
Abstract
Background
Acetoin reductase (Acr) catalyzes the conversion of acetoin to 2,3-butanediol (2,3-BD) with concomitant oxidation of NADH to NAD+. Therefore, intracellular 2,3-BD production is likely governed by the quantities of rate-limiting factor(s) Acr and/or NADH. Previously, we showed that a high level of Acr was beneficial for 2,3-BD accumulation.
Results
Metabolic engineering strategies were proposed to redistribute carbon flux to 2,3-BD by manipulating NADH levels. The disruption of NADH oxidase (YodC, encoded by yodC) by insertion of a formate dehydrogenase gene in Bacillus subtilis was more efficient for enhancing 2,3-BD production and decreasing acetoin formation than the disruption of YodC by the insertion of a Cat expression cassette. This was because the former resulted in the recombinant strain AFY in which an extra NADH regeneration system was introduced and NADH oxidase was disrupted simultaneously. On fermentation by strain AFY, the highest 2,3-BD concentration increased by 19.9 % while the acetoin titer decreased by 71.9 %, relative to the parental strain. However, the concentration of lactate, the main byproduct, increased by 47.2 %. To further improve carbon flux and NADH to 2,3-BD, the pathway to lactate was blocked using the insertional mutation technique to disrupt the lactate dehydrogenase gene ldhA. The resultant engineered strain B. subtilis AFYL could efficiently convert glucose into 2,3-BD with little acetoin and lactate accumulation.
Conclusions
Through increasing the availability of NADH and decreasing the concentration of unwanted byproducts, this work demonstrates an important strategy in the metabolic engineering of 2,3-BD production by integrative recombinant hosts.
Similar content being viewed by others
Background
2,3-Butanediol (2,3-BD) is an important platform chemical due to its wide industrial applications [1–3]. Significantly, 2,3-BD is a potentially valuable fuel additive with a heating value of 27.2 kJ/g, comparable to that of other liquid fuels (e.g., ethanol 29.055 kJ/g, methanol 22.081 kJ/g) [4]. Because of the shortage of fossil fuels and the development of biorefineries from renewable resources, microbial production of 2,3-BD has attracted growing attention [1, 2].
A number of microorganism genera are employed to produce 2,3-BD, including Klebsiella, Enterobacter, Bacillus, and Serratia [5]. Among these strains, B. subtilis, generally regarded as safe (GRAS) [6], is often used as a platform organism to engineer Gram-positive bacterial pathways for industrial production of various secondary metabolites [7]. In such bacterial metabolism, glucose is first converted to pyruvate before generation of major products. In addition to 2,3-BD, the pyruvate produced from glucose is channeled into a mixture of ethanol, acetoin, formate, acetate, and lactate, through the mixed acid-2,3-BD fermentation pathway [1, 2, 5]. In previous studies, several strategies were used to improve 2,3-BD production, such as optimizing culture conditions, and establishing mathematical models [8–10].
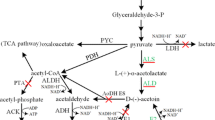
The 2,3-BD pathway takes part in regulating the intracellular NAD+/NADH ratio [1, 2]. Because 2,3-BD, ethanol, and lactic acid are NADH-dependent products involved in the 2,3-BD pathway (Fig. 1), NADH availability and its proportion in the active form (i.e. as NADH rather than NAD+) play a key role in dictating the entire process yield of 2,3-BD. 2,3-BD production could be further improved through manipulation of the NADH-dependent pathways. To block a pathway that competitively consumes NADH and to redirect carbon flux towards 2,3-BD production, many investigators [11–14] have attempted to construct different lactate dehydrogenase (LdhA; encoded by the ldhA gene) inactivated strains. They found that inactivation of LdhA resulted in improving 2,3-BD production, while suppressing lactate and acetoin formation.

