
Abstract
The combination of genetics and genomics in Parkinson´s disease has recently begun to unveil molecular mechanisms possibly underlying disease onset and progression. In particular, catabolic processes such as autophagy have been increasingly gaining relevance as post-mortem evidence and experimental models suggested a participation in neurodegeneration and alpha-synuclein Lewy body pathology. In addition, familial Parkinson´s disease linked to LRRK2 and alpha-synuclein provided stronger correlation between etiology and alterations in autophagy. More detailed cellular pathways are proposed and genetic risk factors that associate with idiopathic Parkinson´s disease provide further clues in dissecting contributions of single players. Nevertheless, the fine-tuning of these processes remains elusive, as the initial stages of the pathways are not yet clarified.
In this review, we collect literature evidence pointing to autophagy as the common, downstream target of Parkinsonian dysfunctions and augment current knowledge on the factors that direct the subsequent steps. Cell and molecular biology evidence indicate that p38 signaling underlies neurodegeneration and autoptic observations suggest a participation in neuropathology. Moreover, alpha-synuclein and LRRK2 also appear involved in the p38 pathway with additional roles in the regulation of GTPase signaling. Small GTPases are critical modulators of p38 activation and thus, their functional interaction with aSyn and LRRK2 could explain much of the detailed mechanics of autophagy in Parkinson´s disease.
We propose a novel hypothesis for a more comprehensive working model where autophagy is controlled by upstream pathways, such as GTPase-p38, that have been so far underexplored in this context. In addition, etiological factors (LRRK2, alpha-synuclein) and risk loci might also combine in this common mechanism, providing a powerful experimental setting to dissect the cause of both familial and idiopathic disease.
Similar content being viewed by others

Background
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder [1] and is defined by parkinsonism, a motor syndrome including resting tremor, bradykinesia, rigidity and postural instability. Non-motor symptoms (dementia, depression, sleep disorders) also manifest and can arise even before motor diagnosis. The pathology is associated with a profound loss of dopaminergic neurons in the Substantia Nigra pars compacta (SNc) [2, 3] accompanied by filamentous protein inclusions termed Lewy bodies (LBs) in the surviving neurons. LBs are mainly composed of alpha-synuclein (aSyn) and are found in both idiopathic and autosomal dominant, familial PD patients. These inclusions are the hallmark pathology not only in PD, but also a group of neurological disorders called synucleinopathies. Their pathogenic relevance is, however, still under debate [4]. Therapies that are capable of slowing down the progression of this disabling neurodegenerative disorder are currently not available and the mainstay of symptomatic treatment remains the pharmacological replacement of dopamine (DA) using its precursor L-DOPA [5]. Until 1997, the etiology of PD was essentially considered the result of several environmental factors such as viral infections, lifestyle and environmental pollution [6]. With the discovery that mutations and multiplications in the SNCA gene (coding for aSyn) are linked to familial PD, the first evidence of a genetic contribution to lifetime risk of PD was uncovered [7, 8]. These genetic alterations can also interact with environmental factors to contribute to disease onset [9]. The “genetics revolution” led to a redefinition of PD as a heterogeneous disorder comprising genetic implications (albeit in a limited percentage of patients), and prompted efforts on the investigation of the roles played by the genes involved. In addition to sequencing strategies in families with a history of PD, genome-wide association studies (GWAS) nominate several risk loci [10, 11]. The combination of all these molecular genetic methods holds promises to discover mechanisms underlying disease onset and/or progression. PD is a multifactorial disorder, the vast majority of cases are classified as idiopathic (i.e. of unknown etiology) and familial PD accounts for only ~10% of all PD cases worldwide [12]. Nevertheless, the study of gene mutations that segregate in families with PD offers a valuable opportunity to better understand molecular mechanisms, improve clinical diagnosis and inform trial design and neuroscientific modelling. Most of the past and recent discoveries in PD research point out three major cellular processes altered in the disease state: vesicle trafficking and synaptic transmission, autophagy and mitochondrial quality control. PD-related mutations are present since the embryonal life, but their pathophysiological effects are not evident until later in life (ranging between ~30 to 60 years of age, including early- and late-onset PD) and only in those patients who will develop PD [13, 14]. We speculate that cellular processes mentioned above might be part of a putative compensatory system and would be compromised with age. This view underlines the importance of understanding where and when these processes become dysfunctional. Notably, age is the major overall risk factor for PD [3]. This understanding could help to intervene in the very early stages of the disease and develop a disease-modifying treatment.
Main text
In this review, we will focus on mechanisms involving aSyn and Leucine-Rich Repeat Kinase 2 (LRRK2), their impact on autophagy and we propose a novel perspective in the upstream modulation through Ras/Mitogen-activated Protein Kinase (MAPK) signaling. aSyn takes on a preponderant role in PD as a cause of familial disease, a genetic risk factor for idiopathic PD and the hallmark neuropathology. Mutations in LRRK2 are amongst the most common causes of familial disorder [15], displaying a clinical presentation that is mostly indistinguishable from idiopathic PD, and are found in 2-4% of idiopathic patients [16]. Both proteins play key functions in molecular mechanisms that are fundamental for the maintenance of neuronal viability, such as protein handling/degradation. These common pathways are coordinated, not only by familial genes, but also candidate genes predicted from identification in GWAS risk loci, suggesting alterations in more than one factor could addictively contribute to development of the disorder. Nevertheless, the same observation could indicate that the relative pathway per se is critical for pathogenesis, and individual perturbations along that pathway could be causal for the disease. Extensive research efforts have been conducted in the study of autophagy and elimination of aSyn aggregates, but no clear molecular pathway has been elucidated yet. Basing on genetics, genomics and functional biology, we specifically propose a novel hypothesis aimed at investigating an underexplored GTPase-p38 MAPK signaling in the modulation of autophagy that might regulate these cellular processes.
The autophagy-lysosome pathway: molecular mechanisms and implication in neurodegenerative diseases
As mentioned above, LBs, together with loss of DA neurons, represent the main pathological hallmark of PD [7, 17]. Although they are mainly composed of aggregated aSyn, other proteins like Tau, ubiquitine or amyloid-β (Aβ) are sometimes found at the neuropathological examination, leading to classification of PD as a proteinopathy. In healthy brains, these proteins are mostly present in a monomeric form, while in PD brains a conformational change occurs triggering the formation of small oligomers, which would eventually evolve into higher order structures. The moment when they become cytotoxic and the exact pathologic conformational shape are still controversial, but a growing body of evidence indicates insoluble aSyn as the pathogenic/pathologic molecular species [18, 19].
Generally, the steady-state levels of cellular proteins is controlled by their rates of production and degradation, but maintaining protein homeostasis in specialized secretory cells appears more challenging since the necessary and continuous synthesis of new proteins can cause cellular stress, eventually leading to proteotoxicity [20]. Therefore, either reducing production or stimulating degradation appeared to be promising strategies to prevent or revert uncontrolled aSyn accumulation [21]. An integrative view of quality control mechanisms in PD is emerging and centering the autophagy-lysosome pathway (ALP) as the process mostly involved in the clearance of pathologic aSyn [22, 23].
Autophagy is a powerful evolutionarily conserved catabolic process that mediates the degradation of unnecessary and dysfunctional proteins or organelles. Three types of autophagy have been identified: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). Macroautophagy remains the best characterized and will be referred to as autophagy hereafter. Microautophagy follows the same pathway as macroautophagy except from the direct sequestration of the cytoplasmic content by the lysosome. Finally, CMA is a highly selective process in which proteins with a specific amino acid motif are recognized and transported directly to the lysosome.
The process of autophagy begins with the formation of a double-membrane vesicle, termed autophagosome, through the extension of an isolation membrane (phagophore). This process can be broadly divided in two major steps regulated by distinct sets of autophagy-related genes (Atg): nucleation and elongation of the isolation membrane. The ULK (Unc-51 Like Kinase)/Atg1 kinase complex and the autophagy-specific phosphatidylinositide 3-kinase complex are important for the nucleation step, while Atg12- and Atg8/LC3-conjugation systems are essential for the elongation step [24]. Once Atg8/LC3 is lipidated and incorporated into the phagophore membrane, the growing membrane engulfs cytoplasmic content destined for degradation and closes in on itself. Then, the autophagosome may fuse with endosomes or directly combine with lysosomes to form autolysosomes, where hydrolases will dismantle the cargo into amino acids and other small molecules.
In neurons, autophagy appears highly compartmentalized. Within the soma, there is a population of autophagic vesicles derived from axons and synapses and a second population of vesicles generated in the cell body [25]. While the latter are created directly where their action will take place, the former are created at the distal tip of axons and are then transported by dynein towards the cell body following a retrograde transport [26]. The regulation of autophagy in cell models is well studied [27], but how the mechanisms work at the synapse, even more than a meter away from the cell soma, is still under investigation. Soukup and colleagues recently showed that the necessary proteins and mechanisms for autophagy are present at the synapse. EndophilinA (EndoA) is a presynaptic protein involved in vesicle endocytosis and colocalizes with autophagosomal markers. Phosphorylation of EndoA induces changes in the protein structure and in vitro this leads to formation of highly curved membrane zones, which are required at the elongation step of autophagosome formation [28].
In recent years, autophagy has been accepted as a cellular process deeply involved in the pathogenesis of both familial and idiopathic PD. Consistently, deletion of autophagic genes in mice causes locomotor defects, accumulation of polyubiquitinated proteins and neurodegeneration [29]. In addition, post-mortem evidence shows perturbation of autophagy markers in different neurodegenerative disorders [30,31,32]. Moreover, an important role for autophagy in brain is highlighted by the observation that this organ is the most severely affected in lysosomal storage disorders [33]. The most significant evidence of the negative impact of lysosome dysfunction emerged from the finding that heterozygous mutations in the GBA gene, encoding the lysosomal hydrolase Glucocerebrosidase (GCase), compromise its function of glucosylceramide cleavage and augment the risk of developing PD by approximately 20-fold [34]. Moreover, deletion of neuronal GCase in Drosophila causes dramatic lysosomal-autophagic defects [35]. Autophagosome number is increased and the level of the fly LC3 homologue is augmented, suggesting a block of the autophagic flux, consistent with observations in the SN of PD brains and associated to aSyn pathology [36]. Additionally, the autophagic machinery becomes less efficient with ageing in the healthy brain and stimulation of autophagy has been proposed to promote longevity [37, 38]. Induction of autophagy is an accepted experimental approach in PD and in neurodegenerative proteinopathies [39, 40], but might not always be sufficient. Neuropathological inclusions are capable of escaping degradation and impair the correct progression of the autophagic pathway [41]. A recent report also shows that accumulation of autophagosomes during lysosomal blockade leads to cytotoxicity [42], but this could also be a consequence of the impaired process as a whole.
Under physiological conditions, autophagy occurs at low levels, while this process is triggered under cellular stress, amino acid deprivation and presence of protein aggregates [24, 43], leading to both compromised autolysosomal activity and increased autophagosome synthesis [44]. However, defects in autophagosome-lysosome fusion alone are not sufficient to induce cellular toxicity, but in combination with increased autophagosome synthesis, they cause deleterious effects on cell viability. Consistently, increased number of autophagosomes has been observed in cultured cells challenged with parkinsonian neurotoxins, such as MPP+ (1-methyl-4-phenylpyridinium), rotenone and 6-OHDA (6-hydroxydopamine) [45].
Tackling the mechanisms that are altered and lead to a “pathway to disease” is the only option to progress to a real cure. Despite great advances in the understanding of autophagy, specific knowledge of the events taking place in a specific condition (such as PD) is critical and still lacking. In the following section, we discuss the role played by p38 MAPK in autophagy and neurodegenerative diseases, as this player has mostly been considered a general regulator of apoptosis but could hold novel insights for PD pathogenesis.
Involvement of p38 MAPK in autophagy and neurodegeneration
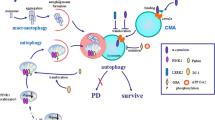
The p38 enzyme belongs to the MAPK family, together with ERK1/2 (Extracellular signal-Regulated Kinase), ERK5 and JNK (c-Jun N-terminal Kinase). They all signal through three modules: MAPK kinase kinases (MAPKKKs), MAPK kinases (MAPKKs) and MAPKs and are activated through dual phosphorylation of a tyrosine and a threonine residue (p-p38, pERK, pJNK) [46]. In mammals, four isoforms of p38 exist: α, β, γ, δ. They are divided into two groups, on one hand p38α and p38β, on the other p38γ and p38δ, mainly based on sequence homology and susceptibilities to different inhibitor compounds [47]. p38 MAPKs are primarily activated by environmental stresses and cytokines, but also growth factors. Two important kinases phosphorylating and thus activating p38 are Mitogen-activated Protein Kinase Kinase 3 and 6 (MKK3 and MKK6) [48] which are in turn activated by different MAPKKKs depending on the stimuli (Fig. 1). Upstream of MAPKKKs are small GTPases from the Rho subfamily (Rac1, Cdc242, Rho) and the Ras-like Rit subfamily [49,50,51].

Molecular interactions between LRRK2, aSyn and p38 MAPK in the regulation of apoptosis and autophagy. Apoptosis and autophagy are the main cellular processes modulated by p38 MAPK. Upstream regulation appears similar, with membrane receptor stimulation coupling to Rho/Ras signaling, activation of MKKs and of p38. In the apoptotic pathway, p38 mainly targets p53 that, when phosphorylated, translocates to the nucleus and promotes gene transcription of the apoptosis mediators Bax and Puma. On the other hand, p38 could also modulate ALP with consequences on protein aggregate degradation (aSyn, Tau). LRRK2 and aSyn probably intervene in this pathway redirecting to one side or the other, depending on the stimuli. LRRK2 can phosphorylate MKKs and/or p38 directly, while aSyn might affect p38 activation through different mechanisms. The initial stages of the cascade, i.e. the specific GTPase (sub)family, could participate as director of the cellular pathway to be activated down the line
The p38 MAPK signaling is known to be implicated in different neurodegenerative diseases through its regulatory action on apoptosis [52, 53]. In vivo, a p38 inhibitor prevents phosphorylation and translocation of p53 to the nucleus where it mediates transcription of Bax and Puma genes, which are known regulators of apoptosis. In vitro, p38 inhibition also prevents nuclear translocation of p53, similarly resulting in an enhancement of cell survival. [54].
In addition, recent research is shedding more light on the role of p38 MAPK in cellular processes other than apoptosis and suggests a role in autophagy. Available in vitro data do not consistently indicate the same direction of effect, which appears greatly dependent on the model system that is used (Table 1). Tang and colleagues show that p38 induces autophagy in astrocytes following accumulation of glial fibrillary acidic protein (GFAP), which is a principle filament protein in these cells. Mutations in the GFAP-coding gene cause Alexander disease, a neurodegenerative disorder affecting the brain white matter. Symptoms are mainly caused by defects in the myelin sheaths triggered by the accumulation of astrocytic GFAP [55, 56]. This accumulation is capable of activating p38, which in turn reduces mTOR phosphorylation and, as a consequence, leads to induction of autophagy [57]. This data suggests a cross-regulatory role on inflammation via autophagic modulation. A recent report indicates that p38 mediates microglial activation, but via a reduction of autophagy through phosphorylation-dependent inhibition of ULK1 [58]. Thus, an opposite effect on autophagy has been reported in microglia cells and astrocytes. In addition, it has been observed that hypo-osmotic conditions in hepatocytes activate p38, with a downstream suppression of the autophagic machinery [59]. Moreover, Webber et al show that depletion of p38α in HEK293 cells leads to a reduction in autophagy [60], whereas its activation in mouse embryonic fibroblasts produces an inhibition of the autophagosome-lysosome fusion [61]. These discrepancies might be caused by the cellular context in which studies are performed and thus, the direction of p38-mediated regulation of autophagy could be cell type-specific (Table 1).
In early stages of Alzheimer´s disease (AD), defects in the ALP can be observed. Schnöder and colleagues were the first to show in vivo data on the possible impact of p38α on autophagy in AD, where Aβ peptide is produced through several digestive steps of amyloid precursor protein by the enzyme BACE1. The inhibition of BACE1 reduces Aβ load and therefore gained increasing attention for therapeutic drug development. Downregulation of p38α induces lysosomal degradation of BACE1 and a consequent reduction of Aβ generation. In addition, pharmacological inhibition of p38α increases autophagy [62, 63]. Thus, in AD activation of neuronal p38 seems to be detrimental for autophagy and underlie neuropathology. Activity of p38 MAPK is also under investigation in Huntington’s disease, where polyglutamine-expanded huntingtin activates p38 signaling in neurons and the overexpression of Mitogen-activated Protein Kinase/Dual-specificity Phosphatase 1 (MKP-1) prevents this activation, possibly having an impact on neuroprotection [64]. Of interest, immunohistochemical analysis of patient brain samples from PD and Dementia with Lewy Bodies reveals positive, granular and inclusion-like p-p38 staining, whereas control samples display only weak diffuse staining of p-p38. In addition, granular p-p38 staining is found in neurons of the SNc containing LBs or diffuse aSyn deposition [65]. Consistently, multiple evidence demonstrate that activation of p38 underlies the toxic effects of 6-OHDA and MPTP on DA neurons [54, 66,67,68,69,70]. These examples might indicate that p38 activity could underlie cellular damage when occurring in neurons. In this regard, it is important to bear in mind that p38 probably acts as an “effector”. We speculate that downstream consequences could also depend on the initial neurotoxic/neuropathological trigger (which would constitute the basis of an etiologic mechanism).
Based on these observations, we hypothesize p38 could participate in neurodegenerative disease pathogenesis also through modulation of autophagy. Nevertheless, how it could be involved specifically in PD etiology remains unclear and unexplored. We mentioned familial PD (especially autosomal dominant) could serve as a starting point to understand idiopathic PD and thus, in the following sections, we will discuss the possible relation of aSyn and LRRK2 with autophagy and p38 signaling.
Alpha-synuclein, autophagy and MAPK modulation
SNCA mutations (A30P, E46K, H50Q, G51D and A53T) and gene multiplications (duplication and triplication) have been identified by linkage analysis to cause rare forms of familial PD, at a rate of about 1% in different populations [8, 71,72,73,74]. Patients carrying SNCA mutations mostly develop early-onset PD and have a good response to L-DOPA in the first stages of the disease [75]. Disease progression of SNCA duplication patients is more similar to idiopathic PD cases [76], whereas SNCA triplication carriers usually have earlier onset and a more severe disease progression and these phenotypes appear correlated with aSyn protein levels. Recent GWAS studies confirmed strong association of the SNCA locus, supporting a role in the idiopathic disease as well [10, 11], with several polymorphisms across the whole locus that exhibit association to increased risk of PD [77].
The aSyn protein is ubiquitously expressed in the brain [78], but its precise function is largely unclear. It has been proposed to interact with the Soluble N-ethylmaleimide-sensitive factor (NSF) Attachment Protein Receptor (SNARE) complex and associate with synaptic vesicles in axon terminals, regulate their trafficking and neurotransmitter release [79]. Furthermore, aSyn participates in cargo sorting and protein degradation as it is involved in endosome trafficking, autophagy and CMA [8, 80,81,82]. In line with this, aSyn overexpression is known to have an impact on the autophagic machinery with an increase of autophagosomes, which is not paralleled by a matched increase in degradative capacity but rather an impairment [41, 83]. Moreover, literature evidence show that pathological aSyn reduces autophagy and causes lysosomal depletion in PD brains, cell and animal models [22, 36]. Physiologically, aSyn is degraded by both the ubiquitin proteasome system (UPS) and ALP [84, 85], but autophagy-mediated degradation appears to be reserved for higher molecular weight species, such as aSyn aggregates, that are not able to enter the proteasome [23]. This evidence tightly link lysosome biology and aSyn inclusions in PD pathophysiology, with the lysosome as the specific site where pathologic aSyn is delivered for degradation [86]. Hence, any dysfunction at this organelle might cause an accumulation of aSyn in the nervous system, which reflects what has been observed in knockout mice lacking cathepsin D, one of the major lysosomal proteases. PD caused by GBA mutations (see previously) also supports this view, as dysfunctional GCase enhances aSyn neuropathology [87].
In neurodegenerative diseases like PD, monomeric aSyn assembles into soluble oligomers or protofibrils, a process shown to be enhanced by the A30P and A53T mutations [88]. The formation of these protofibrils ultimately leads to the generation of insoluble amyloid fibrils. In addition it has been shown that aSyn oligomers are prone to be secreted via exosomes, a possible mechanism responsible for the abovementioned spreading of LB pathology [89] as cells and neurons can secrete aSyn via exocytosis [90, 91]. The exact underlying mechanisms are still under deep investigation, but this cell-to-cell transfer has been proposed to explain the spreading of LB pathology in experimental models and support the prion-like hypothesis of PD [92,93,94].
Overexpression of aSyn is cytotoxic in cell lines, but not necessarily causes the formation of inclusions [95]. At the same time, an effect on activation of MAPKs, such as p38, has been reported as overexpression leads to a reduction of their phosphorylation. At the same time aSyn overexpressing cells are more vulnerable to oxidative stress, which triggers the activation of p38 signaling. This is blocked by aSyn itself and thus leading to reduced cell viability [96]. Of note, Polo-like kinase 2 (Plk2) phosphorylates the Ser129 residue of aSyn in vitro, in primary neurons and in vivo. This result was confirmed by the reduction of pSer129-aSyn levels following knock-down of Plk2 [97]. When expressed at high levels, aSyn inhibits Plk2 function, which in turn leads to lower phosphorylation of the downstream substrates of Plk2, such as Rho guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs), but also aSyn itself. In addition, aSyn inhibits p38 phosphorylation [96] and the dysregulated interaction between Plk2 and Rho GEFs/GAPs could result in reduced phosphorylation of p38 [98].
The amount of DA, produced by SNc neurons, is dramatically decreasing at a certain stage of the disease and accounts for motor symptoms. On the other hand, DA can exert toxic effects and is thought to contribute to neuronal loss [99, 100]. In a cell-based study, DA has been shown to induce a decrease in cell viability and an activation of p38 MAPK. In addition, aSyn expression was increased in this experimental setting and the autophagic machinery activated, which could be reversed by a p38 inhibitor. However, contrasting results have also been reported. Recently, neuronal overexpression of aSyn has been proposed not to activate p38, JNK or ERK1/2 [101], but replication of such data in a more quantitative way is needed.
In recent years, research focused on inflammatory processes that might be involved in PD pathogenesis. The contribution of glial cells and their release of inflammatory mediators is under current investigation. Interestingly, rapamycin- and trehalose-induced autophagy modulates phosphorylation of p38, which is activated by aSyn and required for inflammation-linked processes (which contribute to neurodegeneration [102]). Notably, both rapamycin and trehalose (autophagy enhancers) could reduce aSyn-dependent phosphorylation of p38 [103]. This collection of recent evidence suggests a novel link may be drawn between aSyn and p38 modulating cell physiology and survival via autophagy and not exclusively apoptosis [104] (Fig. 1). Additional experimental evidence is required to pinpoint the machinery of these interactions and the direction(s) of modulation.
LRRK2 in autophagy and MAPK signaling
LRRK2 is a large, multifunctional protein with a central catalytic GTPase/kinase core flanked by several protein-binding domains. Seven missense mutations, clustered in the Ras-of-complex (ROC) GTPase domain, C-terminal-of-ROC (COR) and kinase domains, segregate with PD in families [105]. These mutations show mild differences in their clinical presentation, but altogether, LRRK2 PD is basically indistinguishable from the idiopathic disorder, proposing familial LRRK2 PD as a window on idiopathic PD pathogenesis [15]. The G2019S substitution within the highly conserved kinase domain is associated to 4% of the autosomal dominant forms of familial PD and 1% of sporadic disease worldwide. At the adjacent residue, within the activation loop, a second mutation (I2020T) was discovered [106]. Both mutations are associated with a toxic gain of function, resulting in an increased kinase activity [107]. Three other confirmed pathogenic mutations have been described in the ROC domain affecting the same Arginine residue (R1441C/G/H), defining this codon as a hotspot for PD [108]. The majority of studies have reported these mutations to decrease GTP hydrolysis, increasing the residence time of the GTP-bound (active) state and indirectly enhancing kinase activity [109]. Nonetheless, the exact mechanism of the functional relationship between kinase and GTPase functions is still unresolved and one of the main controversial issues about LRRK2 biology [110].
Several substrates have been proposed to be directly phosphorylated by LRRK2, including LRRK2 itself, which complicated definition of a single cellular role [110]. Likely, LRRK2 acts in several physiological processes, of which vesicular trafficking and protein degradation have attracted specific research interest [111, 112]. Recent work revealed that members of the Ras analog in brain (Rab) family of small GTPases, involved in all forms of intracellular vesicular trafficking and sorting events, are key physiological phosphorylation substrates for LRRK2 [113, 114]. This is consistent with previous reports highlighting a role for LRRK2 in vesicle trafficking [115, 116], endolysosomal function and autophagy [117], and pointing out an important contribution to protein sorting and degradation.
Several studies show an involvement of LRRK2 in the regulation of autophagy and demonstrating that kinase activity modulates these effects, with consequences on PD pathogenesis and neuropathology [112, 118]. Of note, inhibition of LRRK2 kinase activity stimulates autophagy and regulates autophagosome formation [119]. At the presynaptic site, LRRK2 phosphorylates EndoA critically orchestrating the autophagic machinery in this neuronal compartment [40, 41]. Increased kinase activity due to the G2019S mutation also affects EndoA phosphorylation levels and, consequently, the regulation of synaptic autophagy [39]. Consistently, pathogenic LRRK2 mutations perturb lysosomal biology and morphology in a kinase-dependent manner. Overexpression of mutant LRRK2 in astrocytes causes an increase in lysosome size, number and function, a phenotype conserved in other cell types such as neuronal cell lines and human fibroblasts obtained from G2019S PD patients [120,121,122,123]. Conversely, Schapansky et al show a reduction in lysosomal size in G2019S KI primary neurons. These opposite results could be explained by the different models used, ranging from human non-neuronal cells to murine cultured neurons (mostly non-midbrain neurons) [124]. However, the average number of lysosomes per cell appears similarly changed, since it has been observed to increase in all cell models analyzed; a plausible explanation might be that upregulation of lysosome biogenesis serves as a compensatory mechanism. These data suggest that LRRK2 positively regulates the lysosomal system, but we cannot exclude that these changes might represent compensation to an insult in the pathway (e.g. autophagy inhibition). Nevertheless, the pathogenic G2019S mutation seems to exacerbate this function, but ultimately leading to lysosomal impairment [120].
As previously mentioned, the sometimes-contradictory literature and the complex nature of LRRK2 architecture complicate the definition of a role for LRRK2 and a conclusive demonstration of its cellular function(s). A physical and genetic interaction with Rab GTPases, helping to direct cytoplasmic LRRK2 to lipid membranes, has recently been uncovered, supporting the hypothesis of LRRK2 as a complexing scaffold [116]. Rab7L1, indicated by GWAS as a risk locus for idiopathic PD, functionally interacts with LRRK2 and regulates neurite outgrowth and trafficking of vesicles to the Golgi apparatus [122]. At the basis of this interaction, Beilina et al. further propose a complex able to function properly only when its components are present at the right stoichiometric ratio. These members are Rab7L1, Bcl-2 associated athanogene domain chaperone (BAG5) and cyclin G-associated kinase (GAK, notably another GWAS hit) and, when correctly combined with LRRK2, they promote autophagy-mediated clearance of Golgi-derived vesicles. In healthy individuals, a perfect orchestration of these factors is required and the impairment of a single component can affect the entire machinery. Mutations of LRRK2 gene do not affect the binding with its protein partners, but the overall function of the complex is compromised. Similarly, the formation of a partial complex is not sufficient for its correct function, suggesting a specific role for each protein within the complex [125]. This evidence strongly support the hypothesis that physical interactions between different PD-causing genes (e.g. LRRK2) and/or PD-associated factors (e.g. Rab7L1 and GAK) influence intracellular trafficking and autophagy to modulate disease expression (onset and progression). Additionally, other classical Rho GTPases (such as Cdc42, RhoA, Rac1) were found to bind LRRK2, with the strongest specificity of interaction shown by selective co-immunoprecipitation of LRRK2 with Rac1 [126]. Co-expression of LRRK2 and Rac1 increases the activity of the latter, directing it to its site of action. Rac1 is important in modulating actin cytoskeletal dynamics, which is required for the maintenance of neurite morphology.
Thus, similar to aSyn, LRRK2 appears to be involved in GTPase signaling to regulate downstream cellular processes. This could be a direct process (i.e. PD-related mutations directly alter GTPase regulation), but the possibility exists that other pathways and/or upstream regulators are present. Effects of LRRK2 also appear dependent on the cell type under investigation and likely additional, more indirect mechanisms might be involved. Several studies have identified pathway candidates to be activated by LRRK2, which is also reported to play a role in MAPK signaling cascades, not acting as the classical modulator of MAPK phosphorylation activity, but rather inducing a series of events leading to changes in cell physiology (Fig. 1). Binding of LRRK2 to MKK-3/6 and MKK-4/7 was found to mildly activate p38 and JNK pathways [127,128,129]. LRRK2 variants associated with a gain in kinase activity, such as G2019S, display a 3-4 fold increase of MKK phosphorylation compared to wild-type (WT) LRRK2. In addition, treatment with a selective ERK inhibitor suppresses the upregulated transcription of aSyn observed in WT LRRK2 overexpressing cell lines [130]. Consistently, application of a MEK inhibitor blocks the pathologic effect on autophagy and neurite shortening reported in several LRRK2 mutations [131]. Importantly, LRRK2 mediates the cytotoxic increase in p-p38 and the G2019S mutation causes sustained p38 activation and neurodegeneration in vivo [132].
We hypothesize that MAPKs provide a group of potential targets and/or interactors that could link neuronal toxicity, cytoskeletal dynamics and vesicular transport of mutant LRRK2 to molecular mechanism of autophagy. This pathway could well overlap or act in parallel to other, more established mechanisms such as regulation of Rab GTPases. In the following section, we discuss further details on common mechanisms and how they could impinge on protein degradation and underlie PD onset.
Interactions of aSyn and LRRK2 with autophagy and convergence on GTPase-p38 MAPK: a new working hypothesis for PD?
These numerous examples suggest that ALP could constitute a “unifying theme” in PD [86]. Nevertheless, the upstream regulation of ALP and the level at which the dysfunction is triggered is far from being elucidated. Novel mechanisms are being explored, combining different PD-related proteins into a common pathway. For example, LRRK2 mutations enhance protein levels of the lysosomal cation pump ATP13A2 [120]. Loss of function mutations in its coding gene are associated with parkinsonian Kufor-Rakeb syndrome, trigger alkalinisation of lysosomes and diminished efficiency of hydrolysis [133]. The G2019S mutation induces lysosomal deficits in different cell models [134] and upregulation of ATP13A2 could represent a compensatory mechanism as overexpression of ATP13A2 is thought to rescue lysosome dysfunction [135]. This evidence suggest a minimum cell-autonomous mechanism where LRRK2 pathogenic mutations alter lysosome function and aSyn homeostasis in cultured neurons [124].
More detailed interactions begun to unveil, as the role of LRRK2 in aSyn pathology gained strong interest following the observation that neurodegeneration elicited by virally delivered aSyn is alleviated by LRRK2 deletion [136] and kinase inhibition [137]. In addition, overexpression of the pathogenic LRRK2-G2019S enhances aSyn pathology triggered by synthetic aSyn pre-formed fibrils (PFFs) [138], which is reduced by acute LRRK2 silencing in vivo [139]. Interestingly, aSyn and LRRK2 show pro-inflammatory activity in microglial cells [140, 141], where p38-dependent inhibition of autophagy also mediates inflammatory actions (as previously discussed). Prolonged LRRK2 kinase inhibition in astrocytes leads to autophagy induction via hyperphosphorylation of ULK1 [142] on a distinct residue than the ones phosphorylated by p38 MAPK in microglia [58]. These observations add additional convergence points to the hypothesized pathway.
Collectively, this evidence show that aSyn pathology can be modulated by LRRK2 by an unknown mechanism. The common impingement on endosomal trafficking and autophagy, added to the prominent role of the latter in aggregate clearance, strongly points to regulation of protein degradation pathways as the cellular substrate where they probably meet. However, the fine modulation of such mechanisms is largely unexplored and the contribution of other players is still missing. Clarifying these still obscure details much likely will hold novel insights for pathogenesis and disease-modifying treatments. In the previous sections, we presented evidence that both LRRK2 and aSyn interact with MAPKs and p38 but, despite promising, these observations do not provide information on the mechanisms of activation and regulation of signaling. Other players must be involved and, following the directions suggested by genetics and genomics interactions detailed in the previous sections, we hypothesize genes/loci indicated in PD GWAS could shed light on these processes. We speculate that Ras/Rho-p38 MAPK signaling might play a decisive role in connecting dysfunctional proteins linked to familial PD and ALP in one pathway to disease. In addition, the contribution of genetic risk factors might explain pathogenesis in idiopathic disease as well. In this context, we dissected the implication of each GWAS hit in both MAPK pathway and ALP. In Table 2 we summarized the possible interaction of each most recently associated candidate gene [10, 11]. Most of these are of no clear cellular function and implication in p38/MAPK signaling has been scarcely investigated. Nevertheless, a few genes are directly linked to p38 signaling, supporting this as the connecting link between aSyn and LRRK2. With this hypothesis in mind, further upstream regulation could delineate the whole process and, at this level, GWAS could provide novel indications.
Involvement of small GTPases in PD pathogenesis is a current focus of research that has indicated their participation in several cellular processes that are altered in PD (for a recent review see [143]). With regards to our focus, LRRK2 phosphorylates two specific Rab GTPases [113]. Rabs are essential regulators of the endosome system, which mediates intracellular trafficking and autophagy [144]. Their role in endoplasmic reticulum-Golgi trafficking has also been investigated in PD with reports of alterations caused by aSyn [145, 146]. Previously, we mentioned that the Rac1 member of the Rho family of GTPases interacts with LRRK2. Recently, activity of Rac1 has been reported to be critical in the maintenance of DA neurons and to counteract pathologic aSyn via modulation of autophagy [147]. We also described how aSyn dysregulates Plk2-dependent modulation of Rho GEFs and GAPs. This could also impact aggregation of aSyn as Plk2 has been nominated as the kinase responsible for the Ser129 phosphorylation of aSyn in the same study [98]. These examples illustrate the multifaceted connection of GTPases to PD pathogenic processes.
We have recently observed that LRRK2 and aSyn dysregulate expression of the RIT2 gene (Obergasteiger et al., in preparation), which could underlie novel cellular mechanisms to be explored. Genomic variability in its locus has been recently reported [10, 148] to increase risk for PD. The RIT2 gene codes for the protein Rin that, together with Rit, is a member of the Rit subfamily of Ras-like small GTPases. Their cellular functions have been poorly investigated, but an emerging interest for the processes under examination might emerge. Indeed, Rin is involved in neuronal differentiation, p38 and ERK signaling pathways downstream of nerve growth factor (NGF) signaling, but its role in PD is still unknown [149]. Rin is expressed selectively in neural tissue [150], is enriched in rodent DA neurons [151] and shows reduced expression in the remaining SNc of PD brains [152]. Both Rit-like GTPases (Rit and Rin) were shown to regulate p38 MAPK signaling in cells in response to oxidative stress [153] and Rin specifically activates neurotrophin-mediated p38α MAPK signaling [150]. Preliminary results on Rin function and its connection to p38 are available, but definitive interpretation is still prevented by the paucity of the published data. In particular, further studies would need to causally join Rin-p38 activation with LRRK2 and/or aSyn function in the modulation of protein degradation. Moreover, more suitable cellular models, like primary or iPSC-derived neurons, are required to bear a stronger impact in the context of PD.
Conclusions
From our literature analysis, we hypothesize that p38 MAPK signaling might be a key process involved in PD and could be at least one of the “missing links” between most known cellular players and autophagy (Fig. 1). However, its specific role in disease pathogenesis needs further clarification and targeted experimental validation to dissect its contribution from the other MAPKs (if any). We propose p38 MAPK signaling may regulate autophagy, which has gained a priority role in PD research [11]. We also speculate that the Rin protein, upstream of p38 MAPK, could contribute to the orchestration of the autophagic machinery. The further investigation of autophagy- and/or MAPK-related GWAS genes could provide additional information on novel cellular processes contributing to PD onset and progression, which can eventually be targeted for PD therapy.
Abbreviations
- 6-OHDA:
-
6-hydroxydopamine
- AD:
-
Alzheimer´s disease
- ALP:
-
Autophagy lysosome pathway
- aSyn:
-
Alpha-synuclein
- