
Abstract
Background
Yarrowia lipolytica, one of the most widely studied “nonconventional” oleaginous yeast species, is unable to grow on cellulose. Recently, we identified and overexpressed two endogenous β-glucosidases in Y. lipolytica, thus enabling this yeast to use cello-oligosaccharides as a carbon source for growth. Using this engineered yeast platform, we have now gone further toward building a fully cellulolytic Y. lipolytica for use in consolidated bioprocessing of cellulose.
Results
Initially, different essential enzyme components of a cellulase cocktail (i.e,. cellobiohydrolases and endoglucanases) were individually expressed in Y. lipolytica in order to ascertain the viability of the strategy. Accordingly, the Trichoderma reesei endoglucanase I (TrEG I) and II (TrEG II) were secreted as active proteins in Y. lipolytica, with the secretion yield of EG II being twice that of EG I. Characterization of the purified His-tagged recombinant EG proteins (rhTrEGs) revealed that rhTrEG I displayed higher specific activity than rhTrEG II on both cellotriose and insoluble cellulosic substrates, such as Avicel, β-1, 3 glucan, β-1, 4 glucan, and PASC. Similarly, cellobiohydrolases, such as T. reesei CBH I and II (TrCBH I and II), and the CBH I from Neurospora crassa (NcCBH I) were successfully expressed in Y. lipolytica. However, the yield of the expressed TrCBH I was low, so work on this was not pursued. Contrastingly, rhNcCBH I was not only well expressed, but also highly active on PASC and more active on Avicel (0.11 U/mg) than wild-type TrCBH I (0.065 U/mg). Therefore, work was pursued using a combination of NcCBH I and TrCBH II. The quantification of enzyme levels in culture supernatants revealed that the use of a hybrid promoter instead of the primarily used TEF promoter procured four and eight times more NcCBH I and TrCBH II expressions, respectively. Finally, the coexpression of the previously described Y. lipolytica β-glucosidases, the CBH II, and EG I and II from T. reesei, and the N. crassa CBH I procured an engineered Y. lipolytica strain that was able to grow both on model cellulose substrates, such as highly crystalline Avicel, and on industrial cellulose pulp, such as that obtained using an organosolv process.
Conclusions
A Y. lipolytica strain coexpressing six cellulolytic enzyme components has been successfully developed. In addition, the results presented show how the recombinant strain can be optimized, for example, using artificial promoters to tailor expression levels. Most significantly, this study has provided a demonstration of how the strain can grow on a sample of industrial cellulose as sole carbon source, thus revealing the feasibility of Yarrowia-based consolidated bioprocess for the production of fuel and chemical precursors. Further, enzyme and strain optimization, coupled to appropriate process design, will undoubtedly lead to much better performances in the future.
Similar content being viewed by others
Background
The production of second-generation biofuels and platform molecules for the chemical industry from lignocellulosic biomass (LCB) is viewed as crucial part of the bioeconomy [1]. Cellulose, the main component of LCB, is a polymer composed of β-1, 4-linked glucose subunits usually embedded in an amorphous matrix of hemicellulose and lignin [2]. It may exist in two forms, a tightly packed crystalline form where individual chains are organized in microfibrils via hydrogen bonding and van der Waals interactions, or a less-ordered amorphous form [3, 4]. The depolymerization of cellulose requires a variety of enzymes, including endoglucanases (EGs) (EC 3.2.1.4) that hydrolyze internal β-glucosidic bonds, cellobiohydrolases (CBHs) (EC 3.2.1.91) that remove cello-oligosaccharides in a processive manner from chain termini, and β-glucosidases (BGLs) (EC 3.2.1.21) that degrade cello-oligosaccharides to glucose [2]. However, the condensed structure of crystalline cellulose and its intimate proximity with hemicelluloses and lignin combine to make LCB very resistant to enzymatic hydrolysis [5, 6] and render biochemical processing of raw LCB economically unviable. To overcome this recalcitrance, biomass pretreatment is necessary. However, this step constitutes a significant cost driver in the overall economics of LCB biorefinery processes [7,8,9]. Presently, it is widely recognized that significant technological advances, including better pretreatments, lower-cost enzymes, and efficient process design will be required in order to make LCB biorefining competitive within the current economic framework [2].
The pretreatment process operated by CIMV S.A. belongs to the so-called organosolv technology family, because it uses organic solvents to dissolve lignin and hemicelluloses, and yields a pure and relatively amorphous cellulose fraction that is quite amenable to enzyme action [10, 11]. Moreover, the CIMV process produces functionalized lignins (Biolignintm) and an essentially furfural-free pentose-rich fraction, making this pretreatment technology an interesting platform for the design of a new LCB biorefinery concept.
Consolidated bioprocessing (CBP), featuring microbial enzyme production and concomitant microbial conversion of suitable feedstock into value-added products in a single step, offers great potential for cost-effective lignocellulosic bioconversion [1, 12]. This is because it is predicted that CBP will reduce both capital investment and operating costs, and possibly procure higher enzymatic hydrolysis rates [13, 14]. To date, only a few naturally occurring species of bacteria within the genus Clostridium have been described with such capabilities. For instance, Clostridium lentocellum is able to convert cellulose to acetic acid, and Clostridium thermocellum can ferment cellulose for the production of ethanol [15, 16]. Besides, to develop other purpose built microorganisms that will simultaneously convert cellulose pulp into sugars and ferment these to target products is still highly desired [1]. Nevertheless, using microbial engineering approaches and focusing on hosts such as Escherichia coli [17], Saccharomyces cerevisiae (reviewed in [18]) and Kluyveromyces marxianus [19], considerable progress has been made, although so far most studies have used model cellulose substrates and achieved relatively low titers of product. Importantly, none of the engineered strains reported so far have convincingly hydrolyzed the cellulose feedstock, and currently no commercially viable CBP organism has been reported [20].
The so-called oleaginous yeast Yarrowia lipolytica can accumulate lipids up to 50% of its dry weight depending on culture conditions, making this a promising platform for the production of biodiesel precursors [21, 22]. Advantageously, Y. lipolytica is already widely used in detergent, food, pharmaceutical, and environmental industries and has been classified by the Food and Drug Administration (FDA) as “generally recognized as safe” (GRAS) for numerous processes [23]. Moreover, Y. lipolytica is a suitable host for heterologous expression, since it displays high secretion ability and performs a wide-range of posttranslational modifications [24, 25]. However, regarding LCB biorefining, Y. lipolytica is unable to metabolize cellulose.
