
Abstract
The title compounds, 2-azaspiro[4.5]deca-1-one, C9H15NO, (1a), cis-8-methyl-2-azaspiro[4.5]deca-1-one, C10H17NO, (1b), and trans-8-methyl-2-azaspiro[4.5]deca-1-one, C10H17NO, (1c), were synthesized from benzoic acids 2 in only 3 steps in high yields. Crystallization from n-hexane afforded single crystals, suitable for X-ray diffraction. Thus, the configurations, conformations, and interesting crystal packing effects have been determined unequivocally. The bicyclic skeleton consists of a lactam ring, attached by a spiro junction to a cyclohexane ring. The lactam ring adopts an envelope conformation and the cyclohexane ring has a chair conformation. The main difference between compound 1b and compound 1c is the position of the carbonyl group on the 2-pyrrolidine ring with respect to the methyl group on the 8-position of the cyclohexane ring, which is cis (1b) or trans (1c). A remarkable feature of all three compounds is the existence of a mirror plane within the molecule. Given that all compounds crystallize in centrosymmetric space groups, the packing always contains interesting enantiomer-like pairs. Finally, the structures are stabilized by intermolecular N–H···O hydrogen bonds.
Similar content being viewed by others


Introduction
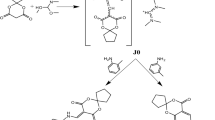
γ-Spirolactams of the general structure 1 (Fig. 1) are interesting organic heterocycles and have attracted much attention as potential medicinal agents [1]. They can be synthesized by various methods, but over many steps [2,3,4]. Cyclohexane-based γ-spirolactams 1 (n = 2) have been described to bind to opioid receptors and as enzyme inhibitors in two patents [5, 6]. Very recently, we developed an easy entry to such compounds by Birch reduction of the corresponding benzoic acids 2 (R = H, Me) and subsequent alkylation and hydrogenation (Fig. 1) [7].

Examples of γ-spirolactams 1 and their synthesis from benzoic acids 2
During the synthesis of γ-spirolactams 1, we obtained different regio- and stereoisomers, some configurations have been already established in our previous studies [7]. However, in the 8-methyl derivatives 1b cis and trans isomers have been isolated, with functional groups too far away from each other to apply NOE NMR experiments for structure elucidation. Therefore, we had to obtain single crystals to determine the structures unequivocally by X-ray diffraction. Besides the configurations, the conformations of γ-spirolactams 1 are interesting, since the six-membered ring will adopt a chair form, which might influence the more flexible five-membered lactam ring. Indeed, crystal structures of simple γ-butyrolactams indicate that the substitution by an additional methyl group influences the conformations remarkably [8,9,10]. Herein, we report three new crystal structures of cyclohexane-based γ-spirolactams 1a, cis-1b and trans-1b. We could determine their configurations and conformations, which will be discussed in detail. Our results should be interesting in comparison with the similar Gabapentin lactam 3 (Fig. 2), whose conformation has already been analyzed in the solid state [11] and which has been found to be neuroprotective in retinal ischemia [12].

Herein investigated γ-spirolactams 1a, cis-1b and trans-1b and known Gabapentin lactam 3
Results and discussion
γ-Spirolactams 1 have been synthesized from the corresponding benzoic acids 2 in only few steps in high yields (Fig. 1, Experimental section). Single crystals have been obtained by crystallization from n-hexane. We started our investigations with the unsubstituted lactam 1a, which crystallizes in the monoclinic space group C2/c, with 32 molecules per unit cell (Fig. 3).

