
Abstract
Background
Forty per cent of critically ill patients are affected by intensive care unit-acquired weakness (ICU-AW), to which skeletal muscle wasting makes a substantial contribution. This can impair outcomes in hospital, and can cause long-term physical disability after hospital discharge. No effective mitigating strategies have yet been identified.
Application of a repetitive vascular occlusion stimulus (RVOS) a limb pressure cuff inducing brief repeated cycles of ischaemia and reperfusion, can limit disuse muscle atrophy in both healthy controls and bed-bound patients recovering from knee surgery. We wish to determine whether RVOS might be effective in mitigating against muscle wasting in the ICU. Given that RVOS can also improve vascular function in healthy controls, we also wish to assess such effects in the critically ill. We here describe a pilot study to assess whether RVOS application is safe, tolerable, feasible and acceptable for ICU patients.
Methods
This is a randomised interventional feasibility trial. Thirty-two ventilated adult ICU patients with multiorgan failure will be recruited within 48 h of admission and randomised to either the intervention arm or the control arm. Intervention participants will receive RVOS twice daily (except only once on day 1) for up to 10 days or until ICU discharge.
Serious adverse events and tolerability (pain score) will be recorded; feasibility of trial procedures will be assessed against pre-specified criteria and acceptability by semi-structured interview. Together with vascular function, muscle mass and quality will be assessed using ultrasound and measures of physical function at baseline, on days 6 and 11 of study enrolment, and at ICU and hospital discharge. Blood and urine biomarkers of muscle metabolism, vascular function, inflammation and DNA damage/repair mechanism will also be analysed. The Health questionnaire will be completed 3 months after hospital discharge.
Discussion
If this study demonstrates feasibility, the derived data will be used to inform the design (and sample size) of an appropriately-powered prospective trial to clarify whether RVOS can help preserve muscle mass/improve vascular function in critically ill patients.
Trial registration
ISRCTN Registry, ISRCTN44340629. Registered on 26 October 2017.
Similar content being viewed by others


Background
Increasing numbers of patients are being admitted to intensive care units (ICUs) and mortality rates have fallen [1,2,3]. However, survival comes at the expense of increased dependency and debility: 50% of patients of working age do not return to work; 70% require assistance with daily living activities in the year following discharge; and physical disability can persist for many years [4, 5]. The UK’s National Institute of Health and Care Excellence (NICE) has declared such post-ICU debility to be a public health issue [6].
Impaired quality of life and functional limitation partly result from ICU-acquired weakness (ICU-AW) [7,8,9,10]. This affects approximately 40% of adult ICU patients [11], with long-term ventilated or severely septic patients being most commonly and severely affected [12,13,14,15,16]. ICU-AW is associated with poorer outcomes: prolonged mechanical ventilation, increased ICU and hospital length of stay, increased hospital and post-discharge mortality rates, chronic functional disability and reduced quality of life among survivors [8, 17,18,19,20]. As a consequence, hospital health-care costs are 30% higher in those affected, with further excess costs relating to the need for rehabilitation, frequent re-admissions and social care upon discharge [18].
The pathophysiology of ICU-AW is multifactorial [10, 21,22,23]. Whilst neuropathies may occur [24], skeletal muscle wasting plays a central role [25]. The cross-sectional area of the rectus femoris (thigh) muscle (RFCSA) falls by an average of 18% in just 10 days in ventilated ICU patients, and to the greatest degree in those with the largest burden of organ failure [21]. This is underpinned by impaired skeletal muscle protein synthesis and, simultaneously, increased protein breakdown [21, 26], to which altered muscle metabolism [27] and disuse may contribute [23, 28]. The putative role of drugs such as corticosteroids, neuromuscular junction blocking agents and aminoglycoside antibiotics is contested [16, 29,30,31].
To date, no therapies exist for prevention or treatment of ICU-AW [32, 33]. Early mobilisation and physical activity regimes have proven ineffective [34,35,36,37,38], perhaps because muscle wasting has already occurred. There is thus growing interest in the early application of non-volitional interventions such as neuromuscular electrical stimulation, although proof of benefit remains lacking [38,39,40,41,42].
Application of a repetitive vascular occlusion stimulus (RVOS) shows promise as a mitigating intervention. Repeated inflation/deflation of a blood pressure cuff around a limb to above arterial pressure (~ 200 mmHg) elicits brief bouts (~ 5 min) of limb ischaemia/reperfusion [43]. In healthy subjects, RVOS can improve exercise performance [44,45,46,47], whilst vascular occlusion during low-intensity exercise (blood flow restriction exercise) can enhance hypertrophic and strength responses in skeletal muscle [48] of healthy controls [49,50,51,52], athletes [53] and the elderly [54,55,56,57,58,59,60], and seems to improve physical function and health-related quality of life in patients with inflammatory muscle disease [61,62,63]. Moreover, RVOS may mitigate against atrophy induced by immobilisation and unloading: patients recovering from ligament reconstruction surgery who received two sessions of RVOS (five cycles of vascular occlusion for 5 min and release for 3 min) to the proximal thigh daily for > 10 days after surgery had 50% less disuse knee extensor muscles mass loss [43]. Similarly, in healthy volunteers with experimentally induced limb immobilisation, RVOS alone or in combination with exercise preserved muscle strength and mass [64, 65].
The mechanism by which RVOS prevents muscle atrophy remains unclear. Application of RVOS enhanced a murine mTOR signalling pathway involved in protein synthesis [66], whilst increased skeletal muscle oxidative capacity is also implicated [67,68,69,70].
In addition to impacts on muscle mass, RVOS improves local and systemic endothelial function and microcirculation in healthy controls [71, 72], perhaps through increases in plasma concentration of vascular endothelial growth factor (VEGF) and endothelial progenitor cells [73]. However, it is unknown whether RVOS can prevent or offset macrovascular and microvascular dysfunction observed in bed-rest/ immobilised individuals [74,75,76] and critically ill patients [77,78,79].
Of note, such benefits are seen locally (i.e. in the muscle distal to the occlusion), but also remotely—in tissue distant from the site of occlusion (so-called ‘remote preconditioning’). RVOS provides protection against myocardial and renal tissue ischaemic injury at the time of vascular intervention or cardiac surgery [80,81,82,83,84,85]. Unilateral daily arm RVOS over a week improves bilateral vascular function [71], while a longer (300 consecutive days), more frequent (twice daily), protocol improves cerebral perfusion and reduces recurrent strokes by 30% in patients with intracranial arterial stenosis [86]. The mechanism of such remote benefit has yet to be described [87].
Given its remote effects, application of RVOS to a single limb could potentially prevent muscle wasting and vascular dysfunction at both local and remote sites in critically ill patients, thus mitigating against the development of ICU-AW. Evidence suggests that this approach would be safe, with no reports of elevation of muscle damage markers, such as creatinine kinase and myoglobin, and oxidative stress markers, such as lipid peroxide, during blood flow restriction exercise [50, 52, 88, 89]. Moreover, RVOS pre or post exercise seems to be protective against exercise-induced muscle damage [90, 91]. Amongst 12,642 recipients (including healthy subjects, older subjects and individuals with clinical conditions), attributable serious side effects were few: venous thrombus (0.055%), pulmonary embolism (0.008%) and rhabdomyolysis (0.008%) [92]. No negative impact on haemodynamic, haemostatic and inflammatory responses has been observed in healthy young, older and clinical populations [61, 93,94,95]. In addition to being safe, RVOS also appears well tolerated, with a pain score of 3.6 ± 3.4 (mean ± SD) out of 10 during application to the lower limb in patients with aneurysmal subarachnoid haemorrhage [96].