Recently, several reports have illustrated how cellulolytic capability can be conferred to Y. lipolytica, with single gene expression and coculturing being used as a pragmatic way to design a Y. lipolytica-based CBP system [26, 27]. However, the use of coculturing is not a feasible solution for industrial implementation, and a fully viable system can only be achieved if BGL activity is present [27]. In this respect, we recently described the overexpression of endogenous BGLs in Y. lipolytica and the use of cello-oligosaccharides by the recombinant yeast strain to support growth [28]. In pursuit of a more ambitious goal, in this work, we have built on this platform strain, adding other cellulolytic enzyme-encoding genes and exploring different combinations in order to procure a Y. lipolytica strain that is able to grow on cellulose.
Results and discussion
Expression of T. reesei endoglucanases in Y. lipolytica
The extensively studied cellulolytic secretome of the soft-rot fungus T. reesei (syn. Hypocrea jecorina) contains four endoglucanases, Cel7B (EG I), Cel5A (EG II), Cel12A (EG III), and Cel45A (EG V). Among these, EG I and EG II are the main endo-acting enzymes, representing approximately 15 and 10% of the total amount of cellulases (w/w) respectively, while the other two represent less than 1% each [29]. Therefore, the first step toward the construction of a fully cellulolytic Y. lipolytica strain was the introduction of sequences encoding T. reesei EG I and EG II under the control of TEF promoter and the Y. lipolytica lipase 2 pre–pro region into the Y. lipolytica zeta strain [30]. Subsequent screening of transformants producing either EG I or EG II with or without His6 revealed the presence of clones possessing the ability to hydrolyze Azo-CMC in solid agar medium (Additional file 1: Figure S1).
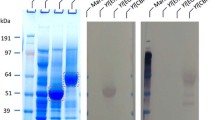
To further confirm the successful expressions of EG I and II, positive clones were grown on YTD, and the activities of EGs in culture supernatant were measured. Accordingly, CMCase activity in culture supernatant steadily increased over a 48-h period until the glucose in the culture medium was completely consumed by the yeast, with rhTrEG II activity (0.78 U/mL) at the end of the period being twice that of rhTrEG I (0.39 U/mL) (Fig. 1a). Furthermore, SDS-PAGE analysis of culture supernatants containing either rhTrEG I or rhTrEG II (Fig. 1b), compared to that of a control culture, revealed in the first case the presence of a smear (70–200 kDa) and in the latter case a discrete species migrating to a position corresponding to an approximate Mw of 55 kDa, which is a little high compared to the actual expected Mw of EG II (47 kDa). In the case of both putative rhTrEG I and II, the anti-His antibody confirmed that these protein species were His-tagged (Fig. 1c). Taken together, these observations suggest that the recombinant proteins are glycosylated forms of EG I and II, in agreement with previous results [26, 29, 31] and with the findings of a bioinformatics study (http://www.imtech.res.in/raghava/glycoep/) [32] that predicted that EG I bears six potential N-glycosylation sites, while EG II possesses only one. To confirm the presence of N-glycosylation, EndoH treatment and then Western blot analysis were performed (Fig. 1c). This revealed that the EndoH-treated protein samples were still detected using anti-His antibodies, but for rhTrEG I, a discrete protein species of approximately 56 kDa appeared in place of the smear, which is slightly higher than the actual expected Mw (49 kDa). Likewise, the migration of EndoH-treated rhTrEG II was slightly modified, consistent with that of a lowered Mw.

Production of rhTrEG I and rhTrEG II in Y. lipolytica a enzyme production on YTD versus time, b SDS-PAGE analysis of the culture supernatant of Y. lipolytica transformants compared with the control, and c Western blot analysis; lanes 1 and 3, culture supernatant of transformant ylEGI and ylEGII, respectively; lanes 2 and 4, culture supernatant of transformant ylEGI and ylEGII treated by endo-H, respectively
Characterization of the recombinant endoglucanases expressed in Y. lipolytica
Attempts to develop optimized cellulase cocktails have revealed that the efficient hydrolysis of pretreated LCB requires that EG I represents 25–35% of the total amount of cellulases (w/w) [33,34,35]. Moreover, such studies have underlined the usefulness of EG II for the rapid reduction of viscosity of acid pretreated wheat straw [36]. Therefore, it is important to understand the specific role of each EG in cellulose degradation in order to optimize the composition of cellulases expressed in Y. lipolytica. Accordingly, rhTrEG I and rhTrEG II were purified and characterized. For the purification of rhTrEG II, a one-step affinity method procured good overall yield (>60%), whereas the yield of rhTrEG I was lower (18%), probably due to the hyper-glycosylated state of this protein (Fig. 2).

SDS-PAGE analysis of the purified rhTrEG I and rhTrEG II produced in Y. lipolytica JMY1212 transformants; lanes 1 and 3, purified rhTrEG I and rhTrEG II, respectively; lanes 2 and 4, endo-H treated rhTrEG I and rhTrEG II, respectively
Subsequently, the hydrolytic activities of purified rhTrEGs were measured on various cellulosic substrates (Table 1). Significantly, compared to rhTrEG II (0.1 µmol/min/mg), rhTrEG I not only displayed 10 times higher specific activity on soluble cellotriose (1.1 µmol/min/mg), but also exhibited higher activity on Avicel, β-1, 3 and β-1, 4 glucans, and phosphoric acid-swollen cellulose (PASC). On the other hand, the specific activities of the two proteins on soluble carboxymethyl cellulose (CMC) were similar. Moreover, it is noteworthy that rhTrEG I exhibited highest hydrolytic activity on β-1, 4 glucan, while CMC was the preferred substrate for rhTrEG II. Similar substrate preferences have been reported for both the native EGs and the catalytic domains of the two proteins expressed in E. coli [34, 37]. However, the specific activities of rhTrEG I and II expressed in Y. lipolytica were higher than previously reported values for the native EGs, although these differences could be linked to the quantification methods employed in each case [34, 35].
The amount of rhTrEG I and rhTrEG II secreted by Y. lipolytica in YTD medium during flask batch culture, as calculated from the total CMCase activity of the culture supernatant and specific activity of the enzyme, were approximately 29 and 67 mg/L, respectively. The secretion yield of rhTrEG II obtained in this study was comparable to that reported in the literature [26, 29]. However, the amount of secreted rhTrEG I was higher than the previously reported value (5 mg/L). In this respect, it is important to note that in the previous work, EG I was produced with the pre-pro region of XPR2 and under the control of the XPR2 promoter [31]. Nevertheless, we were unable to determine why the secretion yield of rhTrEG I was approximately 60% lower than that of rhTrEG II. The identification of key factors that affect protein expression levels will be important for future use of Y. lipolytica as an efficient expression host.