Molecular structure of 1a, showing the atom labeling and 30% probability ellipsoids. H atoms as small circles of arbitrary radius. The asymmetric unit contains four molecules (A–D), only one molecule is drawn
The lactam ring adopts an envelope conformation, whereas the cyclohexane ring has an almost ideal chair conformation. There are four molecules in the asymmetric unit of γ-spirolactam 1a, which show small conformational differences. The bond lengths within the cyclohexane ring lie in the expected ranges [1.514(3)–1.549(2) Å], whereas one bond to the spiro atom C4 is somewhat longer than the other ones. The bond angles range from 109.56(13)° to 112.48(15)°. The smallest angle occurs at the spiro junction. However, the bond angle is the biggest one on that carbon atom, which is adjacent to the spiro atom and bound to that with the longest bond. The conformations of the cyclohexane rings only slightly deviate from the ideal chair [θ = 177.9(2)°/180.00(19)°/178.09(19)°/178.6(2)° for molecules A–D, respectively]. The puckering amplitudes q are 0.566(2), 0.5691(19), 0.564(2) and 0.568(2) Å. The displacement parameters φ are 313(7)°, 293(10)°, 66(8)° and 309(8)°. The torsion angles slightly deviate (maximal deviation: 2.46°) from the value observed in cyclohexane (55.9°). The dihedral angles between the cyclohexane rings and the pyrrolidine rings are 79.931(68)°, 80.112(68)°, 79.975(66)° and 80.034(70)°, whereby the spiro atom C4 has a maximum deviation from the mean plane of the five-membered ring [A: 0.0331(17), B: 0.0276(13), C: − 0.0301(17), D: − 0.0342(14) Å]. The twisting of both rings to each other can also be expressed by the torsion angles including the spiro atom C4 (for details see Additional file 1).
Importantly, the carbonyl group of lactam 1a occupies the pseudo equatorial position in the cyclohexane ring, because it is sterically more demanding than the methylene group of the lactam ring, which is axial. The preference of bulky substituents in the equatorial position of cyclohexanes is well-known [13, 14]. Furthermore, our X-ray structure is in accordance to Gabapentin lactam 3 in the solid state [11], which is a regioisomer of γ-spirolactam 1a (Fig. 2), showing the sterically more demanding carbonyl group in the pseudo equatorial position as well.
The lactam rings adopt a slightly distorted envelope conformation, characterized by the puckering parameters q = 0.2800(19)°/0.2723(19)°/0.2820(19)°/0.2787(19)° Å and φ = 104.6(4)°/104.6(4)°/284.2(4)°/104.7(4)° for molecules A–D (theoretical values for envelope: 108/288°). The torsion angles around the ring bonds deviate up to 27.18(18)° (C1C–C4C–C3C–C2C) from 0° of a perfectly planar ring system. As expected, around the amide bond N1–C1 is the smallest torsion angle of − 1.3(2)° (C2A–N1A–N1A–C4A). The C–C–C bond angles within the five-membered rings are 101.84(13)°–108.84(14)° (A), 102.19(13)°–108.38(16)° (B), 101.75(13)°–108.73(14)° (C), and 102.22(13)°–108.41(16)° (D). The smallest values occur including the spiro atom, the biggest at the carbonyl group. The bond angles including the nitrogen atom range from 114.15(14)° (A) to 114.61(15)° (B). The amide (N1–C1) bond lengths from 1.330(2) to 1.336(2) Å indicate partial double-bond character.
Interestingly, a mirror plane can be put through the atoms C4 and C7, resulting in two mirror images (Fig. 4), with exactly opposite orientation of the lactam rings like in enantiomers. Such a behavior can be described as dynamic stereoisomerism [15] and is known from X-ray structures of simple γ-butyrolactam as well [10]. Therefore, although the lactam ring is in solution very flexible and can adopt different envelope conformations, it is fixed in the solid state by hydrogen bonds.

Mirror plane (brown) and the orientation of the atoms O1, N1, and C2 (molecule A) with respect to this plane in two conformers of γ-spirolactam 1a
The maximal deviations from that plane are 0.0276(13) to − 0.0342(14) Å (C4). Due to the opposite positions of the other lactam ring atoms (O1, N1, C2), with respect to that plane, pairs always occur. In accordance with the envelope conformation, the distances to the plane are significant [O1: 0.2927(27) (B) to 0.2988(32) Å (A), N1: − 0.4394(21) (B) to − 0.4651(21) Å (D), C2: − 0.7110(24) (B) to − 0.7412(24) Å (D)]. The packing is stabilized by N–H···O hydrogen bonds, which are located approximately along the crystallographic c axis as well as in between a and c forming a 2D-network with double strands in the a direction (Fig. 5; for details of the hydrogen-bonding geometry, see Table 1).

Crystal packing of 1a, illustrating the hydrogen bonds (dashed lines). View along b
We investigated γ-spirolactams 1b next, which crystallize in the monoclinic space group P21/c, with one molecule per asymmetric unit. Indeed, it was now possible to determine the relative configurations and assign cis and trans isomers unequivocally (Fig. 6). Similar to derivative 1a, a mirror plane can be found between atoms C4, C10, H7, and H10, with the lactam ring slightly out of this plane in different envelope conformations. Interestingly, in both isomers the methyl group occupies the equatorial position in the cyclohexane ring, due to its steric demand [13, 14]. The conformation of the lactam is more flexible and therefore controlled by this methyl group. Thus, the carbonyl group is forced pseudo axial in cis-1b and pseudo equatorial in trans-1b (Fig. 6).

Molecular structures of cis-1b (a) and trans-1b (b), showing the atomic labeling and 30% probability ellipsoids. H atoms as small circles of arbitrary radius
The crystal packings of the two lactams 1b are different (Fig. 7). The molecules of cis-1b crystallize as centrosymmetric dimers, where the molecules are held together by two N–H···O hydrogen bonds running along the crystallographic a axis. The central unit of trans-1b forms 1D-chains along the crystallographic c axis, fixed by hydrogen bonds (Table 1, Fig. 7). In comparison with 1a, we can find stronger hydrogen bonds in cis-1b and trans-1b, in conclusion of the shorter D···A distances and the D–H···A angles, which are closer to 180°. These findings are in accordance with the N–H stretching frequencies in IR spectra—usually observed from 3500 to 3200 cm−1 for non-associated N–H–but here shifted to lower frequencies due to hydrogen bonds (1a: 3292 cm−1), but even lower in cis-1b and trans-1b (3201 and 3211 cm−1).

Crystal packing of cis-1b (a) and trans-1b (b), illustrating the hydrogen bonds (dashed lines)
The structural features of 1b are closely related to those in 1a. Table 2 lists the geometric parameters in detail. The biggest deviation from the ideal chair conformation of the cyclohexane ring can be observed in cis-1b.
Conclusions
In conclusion, we could easily synthesize three different γ-spirolactams from benzoic acids. Their configurations could not be determined by NMR spectroscopy, and thus X-ray analysis was the method of choice. We could unequivocally establish cis- and trans-isomers for the first time. Despite the configurations, we could additionally determine the conformations of the γ-spirolactams in the solid state. Thus, the cyclohexane rings adopt almost ideal chair forms, although the spiro junction leads to some distortion. Importantly, the steric demand of the methyl groups, which are always oriented pseudo equatorial, controls the conformations. Finally, we found an interesting behavior in the crystal packing, controlled by intermolecular hydrogen bonds between the acidic NH protons and the basic C=O groups. Thus, mirror planes were found between two crystallized molecules, affording enantiomer-like pairs. Since the herein investigated γ-spirolactams are very similar to Gabapentin lactam, which is neuroprotective in retinal ischemia, our studies might be interesting for biological or medical applications.
Experimental section
General synthetic
Melting points (m.p.) were determined by using a Mel-Temp from Electrothermal. TLC was performed using TLC Silica gel 60 F254 aluminium sheets from Merck. 1H NMR and 13C NMR spectra were measured by using a Bruker Avance 500 (500 MHz, 125 MHz) or a Bruker Avance 600 (600 MHz, 150 MHz) NMR spectrometer. Signals were assigned by two-dimensional methods (COSY, HMQC, HMBC). The IR spectra were recorded in KBr pellets by using a Nicolet 6700 FT-IR spectrometer from Thermo Electron Corporation. Elemental analysis was performed on a Vario EL III elemental analyzer. All starting materials were used as purchased without further purification.
γ-Spirolactams 1a, cis-1b, and trans-1b
Benzoic acid (2a) (12.21 g, 100 mmol) or p-toluic acid (13.62 g, 100 mmol) were introduced into a three-necked flask (500 mL), equipped with a dry-ice condenser and cooled to − 78 °C by a dry-ice acetone bath. Ammonia (200 mL) was condensed into the three-necked flask, and lithium (1.39 g, 200 mmol) was added in small pieces to the solution at − 78 °C, until it remained blue. After stirring for 1 h at − 78 °C, chloroacetonitrile (10.0 mL, 160 mmol) was added via syringe within 2 min and the ammonia was allowed to evaporate overnight at RT. The solid residue was dissolved in water (70 mL), cooled to 0 °C, acidified with 6 M HCl to pH 2 and extracted with dichloromethane (3 × 50 mL). The combined organic layers were dried over sodium sulfate, filtered over a small pad of silica gel, and the solvent was removed in vacuo. The crude products were crystallized from n-hexane to obtain the intermediate cyclohexadienes 4a (15.7 g, 96%) or 4b (16.3 g, 92%) as white solids. Cyclohexadiene 4b was obtained as a 55:45 mixture of diastereomers, which were separated by column chromatography (silica gel, hexane/ethyl acetate/methanol 3:1:0.25) to afford 4.9 g (28%) of cis-4b, 5.2 g (29%) of trans-4b and 5.9 g (33%) of a mixture fraction. This fraction was separated again under same conditions to afford additionally 2.5 g (14%) of cis-4b and 3.0 g (17%) of trans-4b in analytically pure forms.