We thus wish to determine whether the application of RVOS might help maintain muscle mass and improve vascular function in ICU patients. We here describe a trial designed to assess the feasibility of performing such an appropriately-powered study.
Methods
Primary objectives
The primary objective of the trial is to determine whether it is safe and tolerable to apply RVOS to a proximal lower limb of ICU patients.
Secondary objectives
Our secondary objectives are to determine the feasibility of screening; obtaining consent and recruiting; randomising; retaining patients; delivering the trial intervention; performing outcome assessments and collecting data; assessing acceptability for patients, personal consultees (next of kin) and staff; and to determine outcome data characteristics to design a future larger trial.
In addition, we will explore any impacts on measures of muscle mass, quality and function (strength and physical function measures); vascular function; blood and urine biomarkers of muscle metabolism; vascular function; inflammation and DNA damage/repair mechanisms; and clinical outcomes to inform powering of any future prospective trial.
Study design
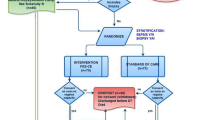
ROSProx is a partially blinded interventional feasibility trial with randomisation. Eligible patients will be randomised to an intervention arm or a control arm. Patient randomised to the control arm will receive standard care, while intervention patients will receive RVOS treatment in addition to all other standard care. The study protocol has been developed in accordance with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines (see Additional file 1 for the SPIRIT checklist). The study flow diagram is shown in Fig. 1.

Study flow diagram. ICU intensive care unit, SOFA Sequential Organ Failure Assessment
Study setting
The study will take place within the ICUs of two hospitals in England: Royal Surrey County Hospital NHS Trust (RSCH); and Ashford and St Peter’s Hospital (ASPH) NHS Trust.
Sample size rationale
A total of 30 participants is recommended for pilot and feasibility trials [97]. To allow for balance in the stratification factors (gender and study site), a sample size of 32 participants was chosen, which will allow us to estimate a failure to deliver RVOS in the intervention arm of 30% to within a 95% confidence interval of ± 15.9%.
Based on an anticipated average rate of recruitment of 6 patients/month, 50% screening failure due to weekends, 20% decline of consent and 30% death before trial completion, we aim to recruit 1.6 patients per site per month. Recruiting from two sites, one starting 2 months after the other, is expected to yield 47 patients over 16 months. Recruitment will continue until 32 ICU survivors have been studied (16 patients in each group).
Study population
Patients that meet the eligibility criteria (Table 1) will be recruited from each participating ICU over a 16-month period.
Recruitment and randomisation process
All sequential ICU admissions will be screened for eligibility. Eligible patients with mental capacity for informed consent will be approached, the risks and benefits of participation explained and written informed consent will be obtained before enrolment by the investigator physician. However, we envision that the majority of eligible patients will be receiving invasive mechanical ventilation and requiring sedation, and thus lacking capacity to consent. In this instance, declaration of agreement will be sought from the patient’s ‘Personal Consultee’ who may be a representative, partner or close friend. Once the participant recovers and is capable of understanding the details of the trial, they will be approached to provide their informed consent retrospectively. If the patient chooses to withdraw from the trial, they will be given the choice of having their existing data and samples destroyed or excluded from final analysis.
Upon informed consent or declaration of agreement, eligible patients will be randomised to either the intervention arm or the control arm in a 1:1 ratio. Separate randomisation lists have been prepared for each site by an independent statistician and uploaded to the study electronic data capture system PROMASYS (Surrey Clinical Trials Unit). In addition, the randomisation list and corresponding envelopes containing randomisation assignments will be stored in the trial file. Randomisation, outcome measure assessments and RVOS intervention will be performed by a research assistant unblinded to patient allocation.
Study protocol
Figure 2 illustrates the schedule of study procedures and outcome measure assessments. Briefly, within 24 h of enrolment and randomisation, baseline assessments will be completed. These will include ultrasound assessment of muscle and arterial vascular function; blood and urine sample collection and physical function assessment using the ICU mobility score; recording of clinical characteristics such as medical history, diagnosis and severity of illness (assessed using the Sequential Organ Failure Assessment (SOFA) score); documentation of physical function (assessed using the Katz Index of Independence in Activities of Daily Living score) and nutritional status (assessed using the Malnutrition Screening Tool) prior to illness.

Schedule of study procedures and outcome measure assessments. DVT deep vein thrombosis, EDTA ethylenediamine tetraacetic acid, ICU intensive care unit, RVOS repetitive vascular occlusion stimulus, SF-12 12-Item Short Form Health Questionnaire
If the participant is randomised to the intervention, one RVOS session will be carried out after baseline assessment on day 1. Arterial blood samples will be collected 3 and 24 h after RVOS and at the same time points after baseline assessments in control participants. From day 2, intervention participants will receive two RVOS sessions per day until day 10 of enrolment or until ICU discharge if this occurs before day 10.
Outcome measure assessments of muscle mass and quality, arterial function, muscle strength, physical function, and blood and urine biomarkers will be repeated on days 6 and 11 of study enrolment. Clinical data including blood results, nutritional and organ support, severity of illness, fluid balance, physiotherapy interventions and specific drug treatment (neuromuscular blocking drugs, corticosteroids, statins and propofol) will be recorded daily from days 1 to 11 of enrolment. At ICU and hospital discharge, physical function and muscle strength assessments will be repeated. In addition, at hospital discharge, an acceptability interview will be conducted with the participant and personal consultee, and blood and urine samples collected. Study participation will end upon completion (by telephone) of the SF-12 health questionnaire 3 months after hospital discharge.
Repetitive vascular occlusion stimulus (RVOS) intervention
Participants in the intervention arm will receive one session of RVOS treatment on day 1 and two sessions per day, at least 4 h apart, from day 2 to 10 of study enrolment or until ICU discharge if this occurs before day 10. For safety, the intervention leg will be examined daily before the first RVOS session for deep vein thrombosis (DVT) from the common femoral to popliteal vein using ultrasound (CX50 Philips Ultrasound with L12–3 linear array transducer). If DVT is suspected, RVOS or arterial assessment will not be performed and the clinical team will be notified immediately.
During the RVOS session, the participant will lie supine in a semi-recumbent position with the lower limbs extended. A pressure cuff (SC12LTM segmental pressure cuff; Hokanson, Bellvue, WA, USA) will be applied around the proximal right thigh with a bandage wrapped underneath to avoid skin irritation and bruising. Each RVOS session will last for 40 min and will include four cycles of 5 min cuff inflation to 50 mmHg above the average systolic blood pressure to completely occlude arterial flow distally to the limb [98], followed by 5 min of complete deflation (0 mmHg) (Fig. 3). Pressure in the pneumatic cuff will be controlled by an inflator and air source device (E20 Rapid Cuff Inflator and AG101 Cuff Inflator Air Source; Hokanson). Maximum cuff pressure will be 200 mmHg.

Protocol for repetitive vascular occlusive stimulus (RVOS). Each session of RVOS includes four repetitions of 5-min inflation of pneumatic cuff to 50 mmHg supra-systolic blood pressure (SBP) followed by 5 min of complete (0 mmHg) cuff deflation
Vital signs such as blood pressure, heart rate and the saturation of peripheral arterial blood with oxygen will be monitored before and during RVOS, and the session will be terminated if the defined criteria are met (Table 2).
A clinical bedside nurse will be present throughout the RVOS session and will monitor facial expression, limb movement and ventilation compliance for any sign of distress in the sedated participants. In the event that the patient experiences significant discomfort, the cuff pressure will be lowered to a pressure that is tolerated by the participant, and such adjustments will be recorded. If conscious, tolerability of the intervention will be evaluated by asking the participant to rate the pain of the procedure on a visual analogue scale (VAS). After each session, the intervention leg will be examined for signs of arterial insufficiency or tissue injury. In addition, serum creatinine kinase levels will be monitored daily as an indicator of muscle damage.
Primary outcome measures
Safety
The number of serious adverse events (SAEs; DVT, pulmonary embolism and elevated serum creatine kinase concentration (creatine kinase > 3800 IU/L)) will be compared between the control and intervention groups.
Tolerability
The VAS pain score will be used to assess the tolerability of RVOS.
Secondary outcome measures
Feasibility
Table 3 presents the pre-specified feasibility criteria that will be assessed.
Acceptability
A semi-structured interview conducted with the participant, personal consultee and clinical staff will be used to determine the acceptability of trial experience and RVOS intervention. Questions will relate to key trial parameters such as experience of recruitment and randomisation and participant/staff burden (see Additional file 2 for acceptability questions).
Muscle mass and quality
To assess muscle mass, the rectus femoris (RF) muscle of both legs (in intervention participants) and only the right leg (control participants) will be imaged using B-mode ultrasound as previously described [21, 99] at baseline and on days 6 and 11 of study enrolment. Briefly, the participant will lie in a semi-recumbent position and the RF muscle will be scanned in the longitudinal plane on the anterior aspect of the thigh, two-thirds of the distance from the anterior superior iliac spine to the superior patella border. RF ultrasound images will be analysed using ImageJ software (NIH, Bethesda, MD, USA) by blinded investigators. Muscle mass will be quantified by assessing the RFCSA, and echogenicity will be measured to evaluate the changes in muscle quality as reported previously [100, 101]. Change over time in these measures will be compared between intervention and control participants, and changes in the treated leg and the untreated leg compared in those receiving the RVOS intervention.
Arterial vascular measures
Arterial vascular function will be assessed at the same time points as muscle evaluation, using techniques previously described [102]. The diameter and blood flow velocity of the right superficial femoral artery at rest and immediately following 5 min of ischaemia will be measured using B-mode and pulse Doppler ultrasound imaging, respectively. Ischaemia will be induced by application of a pneumatic cuff to the right thigh and inflation to 200 mmHg cuff pressure. Blinded investigators will analyse the diameter and blood flow velocity using the commercially available software Brachial Analyzer (Vascular Research Tools 5; Medical Imaging Applications, LLC, Coralville, IA, USA) to assess flow-mediated dilation (FMD) and reactive hyperaemic response, surrogate markers of vascular health. The reactive hyperaemic response is a transient increase in blood flow in response to ischaemic stimulus, and a marker of vasodilator capacity of resistance vessels. FMD assesses endothelial-dependent vasodilation of the conduit artery in response to increased blood flow and internal wall shear stress. The resting SFA diameter and blood velocity, reactive hyperaemic response and FMD will be compared between intervention and control participants at each time point.
Muscle strength
On days 6 and 11 and at ICU and hospital discharge, the grip strength of the dominant hand will be assessed using a handgrip dynamometer (Takei Scientific Instruments Co. Ltd, Niigata City, Japan) and manual muscle strength evaluation of the bilateral six upper and lower limb muscle groups (shoulder abductors, elbow flexors, wrist extensors, hip flexors, knee extensors and foot dorsiflexors) performed using the Medical Research Council Sum Score (MRC-SS) [103]. Strength assessments will only be performed if the participant is sufficiently alert and free from delirium.
Physical function
Mobility will be assessed in the ICU on days 1, 6 and 11 of study enrolment (or at ICU discharge, whichever is earlier) and at hospital discharge, using the ICU Mobility Scale, a validated measure with excellent clinometric properties [104, 105]. In addition, functional assessment (‘timed up and go’ and ‘sit to stand’) will be performed at hospital discharge. ‘Timed up and go’ will involve measuring the time taken to stand up from a standard chair, walk a 3-m distance at normal pace and return to the chair; for ‘sit to stand’, the number of times that the participant is able to completely stand upright from a standard chair and sit back down fully within 30 s will be assessed. Due to the study population being frail, use of aids will be permitted and documented.
Clinical outcomes
Clinical outcomes between participants in the intervention and control arms will be compared: incidence and duration of delirium; incidence, duration and severity of acute kidney injury (assessed using the AKIN classification) [106]; duration of specific organ support (mechanical ventilation, renal replacement therapy, mechanical or pharmacological cardiovascular support); days alive and without a ventilator in the first 28 days of hospital stay; length of ICU and hospital stay; and hospital and 3-month mortality.
Laboratory assessment
A 10-ml urine sample (collected from an indwelling urinary catheter) and 1 × 10 ml EDTA, 3 × 4 ml EDTA, 1 × 4 ml serum, 1 × 3 ml Tempus™ RNA blood tube samples will be collected on days 1, 6 and 11 of study enrolment from indwelling arterial cannulae, and via venepuncture at hospital discharge. In addition, 1 × 4 ml EDTA blood samples will be taken at 3 and 24 h after the first RVOS session in patients randomised to the intervention arm and similarly at 3 and 24 h after baseline assessment in control participants. Plasma and serum will be separated from the EDTA and serum tubes, and the urine sample and Tempus™ blood RNA will be stored at − 80 °C. The, 1 × 10 ml EDTA will be processed to isolate peripheral blood mononuclear cells (PBMCs) using the Optiprep™ protocol [107], which will be stored at − 80 °C until analysis.
Samples will be stored in accordance with the Human Tissue Act (2004) beyond the end of the trial to allow for various laboratory analyses to evaluate mechanisms behind the effect of RVOS. Biomarkers that will be analysed are presented in Table 4. Further biomarkers may be studied to evaluate mechanisms of effect as scientific evidence dictates.
Statistical analysis
The statistical analysis addresses the objectives of demonstrating the safety and tolerability of applying RVOS to the lower proximal limb of ICU patients, and assessing the feasibility of undertaking a larger-scale study to demonstrate efficacy. As a feasibility trial, the statistical analysis will primarily be exploratory in nature, and thus hypothesis generating rather than confirmatory.
The primary objective of safety will be evaluated based on the frequencies and percentages of adverse events categorised by severity, group and site. The tolerability will be evaluated by assessing VAS pain scores of the intervention arm at each time point in terms of means and standard deviations.
The main feasibility outcome of recruitment will be evaluated as the percentage of randomised participants out of total eligible patients. The number of participants randomised as a percentage of targeted recruitment will also be computed. If data are available, the average number of research assistants’ hours spent on recruiting one participant will also be computed. Retention will be evaluated as the proportion of participants remaining at each time point compared to the number recruited in each arm.
Descriptive statistics will be used to summarise all response variables by site, time point and treatment group. Means and standard deviations will be computed for continuous outcomes and frequencies and percentages will be computed for categorical/dichotomous outcomes. The relationship with potential confounding factors—such as site, demographic data, delirium during ICU stay, severity of illness, length of ICU stay, sepsis (defined using SOFA score), and use of drug treatment (such as corticosteroids, neuromuscular blocking drugs, statins and propofol)—will be explored.
The following inferential statistics will be performed strictly for the purpose of identifying the directions and magnitudes of effects rather than to evaluate the statistical significance of the results due to inadequacy of statistical power.
Outcome measures recorded at a time point (such as ‘timed up and go’ and ‘sit to stand’ assessment) and clinical outcomes (such as duration of mechanical ventilation and length of ICU and hospital stay) will be compared between the two arms using an independent-sample t test.
