
Abstract
Background
Angiostrongylus mackerrasae is a metastrongyloid nematode endemic to Australia, where it infects the native bush rat, Rattus fuscipes. This lungworm has an identical life cycle to that of Angiostrongylus cantonensis, a leading cause of eosinophilic meningitis in humans. The ability of A. mackerrasae to infect non-rodent hosts, specifically the black flying fox, raises concerns as to its zoonotic potential. To date, data on the taxonomy, epidemiology and population genetics of A. mackerrasae are unknown. Here, we describe the mitochondrial (mt) genome of A. mackerrasae with the aim of starting to address these knowledge gaps.
Methods
The complete mitochondrial (mt) genome of A. mackerrasae was amplified from a single morphologically identified adult worm, by long-PCR in two overlapping amplicons (8 kb and 10 kb). The amplicons were sequenced using the MiSeq Illumina platform and annotated using an in-house pipeline. Amino acid sequences inferred from individual protein coding genes of the mt genomes were concatenated and then subjected to phylogenetic analysis using Bayesian inference.
Results
The mt genome of A. mackerrasae is 13,640 bp in size and contains 12 protein coding genes (cox1-3, nad1-6, nad4L, atp6 and cob), and two ribosomal RNA (rRNA) and 22 transfer RNA (tRNA) genes.
Conclusions
The mt genome of A. mackerrasae has similar characteristics to those of other Angiostrongylus species. Sequence comparisons reveal that A. mackerrasae is closely related to A. cantonensis and the two sibling species may have recently diverged compared with all other species in the genus with a highly specific host selection. This mt genome will provide a source of genetic markers for explorations of the epidemiology, biology and population genetics of A. mackerrasae.
Similar content being viewed by others


Background
The rat lungworm, Angiostrongylus cantonensis, the cause of neural angiostrongyliasis in humans and animals has been described from most inhabited continents, including Australia. Another two of the 19 species of this genus are neurotropic, namely A. malaysiensis, a parasite of the forest rat, Rattus tiomanicus [1–3] in Southeast Asia, and A. mackerrasae, a parasite of the native bush rats, Rattus fuscipes and R. Leucopus of Australia. The latter species of Angiostrongylus appears to occur in sympatry with A. cantonensis in Australia. To date, the genetic identity of the Australian native species of the rat lungworm, A. mackerrasae, has not been explored and there is no sequence data available for this species. Despite small morphological differences between the two species of Angiostrongylus present in Australia [4], it is uncertain if the morphological differences are accompanied by sufficient genetic divergence so as to support the concept that the two are indeed distinct species. Although A. mackerrasae is not known to infect humans, the ability of the parasite to produce patent infections in the lungs of the black flying fox (Pteropus alecto) [5], also raises questions as to the pathogenicity of this species in non-permissive hosts, including humans.
Angiostrongylus mackerrasae has been distinguished from the sympatric A. cantonensis, on the basis of distinct morphology of the reproductive system. For adult male A. cantonensis, the average length of the copulatory spicules is 1.24 mm, compared with 0.49 mm for A. mackerrasae [4]. A morphometric analysis of 51 adult females of A. cantonensis and 64 adult females of A. mackerrasae by Bhaibulaya [4] revealed that the mean length of the vagina of A. cantonensis was 2.10 mm, whereas for A. mackerrasae it was 1.39 mm. However, there is an overlap in the range of vaginal length between the two species, making it difficult to identify the species by examining only the adult female [6]. Additionally, the adult female of A. mackerrasae possesses a minute terminal projection at the tip of the tail, which in A. cantonensis is absent. Despite these morphological (phenetic) differences, little information is available on the epidemiology of these sympatric species in Australia. Much remains to be investigated, including their host range, whether mixed species infections in the definitive hosts occur and, if so, whether they are capable of producing hybrids in nature.
An important advance in understanding the epidemiology of Angiostrongylus species in Australia would arise from better understanding on genetic divergence of A. mackerrasae and A. cantonensis. Genetic markers, together with morphological characters, could be used to identify parasites associated with disease in humans, domestic and wild animals, as well as investigate the geographical distribution and host selection of A. mackerrasae and A. cantonensis in the large diversity of Rattus species that occur in Australia [7] and their intermediate hosts [8], areas hitherto unexplored. In the present study, we took a first step towards addressing some of these areas by characterising the mt genome of A. mackerrasae as a rich source of genetic markers. We also genetically compared, for the first time, A. mackerrasae with its very closely related congener, A. cantonensis.
Methods
Sample collection and DNA extraction
Ethical approval
All animal experiments were approved by the Animal Ethics Committee of the QIMR Berghofer Medical Research Institute (project P1457) and ratified by the University of Queensland Animal Welfare Unit. Specimens of Rattus fuscipes were collected from the Department of Environment and Heritage Protection of the Queensland Government (permit WIS12109412). Specimens of Rattus fuscipes were trapped in Brisbane and surrounding regions using Eliot traps, baited with peanut butter and rolled oats. Rat faeces were directly examined by light microscopy for the presence of larvae consistent with Angiostrongylus sp. [3]; rats harbouring the parasite were euthanized with an overdose of CO2 in a portable chamber for subsequent transport to the laboratory. Specimens of Angiostrongylus recovered from the pulmonary arteries of infected rats were identified to species morphologically [4] and washed extensively in physiological saline. Genomic DNA was isolated from amid-body section (to avoid ovaries) of an individual adult female worm using the QIAGEN DNeasy blood and tissue extraction kit, according to manufacturer’s instructions (Qiagen, Germany).
Long PCR amplification
The complete mt genome of a single A. mackerrasae female worm was amplified by long-PCR using a high fidelity PCR enzyme (BD Advantage 2, BD Biosciences) as two overlapping amplicons (~8 kb and 10 kb) as described [9], using modified primers (Table 1) and an optimised annealing temperature (58 °C), employing a suitable positive (A. cantonensis DNA recovered from Australian Rattus rattus) and negative (i.e. no template) controls. Individual PCR products were resolved in separate lanes on an agarose gel (1 % w/v) in TBE buffer (Tris/Borate/EDTA) and stained with SYBR®Safe gel stain (Life Technologies). Individual PCR products (~8 kb and 10 kb) were excised from the gel and purified using the QIAquick gel extraction kit (QIAGEN).
Sequencing and data analyses
Short-insert libraries (100 bp) were constructed from the purified products and then sequenced using Mi-seq technology (Illumina platform; Yourgene, Taiwan). FastQC (Babraham Bioinformatics: www.bioinfomatics.babraham.ac.uk) was utilised to assess the quality of sequence data and the paired-end reads were filtered using Trimmomatic (http://www.usadellab.org/cms). De novo assembly of the sequences was performed using SPAdes 3.0.0 Genome Assembler (http://bioinf.spbau.ru/en/spades). The program was run for all odd k-mer sizes between 21 and 125 (inclusive). The k-mer size providing the largest scaffold was selected for further analysis.
Following assembly, the mt genome of A. mackerrasae was annotated using a semi-automated bioinformatic pipeline [10]. Each protein coding mt gene was identified by local alignment comparison (performed in all six frames) using amino acid sequences from corresponding genes from mt genomes of A. vasorum, A. cantonensis and A. costaricensis; accession nos.NC_018602, GQ398121 and GQ398122, respectively [11, 12]. The large and small subunits (rrnL and rrnS) of mt ribosomal RNA genes were identified by local alignment, and all transfer RNA (tRNA) genes were predicted and annotated based on available data from selected nematode superfamilies, (the Metastrongyloidea, Trichostrongyloidea, Ancylostomatidea and Strongyloidea). Annotated sequence data were imported using the program SEQUIN (available via http://www.ncbi.nlm.nih.gov/Sequin/) for the final verification of the mt genome organisation and subsequent submission to the GenBank database. The amino acid sequences translated from individual genes of the mt genome of A. mackerrasae were then concatenated and aligned to sequences for 18 species for which mt genomic data sets were available using the program MUSCLE [13].
Phylogenetic analysis of amino acid sequence data was conducted by Bayesian inference (BI) using Monte Carlo Markov Chain analysis in the program MrBayes v.3.2.2 [14]. Bayesian analysis is more widely accepted and more accurate than the other methods due to the integration of Markov chain monte carlo algorithm. The optimal model of sequence evolution was assessed using a mixed amino acid substitution model, with four chains and 200,000 generations, sampling every 100th generation; the first 25 % of the generations sampled were removed from the analysis as burn-in. In addition, a sliding window analysis was performed on the aligned, complete mt genome sequences of the three Angiostrongylus species using the program DnaSP v.5 (http://www.ub.edu/dnasp/).
A sliding window of 300 bp (steps of 10 bp) was used to estimate nucleotide diversity (π) over the entire alignment; indels were excluded using DnaSP. Nucleotide diversity for the entire alignments was plotted against midpoint positions of each window, and gene boundaries were defined. Pairwise analyses were also performed using amino acid sequences predicted from protein coding genes of the four Angiostrongylus species to identify regions of different magnitudes of amino acid diversity.
Results
Characteristics of mt genome of A. mackerrasae
The circular mt genome of A. mackerrasae is 13,640 bp in length (Fig. 1), similar in length to those of A. cantonensis (13,497 bp), A. costaricensis (13,585 bp) [12] and A.vasorum (13,422 bp) [11]. Consistent with the pattern seen in other metastrongyloids [11, 12, 15], the mt genome of A. mackerrasae is AT-rich, with T being the most frequent and C being the least frequent nucleotides. The nucleotide composition of the mt DNA of A. mackerrasae was 24.42 % for A, 20.81 % for G, 6.35 % for C and 48.42 % for T (Table 2). The mt genome contains 12 protein coding genes (cox1-3, nad1-6, nad4L, atp6and cob), as well as two ribosomal RNA (rRNA) and 22 transfer RNA (tRNA) genes. All of the 36 genes are transcribed in the same direction (5’ > 3’) (Fig. 1).

Schematic representation of the circular mitochondrial genome of Angiostrongylus mackerrasae. Each transfer RNA gene is identified by one letter amino acid code on the outer side of the map. All genes are transcribed in the clockwise direction
Protein genes
The initiation and termination codons were predicted for protein-encoding genes of A. mackerrasae and were then compared with those of A. cantonensis, A. costaricensis and A. vasorum (Table 3). The most common start and stop codons for A. mackerrasae was TTG (for 6 of the 12 proteins) and TAG (for 6 of the 12 proteins). The codon usage of the 12 protein coding genes was compared with A. cantonensis, A. costaricensis and A. vasorum (Table 4). The most frequently used codon was TTT (Phe) and TTG (Leu), similar to those in mt genomes of A. cantonensis, A. vasorum and A. costaricensis. In addition, the least frequently used codons in the mt genome of A. mackerrasae were ATC(Ile) and ACC (Thr) and CTC (Leu), whereas it was TCC (Ser) for A. vasorum, TGC (Cys) for A. cantonensis and TGC (Cys), GAC (Asp), CTC (Leu) and ACC (Thr) for A. costaricensis. Of the 64 possible codons, 62 were used in mt genome of A. mackerrasae. Codons TCC (Ser) and CGC (Arg) were not used.
Transfer RNA and Ribosomal RNA genes
Twenty two tRNA genes were located in the mt genome of A. mackerrasae. The gene sequences ranged between 52 to 61 nt in length, identical to A. vasorum [11].
The rrnS and rrnL genes of A. mackerrasae were determined by sequence comparison with A. cantonensis, A. costaricensis and A. vasorum. As previously described for A. vasorum [11], the two genes were separated from each other by protein-encoding genes, including nad3, nad5 and nad4L (Fig. 1). The size of rrnS gene of A. mackerrasae was 696 bp and the rrnL was 961 bp. The size of both genes was identical to those described for A. vasorum [11] and Bunostomum trigonocephalum [16] and very similar to the size of rRNAs described previously for other nematodes (Table 5).
Genetic comparison between A. mackerrasae and other Angiostrongylus species, as well as other strongylid nematodes
The analysis of nucleotide variation across the mt genomes between or among A. mackerrasae, A. vasorum, A. cantonensis and A. costaricensis showed the most diversity in the rrnL, nad6 and atp6 genes and in the 5’-end of nad5 and 5’- and 3’-ends of nad4. Least diversity was observed in the cox1, cox2 and rrnS genes (Fig. 2).

Sliding window analysis of complete mitochondrial genome of Angiostrongylus mackerrasae. The black line indicates nucleotide diversity in a window size of 300 bp
Pairwise comparisons of the concatenated amino acid sequences of A. mackerrasae, A. cantonensis, A. costaricensis and A. vasorum ranged from 70.63 to 99.57 % between different protein coding genes and showed higher identity between A. mackerrasae and A. cantonensis, ranging between 92.9 % (nad4L) and 99.57 % (cox2) (an average of 2.4 % difference between the two). The sequence identity revealed that cox1 was the most conserved protein among the four, while nad2, nad3 and nad6 were the least conserved proteins (Table 6). In addition, pairwise comparison of amino acid sequences among closely related species of strongylid nematodes showed that A. mackerrasae and A. cantonensis are the most closely related to congeners, followed by Oesophagostomum quadrispinulatum and O. dentatum (3.2 % difference) as well as Ancylostoma caninum and A. duodenale (4.0 % difference).