Cyclohexadiene 4a
Rf = 0.55 (CH2Cl2/MeOH/HOAc 9:1:0.1); m.p. 122–123 °C; 1H NMR (500 MHz, CDCl3): δ 10.08 (brs, 1 H, O–H), 6.13 (dt, J = 10.3, 3.3 Hz, 2 H, 3-H, 5-H), 5.75 (dt, J = 10.3, 1.9 Hz, 2 H, 2-H, 6-H), 2.72–2.79 (m, 2 H, 4-H), 2.74 (s, 2 H, CH2CN); 13C NMR (125 MHz, CDCl3): δ 26.2 (t, CH2CN), 28.4 (t, C-4), 46.0 (s, C-1), 116.5 (s, CN), 123.5 (d, C-2, C-6), 129.3 (d, C-3, C-5), 177.7 (s, CO2H); IR (KBr) \(\widetilde{v}\) = 3164, 2965, 2270, 1724, 1227, 754 cm−1; elemental analysis calcd (%) for C9H9NO2 (163.18): C 66.25, H 5.56, N 8.58; found: C 65.93, H 5.66, N 8.56.

Cyclohexadiene cis-4b
Rf = 0.58 (CH2Cl2/MeOH/HOAc 9:1:0.1); m.p. 143–144 °C; 1H NMR (500 MHz, CDCl3): δ 10.32 (brs, 1 H, O–H), 6.04 (dd, J = 10.3, 3.4, Hz, 2 H, 3-H, 5-H), 5.69 (dd, J = 10.3, 2.0 Hz, 2 H, 2-H, 6-H), 2.83–2.89 (m, 1 H, 4-H), 2.73 (s, 2 H, CH2CN), 1.14 (d, J = 7.4 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 21.0 (q, CH3), 28.2 (t, CH2CN), 30.7 (t, C-4), 46.1 (s, C-1), 116.6 (s, CN), 122.3 (d, C-2, C-6), 135.6 (d, C-3, C-5), 177.8 (s, CO2H); IR (KBr) \(\widetilde{v}\) = 3140, 2939, 2272, 1726, 1227, 755 cm−1; elemental analysis calcd (%) for C10H11NO2 (177.20): C 67.78, H 6.28, N 7.90; found: C 67.74, H 6.06, N 7.90.

Cyclohexadiene trans-4b
Rf = 0.52 (CH2Cl2/MeOH/HOAc 9:1:0.1); m.p. 161–162 °C; 1H NMR (500 MHz, CDCl3): δ 11.42 (brs, 1 H, O–H), 6.06 (dd, J = 10.3, 3.3, Hz, 2 H, 3-H, 5-H), 5.70 (dd, J = 10.3, 2.0 Hz, 2 H, 2-H, 6-H), 2.85–2.92 (m, 1 H, 4-H), 2.78 (s, 2 H, CH2CN), 1.22 (d, J = 7.4 Hz, 3H, CH3); 13C NMR (125 MHz, CDCl3): δ 21.4 (q, CH3), 28.5 (t, CH2CN), 30.8 (t, C-4), 46.3 (s, C-1), 116.7 (s, CN), 122.4 (d, C-2, C-6), 135.6 (d, C-3, C-5), 176.6 (s, CO2H); IR (KBr) \(\widetilde{v}\) = 3164, 2965, 2270, 1725, 1227, 754 cm−1; elemental analysis calcd (%) for C10H11NO2 (177.20): C 67.78, H 6.28, N 7.90; found: C 67.66, H 6.21, N 7.94.
The cyclohexadienes 4a (3.26 g, 20 mmol), cis-4b (3.54 g, 20 mmol), or trans-4b (3.54 g, 20 mmol) were dissolved in methanol (300 mL) at RT and platinum(iv) oxide (50 mg, 1 mol%) and 37% HCl (1.5 mL) was added. The solution was purged with hydrogen gas for 5 min, equipped with a balloon filled with hydrogen gas and hydrogenated under stirring for 48 h. The solution was filtered through a pad of Celite, washed with methanol (2 × 50 mL) and the solvent was removed in vacuo. The residue was dissolved in pyridine (500 mL) and heated for 8 h under reflux. The pyridine was removed in vacuo and the residue was dissolved in dichloromethane (200 mL), extracted with 1 N HCl (100 mL), dried over sodium sulfate, and concentrated in vacuo. The solid γ-spirolactams 1 crystallized from n-hexane in analytically pure form and afforded single crystals for X-ray measurements.

γ-Spirolactam 1a
White solid (3.00 g, 98%). Rf = 0.21 (EtOAc); m.p. 107–108 °C; 1H NMR (500 MHz, CDCl3): δ 7.45 (brs, 1 H, N–H), 3.24 (t, J = 7.0 Hz, 2 H, 3-H), 1.94 (t, J = 7.0 Hz, 2 H, 4-H), 1.18–1.70 (m, 10 H, 6-H–10-H); 13C NMR (125 MHz, CDCl3): δ 22.4 (t, C-7, C-9), 25.6 (t, C-8), 31.7 (t, C-4), 32.2 (t, C-6, C-10), 39.2 (t, C-3), 44.2 (s, C-5), 183.7 (s, C-1); IR (KBr) \(\widetilde{v}\) = 3292, 2928, 1686, 1650, 1279, 1071, 739 cm−1; elemental analysis calcd (%) for C9H15NO (153.22): C 70.55, H 9.87, N 9.14; found: C 70.29, H 9.99, N 9.07.

γ-Spirolactam cis-1b
White solid (3.20 g, 96%). Rf = 0.26 (EtOAc); m.p. 128–129 °C; 1H NMR (600 MHz, CDCl3): δ 7.69 (brs, 1 H, N–H), 3.22 (t, J = 7.0 Hz, 2 H, 3-H), 1.89 (t, J = 7.0 Hz, 2 H, 4-H), 1.54–1.40 (m, 4 H, 7-Hax., 9-Hax., 6-Hequ., 10-Hequ.), 1.37–1.41 (m, 2 H, 7-Hequ., 9-Hequ.), 1.27–1.34 (m, 1 H, 8-H), 0.89 (ddd, J = 15.7, 9.8, 4.7 Hz, 2 H, 6-Hax., 10-Hax.), 0.81 (d, J = 6.7 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3): δ 22.6 (q, CH3), 31.1 (t, C-7, C-9), 31.3 (t, C-4), 31.9 (d, C-8), 32.1 (t, C-6, C-10), 39.2 (t, C-3), 44.0 (s, C-5), 184.0 (s, C-1); IR (KBr) \(\widetilde{v}\) = 3201, 2918, 1680, 1287, 753 cm−1; elemental analysis calcd (%) for C10H17NO (167.25): C 71.81, H 10.24, N 8.38; found: C 71.60, H 10.31, N 8.44.