Expression of cellobiohydrolases in Y. lipolytica
CBH I and CBH II (Cel7A and Cel6A) are the major exo-acting components of the T. reesei cellulolytic secretome, representing 50 and 20% of the total amount of the protein respectively (w/w) [31]. However, type I cellobiohydrolases from various fungal sources have so far proved to be difficult to express in heterologous hosts, such as S. cerevisiae and Y. lipolytica, probably due to the improper folding and/or unnatural post-translation patterns [27, 38, 39]. Taking this into account, we attempted to express in Y. lipolytica three different CBH I from T. reesei (TrCBH I), Penicillium funiculosum (PfCBH I) and Neurospora crassa (NcCBH I). It is noteworthy that the latter two have been shown to possess quite potent cellulose-degrading activities [40,41,42]. Accordingly, TrCBH I, PfCBH I and NcCBH I were produced in Y. lipolytica as His-tagged proteins (rhTrCBH I, rhPfCBH I and rhNcCBH I) using the TEF promoter and the Y. lipolytica lipase 2 pre-pro region. Subsequent SDS-PAGE and anti-His Western blot analyses of the culture supernatants indicated that rhTrCBH I could only be detected as a faint band, consistent with previous results [27] (data not shown). Contrastingly, the successful expression of rhNcCBH I and rhPfCBH I was confirmed by the clear presence of new protein species. In the case of rhNcCBH I a Mw of approximately 75 kDa was determined, while expression of rhPfCBH I produced a smear in the Mw range 70–200 kDa. Since the theoretical Mw of PfCBH I and NcCBH I is 52 kDa each, it was possible to deduce that rhPfCBH I and rhNcCBH I are glycosylated (Fig. 3a). Therefore, along with rhTrCBH I, rhPfCBH I and rhNcCBH I were submitted to EndoH-mediated deglycosylation. This yielded protein products with Mw of approximately 75 kDa, suggesting that the enzymes might bear other post-translational modifications other than N-glycosylation (Fig. 3a). Further analysis using an activity assay revealed that the Avicelase activity in the culture supernatant of the transformant ylNcCBH I (0.01 U/mL) was 10 times higher than that in the supernatant of ylPfCBH I. Avicelase activity lower than the minimum detectable limit (0. 001 U/mL) was found in the case of ylTrCBH I. Based upon this simple screening approach, NcCBH I was retained as the CBH I component for future work.

Western blot analysis of the heterologous CBH proteins produced by Y. lipolytica a lanes 1 and 3, rhNcCBH I and rhPfCBH I, respectively; lanes 2 and 4, corresponding rhCBH I treated by endo-H; b lanes 1 and 2, rhTrCBH II and endo-H treated rhTrCBH II, respectively
Regarding the requirement for CBH II, the T. reesei enzyme was chosen, because previous work has already shown that this enzyme can be satisfactorily produced in Y. lipolytica [26, 27]. In the present work, Western blot analysis indicated that rhTrCBH II bears post-translational modifications, since its apparent Mw is 75 kDa, which is higher than the theoretical value of 48 kDa. Deglycosylation of rhTrCBH II using EndoH revealed that this protein contains N-linked glycosylation, yet, its Mw remains slightly superior to 48 kDa after this enzymatic treatment (Fig. 3b). This could possibly be related to the presence of linker regions, rich in serine and threonine which are often highly O-glycosylated, connecting T. reesei CBH II catalytic domain and carbohydrate-binding module. Finally, it is noteworthy that the expression of the different rCBHs without the His-tag yielded similar activities on Avicel and PASC when culture supernatants were tested (i.e,. comparison of unpurified protein preparations).
Characterization of the recombinant cellobiohydrolases expressed in Y. lipolytica
In order to investigate the cellulose-degrading abilities of rhNcCBH I and rhTrCBH II, the recombinant CBHs were purified and characterized. Purification was achieved in a single step using IMAC with both proteins being obtained in good yields (>60% of the expressed protein) (Fig. 4). Afterward, each enzyme was tested for its ability to hydrolyze various substrates (Table 2). The specific activities of rhNcCBH I on Avicel and PASC were two and four times that of the reported values for native TrCBH I, respectively [43], that is to say two times the amount of the only cellobiohydrolase of family I expressed in Y. lipolytica [27]. In contrast, the comparison of native and rhTrCBH II revealed similar specific activities on these substrates, consistent with the previously reported values [26, 43]. The high Avicelase activity of rhNcCBH I is noteworthy, because Avicel is known to be crystalline and quite recalcitrant to enzyme hydrolysis. Likewise, the specific activity of rhNcCBH I on amorphous PASC is four times that of native TrCBH I and CBH II (Table 2). A recent study has illustrated that TrCBH I remains poorly active on cellulosic substrates until amorphous regions in the cellulose substrate are removed by TrCBH II [44]. Nevertheless, in the case of a cocktail containing NcCBH I, this limitation might be somehow attenuated by the superior activity of this CBH I on amorphous cellulose. Because it is known that the exo–exo synergy between type I and type II CBHs is crucial for efficient cellulose degradation, coexpression of NcCBH I and TrCBH II is expected to satisfy this criterion [45].

SDS-PAGE analysis of the purified rhNcCBH I (lane 1) and rhTrCBH II (lane 2) produced by Y. lipolytica JMY1212
Finally, based on the measurement of total PASCase activities in the culture supernatant and specific activities of the rhCBH enzymes, it was possible to determine that the amount of rhNcCBH I and rhTrCBH II secreted by Y. lipolytica in YTD medium was approximately 24 and 75 mg/L, respectively. Regarding rhTrCBH II, these results are consistent with those of a previous study in which it was expressed without a His-tag in Y. lipolytica [26, 27].
Enhancement of cellobiohydrolase production in Y. lipolytica using hybrid promoter
Several studies have shown that it is important to ensure that a sufficient amount of cellobiohydrolases are present in LCB-active cocktails in order to promote synergy with EGs and BGLs [33,34,35]. Therefore, to enhance the expression of rTrCBH II and rNcCBH I, their encoding sequences were placed under the control of the hybrid promoter (HTEF), which is composed of four tandem copies of upstream activation sequences (UAS) and the core promoter element of TEF [46]. Monitoring PASCase activity and using SDS-PAGE revealed that while the TEF-controlled production of rNcCBH I was highly cell growth-dependent, reaching its highest level at the beginning of stationary phase (Fig. 5a, b), HTEF-controlled production was much higher and continued to increase over the 5-day growth period. Importantly, when using the HTEF promoter, the final yield of rhNcCBH I (95 mg/L) was 4 times higher than that obtained using TEF and was much higher than previously reported yields [27]. Similarly, when rhTrCBH II was produced under the control of HTEF, the yield of this protein was eightfold higher (600 mg/L) than that obtained when using the TEF promoter (Fig. 5c). This significant increase was also evidenced upon SDS-PAGE (Fig. 5d). In this regard, although previous studies have already demonstrated the benefits of using the hybrid promoter to enhance protein production in Y. lipolytica, studies so far have only been focused on intracellular proteins [46]. Here we supply two examples of enhanced extracellular production and further reveal that, based on the findings of Blazeck et al. [46], the production level of TrCBH II surpasses expectations, with the use of four tandem copies of UAS yielding a fourfold increase in protein production.

Production of rhNcCBH I and rhTrCBH II under the control of TEF or 4UASTEF promoter in Y. lipolytica a and c enzyme production on YTD vs. time; b and d SDS-PAGE analysis of the culture supernatant of Y. lipolytica transformants
Construction of recombinant Y. lipolytica strains expressing different combinations and ratios of cellulases
To confer optimal cellulose-degrading ability to Y. lipolytica, several strains were constructed, using the previously developed BGL-producing strain as a platform [28]. Moreover, to identify the best configuration, strains were built using different enzyme combinations and protein production ratios (Table 3). The success of introduction of different cellulase-encoding genes into Y. lipolytica was verified by PCR (Additional file 1: Figure S2). The positive clones were chosen based on the results of enzymatic activity assays (data not shown). Subsequently, these selected strains were cultivated in YTD medium and the hydrolytic activities of the total secretory secreted proteins were analyzed on cellulosic substrates CMC, PASC and Avicel (Fig. 6).

Comparison of the hydrolytic activities of the total secreted cellulases produced by different cellulolytic Y. lipolytica strains cultivated in Y1T2D5 media after 120 h on various cellulosic substrates
The strain YLC3, expressing two rTrEGs, displayed higher CMCase activity than YLC1 and YLC2, which express either rTrEG I or rTrEG II, alone (Fig. 6). Moreover, quite predictably, strain YLC1 expressing only rTrEG I and two CBHs, was more active on Avicel than YLC2, which is a homolog that expresses rTrEG II instead of rTrEG I. Consistently, YLC2 displayed better activity on PASC. When both rTrEG I and II are present (strains YLC3-6), the hydrolyses of Avicel and PASC were enhanced, demonstrating that the presence of both EGs is necessary to achieve optimal cellulolytic activity. In contrast, the ability of strain YLC3 to hydrolyze CMC was similar to that of strains YLC4-6, implying that the basic combination of the six enzymes is sufficient to achieve optimal results with this substrate. On the other hand, increasing the expression level of either rTrCBH II (YLC4) or rNcCBH I (YLC5) using the HTEF promoter resulted in improved Avicel and PASC hydrolyses. Further increases of Avicel and PASC hydrolyses were achieved by enhancing the expression levels of both NcCBH I and TrCBH II (YLC6). The presence and the abundance of each expressed enzyme in YLC6 were confirmed by proteomics analysis (Additional file 2), which revealed that the ratio between each cellulase component in the secretome of YLC6 is consistent with the ratio of respective protein individually produced by recombinant Y. lipolytica (Plateforme Protéomique de la Génopole Toulouse Midi-Pyrénées, IPBS, France). Overall, the performance of YLC6 was rather encouraging. However, accounting for the fact that Avicel and PASC are model substrates, our conclusions cannot be further extrapolated to predict the aptitude of YLC6 for use on industrial cellulose pulps, because these are generally of a much more complex nature [47].
CIMV-cellulose and Avicel fermentation with recombinant Y. lipolytica strains
In order to explore the potentiality of the Y. lipolytica strains developed in this study for use as CBP microorganisms, these were grown in defined minimum medium containing either industrial cellulose pulp (CIMV, cellulose content ≈91%) or Avicel as the carbon source. Gratifyingly, all of the strains consumed CIMV-cellulose, although Avicel was less amenable to hydrolysis. The strain YLC3 expressing all the essential cellulase components used 40% of the CIMV-cellulose provided (27.5 g/L, initial concentration, i.e,. 25 g/L cellulose) and produced 0.38 g DW cells/(g substrate). These values increased to approximately 50% and 0.40 g DW cells/(g substrate) in the case of YLC4 and 5, and to 59% and 0.41 g DW cells/(g substrate) in the case of YLC6. Overall, these results demonstrate that cellulolytic capacity had been successfully conferred to Y. lipolytica and support findings that CIMV-cellulose is mainly amorphous and highly amenable to enzyme-mediated cellulolysis. However, our results also highlight the highly recalcitrant nature of Avicel, since the final consumption of this substrate by strains YLC4-6 (26–30%) was not much higher than that achieved by YLC3 (22%) or that procured by a coculturing approach (23%) [27], where each cellulase component was produced separately (Table 4). Significantly, even prolonged growth periods did not enhance Avicel degradation, indicating that the cellulase combinations used in this study are inadequate for the hydrolysis of crystalline cellulose. By contrast, as an attractive microorganism for CBP, C. thermocellum displays remarkable capacity toward the hydrolysis of crystalline cellulose for which the highest Avicel cellulose consumption rate of 0.5 g/L/h was reported recently [16], which is 7 times higher than the one obtained in YLC6. In addition to substrate crystallinity, it is clear that the performance of the strains was also hampered by the amounts of enzyme activity available in the culture medium, which were lower than those achieved when using richer YTD medium (data not shown). Beyond the penalizing effect of the minimal medium on protein expression, it is important to note that the enzymes were also operating in suboptimal pH and temperature conditions. This is because they were deployed in a simultaneous hydrolysis and fermentation reaction that was necessarily conducted at the optimal conditions for yeast growth. Therefore, in future work it will be necessary to address this issue, perhaps through the use of enzymes that are better adapted to low temperature that characterize the optimal growth conditions of Y. lipolytica.
Conclusions
In this article, we have provided a clear demonstration of how cellulolytic activity can be conferred to Y. lipolytica, thus opening the way toward a consolidated bioprocess. Using the panoply of available tools, including strong artificial promoters and selected cellulase components, it has been possible to show that Y. lipolytica can achieve high protein expression levels and that even when the enzymes function in suboptimal temperature conditions, their production and activity are sufficient to allow cellulose hydrolysis. In particular, it is noteworthy that the heterologous expression of CBH I from N. crassa was particularly successful and that this enzyme is more active on Avicel than its T. reesei counterpart. This is significant because CBH I is essential for the degradation of recalcitrant LCB.
Options for future improvements to the strains described herein include the introduction of lytic polysaccharide monooxygenases, the adaptation of the operating conditions of the enzymes to suit the growth conditions of Y. lipolytica or, alternatively, the enhancement of the thermal resistance of the host yeast strain. Overall, these encouraging findings confirm that the creation of an efficient, engineered cellulolytic Y. lipolytica strain is achievable. Moreover, the good performance of our prototype strain on a sample of industrial cellulose substrate reveals that such a strain could be a useful starting point for the development of an advanced generation biorefinery process for the production of bioenergy and valuable chemicals. In addition to the future steps described above, a further action will be to demonstrate that a cellulolytic Y. lipolytica strain can produce lipids when growing on cellulose as the sole carbon source.
Methods
Strains and media
The genotypes of the microbial strains used in the present study are summarized in Table 3. E. coli DH5 were purchased from Invitrogen (Paisley, UK) and used for plasmid construction. The Y. lipolytica strains were routinely cultivated in a medium composed of 10 g/L yeast extract, 20 g/L Bacto peptone, and 20 g/L glucose (YPD). Transformants were selected on solid YNB medium (1.7 g/L YNB, 10 g/L glucose or cellobiose, 5 g/L ammonium chloride, with (for Ura+) or without (for Leu+) 2 g/L casamino acids, and 50 mM sodium–potassium phosphate buffer, pH 6.8), supplemented with uracil (440 mg/L) or leucine (440 mg/L) depending on the auxotrophic requirements. Solid media contained 1.5% agar. The detection of endoglucanase activity in solid YNBcasa medium was achieved by incorporating 2 g/L Azo-CM-Cellulose (Megazyme). For cellulase characterization, enzymes were produced in YTD medium (10 g/L, 20 g/L tryptone, 50 g/L glucose, and 100 mM phosphate buffer, pH 6.8). To evaluate the growth of the engineered cellulolytic Y. lipolytica on cellulose, transformants were aerobically cultivated in defined medium containing vitamins, trace elements [48], and salts, including 3.5 g/L (NH4)2SO4, 3.0 g/L K2HPO4, 3.0 g/L NaH2PO4, and 1.0 g/L MgSO4·7H2O with 27 g/L CIMV-cellulose (cellulose content ≈91%, provided by CIMV S.A.) or 25 g/L Avicel PH-101 (Sigma).
Plasmid constructions
The plasmids constructed in the present study are summarized in Table 5, and all primers are listed in Table 6. In brief, the total RNA from 5-day-cultured T. reesei QM9414 was isolated using RNeasy Plus Mini Kit (QIAGEN), and reverse transcription was performed using iScript™ cDNA Synthesis Kit (BIO-RAD) according to the manufacturer’s instructions. For the expression of wild-type proteins, EG I (GenBank accession code: XM_006965612.1), EG II (GenBank accession code: XM_006965612.1), and CBH II (GenBank accession code: XM_006962518.1) were amplified from the cDNA of T. reesei by PCR using F (1-3) as forward primers and R (1-3) as reverse primers, respectively. A 15-base pair homologous sequence of the target plasmid was introduced into the end of each gene during PCR amplification. Afterward, the genes encoding EG I, EG II, and CBH II were fused with the PCR fragment (primers JMP1F/JMP1R) of secretion vector JMP62UraTEF or JMP62LeuTEF under the control of TEF promoter and the pre-pro sequence of lipase 2 (33N-terminal amino acids of Lipase 2, Genbank accession number: Q9P8F7) of Y. lipolytica by In-Fusion Cloning Kits (Clontech). For the expression of His-tagged proteins, EG I, EG II, and CBH II were cloned by PCR with F4 as forward primer and R (4-6) as reverse primers using the expression vectors constructed in last step as template, and fused with PCR fragment (primers JMP2F/JMP2R) of the vector JMP62UraTB1his [28].
Considering the challenge to express CBH I proteins in yeast, the gene coding the sequences of CBH I from T. reesei, P. funiculosum, and N. crassa were codon-optimized based on the codon bias of Y. lipolytica, and were synthesized by GenScript (USA) and cloned into the plasmid JMP62UraTEF, JMP62LeuTEF, or JMP62UraTB1his under the control of TEF promoter. These codon-optimized nucleotide sequences can be found in Additional file 3.
In addition, an enhancer comprising four tandem copies of upstream activation sequences (4UASs) was cloned by PCR with primers HTF and HTR using vector PUB4-CRE as template [49]. The fusion of this DNA fragment with the PCR product (primers JMP3F/JMP3R) of vector JMP62UraTEF and JMP62LeuTEF yielded vectors JMP62UraHTEF and JMP62LeuHTEF, respectively. To enhance the expression level of CBH proteins, the plasmids, JMP62UraCBHI and JMP62LeuCBHII, were digested using BamHI/AvrII, and the NcCBH I and TrCBH II genes were, respectively, recovered and then inserted into the corresponding sites of the plasmids, JMP62UraHTEF and JMP62LeuHTEF.
After construction, all expression vectors were verified by DNA sequencing (GATC Biotech, Konstanz, Germany). For Y. lipolytica transformation, vectors were digested using NotI, thus generating a linear DNA with Zeta sequences at both extremities. Then, the gel-purified expression cassettes were introduced into the Zeta docking platform of Y. lipolytica JMY1212 Zeta for the expression of single cellulase, or randomly into the genome of ∆poxB12 strain, for coexpression of multiple cellulases, using the lithium acetate method [50]. For the latter case, the LoxP-Cre recombination system was used for marker rescue and to ensure the multistep insertion of the target genes [49]. The successful multiple integration of the heterologous genes into the genome of Y. lipolytica was verified by PCR using gene-specific primers (Additional file 1: Table S1). In addition, transformants expressing multiple enzymes were tested for growth on cellobiose, and for degradation of Azo-CMC. Clones displaying both activities were retained for further analysis. Table 3 summarizes the expressed cellulase genes and their corresponding Y. lipolytica transformants.