Repeated outcome measures such as the RFCSA and echogenicity, superficial femoral artery measures (resting diameter and blood flow, FMD and reactive hyperaemic response), ICU mobility score, MRC-SS and handgrip strength data will be analysed separately as the response variable in a general linear mixed model using SAS PROC MIXED, with group (the dichotomy of standard care or RVOS), site, baseline measure, time and group × time interaction as fixed effects and subject as random effects. Appropriately selected covariates will be included in the model as fixed effects to adjust for the variability caused by these covariates.
All statistical analyses will be performed by the Trial Statistician at Surrey Clinical Trials Unit (CTU) according to a pre-specified Statistical Analysis Plan, which will be finalised and signed off before the database lock.
Data collection and monitoring
All study data will be collected in compliance with the principles of Good Clinical Practice (GCP) and with the Data Protection Act. Patients’ medical notes will be used as source documents for clinical data, while data related to the RVOS intervention (e.g. time of session, cuff pressure, VAS score) and outcome measure assessments will be recorded in a paper workbook before transcribing to a secure web-based electronic data capture system (Promasys) provided by Surrey CTU. The Surrey CTU data management team and sponsor (RSCH) will jointly oversee the administration of study documentation to ensure that study data are authentic, accurate and complete. The sponsor’s Quality and Assurance Officer will conduct monitoring visits to review the source documentation and electronic case report forms (eCRF) and to evaluate them for accuracy, completeness and compliance with the approved protocol, applicable regulations and GCP standards. The frequency of the monitoring visits will depend on the findings of the first patient-monitoring visit at each site.
Participants’ anonymity will be maintained at all times, and study workbooks and other documents will only be labelled with a non-identifiable study participant number. Paper documents that identify participants (such as consent forms) will be stored in a separate folder in a locked office of the secured department site and maintained by investigators in strict confidence. At the end of the trial, any non-essential confidential documents will be destroyed and essential documents will be kept securely for at least 10 years after study completion.
Trial monitoring groups
Day-to-day management of the trial will be the responsibility of the Trial Management Group (TMG), members of which will include the Chief Investigator and co-investigators, the research assistant, the CTU Trial Statistician and Data Manager, and the sponsor’s Quality and Assurance Officer. The group will monitor all aspects of the conduct and progress of the trial including recruitment figures, data quality and protocol adherence. In addition, experts in the study research field (consultant intensivists, Chief and Principle Investigators, sponsor and CTU representatives) and patient and public involvement (PPI) members have been appointed as members of a Trial Steering Committee (TSC). The TSC will be responsible for overseeing conduct of the trial and will convene every 6 months, will review trial progress, recruitment rates, safety data (reviewed blinded to treatment allocation) and ethical issues, and will make recommendations for protocol modifications. In the case of major safety concerns, the TSC will be able to request unblinded review of the safety data.
In order to safeguard the interests of trial participants, an independent Trial Safety Group (TSG) has been appointed and will be updated with safety data after every five patients recruited or in the event of an SAE occurring. The TSG will be responsible for monitoring any evidence for treatment harm, informing the TSC of any safety concerns and advising with regard to protocol modifications suggested by investigators or sponsors.
Four previous ICU-AW sufferers and their carers who constitute our Project Advisory Group (PAG) were involved in defining the research questions and in aiding development of a patient-friendly study design. The PAG has reviewed and advised on language and content of participant-related study documents. Some members are also TSC members and will be involved in the delivery and management of the trial. At the end of the study, they will also assist in writing a letter thanking participants for their involvement and lay summary of findings.
Serious adverse events
Safety reporting will be performed in accordance with HRA guidelines for non-CTIMP research (not involving investigational medicinal products or medical devices) with adherence to GCP standards. Untoward medical occurrences are expected in critically ill patients and their monitoring and treatment is considered standard care. As a result, only serious adverse events (SAEs) will be reported. These are defined as any untoward occurrence that results in death; is life-threatening; requires hospitalisation or prolongs existing hospitalisation; or results in persistent or significant disability/incapacity; or any other important medical event that required intervention to prevent the outcomes already listed based upon appropriate judgement by the investigators.
SAEs will be recorded from when participant consent is obtained to 3-month follow-up after hospital discharge. SAEs possibly, probably and definitely related to the study, such as DVT, pulmonary embolism and elevated creatine kinase (creatine kinase > 3800 IU/L), will be recorded. For all SAEs, the following data will be recorded in the eCRF and patient notes: description; date and time of occurrence; severity; relationship of SAE to study procedure (determined by the investigator physician); treatment required; and action taken with research procedure. The completed SAE form, signed by the reporting investigator and Principle Investigator of the site, will be submitted to the sponsor within 24 h of becoming aware of the event and all SAEs will be followed-up until resolved. If unexpected serious related event occurs, the Chief Investigator will notify Principle Investigators at local sites and the research ethics committee within 15 days of the event.
In the case of unanticipated concerns of safety to study participants or availability of new data arising from clinical or preclinical studies with this intervention, the study will be paused during review of newly available data prior to a final decision for continuation or termination of the study. If, in the opinion of the Chief Investigator, the clinical events indicate that it is not justifiable to continue the trial, the TSC will terminate the trial following consultation with the sponsor.
Indemnity
The Royal Surrey County Hospital Foundation Trust, as the trial sponsor, holds professional liability insurance to meet the potential legal liability of the sponsor and employees for harm to participants arising from the design and management of the research.
Indemnity to meet the potential legal liability of the investigators/collaborators for the harm of participants arising from the conduct of the research is provided by the NHS Indemnity scheme or through professional indemnity.
Dissemination of results
Study results will be published in peer-reviewed journals and presented at international/national surgical, anaesthetic and perioperative medicine conferences.
In addition, a lay summary of findings will be disseminated to study participants.
Discussion
This study will be the first to assess the safety, tolerability, feasibility and acceptability of RVOS in ICU patients. The effect of RVOS on muscle mass atrophy and weakness, and on arterial dysfunction observed in ICU patients, will also be explored.
Thirty-two ventilated critically ill patients with at least two other organ failures will be recruited to the study. Multiple organ failure patients were chosen as the study population because these patients undergo more pronounced muscle wasting (16% decrease in the RFCSA compared to 3% in single organ failure patients within 7 days of ICU admission) [21].
RVOS will be performed using equipment conventionally and safely used for vascular disease diagnostics and routinely used during vascular and orthopaedic operations. Control participants will receive standard care and no sham treatment (repeated cuff inflation at lower pressure) so as to avoid any detrimental impacts of isolated venous occlusion. Participants will thus not be blinded in the study. However, this is unlikely to introduce bias to the objective measures as muscle and arterial ultrasound images will be anonymised and analysed blinded to subject identity, study time points and group allocation.
The tolerability of RVOS cannot be assessed when participants are ventilated and sedated. Even when participants are extubated and off sedation, delirium (an acute brain dysfunction) is common in critically ill patients and can influence the VAS score. CAM-ICU assessment will be performed and the mental state of the participant during the RVOS session will be recorded to observe any effects of delirium on the VAS score.
Ultrasound assessments of muscle mass and quality and arterial function can be performed when participants are sedated; however, muscle strength and physical function measures are volitional assessments and require the participant’s full attention and cooperation. Therefore, sedation, lack of willingness to participate or lack of mental capacity due to delirium can prevent these outcome assessments. These measures will therefore only be performed when the participant is off sedation and CAM-ICU negative; this could lead to the time points when the strength and physical function assessments are performed varying between participants.
Our study population has a high risk of hospital death and post-discharge mortality. This means that participants could be lost due to death before completion of the study, with previous reports of 30% mortality in a study with similar inclusion and exclusion criteria [109]. We will recruit until we have 32 participants that survive the ICU stay, recognising that subsequent deaths after ICU discharge may reduce the data available at hospital discharge and at 3-month follow-up.