γ-Spirolactam trans-1b
White solid (3.24 g, 97%). Rf = 0.35 (EtOAc); m.p. 114–115 °C; 1H NMR (600 MHz, CDCl3): δ 6.70 (brs, 1 H, N–H), 3.26 (t, J = 6.9 Hz, 2 H, 3-H), 1.89 (t, J = 6.9 Hz, 2 H, 4-H), 1.85 (ddd, J = 13.3, 8.1, 3.0 Hz, 2 H, 6-Hequ., 10-Hequ.), 1.48–1.57 (m, 5 H, 7-H, 8.H, 9-H), 1.24 (ddd, J = 13.3, 8.4, 4.1 Hz, 2 H, 6-Hax., 10-Hax.), 0.96 (d, J = 6.2 Hz, 3 H, CH3); 13C NMR (75 MHz, CDCl3): δ 20.0 (q, CH3), 29.3 (t, C-7, C-9), 29.8 (d, C-8), 31.2 (t, C-6, C-10), 35.3 (t, C-4), 38.8 (t, C-3), 42.2 (s, C-5), 183.0 (s, C-1); IR (KBr) \(\widetilde{v}\) = 3211, 2924, 1677, 1289, 755 cm−1; elemental analysis calcd (%) for C10H17NO (167.25): C 71.81, H 10.24, N 8.38; found: C 71.71, H 10.12, N 8.37.
X-ray structure analysis
The crystals were embedded in perfluoropolyalkylether oil and mounted within a MicroGripper. The data collections were performed at 210 K on a STOE StadiVari diffractometer equipped with a four-circle goniometer (open Eulerian cradle), a Genix Microfocus X-ray source (Mo) with a graded multilayer mirror and a Dectris 200 K detector (Δω = 0.5°; detector distance 60 mm; 1a: 4110 frames, 5 s exposure time per frame; cis-1b: 2488 frames, 10 s exposure time per frame; trans-1b: 2516 frames, 10 s exposure time per frame). The data were corrected for absorption as well as for Lorentz and polarization effects using the program X-Area [16]. The structures were solved by direct methods using SHELXS-2013/2 [17] and refined by full-matrix least squares on F2 using the program SHELXL-2014/7 [18]. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms of the N–H groups were located from the difference Fourier maps and free refined. The other hydrogen atoms were calculated in their expected positions using a riding model with C–H = 0.98 Å (–CH2) and C–H = 0.97 Å (–CH3), allowing for rotation), and Uiso(H) = 1.2Ueq(CH2) and Uiso(H) = 1.5Ueq(CH3). For the visualization the programs ORTEP-3 for windows [19] and DIAMOND [20] were used.
Crystal data of 1a
C9H15NO, M = 153.22, monoclinic, a = 20.4582(6), b = 11.8454(4), c = 28.4142(10) Å, β = 106.017(5)°, V = 6815.2(4) Å3, T = 210(2) K, space group C2/c (no. 15), Z = 32, μ(MoKα) = 0.077 mm−1; 92,264 reflections measured, 5983 unique (Rint = 0.0616) which were used in all calculations. Final R values: wR2(F2) = 0.1079, R1 = 0.0697 (all data); wR2(F2) = 0.0969, R1 = 0.0373 [I > 2σ(I)].
Crystal data of cis-1b
C10H17NO, M = 167.24, monoclinic, a = 14.7946(9), b = 6.4589(2), c = 10.5769(7) Å, β = 105.199(5)°, V = 975.34(10) Å3, T = 210(2) K, space group P21/c (no. 14), Z = 4, μ(MoKα) = 0.073 mm−1; 15,495 reflections measured, 1713 unique (Rint = 0.0236) which were used in all calculations. Final R values: wR2(F2) = 0.0940, R1 = 0.0391 (all data); wR2(F2) = 0.0913, R1 = 0.0336 [I > 2σ(I)].
Crystal data of trans-1b
C10H17N O, M = 167.24, monoclinic, a = 11.1386(5), b = 10.5587(5), c = 8.3892(7) Å, V = 948.34(10) Å3, T = 210(2) K, space group P21/c (no. 14), Z = 4, μ(MoKα) = 0.073 mm−1; 14,814 reflections measured, 1655 unique (Rint = 0.0261) which were used in all calculations. Final R values: wR2(F2) = 0.0847, R1 = 0.0368 (all data); wR2(F2) = 0.0816, R1 = 0.0313 [I > 2σ(I)].
Availability of data and materials
Additional files 1, 2, and 3 include crystallographic information files (CIF). CCDC 1812882 (Ia), CCDC 1812884 (cis-1b), and CCDC 1812883 (trans-1b) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Abbreviations
- NMR:
-
nuclear magnetic resonance
- NOE NMR:
-
nuclear overhauser effect NMR
- 2D:
-
2-dimensional
- FT-IR:
-
Fourier transformation IR
- m.p.:
-
melting point
- TLC:
-
thin layer chromatography
- COSY:
-
correlated spectroscopy
- HMQC:
-
heteronuclear multiple quantum coherence
- HMBC:
-
heteronuclear multiple bond correlation