Metabolic engineering strategies for redistributing the carbon flux to 2,3-BD by manipulating NADH levels. 2,3-BD is an NADH-dependent product; the NADH availability and its proportion in the active form play important roles in 2,3-BD production. But NADH oxidase catalyzes the oxidation of NADH to NAD+. Lactate, one of the main by-products, competes with 2,3-BD for pyruvate as a metabolic intermediate and NADH as a cofactor. To block a pathway that competitively consumes NADH and to redirect carbon flux towards 2,3-BDO production, the NADH oxidase was disrupted by insertion of a formate dehydrogenase gene fdh, and then the pathway to lactate was blocked using the insertional mutation technique to disrupt the lactate dehydrogenase gene ldhA. (3-PGA 3-Phosphate glyceraldehyde, 1,3-BPG 1,3-Bisphosphoglycerate, Red arrows, oxidation of NADH to NAD+; Green arrows, reduction of NAD+ to NADH;  , blocking the pathway; The blue dotted box, introducing extrinsic pathway)
, blocking the pathway; The blue dotted box, introducing extrinsic pathway)
Furthermore, Ji et al. [15] successfully constructed a K. oxytoca mutant deficient in ethanol accumulation by replacing the aldA gene (coding aldehyde dehydrogenase) with a tetracycline resistance cassette. They found that the carbon flux to 2,3-BD was improved by eliminating the ethanol pathway and in the meantime suppressing the formation of another main byproduct, acetoin. A double mutant K. pneumoniae strain with deletion of adhE (coding acetaldehyde dehydrogenase) and ldhA was constructed by Guo et al. [16], which resulted in accelerated fermentation and higher 2,3-BD production. Previously, we introduced extra copies of glyceraldehyde-3-phosphate dehydrogenase (GpdH) and acetoin reductase (Acr) into the GRAS strain B. amyloliquefaciens and found that overexpressing this NADH/NAD+ regeneration system effectively improved 2,3-BD production and inhibited byproduct accumulation [17]. Dai et al. [18] reported that the addition of vitamin C could elevate the NADH/NAD+ ratio, resulting in a remarkable increase of 2,3-BD titer in Paenibacillus polymyxa CJX518.
Acetoin reductase could convert acetoin to 2,3-BD, with concomitant oxidation of NADH to NAD+. Therefore, intracellular 2,3-BD production is likely governed by the quantities of the rate-limiting factor(s) the acetoin dehydrogenase complex AcoABCL (Acr plays the most important role) and/or NADH. Previously, we identified that low levels of Acr and NADH were key factors that limited acetoin degradation and found that increasing the Acr level in B. subtilis was beneficial for 2,3-BD production [17, 19], but there were still large quantities of acetoin and lactate in the culture broth. In this work, metabolic engineering strategies were proposed to redistribute the carbon flux to 2,3-BD by manipulating NADH levels (Fig. 1).
Results
Inactivation of NADH oxidase and its effect on 2,3-BD production in Bacillus subtilis
Because 2,3-BD is an NADH-dependent product, the NADH availability and its proportion in the reduced form play important roles in 2,3-BD production. NADH oxidase catalyzes the oxidation of NADH to NAD+. Previously, we successfully screened a water-forming NADH oxidase (YodC, encoded by yodC, gene ID: 939506) from B. subtilis 168 [20]. Therefore, to prevent NADH oxidation, yodC was disrupted by transforming the plasmid T-yodC::cat into B. subtilis strain ACR. PCR amplification results indicated that the yodC gene was successfully knocked out from B. subtilis ACR, and the activity of YodC was barely detectable in the resulting recombinant strain AY.
The effects of disruption of YodC on cell growth and the intracellular concentrations of NADH and NAD+ were observed. Figure 2 shows that strain AY grew at a similar rate to the parental strain ACR, indicating that the disruption of YodC in B. subtilis ACR did not affect cell growth. In both the parental and engineered strains, intracellular concentrations of NADH, NAD+ and NAD+/NADH ratio continuously changed during the batch fermentation, but no significant difference was observed between the two strains (Fig. 3). This suggests that the redox balance in strain AY was not disturbed, which also explains why cell growth of strain AY was not significantly affected (Fig. 2).