We expect repeated inflation and deflation of the pneumatic cuff during the RVOS to cause minor fluctuations in heat rate and blood pressure. As a result, patients with profound cardiovascular instability (defined by infused vasopressors ≥ 0.5 μg/kg/min of norepinephrine or by opinion of the senior attending physician) will not be studied. Moreover, during the RVOS session, haemodynamic parameters will be monitored closely. Critically ill patients have a 10% incidence rate of DVT [110], whilst an incidence of 0.055% is reported in others who have received blood flow restriction exercise [92]. We will thus exclude patients with a history of DVT and pulmonary embolism (PE) as well as patients who have contraindication to pharmacological venous thromboembolism prophylaxis. Apart from an incidence of minor events such as skin petechiae (4.4%) and subcutaneous bruising (13.1%), no major bleeding risk of RVOS has been reported in the literature [92, 111]. However, as a safety measure, patients with profound coagulopathy (defined by > 2.5 times normal prothrombin time, > 2 times normal APTT, platelet count ≤ 50 or on IV heparin infusion APTR ≥ 2) will not be recruited or studied if this occurs.
If our pilot study shows that RVOS is safe, feasible and acceptable to apply to ICU patients, an appropriately-powered randomised controlled trial will be designed to assess any potential benefit of this procedure.
Trial status
The study was opened to recruitment at the RSCH site in October 2017 and at the ASPH site in January 2018, and the first participant was recruited in January 2018. Study recruitment was completed in Feburary 2019.
Availability of data and materials
The data generated from this study will be made available on request to the corresponding author.
Abbreviations
- APTR:
-
Activated partial thromboplastin time ratio
- APTT:
-
Activated partial thromboplastin time
- CAM-ICU:
-
Confusion assessment method for the ICU
- CPAP:
-
Continuous positive airway pressure
- DVT:
-
Deep vein thrombosis
- FMD:
-
Flow-mediated dilation
- GCP:
-
Good Clinical Practice
- ICU:
-
Intensive care unit
- ICU-AW:
-
Intensive care unit-acquired weakness
- MRC-SS:
-
Medical Research Council Sum Score
- NIV:
-
Non-invasive ventilation
- PAG:
-
Project advisory group
- PBMC:
-
Peripheral blood mononuclear cell
- RFCSA:
-
Rectus femoris cross-sectional area
- RVOS:
-
Repetitive vascular occlusion stimulus
- SAE:
-
Serious adverse event
- SFA:
-
Superficial femoral artery
- SOFA:
-
Sequential Organ Failure Assessment
- TMG:
-
Trial Management Group
- TSC:
-
Trial Steering Committee
- TSG:
-
Trial Safety Group
- VAS:
-
Visual analogue scale
- VEGF:
-
Vascular endothelial growth factor
References
Hutchings A, Durand MA, Grieve R, Harrison D, Rowan K, Green J, et al. Evaluation of modernisation of adult critical care services in England: time series and cost effectiveness analysis. BMJ. 2009;339:b4353.
Milbrandt E, Kersten A, Watson S, Rahim M, Clermont G, Angus D, et al. Rising use of intensive care unit services in Medicare. Crit Care. 2005;9(1):112.
Zimmerman JE, Kramer AA, Knaus WA. Changes in hospital mortality for United States intensive care unit admissions from 1988 to 2012. Crit Care. 2013;17(2):R81.
Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304(16):1787–94.
Herridge M, Tansey C, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med. 2011;364(14):1293–304.
National Institute for Health and Clinical Excellence. Rehabilitation after critical illness: NICE clinical guideline 83. London: National Institute for Health and Clinical Excellence; 2009. Available from: www.nice.org.uk/CG83.
Herridge MS, Cheung AM, Tansey CM, Matte-Martyn A, Diaz-Granados N, Al-Saidi F, et al. One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med. 2003;348(8):683–93.
Wieske L, Dettling-Ihnenfeldt DS, Verhamme C, Nollet F, van Schaik IN, Schultz MJ, et al. Impact of ICU-acquired weakness on post-ICU physical functioning: a follow-up study. Crit Care. 2015;19(1):1–8.
Stevens RD, Marshall SA, Cornblath DR, Hoke A, Needham DM, De Jonghe B, et al. A framework for diagnosing and classifying intensive care unit-acquired weakness. Crit Care Med. 2009;37(Suppl 10):299–308.
Kress JP, Hall JB. ICU-acquired weakness and recovery from critical illness. N Engl J Med. 2014;370(17):1626–35.
Appleton RTD, Kinsella J, Quasim T. The incidence of intensive care unit-acquired weakness syndromes: a systematic review. J Intensive Care Soc. 2015;16(2):126–36.
Hermans G, Van Den Berghe G. Clinical review: intensive care unit acquired weakness. Crit Care. 2015;19(1):274.
Khan J, Harrison TB, Rich MM, Moss M. Early development of critical illness myopathy and neuropathy in patients with severe sepsis. Neurology. 2006;67(8):1421–5.
Lewis A, Riddoch-Contreras J, Natanek SA, Donaldson A, Man WD-C, Moxham J, et al. Downregulation of the serum response factor/miR-1 axis in the quadriceps of patients with COPD. Thorax. 2012;67(1):26–34.
Mirzakhani H, Williams J-N, Mello J, Joseph S, Meyer MJ, Waak K, et al. Muscle weakness predicts pharyngeal dysfunction and symptomatic aspiration in long-term ventilated patients. Anesthesiology. 2013;119(2):1–9.
de Letter M-ACJ, Schmitz PIM, Visser LH, Verheul FAM, Schellens RLLA, Op de Coul DAW, et al. Risk factors for the development of polyneuropathy and myopathy in critically ill patients. Crit Care Med. 2001;29(12):2281–6.
Hodgson C, Bellomo R, Berney S, Bailey M, Buhr H, Denehy L, et al. Early mobilization and recovery in mechanically ventilated patients in the ICU: a bi-national, multi-centre, prospective cohort study. Crit Care. 2015;19(1):81.
Hermans G, Van Mechelen H, Clerckx B, Vanhullebusch T, Mesotten D, Wilmer A, et al. Acute outcomes and 1-year mortality of intensive care unit-acquired weakness: a cohort study and propensity-matched analysis. Am J Respir Crit Care Med. 2014;190(4):410–20.
Kelmenson DA, Held N, Allen RR, Quan D, Burnham EL, Clark BJ, et al. Outcomes of ICU patients with a discharge diagnosis of critical illness polyneuromyopathy: a propensity-matched analysis. Crit Care Med. 2017;45(12):2055–60.
Garnacho-Montero J, Madrazo-Osuna J, García-Garmendia JL, Ortiz-Leyba C, Jimønez-Jimønez FJ, Barrero-Almodóvar A, et al. Critical illness polyneuropathy: risk factors and clinical consequences. A cohort study in septic patients. Intensive Care Med. 2001;27:1288–96.
Puthucheary ZA, Rawal J, McPhail M, Connolly B, Ratnayake G, Chan P, et al. Acute skeletal muscle wasting in critical illness. JAMA. 2013;310(15):1591–600.
Schweickert WD, Hall J. ICU-acquired weakness. Chest. 2007;131:1541–9.
Bodine SC. Disuse-induced muscle wasting. Int J Biochem Cell Biol. 2013;45(10):2200–8.
Bolton CF, Gilbert JJ, Hahn AF, Sibbald WJ. Polyneuropathy in critically ill patients. J Neurol Neurosurg Psychiatry. 1984;47:1223–31.
Lacomis D, Zochodne DW, Bird SJ. Critical illness myopathy. Muscle Nerve. 2000;23:1785–8.
Wollersheim T, Woehlecke J, Krebs M, Hamati J, Lodka D, Luther-Schroeder A, et al. Dynamics of myosin degradation in intensive care unit-acquired weakness during severe critical illness. Intensive Care Med. 2014;40(4):528–39.
Puthucheary ZA, Astin R, McPhail MJW, Saeed S, Pasha Y, Bear DE, et al. Metabolic phenotype of skeletal muscle in early critical illness. Thorax. 2018;73(10):926–35.
Reid MB, Judge AR, Bodine SC. CrossTalk opposing view: The dominant mechanism causing disuse muscle atrophy is proteolysis. J Physiol. 2014;592(24):5345–7.
Fischer JR, Baer RK. Acute myopathy associated with combined use of corticosteroids and neuromuscular blocking agents. Ann Pharmacother. 1996;30(12):1437–45.
Puthucheary Z, Rawal J, Ratnayake G, Harridge S, Montgomery H, Hart N. Neuromuscular blockade and skeletal muscle weakness in critically ill patients: time to rethink the evidence? Am J Respir Crit Care Med. 2012;185(9):911–7.
Weber-Carstens S, Deja M, Koch S, Spranger J, Bubser F, Wernecke KD, et al. Risk factors in critical illness myopathy during the early course of critical illness: a prospective observational study. Crit Care. 2010;14(3):R119.
Shepherd SJ, Newman R, Brett SJ, Griffith DM. Pharmacological therapy for the prevention and treatment of weakness after critical illness: a systematic review. Crit Care Med. 2016;44(6):1198–205.
Hermans G, De Jonghe B, Bruyninckx F, Van den Berghe G. Interventions for preventing critical illness polyneuropathy and critical illness myopathy. Cochrane Database Syst Rev. 2014;1:CD006832.
Burtin C, Clerckx B, Robbeets C, Ferdinande P, Langer D, Troosters T, et al. Early exercise in critically ill patients enhances short-term functional recovery. Crit Care Med. 2009;37(9):2499–505.
Morris PE, Goad A, Thompson C, Taylor K, Harry B, Passmore L, et al. Early intensive care unit mobility therapy in the treatment of acute respiratory failure. Crit Care Med. 2008;36(8):2238–43.
Schweickert WD, Pohlman MC, Pohlman AS, Nigos C, Pawlik AJ, Esbrook CL, et al. Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet. 2009;373(9678):1874–82.
Eggmann S, Verra ML, Luder G, Takala J, Jakob SM. Effects of early, combined endurance and resistance training in mechanically ventilated, critically ill patients: a study protocol for a randomised controlled trial. PLoS One. 2018;13(11):e0207428.
Fossat G, Baudin F, Courtes L, Bobet S, Dupont A, Bretagnol A, et al. Effect of in-bed leg cycling and electrical stimulation of the quadriceps on global muscle strength in critically ill adults. JAMA. 2018;320(4):368.
Fischer A, Spiegl M, Altmann K, Winkler A, Salamon A, Themessl-Huber M, et al. Muscle mass, strength and functional outcomes in critically ill patients after cardiothoracic surgery: does neuromuscular electrical stimulation help? The Catastim 2 randomized controlled trial. Crit Care. 2016;20(1):1–13.
Kho ME, Truong AD, Zanni JM, Ciesla ND, Brower RG, Palmer JB, et al. Neuromuscular electrical stimulation in mechanically ventilated patients: a randomized, sham-controlled pilot trial with blinded outcome assessment. J Crit Care. 2015;30(1):32–9.
Maffiuletti NA, Roig M, Karatzanos E, Nanas S. Neuromuscular electrical stimulation for preventing skeletal-muscle weakness and wasting in critically ill patients: a systematic review. BMC Med. 2013;11(1):137.
Burke D, Gorman E, Stokes D, Lennon O. An evaluation of neuromuscular electrical stimulation in critical care using the ICF framework: a systematic review and meta-analysis. Clin Respir J. 2016;4:407–20.
Takarada Y, Takazawa H, Ishii N. Applications of vascular occlusion diminish disuse atrophy of knee extensor muscles. Med Sci Sports Exerc. 2000;32(12):2035–9.
De Groot PCE, Thijssen DHJ, Sanchez M, Ellenkamp R, Hopman MTE. Ischemic preconditioning improves maximal performance in humans. Eur J Appl Physiol. 2010;108(1):141–6.
Bailey TG, Jones H, Gregson W, Atkinson G, Cable NT, Thijssen DHJ. Effect of ischemic preconditioning on lactate accumulation and running performance. Med Sci Sports Exerc. 2012;44(11):2084–9.
Crisafulli A, Tangianu F, Tocco F, Concu A, Mameli O, Mulliri G, et al. Ischemic preconditioning of the muscle improves maximal exercise performance but not maximal oxygen uptake in humans. J Appl Physiol. 2011;111(2):530–6.
Jean-St-Michel E, Manlhiot C, Li J, Tropak M, Michelsen MM, Schmidt MR, et al. Remote preconditioning improves maximal performance in highly trained athletes. Med Sci Sports Exerc. 2011;43(7):1280–6.
Scott BR, Loenneke JP, Slattery KM, Dascombe BJ. Exercise with blood flow restriction: an updated evidence-based approach for enhanced muscular development. Sports Med. 2015;45(3):313–25.
Sumide T, Sakuraba K, Sawaki K, Ohmura H, Tamura Y. Effect of resistance exercise training combined with relatively low vascular occlusion. J Sci Med Sport. 2009;12(1):107–12.
Abe T, Yasuda T, Midorikawa T, Sato Y, Kearns CF, Inoue K, et al. Skeletal muscle size and circulating IGF-1 are increased after two weeks of twice daily ‘KAATSU’ resistance training. Int J Kaatsu Train Res. 2005;1:6–12.
Patterson SD, Ferguson RA. Increase in calf post-occlusive blood flow and strength following short-term resistance exercise training with blood flow restriction in young women. Eur J App Physiol. 2010;108:1025–33.
Abe T, Kearns CF, Sato Y. Muscle size and strength are increased following walk training with restricted venous blood flow from the leg muscle, Kaatsu-walk training. J Appl Physiol. 2006;100(5):1460–6.
Takarada Y, Sato Y, Ishii N. Effects of resistance exercise combined with vascular occlusion on muscular function in athletes. Eur J Appl Physiol. 2002;86:308–14.
Karabulut M, Abe T, Sato Y, Bemben MG. The effects of low-intensity resistance training with vascular restriction on leg muscle strength in older men. Eur J Appl Physiol. 2010;108(1):147–55.
Ozaki H, Sakamaki M, Yasuda T, Fujita S, Ogasawara R, Sugaya M, et al. Increases in thigh muscle volume and strength by walk training with leg blood flow reduction in older participants. J Gerontol A Biol Sci Med Sci. 2011;66(3):257–63.
Yasuda T, Fukumura K, Fukuda T, Uchida Y, Iida H, Meguro M, et al. Muscle size and arterial stiffness after blood flow-restricted low-intensity resistance training in older adults. Scand J Med Sci Sports. 2014;24(5):799–806.
Patterson SD, Ferguson RA. Enhancing strength and postocclusive calf blood flow in older people with training with blood-flow restriction. J Aging Phys Act. 2011;19(3):201–13.
Vechin FC, Libardi CA, Conceição MS, Damas FR, Lixandrão ME, Berton RPB, et al. Comparisons between low-intensity resistance training with blood flow restriction and high-intensity resistance training on quadriceps muscle mass and strength in elderly. J Strength Cond Res. 2015;29(4):1071–6.