Using mt datasets, based upon pairwise comparisons of concatenated amino acid sequences predicted herein, we found considerable variation in the magnitude of sequence differences between closely related species of trichostrongyloids (14.9 % between Trichostrongylus axei and T. vitrinus); (19.9 % between Haemonchus contortus and Mecistocirrus digitatus), ancylostomatoids (4.0 % between A. caninum and A. duodenale); (11.4 % between B. phlebotomum and B. trigonocephalum), strongyloids (3.2 % between Oe. dentatum and Oe. Quadrispinulatum) and selected metastrongyloids (19.2 % between D. eckerti and D. viviparus; 13.6 % between M. pudendotectus and M. salmi; 16.8 % between A. costaricensis and A.cantonensis and 18.7 % between A. costaricensis and A. vasorum) (Table 7).
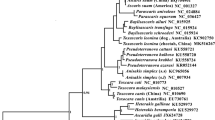
Extending these comparisons, phylogenetic analysis of the amino acid sequences encoded by the 12 mt genes revealed that A. cantonensis is the most closely related to A. mackerrasae among the four Angiostrongylus species. (Posterior probability (pp) =1:00). Metastrongylus spp. (Metastrongylidae) clustered separately from Angiostrongylus species but was the most closely related to the four Angiostrongylus species (pp = 1:00). Other strongylids such as Dictyocaulus spp. (Dictyocaulidae), Trichostrongylus spp. (Trichostrongylidae), Haemonchus contortus (Haemonchidae), Mecistocirrus digitatus (Haemonchidae); Oesophagostomum spp. (Chabertiidae); Ancylostoma spp. (Ancylostomatidae) and Bunostomum spp. (Ancylostomatidae) clustered separately from metastrongyloids (Fig. 3).

Relationship of Angiostrongylus mackerrasae with strongylid nematodes based on a phylogenetic analysis of concatenated amino acid sequence data for the 12 inferred mt proteins. There was absolute support (pp = 1.00) at each individual node
Discussion
Mitochondrial sequences have been used as genetic markers for identification of organisms and interrelationships among diverse taxa [10, 17, 18]. Although nucleotide variation within species of nematodes is relatively high for the mt genes studied [18] and, thus, is not useful for specific identification, this is not the case for the inferred sequences of mt proteins. Amino acid sequence variation within species of nematodes is usually very low (0–1.3 %) [10, 18, 19]. Therefore, amino acid sequences inferred from the mt genomes provide species identification for studying the systematics (taxonomy and phylogeny) of nematodes [10]. Indeed, phylogenetic analysis of mt amino acid datasets usually provides strong statistical support for the relationships of nematodes, which is not achieved using data from short sequence tracts.
The amino acid sequence difference of 2.4 % across the entire predicted mt protein repertoire between A. mackerrasae and A. cantonensis is higher than the upper level of within-species sequence variation estimated to date (1.3 %), and similar to the lowest levels of sequence difference (3.2–4.0 %) between pairs of other closely related strongylid nematodes (i.e. A. caninum and A. duodenale; Oe. dentatum and Oe. quadrispinulatum) [18], providing support for the hypothesis that A. mackerrasae and A. cantonensis are separate species. Experimental hybridization by Bhaibulaya [20] has been shown to produce fertile female but sterile males (F1s), which provides biological evidence to support this proposal.
Similar specific distinctions on genetic grounds have been made for pairs of morphologically similar or identical species of strongylid that show distinct host preferences. Jabbar et al. [21] studied the mt genome of the strongyloid Hypodontus macropi from three different hosts species and concluded that the parasites from the different hosts represent three distinct species of Hypodontus. The lowest sequence difference between two H. macropi isolates from Macropus robustus robustus and Macropus bicolour was 5.8 %. Nonetheless, further study using independent, informative nuclear genetic markers is required to lend additional independent support for the two closely related Angiostrongylus species.