Time profiles of 2,3-BD fermentation with various strains. (ACR, B. subtilis 168 with pMA5-acr; AY, ACR blocked in YodC by insertion of a Cat expression cassette; AFY, Acr blocked in YodC by insertion of fdh gene; AFYL, AFY blocked in LdhA by insertion of a Cat expression cassette. All assays were performed by triplicate cultures, standard deviations of the biological replicates were represented by error bars)

Time profiles of intracellular NADH and NAD+ concentrations of various strains. (ACR, B. subtilis 168 with pMA5-acr; AY, ACR blocked in YodC by insertion of a Cat expression cassette; AFY, Acr blocked in YodC by insertion of fdh gene; AFYL, AFY blocked in LdhA by insertion of a Cat expression cassette. All assays were performed by triplicate cultures, standard deviations of the biological replicates were represented by error bars)
The effects of disruption of YodC on 2,3-BD production were also observed. Figure 2 shows that the glucose consumption rates were very similar between the parental and engineered strains. 2,3-BD production was enhanced in strain AY (Fig. 2), indicating that disruption of YodC increased NADH availability. The highest 2,3-BD concentration was increased by 8.8 % relative to the parental strain, while that of acetoin was decreased by 23.6 % (Fig. 2; Table 1). However, compared with the parental strain, the molar yield of the major unwanted byproduct lactate was 16.7 % higher in the engineered strain AY (Fig. 4; Table 1). This suggests that the ‘‘extra’’ NADH generated by inactivating YodC was utilized in other NADH-dependent pathways (especially in 2,3-BD, lactate pathways), which explains why the knockout of the yodC gene has no significant influence on the NAD +/NADH ratio.

Time profiles of lactate, acetate, and ethanol production with various strains. (ACR, B. subtilis 168 with pMA5-acr; AY, ACR blocked in YodC by insertion of a Cat expression cassette; AFY, Acr blocked in YodC by insertion of fdh gene; AFYL, AFY blocked in LdhA by insertion of a Cat expression cassette. All assays were performed by triplicate cultures: standard deviations of the biological replicates were represented by error bars)
Disruption of NADH oxidase gene yodC and simultaneous introduction of an extra NADH regeneration system in B. subtilis
Although disruption of YodC increased the 2,3-BD yield, the acetoin accumulation in strain AY remained significant. Heterogeneous production of formate dehydrogenase (Fdh, encoded by fdh) in B. subtilis was expected to increase the overall intracellular NADH pool, thereby improving the flux through NADH-dependent pathways (mainly the 2,3-BD pathway). The plasmid T-yodC::fdh was transformed into B. subtilis ACR, resulting in recombinant strain AFY in which an extra NADH regeneration system was introduced and YodC was simultaneously disrupted. PCR amplification results indicated that the yodC gene was successfully knocked out in strain AFY. The Fdh activity was increased by about 15-fold relative to the parental strain. To use the NADH regeneration capability of Fdh, extra sodium formate was needed in the culture medium so that the reaction NaCOOH + NAD+ → CO2 + NADH + Na+ could be catalyzed by Fdh [21]. We found that cell growth of strain AFY was not affected when the concentration of sodium formate was lower than 40 mM (data not shown). Based on the three-stage division of cell growth curves (Fig. 2), 30 mM sodium formate was fed into the cultivation broth of strain AFY at 12, 24, and 48 h after culture inoculation, respectively, to serve as the substrate in the reaction catalyzed by Fdh.
Production of Fdh in B. subtilis AFY led to a slower decrease of both NADH and NAD+ pools than was observed for the parental strain. Meanwhile, there was no remarkable difference in the ratio of NADH/NAD+ between the parental and engineered strains (Fig. 3). These data suggest that the redox balance in B. subtilis AFY producing Fdh was not disturbed, as also observed in K. oxytoca by Zhang et al. [21], which explains why the cell growth of strain AFY was not remarkably affected (Fig. 2). Furthermore, the consumption rate of glucose was not notable altered between the engineered strain AFY and the parental strain Acr (Fig. 2). 2,3-BD was the main product of strain AFY metabolism, followed by lactate and acetoin (Table 1). The maximum 2,3-BD concentration increased by 18.5 % and the acetoin titer decreased by 65.7 % in engineered strain AFY relative to the parental strain (Fig. 2; Table 1). However, the concentration of the main byproduct, lactate, increased by 47.2 % in batch fermentation compared with the corresponding level in strain ACR.