- IR:
-
infrared
References
Saraswat P, Jeyabalan G, Hassan MZ, Rahman MU, Nyola NK (2016) Review of synthesis and various biological activities of spiro heterocyclic compounds comprising oxindole and pyrrolidine moities. Synth Commun 46:1643–1664
Pradhan R, Patra M, Behera AK, Mishra BK, Behera RK (2006) A synthon approach to spiro compounds. Tetrahedron 62:779–828
Kotha S, Deb AC, Lahiri K, Manivannan E (2009) Selected synthetic strategies to spirocyclics. Synthesis. 165:93
Huang X, Liu M, Pham K, Zhang X, Yi WB, Jasinski JP, Zhang W (2016) Organocatalytic one-pot asymmetric synthesis of thiolated spiro-γ-lactam oxindoles bearing three stereocenters. J Org Chem 81(13):5362–5369
Wegert A, Nolte B, Linz K, Harlfinger S, Kögel BY, Ratcliffe P, Theil F, Gröger O, Braun B (2016) Substituierte aza-spiro(4.5)decan-derivate. WO 2016/008583 A1
Chesworth R, Mitchell LH, Campbell JE, Reiter LA, Swinger KK (2016) Arginine methyltransferase inhibitors and uses thereof. WO 2016/044626 A1
Krüger T, Kelling A, Schilde U, Linker T (2017) Simple synthesis of γ-spirolactams by birch reduction of benzoic acids. Eur J Org Chem. 6:1074–1077
Pirilä P, Mutikainen I, Pursiainen J (1999) Structure of the 2-pyrrolidinone monohydrate. Z Naturforsch. 54b:1598–1601
Müller G, Lutz M, Harder S (1996) Methyl group conformation-determining intermolecular C–H···O hydrogen bonds: structure of N-methyl-2-pyrrolidone. Acta Cryst. B52:1014–1022
Goddard R, Heinemann O, Krüger C, Magdo I, Mark F, Schaffner K (1998) A low-temperature phase of pyrrolidone. Acta Cryst. C54:501–504
Ananda K, Aravinda S, Vasudev PG, Raja KMP, Sivaramakrishnan H, Nagarajan K, Shamala N, Balaram P (2003) Stereochemistry of gabapentin and several derivatives: solid state conformations and solution equilibria. Curr Sci 85:1002–1011
Jehle T, Feuerstein TJ, Lagreze WA (2001) The effect of gabapentin and gabapentin lactam on retinal ganglion cell survival in an animal model in acute retina ischemia. Ophthalmol. 98:237–241
Eliel EL, Wilien SH, Mander LN (1994) Stereochemistry of organic compounds. Wiley, New York, pp 686–753
Smith MB (2013) March’s advanced organic chemistry, 7th edn. Wiley, New York, p 180
Wolf C (2007) Dynamic stereochemistry of chiral compounds: principles and applications. The Royal Society of Chemistry, Cambridge
Stoe & Cie (2015) X-Area 1.76. Darmstadt
Sheldrick GM (2013) SHELXS-2013/1, program for the solution of crystal structures. University of Göttingen, Germany
Sheldrick GM (2014) SHELXL-2014/7, program for the refinement of crystal structures. University of Göttingen, Germany
Farrugia LJ (2012) Ortep for windows. 2013.1. University of Glasgow, Glasgow
Brandenburg K (2017) Diamond 4.2.2. Bonn, Crystal Impact
Funding
We acknowledge the support of Deutsche Forschungsgemeinschaft (German Research Foundation) and Open Access Publication Fund of Potsdam University.
Author information
Authors and Affiliations
Contributions
TL has formulated the research idea and prepared the manuscript draft version. TK carried out the synthetic experiments. AK and US collected the X-ray data and performed the structure solution. US prepared the manuscript for submission and coordinated final formulation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1.
Crystallographic information file of 1a.
Additional file 2.
Crystallographic information file of cis-1b.
Additional file 3.
Crystallographic information file of trans-1b.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Krueger, T., Kelling, A., Linker, T. et al. Crystal structures of three cyclohexane-based γ-spirolactams: determination of configurations and conformations. BMC Chemistry 13, 69 (2019). https://doi.org/10.1186/s13065-019-0586-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-019-0586-7