Enzyme production and activity assay
Recombinant protein production by Y. lipolytica was carried in YTD medium in shake flask at 28 °C and 120 rpm for 5 days. PASC was prepared from Avicel PH-101 as previously described [51]. The activities of EGs were measured on CMC (Megazyme), PASC, β-1, 3-glucan from Euglena gracilis (≥99%, Sigma), β-1, 4-glucan from barley (≥95%, Sigma), Avicel PH-101 (Sigma), and cellotriose (Megazyme) using previously described method with slight modifications [52]. In brief, the reaction mixture contained 1% (w/v) insoluble cellulosic substrate, or 5 mM cellotriose, 50 mM citrate buffer (pH 4.8), and proper volume of diluted enzyme solution. The reaction was conducted at 50 °C for 30 min, and then reducing sugars were quantified using the dinitrosalicylic acid (DNS) reagent. A similar method was used to evaluate CBH activity using PASC and Avicel as substrates. One unit of activity (U) was defined as the amount of enzyme required to release 1 μmol of reducing sugars per min. All protein concentrations were measured using the Bradford method and bovine serum albumin as a standard [53]. All enzymatic activity measurements were performed in triplicate unless otherwise stated.
SDS-PAGE and Western blot analysis
SDS-PAGE was conducted using Mini-PROTEAN TGX Stain-Free precast gels (Biorad) according to the manufacturer’s instructions. 15 μL of culture supernatant or enzyme solution was loaded into each well. Western blotting of proteins was performed as described previously [54]. Crude supernatant of Y. lipolytica JMY1212 expressing EGs and CBHs fused with the His6 tag were concentrated 10-fold by ultrafiltration with an Omega™ membrane disk filter at 10 kDa cut off (Pall, France). Blots were sequentially treated with mouse non position-specific His-Tag antibody 1:2500 (THE™ from Genscript, Piscataway, NJ, USA) and the alkaline phosphatase-conjugated goat anti-mouse IgG (1:10000). For the detection, the PVDF membrane was incubated with a mixture of nitro blue tetrazolium chloride and 5-Bromo-4-chloro-3-indolyl phosphate (NBT/BCIP) (Sigma).
Purification of recombinant cellulases and deglycosylation
Yarrowia lipolytica JMY1212 expressing His6-tagged EG I, EG II, CBH I, and CBH II, respectively, was grown in 200 mL YTD medium at 120 rpm, 28 °C for 48 h. After centrifugation (8000×g for 5 min at 4 °C), the supernatant was concentrated 10-fold by ultrafiltration using an Omega™ membrane disk filter at 10 kDa cutoff (Pall, France), and applied to 2 mL of TALON Metal Affinity Resin (Clontech, Takara-Bio, Kyoto, Japan). Subsequently, protein was eluted using imidazole buffer according to the manufacturer’s instructions. Deglycosylation was carried out by treating the purified proteins with endoglycosidase H (New England Biolabs, Beverly, MA, USA) to remove N-linked carbohydrates at 37 °C for 1 h. Protein samples were analyzed by SDS-PAGE and visualized with colloidal coomassie blue staining.
Growth of yeast expressing multiple cellulases on CIMV and Avicel cellulose
Yeast growth on CIMV-cellulose and Avicel was performed in 50 mL of the defined medium containing 27.5 g/L CIMV or 25 g/L Avicel cellulose stored in 250-mL Erlenmeyer flasks. Yeasts were precultivated in defined medium until middle exponential phase, and the cells were collected by centrifugation. After washing with deionized water, the cells were used to inoculate the defined medium to yield an initial biomass concentration of 1.0 g-DCW/L. The cultivations were conducted at 28 °C, and samples were taken at the end of 5 days to determine concentrations of biomass and residual cellulose.
Analysis of residual cellulose and determination of dry cell weight
The quantification of cellulose residues and dry cell matter was conducted as previously described with slight modifications [27]. In brief, the cell pellets mixed with cellulose residues from the above cultures were harvested by centrifugation at 8000×g for 10 min at 4 °C. After 2-time wash with distilled water, the collected cellulose–cell pellet was freeze-dried and weighed. The amount of cellulose that remained was calculated from the total glucose released from enzymatic hydrolysis of the cellulose residues using Cellic® CTec2, and verified by diluted acid-based hydrolysis of the residues with 2.5% sulfuric acid at 121 °C for 1 h. Dry cell weight was deduced by subtracting the amount of cellulose from the weight of cellulose–cell pellet. Glucose was measured by HPLC as described before [28]. The biomass yield was calculated as the ratio of the amount of biomass obtained divided by the amount of carbon source consumed.
Abbreviations
- BGL:
-
β-glucosidase
- CBH:
-
cellobiohydrolase
- CBP:
-
consolidated bioprocessing
- CMC:
-
carboxymethyl cellulose
- DCW:
-
dry cell weight
- EG:
-
endoglucanase
- Endo H:
-
endoglycosidase
- FDA:
-
Food and Drug Administration
- GRAS:
-
generally recognized as safe
- rh:
-
His6-tagged recombinant proteins
- LCB:
-
lignocellulosic biomass
- MW:
-
molecular weight
- Nc :
-
Neurospora crassa
- PAGE:
-
polyacrylamide gel electrophoresis
- PASC:
-
phosphoric acid-swollen cellulose
- Pf :
-
Penicillium funiculosum
- r :
-
recombinant proteins
- Tr :
-
Trichoderma reesei
- yl :
-
Y. lipolytica
References
den Haan R, van Rensburg E, Rose SH, Görgens JF, van Zyl WH. Progress and challenges in the engineering of non-cellulolytic microorganisms for consolidated bioprocessing. Curr Opin Biotechnol. 2015;33:32–8.
Menon V, Rao M. Trends in bioconversion of lignocellulose: biofuels, platform chemicals & biorefinery concept. Progr Energy Combust Sci. 2012;38:522–50.
Parthasarathi R, Bellesia G, Chundawat SPS, Dale BE, Langan P, Gnanakaran S. Insights into hydrogen bonding and stacking interactions in cellulose. J Phys Chem A. 2011;115(49):14191–202.
Mansfield SD, Mooney C, Saddler JN. Substrate and enzyme characteristics that limit cellulose hydrolysis. Biotechnol Prog. 1999;15(5):804–16.