Enhanced 2,3-BD yield through inactivation of lactate dehydrogenase in B. subtilis AFY
Disruption of the NADH oxidase gene and simultaneous introduction of an extra NADH regeneration system in B. subtilis not only enhanced 2,3-BD production and suppressed acetoin accumulation, but also increased byproduct lactate production, which negatively regulates the 2,3-BD yield and increases the costs of downstream separation and purification. Therefore, we also knocked out LDH (gene ldhA) by transforming plasmid T-ldhA::cat into B. subtilis AFY. The PCR amplification results indicated the ldhA gene was successfully knocked out from B. subtilis AFYL, and the activity of LdhA decreased by 90.1 %, which confirmed that ldhA was the major LdhA-producing enzyme in B. subtilis.
The inactivation of LdhA resulted in faster cell growth and shorter fermentation time than for the parental and AFY strains (Fig. 2). Similar stimulatory effects on cell growth and glucose consumption were also observed by Guo et al. [16] in an analogous K. pneumoniae strain. Furthermore, as shown in Fig. 2 and Table 1, the maximum 2,3-BD concentration increased by 25.5 %, while the acetoin titer decreased by 76.4 %, in engineered strain AFYL relative to strain ACR; the lactate concentration decreased by 81.1 % (Fig. 4; Table 1).
Discussion
In the 2,3-butanediol metabolic pathway, ACR catalyzes the conversion of acetoin to 2,3-BD with concomitant oxidation of NADH to NAD+ [7]. NADH oxidase (Nox) catalyzes the oxidation of NADH to NAD+ using molecular oxygen as the electron acceptor. Zhang et al. [20] identified an water-forming NADH oxidase (YodC) in B. subtilis. When YodC was over-expressed in B. subtilis, acetoin production was increased, while the production of 2,3-BD was decreased significantly. Ji et al. [22] and Sun et al. [23] also found that overexpression of NADH oxidase in Klebsiella pneumoniae resulted in a large increase of carbon flux to acetoin and in the ratio of acetoin to 2,3-BD. Because NADH is preferentially used in 2,3-BD synthesis, low levels of this coenzyme may limit the Acr reaction [17, 19]. Therefore, decreasing YodC activity which is possibly beneficial for 2,3-BD production, because NADH oxidase pathway competes with 2,3-BD branch for NADH as a cofactor. Inspired by this idea, we initially disrupted YodC in B. subtilis strain ACR, which increased not only the 2,3-BD yield, but also lactate and ethanol titer. It may be that disruption of YodC decreased metabolism of NADH and increased NADH availability, which resulted in increased fluxes to NADH-dependent pathways (e.g., 2,3-BD, lactate, and ethanol production pathways).
The fluxes through NADH-dependent pathways are determined by the availability of NADH, which could be improved by either decreasing the metabolic activity of branches competing for NADH [24, 25], or by introducing a NADH regeneration system [21]. The most successful NADH regeneration system involves the NAD+-dependent formate dehydrogenase gene fdh from C. boidinii [26]. Ma et al. [27] and Zhang et al. [21] found that introduction of this NADH regeneration system into Klebsiella sp. could efficiently improve glycerol metabolism and promote 1,3-propanediol yield. Therefore, we tried to simultaneously disrupt a NADH metabolic branch and introduce an extra NADH regeneration system into B. subtilis, and observe their effects on 2,3-BD metabolism. As expected, 2,3-BD production was further improved and acetoin accumulation was suppressed, relative to the parental strain. However, the production of byproducts lactate and ethanol was also enhanced; in particular, the lactate concentration was 47 % higher than for the parental strain. If disruption of YodC and introduction of an extra NADH regeneration system indeed increase the available NADH, all of the NADH-dependent pathways, (2,3-BD, lactate, and ethanol production pathways) would benefit from the enhancement.
