIAYOU (5'-CCCAATAAATGACGCTCATA-3'), MWTM-1 (5'-GTATATTGCTAGCGGGTCTTTT-3'), MW0209-115 (5'-AAACTGGTGAACAGATATCAAA-3'), MW4F (5'-GGAGTAAGTTGTAGTAAAGTAGA-3'), MW38R (5'-AGAAAAAGCAATCTCATAAGAA-3'), MW1021-3 (5'-TAAATGGCAGTCTTAGCGTGA-3'), MW5-3F (5'-TTAGATTGTAAATCTAAAGA-3'), MWND4-2F (5'-TTACTATCTGATTTTTTAT-3'), MWND4-1F (5'-TTATTAGTTTGGTTTATTTT-3'), MW6K2F (5'-TTAGGAGAAATTTCCGTAGTTT-3'), MW9K1F (5'-ATCAATTCGGCTTATTATTTAA-3'), MW62R (5'-ATAGCATAGACCATGCCCAAAG-3'), MW62-2F (5'-ACTTTTCAGAAGTTACCTTTTTT-3'), MW51F (5'-TGGTTTTCTTACTTTATTTGTTT-3'), MWND1R (5'-TATCATAACGAAAACGAGG-3'), MW1R (5'-CCAGGAACCAAAATAAAAGA-3'), MWTM62F (5'-TTTCTTACTTTTCAGAAGTT-3'), MWSRNAF1 (5'-TAATCGGCTAGACTTTATAAACT-3'), MWND1-1R (5'-ACAAACCACTCCAAATACAT-3'), MWSRNAR (5'-TAATGAGGGCTCTCAATTACT-3'), and MWSRNAF2 (5'-TTAAAAGAGCAGGAGTAAAGTT-3').
Sequence analyses
Sequences were assembled manually and aligned against the complete mt genome sequence of A. suum (GenBank™ accession number NC001327) using the program Clustal X to identify gene boundaries. The open-reading frames and codon usage profiles of protein-coding genes were analyzed using the program MacVector 4.1.4 (Kodak, version 4.0). Translation initiation and translation termination codons were identified based on comparison with those reported previously for A. suum. The amino acid sequences inferred for the mt genes of three ascaridoids were aligned with those of A. simplex (GenBank™ accession number AY994157) and A. suum using Clustal X. Based on pairwise alignments, amino acid identity (%) was calculated for homologous genes. Codon usage was examined using the relationships between the nucleotide composition of codon families and amino acid occurrence, where the genetic codons are partitioned into AT-rich codons (i.e. those which are AT-rich at the first two codon positions), GC-rich codons (those which are GC-rich at the first two codon positions) and unbiased codons. For analyzing ribosomal RNA genes, putative secondary structures of 22 tRNA genes were identified using tRNAscan-SE [34], or by recognizing potential secondary structures and anticodon sequences by eye by aligning sequences with those of A. simplex and A. suum.
Phylogenetic analyses
Phylogenetic analyses were performed using the five ascaridoid species (T. canis, T. cati, T. malaysiensis, A. suum,A. simplex) as ingroups, and one filarioid species (O. volvulus) serving as outgroup (GenBank™ accession number AF015193), based on amino acid sequences of 12 protein-coding genes. Amino acid sequences for each gene were individually aligned using Clustal X under default setting, and then concatenated into single alignments for phylogenetic analyses. Standard unweighted maximum parsimony (MP) were performed in PAUP* 4.0b10 [35] using heuristic searches with tree-bisection-reconnection branch swapping and 1000 random-addition sequence replicates with 10 trees held at each step. The Dayhoff matrix model was utilized in the analyses of neighbour joining (NJ), implemented by MEGA 3.1 [36], and maximum likelihood (ML) implemented by PhyML 2.1 [37]. Branch supports were estimated by bootstrap analysis of 1000 replicates for NJ and MP trees, and 100 replicates for ML tree.
References
Wolstenholme DR: Animal mitochondrial DNA, structure and evolution. Int Rev Cytol. 1992, 141: 173-216.
Boore JL: Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27: 1767-1780. 10.1093/nar/27.8.1767.
Avise JC: Molecular Markers, Natural History and Evolution. 1994, New York and London, Chapman and Hall, 1-511.
Hu M, Chilton NB, Gassar RB: The mitochondrial genomics of parasitic nematodes of socio-economic importance: recent progress, and implications for population genetics and systematics. Adv Parasitol. 2004, 56: 133-212.
Hu M, Gasser RB: Mitochondrial genomes of parasitic nematodes – progress and perspectives. Trends Parasitol. 2006, 22: 78-84. 10.1016/j.pt.2005.12.003.
Okimoto R, Macfarlane JL, Clary DO, Wolstenholme DR: The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics. 1992, 130: 471-498.
Keddie EM, Higazi T, Unnasch TR: The mitochondrial genome of Onchocerca volvulus : sequence, structure and phylogenetic analysis. Mol Biochem Parasitol. 1998, 95: 111-127. 10.1016/S0166-6851(98)00102-9.
Lavrov DV, Brown WM: Trichinella spiralis mtDNA: a nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAs and has a gene arrangement relatable to those of coelomate metazoans. Genetics. 2001, 157: 621-637.
Hu M, Chilton NB, Gasser RB: The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int J Parasitol. 2002, 32: 145-158. 10.1016/S0020-7519(01)00316-2.
Hu M, Chilton NB, Gasser RB: The mitochondrial genome of Strongyloides stercoralis (Nematoda) – idiosyncratic gene order and evolutionary implications. Int J Parasitol. 2003, 33: 1393-1408. 10.1016/S0020-7519(03)00130-9.
Hu M, Gasser RB, Abs EL-Osta YG, Chilton NB: Structure and organization of the mitochondrial genome of the canine heartworm, Dirofilaria immitis. Parasitology. 2003, 127: 37-51. 10.1017/S0031182003003275.
Veer Van der M, de Vries E: A single nucleotide polymorphism map of the mitochondrial genome of the parasitic nematode Cooperia oncophora. Parasitology. 2004, 128: 421-431. 10.1017/S0031182003004633.
He Y, Jones J, Armstrong M, Lamberti F, Moens M: The mitochondrial genome of Xiphinema americanus sensu stricto (Nematoda: Enoplea): considerable economization in the length and structural features of encoded genes. J Mol Evol. 2005, 61: 819-833. 10.1007/s00239-005-0102-7.
Montiel R, Lucena MA, Medeiros J, Simöes N: The complete mitochondrial genome of the entomopathogenic nematode Steinernema carpocapsae : insights into nematode mitochondrial DNA evolution and phylogeny. J Mol Evol. 2006, 62: 211-225. 10.1007/s00239-005-0072-9.
Kim KH, Eom KS, Park JK: The complete mitochondrial genome of Anisakis simplex (Ascaridida: Nematoda) and phylogenetic implications. Int J Parasitol. 2006, 36: 319-328. 10.1016/j.ijpara.2005.10.004.
Habluetzel A, Traldi G, Ruggieri S, Attili AR, Scuppa P, Marchetti R, Menghini G, Esposito F: An estimation of Toxocara canis prevalence in dogs, environmental egg contamination and risk of human infection in the Marche region of Italy. Vet Parasitol. 2003, 113: 243-252. 10.1016/S0304-4017(03)00082-7.
Minnaar WN, Krecek RC, Fourie LJ: Helminths in dogs from a peri-urban resource-limited community in Free State Province, South Africa. Vet Parasitol. 2002, 107: 343-349. 10.1016/S0304-4017(02)00155-3.
Oliveira-Sequeira TC, Amarante AF, Ferrari TB, Nunes LC: Prevalence of intestinal parasites in dogs from São Paulo State, Brazil. Vet Parasitol. 2002, 103: 19-27. 10.1016/S0304-4017(01)00575-1.
Martínez-Barbabosa I, Vázquez Tsuji O, Cabello RR, Cárdenas EM, Chasin OA: The prevalence of Toxocara cati in domestic cats in Mexico City. Vet Parasitol. 2003, 114: 43-49. 10.1016/S0304-4017(03)00038-4.
Calvete C, Lucientes J, Castillo JA, Estrada R, Gracia MJ, Peribáñez MA, Ferrer M: Gastrointestinal helminth parasites in stray cats from the mid-Ebro Valley, Spain. Vet Parasitol. 1998, 75: 235-240. 10.1016/S0304-4017(97)00182-9.