Libardi CA, Chacon-Mikahil MP, Cavaglieri CR, Tricoli V, Roschel H, Vechin FC, et al. Effect of concurrent training with blood flow restriction in the elderly. Int J Sport Med. 2015;36:395–9.
Takarada Y, Takazawa H, Sato Y, Takebayashi S, Tanaka Y, Ishii N. Effects of resistance exercise combined with moderate vascular occlusion on muscular function in humans. J Appl Physiol. 2000;88:2097–106.
Mattar MA, Gualano B, Perandini LA, Shinjo SK, Lima FR, Sá-Pinto AL, et al. Safety and possible effects of low-intensity resistance training associated with partial blood flow restriction in polymyositis and dermatomyositis. Arthritis Res Ther. 2014;16(1):1–8.
Gualano B, Neves M, Lima FR, Pinto ALDS, Laurentino G, Borges C, et al. Resistance training with vascular occlusion in inclusion body myositis: a case study. Med Sci Sports Exerc. 2010;42(2):250–4.
Loenneke JP, Thiebaud RS, Abe T, Manfro IG, Marin PJ, Duan D, et al. Acute blood flow restricted exercise to treat Duchenne muscular dystrophy: would it be efficacious? Front Physiol. 2013;4(114):1–3.
Kubota A, Sakuraba K, Sawaki K, Sumide T, Tamura Y. Prevention of disuse muscular weakness by restriction of blood flow. Med Sci Sports Exerc. 2008;40(3):529–34.
Cook SB, Brown KA, DeRuisseau K, Kanaley JA, Ploutz-Snyder LL. Skeletal muscle adaptations following blood flow-restricted training during 30 days of muscular unloading. J Appl Physiol. 2010;109(2):341–9.
Nakajima T, Yasuda T, Koide S, Yamasoba T, Obi S, Toyoda S, et al. Repetitive restriction of muscle blood flow enhances mTOR signaling pathways in a rat model. Heart Vessel. 2016;31(10):1685–95.
Cannavino J, Brocca L, Sandri M, Grassi B, Bottinelli R, Pellegrino MA. The role of alterations in mitochondrial dynamics and PGC-1α over-expression in fast muscle atrophy following hindlimb unloading. J Physiol. 2015;593(8):1981–95.
Thaveau F, Zoll J, Rouyer O, Chafke N, Kretz JG, Piquard F, et al. Ischemic preconditioning specifically restores complexes I and II activities of the mitochondrial respiratory chain in ischemic skeletal muscle. J Vasc Surg. 2007;46(3):541–7.
Nakajima T, Koide S, Yasuda T, Hasegawa T, Yamasoba T, Obi S, et al. Muscle hypertrophy following blood flow-restricted low force isometric electrical stimulation in rat tibialis anterior: role for muscle hypoxia. J Appl Physiol. 2018;125:134–45.
Jeffries O, Waldron M, Pattison JR, Patterson SD. Enhanced local skeletal muscle oxidative capacity and microvascular blood flow following 7-day ischemic preconditioning in healthy humans. Front Physiol. 2018;9:463.
Jones H, Hopkins N, Bailey TG, Green DJ, Cable NT, Thijssen DHJ. Seven-day remote ischemic preconditioning improves local and systemic endothelial function and microcirculation in healthy humans. Am J Hypertens. 2014;27(7):918–25.
Jones H, Nyakayiru J, Bailey TG, Green DJ, Cable NT, Sprung VS, et al. Impact of eight weeks of repeated ischaemic preconditioning on brachial artery and cutaneous microcirculatory function in healthy males. Eur J Prev Cardiol. 2015;22(8):1083–7.
Kimura M, Ueda K, Goto C, Jitsuiki D, Nishioka K, Umemura T, et al. Repetition of ischemic preconditioning augments endothelium-dependent vasodilation in humans: role of endothelium-derived nitric oxide and endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2007;27(6):1403–10.
Hamburg NM, Mcmackin CJ, Huang AL, Shenouda SM, Michael E, Schulz E, et al. Physical inactivity rapidly induces insulin resistance and microvascular dysfunction in healthy volunteers. Arterioscler Thromb Vasc Biol. 2007;27(12):2650–6.
Birk GK, Dawson EA, Timothy Cable N, Green DJ, Thijssen DHJ. Effect of unilateral forearm inactivity on endothelium-dependent vasodilator function in humans. Eur J Appl Physiol. 2013;113(4):933–40.
Van Duijnhoven NTL, Green DJ, Felsenberg D, Belavý DL, Hopman MTE, Thijssen DHJ. Impact of bed rest on conduit artery remodeling: effect of exercise countermeasures. Hypertension. 2010;56(2):240–6.
Wexler O, Morgan MA, Gough MS, Steinmetz SD, Mack CM, Darling DC, et al. Brachial artery reactivity in patients with severe sepsis: an observational study. Crit Care. 2012;16(2):R38.
Doerschug KC, Delsing AS, Schmidt GA, Haynes WG. Impairments in microvascular reactivity are related to organ failure in human sepsis. Am J Physiol Heart Circ Physiol. 2007;293:1065–71.
Creteur J, Carollo T, Soldati G, Buchele G, De Backer D, Vincent J-L. The prognostic value of muscle StO2 in septic patients. Intensive Care Med. 2007;33:1549–56.
Cheung MMH, Kharbanda RK, Konstantinov IE, Shimizu M, Frndova H, Li J, et al. Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery. First clinical application in humans. J Am Coll Cardiol. 2006;47(11):2277–82.
Hausenloy DJ, Mwamure PK, Venugopal V, Harris J, Barnard M, Grundy E, et al. Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomised controlled trial. Lancet. 2007;370:575–9.
Ali ZA, Callaghan CJ, Lim E, Ali AA, Nouraei SAR, Akthar AM, et al. Remote ischemic preconditioning reduces myocardial and renal injury after elective abdominal aortic aneurysm repair: a randomized controlled trial. Circulation. 2007;116:98–105.
Hoole SP, Heck PM, Sharples L, Khan SN, Duehmke R, Densem CG, et al. Cardiac Remote Ischemic Preconditioning in Coronary Stenting (CRISP Stent) Study. A prospective, randomized control trial. Circulation. 2009;119(6):820–7.
Zarbock A, Schmidt C, Van Aken H, Wempe C, Martens S, Zahn PK, et al. Effect of remote ischemic preconditioning on kidney injury among high-risk patients undergoing cardiac surgery: a randomized clinical trial. JAMA. 2015;313(21):2133–41.
Zarbock A, Kellum J. Remote ischemic preconditioning and protection of the kidney—a novel therapeutic option. Crit Care Med. 2016;44(3):607–16.
Meng R, Asmaro K, Meng L, Liu Y, Ma C, Xi C, et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology. 2012;79(18):1853–61.
Hausenloy DJ, Yellon DM. Remote ischaemic preconditioning: underlying mechanisms and clinical application. Cadiovasc Res. 2008;79:377–86.
Loenneke JP, Wilson JM, Wilson GJ, Pujol TJ, Bemben MG. Potential safety issues with blood flow restriction training. Scand J Med Sci Sport. 2011;21(4):510–8.
Takarada Y, Nakamura Y, Aruga S, Onda T, Miyazaki S, Ishii N. Rapid increase in plasma growth hormone after low-intensity resistance exercise with vascular occlusion. J Appl Physiol. 2000;88:61–5.
Page W, Swan R, Patterson SD. The effect of intermittent lower limb occlusion on recovery following exercise-induced muscle damage: a randomized controlled trial. J Sci Med Sport. 2017;20(8):729–33.