Given the close genetic identity but biological differences between the two species of Australian angiostrongylids, the origins and divergence of A. mackerrasae and A. cantonensis are interesting questions. It has been suggested that feral rats (species of Rattus rattus and Rattus norvegicus) were introduced to Australia with the first European ships in late 1700s [7]. Presumably, A. cantonensis arrived in Australia on ships travelling from Asia [22]. Considering the ongoing geographical expansion of A. cantonensis in Australia, based on the recent reports of parasite from human, dogs, rats and molluscs from NSW [23–26], a phylogeographical analysis of this species is needed to resolve the question of origin of Australian populations of this species.
Angiostrongylus mackerrasae appears to be mainly specific to native Rattus (R. fuscipes and R. lutreolus) [3]. The native rat, R. fuscipes, is one of a number of Australian species that has been traced back to an invasion event in the final stages of the Pleistocene, when the Australian land mass was linked to Papua New Guinea (PNG) [7]. Molecular analysis of Rattus species in Australia demonstrates strong support for the specific identity of R. fuscipes, indicating that this species has not crossed with other Australian Rattus species, whereas the genetic fidelity of other Rattus species in Australia is less certain [27].
Did A. mackerrasae arise in Australia through natural invasions of a shared ancestor of the two parasite species from PNG? or did the current populations diverge from A. cantonensis populations after more recent introduction with European settlement and feral rat invasion? Bhaibulaya [20] favoured the more ancient, and northern, invasion by A. mackerrasae, explaining the morphologic similarity between the two species by hybridization and species introgression in the wild [20]. A challenge to this hypothesis is the apparent absence of any species of Angiostrongylus in tropical northern Australia. Dunsmore investigated rats on the Gulf of Carpentaria in the Northern Territory, but did not observe angiostrongylids [28] and there are no reports of eosinophilic meningitis in humans or animals from tropical Queensland, Australia. It should be noted however, that Dunsmore did not examine species of Rattus in his survey and may have missed evidence of the parasites. A recent survey of Rattus spp. in northern Queensland was also unable to show the presence of the parasite in tropical Queensland [29].
Anecdotal evidence from rodent trappers suggests that R. fuscipes actively excludes feral rats (Rattus rattus and Rattus norvegicus) from its habitats. Coupled with this, is the finding by Stokes et al. [23] that populations of A. cantonensis and A. mackerrasae were found in rats in different zones of forests of Jervis Bay, NSW. The evidence thus tentatively leans towards the view that despite their close genetic identity, the populations of A. cantonensis and A. mackerrasae are populations recently introduced into Australia. Accidental infection and establishment of populations in R. fuscipes has led to the two populations becoming isolated in terms of geographic habitat and host selection.
The occurrence of A. mackerrasae in Australia indicates a need to develop a molecular tool for the accurate/specific diagnosis of neural angiostrongyliasis in humans. Although A. mackerrasae has not been detected in humans, it has recently been recovered from a flying fox (Pteropus alecto) [5]. This raises questions as to the ability of A. mackerrasae to infect and cause disease in non-permissive hosts. There is even a possibility that A. mackerrasae is responsible for a portion of Angiostrongylus infections in humans in Australia. Yet, the focus of most studies of Angiostrongylus has been on A. cantonensis as it occurs in feral rats which live close to human dwellings. However, the expansion and encroachment of residential areas in Australia on forests has resulted in the native rats (e.g. Rattus fuscipes) being found in relatively close proximity to human habitation, potentially implicating A. mackerrasae as a potential zoonosis in these peri-urban regions. Moreover, current immunological [30] and molecular-based tools [31] for the detection of larvae in tissue target only A. cantonensis. If there is considerable divergence in protein sequence and immunological profiles of the two species, tools for diagnosis of neural angiostrongyliasis may not detect cases caused by A. mackerrasae.
The complete mt genome described here, now provides enough information to develop highly specific PCR-based tests to screen archival tissues of humans and dogs diagnosed with eosinophilic meningitis in order to distinguish the species of Angiostrongylus responsible for the infection. Genes such as nad4L showed a higher diversity between A. cantonensis and A. mackerrasae and could be a good region to be used in order to distinguish the two species.
The outcome of sliding window analysis in this study, offers valuable information of the high and low variability regions within the inter-species mt genome, providing useful data for population genetic studies and adds to the previously performed phylogenetic study of Angiostrongylus taxa by Eamsobhana et al. [32] which was restricted to the cox1 region of mt DNA and did not include A. mackerrasae.