Labarthe N, Serrão ML, Ferreira AM, Almeida NK, Guerrero J: A survey of gastrointestinal helminths in cats of the metropolitan region of Rio de Janeiro, Brazil. Vet Parasitol. 2004, 123: 133-139. 10.1016/j.vetpar.2004.06.002.
Despommier D: Toxocariasis: clinical aspects, epidemiology, medical ecology, and molecular aspects. Clin Microbiol Rev. 2003, 16 (2): 265-272. 10.1128/CMR.16.2.265-272.2003.
Fisher M: Toxocara cati : an underestimated zoonotic agent. Trends Parasitol. 2003, 19: 167-170. 10.1016/S1471-4922(03)00027-8.
Vidal JE, Sztajnbok J, Seguro AC: Eosinophilic meningoencephalitis due to Toxocara canis : case report and review of the literature. Am J Trop Med Hyg. 2003, 69: 341-343.
Zhu XQ, Jacobs DE, Chilton NB, Sani RA, Cheng NA, Gasser RB: Molecular characterization of a Toxocara variant from cats in Kuala Lumpur, Malaysia. Parasitology. 1998, 117: 155-164. 10.1017/S0031182098002856.
Gibbons LM, Jacobs DE, Sani RA: Toxocara malaysiensis n. sp. (Nematoda: Ascaridoidea) from the domestic cat (Felis catus, Linnaeus, 1758). J Parasitol. 2001, 87: 660-665.
Ikemura T: Correlation between the abundance of yeast transfer RNAs and occurrence of respective codons in protein genes: differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J Mol Evol. 1982, 158: 573-597.
Zhang DX, Hewitt GM: Insect mitochondrial control region. A review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Evol. 1997, 25: 99-120. 10.1016/S0305-1978(96)00042-7.
Hartwich G: Keys to genera of the Ascaridoidea. CIH keys to the nematode parasites of vertebrates. Edited by: Anderson RC, Chabaud AG, Willmott S. 1974, Farnham Royal, Bucks, Commonwealth Agricultural Bureaux, 2: 1-15.
Zhu XQ, Gasser RB, Jacobs DE, Hung GC, Chilton NB: Relationships among some ascaridoid nematodes based on ribosomal DNA sequence data. Parasitol Res. 2000, 86: 738-744. 10.1007/PL00008561.
Li MW, Lin RQ, Chen HH, Sani RA, Song HQ, Zhu XQ: PCR tools for the verification of the specific identity of ascaridoid nematodes from dogs and cats. Mol Cell Probes. 2007, 21: 349-354. 10.1016/j.mcp.2007.04.004.
Li MW, Zhu XQ, Gasser RB, Lin RQ, Sani RA, Lun ZR, Jacobs DE: The occurrence of Toxocara malaysiensis in cats in China, confirmed by sequence-based analyses of ribosomal DNA. Parasitol Res. 2006, 99: 554-557. 10.1007/s00436-006-0194-z.
Hu M, Chilton NB, Gasser RB: Long PCR-based amplification of the entire mitochondrial genome from single parasitic nematodes. Mol Cell Probes. 2002, 16: 261-267. 10.1006/mcpr.2002.0422.
Lowe TM, Eddy SR: tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25: 955-964. 10.1093/nar/25.5.955.
Swofford DL: PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods). 2002, Sunderland, MA., Sinauer Associates
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004, 5: 150-163. 10.1093/bib/5.2.150.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Acknowledgements
Project support was provided to XQZ in part by grants from the Program for Changjiang Scholars and Innovative Research Team in University (grant no. IRT0723), the Special Funds for the Training of PhD Students from the Ministry of Education, China (grant no. 20040564008), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, the Ministry of Education, China (grant no. [2003] 406). Funding to pay the Open Access publication charges for this article was provided by the College of Veterinary Medicine, South China Agricultural University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MWL performed the majority of the study and analyzed the data, and contributed to drafting of the manuscript. RQL and HQS performed part of the study, and provided technical assistance. XYW contributed to the analysis of the data and helped in revising the manuscript. XQZ conceived and designed the research plan, participated in all aspectes of the study, provided funds, supervised the research, and took the lead on drafting the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Li, MW., Lin, RQ., Song, HQ. et al. The complete mitochondrial genomes for three Toxocara species of human and animal health significance. BMC Genomics 9, 224 (2008). https://doi.org/10.1186/1471-2164-9-224
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-9-224