Lactate is produced as a main byproduct during B. subtilis fermentation. Decreasing the production of this byproduct can be beneficial for 2,3-BD production, because lactate competes with 2,3-BD for pyruvate as a metabolic intermediate and NADH as a cofactor. Jung et al. [28] found that deleting the ldhA gene from Enterobacter aerogenes increased 2,3-BD production. In this study, we successfully deleted the ldhA gene from B. subtilis strain AFY to construct the recombinant strain AFYL. As expected, deletion of ldhA increased 2,3-BD production, by 25.5 %, and decreased the acetoin titer, by 76.4 %; significantly, the concentration of the principal byproduct lactate decreased by 81.1 % (all relative to strain ACR). These results for strain AFYL can be explained by the NADH consumption [12]: by blocking the lactate production pathway, the cell needed to consume NADH through other pathways; thus, the carbon flux toward the 2,3-BD pathway, and hence 2,3-BD biosynthesis, increased. More interestingly, the deletion of ldhA resulted in an increase in microbial cell growth. A similar stimulatory effect on cell growth was observed by Guo et al. [16] and Kim et al. [12] in K. pneumoniae and Jung et al. [28] in E. aerogenes. Generally, a mutant strain has a reduced growth rate compared to its parental strain. The main reason for the improved cell growth of the mutant could be that decreasing lactate production lowered the acidification rate of the culture medium [28].
Conclusion
The GRAS engineered strain B. subtilis AFYL with yodC and ldhA deletion and fdh overexpression was constructed to redistribute carbon flux towards 2,3-BD biosynthesis by manipulation of the NADH levels. Strain AFYL exhibited elevated 2,3-BD production and reduced lactate and acetoin production. In this study, we demonstrated the construction of a target microorganism with its carbon flux redistributed towards 2,3-BD production through metabolic engineering.
Materials and methods
Bacterial strains, primers, and plasmids
All bacterial strains, primers, and plasmids used and constructed in this study are described in Table 2.
Culture conditions
Bacillus subtilis and Escherichia coli were cultured in Luria–Bertani medium at 37 °C on a rotary shaker at 180 rpm. When necessary, 100 mg/L ampicillin, 25 mg/L chloromycetin, 50 mg/L kanamycin, or 900 mg/L formate [29] was added into the culture medium. Well-grown cells were selected for further positive colony selection.
For 2,3-BD fermentation, the seed culture was agitated in 250 mL shaken flasks for 10 h at 180 rpm and 37 °C. Then 1 mL seed culture (OD600 = 4.0–5.0) was inoculated into fermentation medium at 180 rpm and 37 °C. Seed culture medium contained 40 g/L glucose, 10 g/L peptone, 5 g/L yeast extract, and 10 g/L NaCl. The medium was sterilized at 121 °C for 20 min. The 2,3-BD fermentation medium (pH 6.5) contained 100 g/L glucose, 25 g/L corn steep liquor, 5 g/L ammonium citrate, 3 g/L K2HPO4, 0.6 g/L succinic acid, and 0.3 g/L MgSO4·7H2O. For preparing the 2,3-BD fermentation medium, glucose and the other supplements were separately sterilized at 121 °C for 20 min, and then the two solutions were mixed.