Franz A, Behringer M, Harmsen JF, Mayer C, Krauspe R, Zilkens C, et al. Ischemic preconditioning blunts muscle damage responses induced by eccentric exercise. Med Sci Sports Exerc. 2018;50(1):109–15.
Nakajima T, Kurano M, Iida H, Takano H, Oonuma H, Morita T, et al. Use and safety of KAATSU training: results of a national survey. Int J KAATSU Train Res. 2006;2(1):5–13.
Madarame H, Kurano M, Fukumura K, Fukuda T, Nakajima T. Haemostatic and inflammatory responses to blood flow-restricted exercise in patients with ischaemic heart disease: a pilot study. Clin Physiol Funct Imaging. 2013;33(1):11–7.
Staunton CA, May AK, Brandner CR, Warmington SA. Haemodynamics of aerobic and resistance blood flow restriction exercise in young and older adults. Eur J Appl Physiol. 2015;115(11):2293–302.
Clark BC, Manini TM, Hoffman RL, Williams PS, Guiler MK, Knutson MJ, et al. Relative safety of 4 weeks of blood flow-restricted resistance exercise in young, healthy adults. Scand J Med Sci Sport. 2011;21(5):653–62.
Koch S, Katsnelson M, Dong C, Perez-Pinzon M. Remote ischemic limb preconditioning after subarachnoid hemorrhage: a phase Ib study of safety and feasibility. Stroke. 2011;42(5):1387–91.
Lancaster GA, Dodd S, Williamson PR. Design and analysis of pilot studies: recommendations for good practice. J Eval Clin Pract. 2004;10(2):307–12.
Sharma V, Cunniffe B, Verma AP, Cardinale M, Yellon D. Characterization of acute ischemia-related physiological responses associated with remote ischemic preconditioning: a randomized controlled, crossover human study. Physiol Rep. 2014;2:11.
Seymour JM, Ward K, Sidhu PS, Puthucheary Z, Steier J, Jolley CJ, et al. Ultrasound measurement of rectus femoris cross-sectional area and the relationship with quadriceps strength in COPD. Thorax. 2009;64(5):418–23.
Sarwal A, Parry SM, Berry MJ, Hsu FC, Lewis MT, Justus NW, et al. Interobserver reliability of quantitative muscle sonographic analysis in the critically ill population. J Ultrasound Med. 2015;34(7):1191–200.
Parry SM, El-Ansary D, Cartwright MS, Sarwal A, Berney S, Koopman R, et al. Ultrasonography in the intensive care setting can be used to detect changes in the quality and quantity of muscle and is related to muscle strength and function. J Crit Care. 2015;30(5):1151.
Hunt JEA, Galea D, Tufft G, Bunce D, Ferguson RA. Time course of regional vascular adaptations to low load resistance training with blood flow restriction. J Appl Physiol. 2013;115:403–11.
Hermans G, Clerckx B, Vanhullebusch T, Segers J, Vanpee G, Robbeets C, et al. Interobserver agreement of Medical Research Council sum-score and handgrip strength in the intensive care unit. Muscle Nerve. 2012;45(1):18–25.
Hodgson C, Needham D, Haines K, Bailey M, Ward A, Harrold M, et al. Feasibility and inter-rater reliability of the ICU Mobility Scale. Hear Lung. 2014;43(1):19–24.
Tipping CJ, Bailey MJ, Bellomo R, Berney S, Buhr H, Denehy L, et al. The ICU Mobility Scale has construct and predictive validity and is responsive: a multicenter observational study. Ann Am Thorac Soc. 2016;13(6):887–93.
Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31.
de Roos B, Duthie SJ, Polley AC, Mulholland F, Bouwman FG, Heim C, et al. Proteomic methodological recommendations for studies involving human plasma, platelets, and peripheral blood mononuclear cells. J Proteome Res. 2008;7(6):2280–90.
van de Worp WRPH, Theys J, van Helvoort A, Langen RCJ. Regulation of muscle atrophy by microRNAs: ‘AtromiRs’ as potential target in cachexia. Curr Opin Clin Nutr Metab Care. 2018;21(6):423–9.
Harvey SE, Parrott F, Harrison DA, Bear DE, Segaran E, Beale R, et al. Trial of the route of early nutritional support in critically ill adults. N Engl J Med. 2014;371(18):1673–84.
Cook D, Crowther M, Meade M, Rabbat C, Griffith L, Schiff D, et al. Deep venous thrombosis in medical-surgical critically ill patients: prevalence, incidence, and risk factors. Crit Care Med. 2005;33(7):1565–71.
Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, Kolvekar S, et al. Remote ischemic preconditioning and outcomes of cardiac surgery. N Engl J Med. 2015;373(15):1408–17.
Acknowledgements
The authors would like to thank members of the Trial Safety Group, Trial Steering Committee and Project Advisory Group for their continued invaluable support and advice; local investigators and clinical staff in the participating sites; and the sponsor (Royal Surrey County Hospital NHS Foundation Trust, Guildford, UK), funders (NIHR Research for Patient Benefit and Intensive Care Foundation) and collaborators of the University of Surrey and Surrey CTU.
HEM and ZAP are supported by the NIHR BRC of University College London Hospitals, to whom their thanks are expressed.
Protocol version
This manuscript is based on the most recent protocol version (4.3, March 2018).
Funding
This paper presents independent research funded by the National Institute for Health Research (NIHR) under its Research for Patient Benefit (RfPB) Programme (Grant Reference Number PB-PG-0815-20029) and Intensive Care Foundation New Investigator Award to BCC-B. The views expressed are those of the authors and not necessarily those of the NIHR, or the Department of Health and Social Care or Intensive Care Foundation.
Author information
Authors and Affiliations
Contributions
IC prepared the manuscript’s first draft, and is responsible for set-up and day-to-day conduct of study at the RSCH and ASPH sites. JEAH helped design and draft the study protocol, obtained NIHR Research for Patient Benefit funding together with BCC-B, trained IC in ultrasound assessment and was involved in manuscript preparation. BCC-B helped design and draft the study protocol, obtained NIHR Research for Patient Benefit funding together with JEAH, obtained Intensive Care Foundation New Investigator Award funds, identified study sites and local principle investigators and facilitated set-up of study at sites, is responsible for overall study conduct at the RCSH site and was involved in manuscript preparation. JRM prepared the Statistical Analysis Plan, was involved in manuscript preparation and will be involved in the statistical analysis of the trial data. SDP, ZAP and HEM contributed to design and study protocol preparation and assisted in authoring the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was reviewed and approved by London Queens Square Ethics Committee (ref. 17/LO/0934) in September 2017. All subsequent substantial amendments to the protocol have been reviewed and approved by the ethics committee and Health Research Authority (HRA) and all non-substantial amendments were notified to the HRA. The trial has been registered in the ISRCTN Registry (trial ID: ISRCTN44340629) and is conducted in compliance with Good Clinical Practice standards and principles of the Declaration of Helsinki (2013).
Written prospective or retrospective informed consent will be obtained from all participants.
Consent for publication
By giving written informed consent, participants have provided their permission for storage of data and publication of the study results. All authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 checklist: recommended items to address in a clinical trial protocol and related documents (DOC 137 kb)
Additional file 2:
Acceptability questions (DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chhetri, I., Hunt, J.E.A., Mendis, J.R. et al. Repetitive vascular occlusion stimulus (RVOS) versus standard care to prevent muscle wasting in critically ill patients (ROSProx):a study protocol for a pilot randomised controlled trial. Trials 20, 456 (2019). https://doi.org/10.1186/s13063-019-3547-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-019-3547-5