Conclusion
In conclusion, the present study emphasizes the importance and utility of the mt genomic datasets for nematodes from rodents, as a basis for the diagnosis of A. mackerrasae and A. cantonensis for ecological and biological studies of these nematodes. Importantly, the study also provides a stimulus to explore, in detail, the population genetics of these taxa across their distributional and host ranges using complete or partial (informative) mt genomic and protein sequence data sets. Although the present study focused on these two taxa, the approach used has important implications for investigating the systematics of a range of parasites (nematodes) from rodents, and defining genetic markers of utility to explore their epidemiology and population genetics. Future studies should focus on comparing multiple adult nematodes of A. mackerrasae and A. cantonensis from different geographical locations such as North and South eastern Australia and Southeast Asia, including PNG, to ascertain that the species sequenced in this study is not a hybrid.
References
Bhaibulaya M, Cross JH. Angiostrongylus malaysiensis (Nematoda: Metastrongylidae), a new species of rat lung-worm from Malaysia. Southeast Asian J Trop Med Public Health. 1971;2(4):527–33.
Kamis AB, Ahmad RA, Badrul-Munir MZ. Worm burden and leukocyte response in Angiostrongylus malaysiensis-infected rats: the influence of testosterone. Parasitol Res. 1992;78(5):388–91.
Spratt DM. Species of Angiostrongylus (Nematoda: Metastrongyloidea) in wildlife: A review. Int J Parasitol Parasites Wildl. 2015;4(2):178–89.
Bhaibulaya M. A new species of Angiostrongylus in an Australian rat Rattus fuscipes. Parasitol. 1968;58:789–99.
Mackie J, Lacasse C, Spratt D. Patent Angiostrongylus mackerrasae infection in a black flying fox (Pteropus alecto). Aust Vet J. 2013;91(9):366–7.
Aghazadeh M, Jones MK, Aland KV, Reid SA, Traub RJ, McCarthy JS, et al. Emergence of neural angiostrongyliasis in eastern australia. Vector Borne Zoonotic Dis (Larchmont, NY). 2015;15(3):184–90.
Robins J, McLenachan P, Phillips M, McComish B, Matisoo-Smith E, Ross H. Evolutionary relationships and divergence times among the native rats of Australia. BMC Evol Biol. 2010;10(1):375.
Stanisic J, Shea M, Potter D, Griffiths O. Australian Land Snails Volume 1: A Field Guide to Eastern Australian Species. Queensland Museum. Riviere des Anguilles, Mauritius: Bioculture Press; 2010.
Hu M, Jex AR, Campbell BE, Gasser RB. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nat Protoc. 2007;2(10):2339–44.
Jex AR, Hall RS, Littlewood DT, Gasser RB. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010;38(2):522–33.
Gasser RB, Jabbar A, Mohandas N, Schnyder M, Deplazes P, Littlewood DT, et al. Mitochondrial genome of Angiostrongylus vasorum: comparison with congeners and implications for studying the population genetics and epidemiology of this parasite. Infect Genet Evol. 2012;12(8):1884–91.
Lv S, Zhang Y, Zhang L, Liu Q, Liu HX, Hu L, et al. The complete mitochondrial genome of the rodent intra-arterial nematodes Angiostrongylus cantonensis and Angiostrongylus costaricensis. Parasitol Res. 2012;111(1):115–23.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–5.
Jabbar A, Mohandas N, Jex AR, Gasser RB. The mitochondrial genome of Protostrongylus rufescens - implications for population and systematic studies. Parasit Vectors. 2013;6(1):263.
Gao JF, Zhao Q, Liu GH, Zhang Y, Zhang Y, Wang WT, et al. Comparative analyses of the complete mitochondrial genomes of the two ruminant hookworms Bunostomum trigonocephalum and Bunostomum phlebotomum. Gene. 2014;541(2):92–100.
Le TH, Blair D, McManus DP. Mitochondrial genomes of parasitic flatworms. Trends Parasitol. 2002;18(5):206–13.
Hu M, Gasser RB. Mitochondrial genomes of parasitic nematodes--progress and perspectives. Trends Parasitol. 2006;22(2):78–84.
Hu M, Chilton NB, Gasser RB. The mitochondrial genomics of parasitic nematodes of socio-economic importance: recent progress, and implications for population genetics and systematics. Advances Parasitol. 2004;56:133–212.
Bhaibulaya M. Experimental hybridization of Angiostrongylus mackerrasae, Bhaibulaya, 1968 and Angiostrongylus cantonensis (Chen, 1935). Int J Parasitol. 1974;4(6):567–73.
Jabbar A, Beveridge I, Mohandas N, Chilton NB, Littlewood DT, Jex AR, et al. Analyses of mitochondrial amino acid sequence datasets support the proposal that specimens of Hypodontus macropi from three species of macropodid hosts represent distinct species. BMC Evol Biol. 2013;13:259.
Banks PB, Hughes NK. A review of the evidence for potential impacts of black rats (Rattus rattus) on wildlife and humans in Australia. Wildlife Res. 2012;39.
Stokes VL, Spratt DM, Banks PB, Pechc RP, Williams RL. Occurrence of Angiostrongylus species (Nematoda) in populations of Rattus rattus and Rattus fuscipes in coastal forests of south-eastern Australia. Aust J Zoo. 2007;55(3):177–84.
Morton NJ, Britton P, Palasanthiran P, Bye A, Sugo E, Kesson A, et al. Severe Hemorrhagic Meningoencephalitis Due to Angiostrongylus cantonensis Among Young Children in Sydney, Australia. Clin Infect Dis. 2013;57(8):1158–61.
Chan D, Barratt J, Roberts T, Lee R, Shea M, Marriott D, et al. The Prevalence of Angiostrongylus cantonensis/mackerrasae Complex in Molluscs from the Sydney Region. PloS one. 2015;10(5), e0128128.
Walker AG, Spielman D, Malik R, Graham K, Ralph E, Linton M, et al. Canine neural angiostrongylosis: a case–control study in Sydney dogs. Aust Vet J. 2015;93(6):195–9.
Robins JH, Tintinger V, Aplin KP, Hingston M, Matisoo-Smith E, Penny D, et al. Phylogenetic species identification in Rattus highlights rapid radiation and morphological similarity of New Guinean species. PloS one. 2014;9(5), e98002.
Dunsmore JD. Absence of Angiostrongylus cantonensis from the Gulf of Carpentaria in Northern Australia. J Parasitol. 1968;54(4):826.
Aghazadeh M, Reid SA, Aland KV, Restrepo AC, Traub RJ, McCarthy JS, et al. A survey of Angiostrongylus species in definitive hosts in Queensland. Int J Parasitol Parasites Wildl. 2015;4(3):323–8.
Maleewong W, Sombatsawat P, Intapan PM, Wongkham C, Chotmongkol V. Immunoblot evaluation of the specificity of the 29-kDa antigen from young adult female worms Angiostrongylus cantonensis for immunodiagnosis of human angiostrongyliasis. Asian Pac J Allergy Immunol. 2001;19(4):267–73.
Qvarnstrom Y, da Silva ACA, Teem JL, Hollingsworth R, Bishop H, Graeff-Teixeira C, et al. Improved Molecular Detection of Angiostrongylus cantonensis in Mollusks and Other Environmental Samples with a Species-Specific Internal Transcribed Spacer 1-Based TaqMan Assay. Appl Environ Microbiol. 2010;76(15):5287–9.
Eamsobhana P, Lim PE, Zhang H, Gan X, Yong HS. Molecular differentiation and phylogenetic relationships of three Angiostrongylus species and Angiostrongylus cantonensis geographical isolates based on a 66-kDa protein gene of A. cantonensis (Nematoda: Angiostrongylidae). Exp Parasitol. 2010;126(4):564–9.
Acknowledgements
Mahdis Aghazadeh is supported by a University of Queensland Postgraduate Award. This research was supported by grants from the ANZ Queensland Community Foundation – Peter and Mary Ellen Stone Memorial Fund, awarded to Malcolm Jones.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MA carried out the molecular laboratory work, participated in bioinformatic analysis and prepared the draft of the manuscript. RJT participated in the design of the study, provided technical advice, participated in bioinformatic analysis and edited the draft of the manuscript. NM performed the bioinformatic analysis and edited the draft of the paper. KVA carried out field work for the study and edited the draft of the manuscript. SAR participated in the design of the study and edited the draft of the manuscript. JSM participated in the design of the study and provided technical advice. MKJ participated in the design of the study and substantially revised the draft of the manuscript. All authors read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Aghazadeh, M., Traub, R.J., Mohandas, N. et al. The mitochondrial genome of Angiostrongylus mackerrasae as a basis for molecular, epidemiological and population genetic studies. Parasites Vectors 8, 473 (2015). https://doi.org/10.1186/s13071-015-1082-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-015-1082-0