Disruption of the yodC and ldhA genes
For disruption of the yodC gene, plasmids T-yodC::cat and T-yodC::fdh were constructed, respectively. The yodC gene encoding NADH oxidase (YodC) from B. subtilis strain ACR was amplified by PCR using primers P1 and P2 (Table 2). T-yodC was constructed by inserting yodC into the cloning sites of pMD-18T. The CAT expression cassette (encoded by cat) from the vector pBGSC6 was amplified using primers P3 and P4. The fdh gene encoding formate dehydrogenase (Fdh) from Candida boidinii was PCR amplified using primers P5 and P6. The amplified fdh gene was inserted into the Nde I and Mlu I sites of plasmid pMA5-HapII to create pMA5-fdh. The fdh gene plus the Hap II promoter from plasmid pMA5-fdh was then PCR amplified using primers P7 and P8. Then, cat and HapII-fdh were separately inserted into the Eco47 III site of T-yodC to construct plasmids T-yodC::cat and T-yodC:: HapII-fdh. The plasmids T-yodC::cat and T-yodC:: HapII-fdh were transformed into B. subtilis ACR to construct the recombinant strains AY and AFY, respectively. The ldhA gene encoding LDH from B. subtilis ACR was amplified by PCR using primers P9 and P10. T-ldhA was constructed by inserting yodC into the cloning sites of pMD-18T. The ldhA gene was disrupted by the insertion of cat to construct plasmid T-ldhA::cat; then T-ldhA::cat was transformed into B. subtilis AFY to construct the recombinant strain AFYL. The insertions of the Cat expression cassette and HapII-fdh were confirmed by PCR. Plasmids were first transformed into E. coli JM109 cells by the calcium chloride method [18], subsequently isolated from E. coli JM109, and then transformed into B. subtilis using the method described by Vojcic et al. [30].
Determination of NAD+ and NADH concentrations
The intracellular concentrations of NAD+ and NADH were determined by procedures described in our previous study [19].
Enzyme assays
For preparation of crude YodC, the cell pellets collected by centrifugation were suspended and washed with 0.1 M potassium phosphate buffer (pH7.0) at least for three times. Then cells were resuspended in a washing buffer containing 0.15 mM FAD and 62.5 mM l-cysteine-HCl (PPFC-buffer). DNase (2000 units) and phenylmethylsulfonyl fluorid (1 mM) were then added to the suspension. The YodC activity was assayed as described by previous study by measuring the decrease of absorbance at 340 nm at 25 °C [31].
Cell-free extracts used for Fdh activity assay were prepared as previously described [21, 27]. Berríos-Rivera et al. [26] reported that Abs340 measurements are increased by cell extracts as NAD+ is reduced to NADH, which has an absorption maximum at 340 nm. From the resulting linear increase in Abs340, the Fdh activity can be measured. The reaction mixture (5 mL) contained 20 μL cell-free extract, 2.0 mM NAD+, 0.1 M sodium formate, and 100 mM 2-mercaptoethanol in phosphate buffer (10 mM, pH 7.5). The protein concentration of the cell-free extracts was determined by the method of Bradford [32]. All experiments were replicated at least three times.
Analysis of cell growth and metabolites
The cell mass concentration was determined from the OD at 600 nm in an ultraviolet (UV)-visible spectrophotometer (UNICO UV-2000 spectrophotometer, Shanghai, China). The dry cell weight (DCW) could be calculated from OD600 using an equation that DCW (g/L) = 0.237 OD600 − 0.0126. The composition of the fermentation broth (glucose, 2,3-BD, acetoin, acetate, lactate, and ethanol) was determined by high-performance liquid chromatography [33]. All experiments were replicated at least three times.