Yang B, Wyman CE. Effect of xylan and lignin removal by batch and flow through pretreatment on the enzymatic digestibility of corn stover cellulose. Biotechnol Bioeng. 2004;86(1):88–95.
Jeoh T, Ishizawa CI, Davis MF, Himmel ME, Adney WS, Johnson DK. Cellulase digestibility of pretreated biomass is limited by cellulose accessibility. Biotechnol Bioeng. 2007;98(1):112–22.
Stephanopoulos G. Challenges in engineering microbes for biofuels production. Science. 2007;315(5813):801–4.
Gray KA, Zhao L, Emptage M. Bioethanol. Curr Opin Chem Biol. 2006;10(2):141–6.
Mazzoli R. Development of microorganisms for cellulose-biofuel consolidated bioprocessings: metabolic engineers’ tricks. Comput Struct Biotechnol J. 2012;3:e201210007.
Delmas M. Vegetal refining and agrochemistry. Chem Eng Technol. 2008;31:792–7. http://doi.wiley.com/10.1002/ceat.200800052. Accessed 28 Feb 2011.
Delmas M, Benjelloun MB. Process for producing bioethanol by enzymatic hydrolysis of cellulose. E.U. Patent EP 2627775 A2, Aug 21, 2013.
Olson DG, McBride JE, Shaw AJ, Lynd LR. Recent progress in consolidated bioprocessing. Curr Opin Biotechnol. 2012;23:396–405.
Elkins JG, Raman B, Keller M. Engineered microbial systems for enhanced conversion of lignocellulosic biomass. Curr Opin Biotechnol. 2010;21:657–62.
Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev. 2002;66(3):506–77.
Tammali R, Seenayya G, Reddy G. Fermentation of cellulose to acetic acid by Clostridium lentocellum SG6: induction of sporulation and effect of buffering agent on acetic acid production. Lett Appl Microbiol. 2003;37(4):304–8.
Tian L, Papanek B, Olson DG, Rydzak T, Holwerda EK, Zheng T, Zhou J, Maloney M, Jiang N, Giannone RJ, Hettich RL, Guss AM, Lynd LR. Simultaneous achievement of high ethanol yield and titer in Clostridium thermocellum. Biotechnol Biofuels. 2016;9:116.
Ryu S, Karim MN. A whole cell biocatalyst for cellulosic ethanol production from dilute acid-pretreated corn stover hydrolyzates. Appl Microbiol Biotechnol. 2011;91:529–42.
la Grange DC, den Haan R, van Zyl WH. Engineering cellulolytic ability into bioprocessing organisms. Appl Microbiol Biotechnol. 2010;87:1195–208.
Yanase S, Hasunuma T, Yamada R, Tanaka T, Ogino C, Fukuda H, et al. Direct ethanol production from cellulosic materials at high temperature using the thermotolerant yeast Kluyveromyces marxianus displaying cellulolytic enzymes. Appl Microbiol Biotechnol. 2010;88:381–8.
Brethauer S, Studer MH. Consolidated bioprocessing of lignocellulose by a microbial consortium. Energy Environ Sci. 2014;7:1446–53.
Thevenieau F, Nicaud J-M. Microorganisms as sources of oils. OCL. 2013;20(6):1–8.
Blazeck J, Hill A, Liu L, Knight R, Miller J, Pan A, Otoupal P, Alper HS. Harnessing Yarrowia lipolytica lipogenesis to create a platform for lipid and biofuel production. Nat Commun. 2014;5:3131.
Groenewald M, Boekhout T, Neuvéglise C, Gaillardin C, van Dijck PW, Wyss M. Yarrowia lipolytica: safety assessment of an oleaginous yeast with a great industrial potential. Crit Rev Microbiol. 2014;40(3):187–206.
Madzak C, Gaillardin C, Beckerich JM. Heterologous protein expression and secretion in the non-conventional yeast Yarrowia lipolytica: a review. J Biotechnol. 2004;109(1–2):63–81.
Nicaud J-M, Madzak C, van den Broek P, Gysler C, Duboc P, Niederberger P, Gaillardin C. Protein expression and secretion in the yeast Yarrowia lipolytica. FEMS Yeast Res. 2002;2(3):371–9.
Boonvitthya N, Bozonnet S, Burapatana V, O’Donohue MJ, Chulalaksananukul W. Comparison of the heterologous expression of Trichoderma reesei endoglucanase II and cellobiohydrolase II in the yeasts Pichia pastoris and Yarrowia lipolytica. Mol Biotechnol. 2013;54:158–69.
Wei H, Wang W, Alahuhta M, Vander Wall T, Baker JO, Taylor LE II, Decker SR, Himmel ME, Zhang M. Engineering towards a complete heterologous cellulase secretome in Yarrowia lipolytica reveals its potential for consolidated bioprocessing. Biotechnol Biofuels. 2014;7(1):148.
Guo Z, Duquesne S, Bozonnet S, Cioci G, Nicaud JM, Marty A, O’Donohue MJ. Development of cellobiose-degrading ability in Yarrowia lipolytica strain by overexpression of endogenous genes. Biotechnol Biofuels. 2015;8:109.
Chokhawala HA, Roche CM, Kim TW, Atreya ME, Vegesna N, Dana CM, Blanch HW, Clark DS. Mutagenesis of Trichoderma reesei endoglucanase I: impact of expression host on activity and stability at elevated temperatures. BMC Biotechnol. 2015;15:11.
Bordes F, Fudalej F, Dossat V, Nicaud J-M, Marty A. A new recombinant protein expression system for high-throughput screening in the yeast Yarrowia lipolytica. J Microbiol Methods. 2007;70(3):493–502.
Park CS, Chang CC, Ryu DD. Expression and high-level secretion of Trichoderma reesei endoglucanase I in Yarrowia lipolytica. Appl Biochem Biotechnol. 2000;87:1–15.
Chauhan JS, Rao A, Raghava GPS. In silico platform for prediction of N-, O- and C-glycosites in eukaryotic protein sequences. PLoS ONE. 2013;8(6):e67008.
Banerjee G, Car S, Scott-Craig JS, Borrusch MS, Aslam N, Walton JD. Synthetic enzyme mixtures for biomass deconstruction: production and optimization of a core set. Biotechnol Bioeng. 2010;106(5):707–20.
Billard H, Faraj A, Lopes Ferreira N, Menir S, Heiss-Blanquet S. Optimization of a synthetic mixture composed of major Trichoderma reesei enzymes for the hydrolysis of steam-exploded wheat straw. Biotechnol Biofuels. 2012;5(1):9.
Gao DH, Chundawat SPS, Krishnan C, Balan V, Dale BE. Mixture optimization of six core glycosyl hydrolases for maximizing saccharification of ammonia fiber expansion (AFEX) pretreated corn stover. Bioresour Technol. 2010;101(8):2770–81.
Szijártó N, Siika-aho M, Sontag-Strohm T, Viikari L. Liquefaction of hydrothermally pretreated wheat straw at high-solids content by purified Trichoderma enzymes. Biores Technol. 2011;102:1968–74.
Nakazawa H, Okada K, Kobayashi R, Kubota T, Onodera T, Ochiai N, Omata N, Ogasawara W, Okada H, Morikawa Y. Characterization of the catalytic domains of Trichoderma reesei endoglucanase I, II, and III, expressed in Escherichia coli. Appl Microbiol Biotechnol. 2008;81(4):681–9.
Godbole S, Decker SR, Nieves RA, Adney WS, Vinzant TB, Baker JO, Thomas SR, Himmel ME. Cloning and expression of Trichoderma reesei cellobiohydrolase I in Pichia pastoris. Biotechnol Prog. 1999;15:828–33.
Boer H, Teeri TT, Koivula A. Characterization of Trichoderma reesei cellobiohydrolase Cel7A secreted from Pichia pastoris using two different promoters. Biotechnol Bioeng. 2000;69:486–94.
Broda P, Birch PR, Brooks PR, Sims PF. Lignocellulose degradation by Phanerochaete chrysosporium: gene families and gene expression for a complex process. Mol Microbiol. 1996;19(5):923–32.
Hildebrand A, Szewczyk E, Lin H, Kasuga T, Fan Z. Engineering Neurospora crassa for improved cellobiose and cellobionate production. Appl Environ Microbiol. 2015;81(2):597–603.
Taleb F, Radford A. The cellulase complex of Neurospora crassa: cbh-1 cloning, sequencing and homologies. Gene. 1995;161(1):137–8.
Baker JO, Ehrman CI, Adney WS, Thomas SR, Himmel ME. Hydrolysis of cellulose using ternary mixtures of purified cellulases. Appl Biochem Biotechnol. 1998:395–403.
Ganner T, Bubner P, Eibinger M, Mayrhofer C, Plank H, Nidetzky B. Dissecting and reconstructing synergism: in situ visualization of cooperativity among cellulases. J Biol Chem. 2012;287(52):43215–4322.
Boisset C, Fraschini C, Schulein M, Henrissat B, Chanzy H. Imaging the enzymatic digestion of bacterial cellulose ribbons reveals the endo character of the cellobiohydrolase Cel6A from Humicola insolens and its mode of synergy with cellobiohydrolase Cel7A. Appl Environ Microbiol. 2000;66(4):1444–52.
Blazeck J, Liu L, Redden H, Alper H. Tuning gene expression in Yarrowia lipolytica by a hybrid promoter approach. Appl Environ Microbiol. 2011;77(22):7905–14.
Lantz SE, Goedegebuur F, Hommes R, Kaper T, Kelemen BR, Mitchinson C, Wallace L, Ståhlberg J, Larenas EA. Hypocrea jecorina CEL6A protein engineering. Biotechnol Biofuels. 2010;3:20.
Verduyn C, Postma E, Scheffers WA, van Dijken JP. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous culture study on the regulation of respiration and alcoholic fermentation. Yeast. 1992;8:501–17.
Fickers P, Le Dall MT, Gaillardin C, Thonart P, Nicaud J-M. New disruption cassettes for rapid gene disruption and marker rescue in the yeast Yarrowia lipolytica. J Microbiol Methods. 2003;55(3):727–37.
Duquesne S, Bordes F, Fudalej F, Nicaud J-M, Marty A. The yeast Yarrowia lipolytica as a generic tool for molecular evolution of enzymes. Methods Mol Biol. 2012;861:301–12.
Zhang YHP, Cui J, Lynd LR, Kuang LR. A transition from cellulose swelling to cellulose dissolution by o-phosphoric acid: evidence from enzymatic hydrolysis and supramolecular structure. Biomacromolecules. 2006;7:644–8.
Ghose TK. Measurement of cellulase activities. Pure Appl Chem. 1987;59:257–68.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Duquesne S, Bozonnet S, Bordes F, Dumon C, Nicaud J-M, Marty A. Construction of a highly active xylanase displaying oleaginous yeast: comparison of anchoring systems. PLoS ONE. 2014;9(4):e95128.
Authors’ contributions
ZPG, SD, SB, AM and MJD conceived the study and participated in its design. ZPG designed the constructs, carried out all the experiments, and drafted the manuscript. GC participated in the protein purification. SB, SD, JMN, AM, and MJD revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to express their gratitude to Nelly Monties for her help with chromatographic analyses. The authors thank Françoise Ouarne for kindly providing the CIMV-cellulose. The authors also thank the ICEO facility dedicated to enzyme screening and discovery, and part of the Integrated Screening Platform of Toulouse (PICT, IBiSA) for providing access to its protein purification and analytical facilities.
Competing interests
The authors declare that they have no competing interests.
Availability of supporting data
All data generated or analyzed in the present study are included in this published article and a supporting material “Additional files 1, 2, and 3”.
Consent for publication
All authors have given their consent for publication.
Funding
This work was funded by the Agence Nationale de la Recherche (Investissements d’Avenir program; reference ANR-11-BTBR-0003). Dr. Guo is a recipient of a postdoctoral fellowship from the Institut National de la Recherche Agronomique.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
13068_2017_819_MOESM1_ESM.pdf
Additional file 1: Figure S1. Screening of Y. lipolytica expressing EGs on indication plate containing YNBcasa medium supplemented with 0.2% w/vAzo-CM-Cellulose. Figure S2. PCR verification of Y. lipolytica transformants expressing multiple cellulases. Figure S3. Nucleotide sequences of constructs. Table S1. The sequences of the oligonucleotide primers used for PCR verification of yl-transformants.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guo, Zp., Duquesne, S., Bozonnet, S. et al. Conferring cellulose-degrading ability to Yarrowia lipolytica to facilitate a consolidated bioprocessing approach. Biotechnol Biofuels 10, 132 (2017). https://doi.org/10.1186/s13068-017-0819-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-017-0819-8