Abbreviations
- 2,3-BD:
-
2,3-butanediol
- Acr:
-
acetoin reductase
- YodC:
-
NADH oxidase
- LdhA:
-
lactate dehydrogenase
- GRAS:
-
generally regarded as safe
- Fdh:
-
formate dehydrogenase
References
Ji XJ, Huang H, Ouyang PK (2011) Microbial 2,3-butanediol production: a state-of-the-art review. Biotechnol Adv 29(3):351–364
Celinska E, Grajek W (2009) Biotechnological production of 2,3-butanediol–current state and prospects. Biotechnol Adv 27(6):715–725
Syu MJ (2001) Biological production of 2,3-butanediol. Appl Microbiol Biotechnol 55(1):10–18
Flickinger MC (1980) Current biological research in conversion of cellulosic carbohydrates into liquid fuels: how far have we come. Biotechnol Bioeng 22:27–48
Maddox IS (1996) Microbial production of 2,3-butanediol. 2nd ed. In: Rehm HJ, Reed G, Pühler A, Stadler P (ed) Biotechnology. Weinheim: VCH Verlagsgesellschaft
de Boer AS, Diderichsen B (1991) On the safety of Bacillus subtilis and B. amyloliquefaciens: a review. Appl Microbiol Biotechnol 36(1):1–4
Nicholson WL (2008) The Bacillus subtilis ydjL (bdhA) gene encodes acetoin reductase/2,3-butanediol dehydrogenase. Appl Environ Microbiol 74(22):6832–6838
Ma C, Wang A, Qin J, Li L, Ai X, Jiang T, Tang H, Xu P (2009) Enhanced 2,3-butanediol production by Klebsiella pneumoniae SDM. Appl Microbiol Biotechnol 82(1):49–57
Ji XJ, Huang H, Du J, Zhu JG, Ren LJ, Hu N, Li S (2009) Enhanced 2,3-butanediol production by Klebsiella oxytoca using a two-stage agitation speed control strategy. Bioresour Technol 100(13):3410–3414
Ehsani M, Fernandez MR, Biosca JA, Julien A, Dequin S (2009) Engineering of 2,3-butanediol dehydrogenase to reduce acetoin formation by glycerol-overproducing, low-alcohol Saccharomyces cerevisiae. Appl Environ Microbiol 75(10):3196–3205
Kim DK, Rathnasingh C, Song H, Lee HJ, Seung D, Chang YK (2013) Metabolic engineering of a novel Klebsiella oxytoca strain for enhanced 2,3-butanediol production. J Biosci Bioeng 116(2):186–192
Kim B, Lee S, Jeong D, Yang J, Oh MK, Lee J (2014) Redistribution of carbon flux toward 2,3-butanediol production in Klebsiella pneumoniae by metabolic engineering. PLoS One 9(10):e105322
Li L, Li K, Wang Y, Chen C, Xu Y, Zhang L, Han B, Gao C, Tao F, Ma C, Xu P (2014) Metabolic engineering of Enterobacter cloacae for high-yield production of enantiopure (2R,3R)-2,3-butanediol from lignocellulose-derived sugars. Metab Eng 28C:19–27
Nielsen DR, Yoon SH, Yuan CJ, Prather KL (2010) Metabolic engineering of acetoin and meso-2, 3-butanediol biosynthesis in E. coli. Biotechnol J 5(3):274–284
Ji XJ, Huang H, Zhu JG, Ren LJ, Nie ZK, Du J, Li S (2010) Engineering Klebsiella oxytoca for efficient 2, 3-butanediol production through insertional inactivation of acetaldehyde dehydrogenase gene. Appl Microbiol Biotechnol 85(6):1751–1758
Guo X, Cao C, Wang Y, Li C, Wu M, Chen Y, Zhang C, Pei H, Xiao D (2014) Effect of the inactivation of lactate dehydrogenase, ethanol dehydrogenase, and phosphotransacetylase on 2,3-butanediol production in Klebsiella pneumoniae strain. Biotechnol Biofuels 7(1):44
Yang T-W, Rao Z-M, Zhang X, Xu M-J, Xu Z-H, Yang S-T (2013) Improved production of 2,3-butanediol in Bacillus amyloliquefaciens by over-expression of glyceraldehyde-3-phosphate dehydrogenase and 2,3-butanediol dehydrogenase. PLoS One 8(10):e76149
Dai JJ, Cheng JS, Liang YQ, Jiang T, Yuan YJ (2014) Regulation of extracellular oxidoreduction potential enhanced (R, R)-2,3-butanediol production by Paenibacillus polymyxa CJX518. Bioresour Technol 167:433–440
Yang T-W, Rao Z-M, Zhang X, Xu M-J, Xu Z-H, Yang S-T (2013) Effects of corn steep liquor on production of 2, 3-butanediol and acetoin by Bacillus subtilis. Process Biochem 48:1610–1617
Zhang X, Zhang R, Bao T, Rao Z, Yang T, Xu M, Xu Z, Li H, Yang S (2014) The rebalanced pathway significantly enhances acetoin production by disruption of acetoin reductase gene and moderate-expression of a new water-forming NADH oxidase in Bacillus subtilis. Metab Eng 23:34–41
Zhang Y, Huang Z, Du C, Li Y, Cao Z (2009) Introduction of an NADH regeneration system into Klebsiella oxytoca leads to an enhanced oxidative and reductive metabolism of glycerol. Metab Eng 11(2):101–106
Ji X, Xia Z, Fu N, Nie Z, Shen M, Tian Q, Huang H (2013) Cofactor engineering through heterologous expression of an NADH oxidase and its impact on metabolic flux redistribution in Klebsiella pneumoniae. Biotechnol Biofuels 6:7
Sun JA, Zhang LY, Rao B, Shen YL, Wei DZ (2012) Enhanced acetoin production by Serratia marcescens H32 with expression of a water-forming NADH oxidase. Bioresour Technol 119:94–98
Zhang Y, Li Y, Du C, Liu M, Cao Z (2006) Inactivation of aldehyde dehydrogenase: a key factor for engineering 1,3-propanediol production by Klebsiella pneumoniae. Metab Eng 8(6):578–586
Berrios-Rivera SJ, San KY, Bennett GN (2003) The effect of carbon sources and lactate dehydrogenase deletion on 1,2-propanediol production in Escherichia coli. J Ind Microbiol Biotechnol 30(1):34–40
Berríos-Rivera S, Bennett G, San K (2002) Metabolic engineering of Escherichia coli: increase of NADH availability by overexpressing an NAD+-dependent formate dehydrogenase. Metab Eng 4(3):217–229
Ma Z, Shentu X, Bian Y, Yu X (2013) Effects of NADH availability on the Klebsiella pneumoniae strain with 1,3-propanediol operon over-expression. J Basic Microbiol 53(4):348–354
Jung MY, Ng CY, Song H, Lee J, Oh MK (2012) Deletion of lactate dehydrogenase in Enterobacter aerogenes to enhance 2,3-butanediol production. Appl Microbiol Biotechnol 95(2):461–469
Wu Z, Wang Z, Wang G, Tan T (2013) Improved 1,3-propanediol production by engineering the 2,3-butanediol and formic acid pathways in integrative recombinant Klebsiella pneumoniae. J Biotechnol 168(2):194–200
Vojcic L, Despotovic D, Martinez R, Maurer K-H, Schwaneberg U (2012) An efficient transformation method for Bacillus subtilis DB104. Appl Microbiol Biotechnol 94(2):487–493
de Felipe FL, Hugenholtz J (2001) Purification and characterisation of the water forming NADH-oxidase from Lactococcus lactis. Int Dairy J 11(1):37–44
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72(1–2):248–254
Yang T, Rao Z, Zhang X, Lin Q, Xia H, Xu Z, Yang S (2011) Production of 2,3-butanediol from glucose by GRAS microorganism Bacillus amyloliquefaciens. J Basic Microbiol 51(6):650–658
Hua Q, Shimizu K (1999) Effect of dissolved oxygen concentration on the intracellular flux distribution for pyruvate fermentation. J Biotechnol 68(2–3):135–147
Authors’ contributions
TY conceived of the study, performed the data analysis, and coordinated the manuscript draft and revision. GH, ML, YD executed the experimental work and data analysis. ZR, ZX helped to revise and proofread the manuscript. XZ, MX helped with data analysis. SY coordinated the manuscript draft and revision. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the High-tech Research and Development Programs of China (2015AA021004), the National Natural Science Foundation of China (31400082, 21276110), the Fundamental Research Funds for the Central Universities (JUSRP11544), the National Training Programs of Innovation and Entrepreneurship for Undergraduates (201510295071), the Research Project of Chinese Ministry of Education (113033A), the Priority Academic Program Development of Jiangsu Higher Education Institutions,the 111 Project (111-2-06),and the Jiangsu province “Collaborative Innovation Center for Advanced Industrial Fermentation” industry development program.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, T., Rao, Z., Hu, G. et al. Metabolic engineering of Bacillus subtilis for redistributing the carbon flux to 2,3-butanediol by manipulating NADH levels. Biotechnol Biofuels 8, 129 (2015). https://doi.org/10.1186/s13068-015-0320-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-015-0320-1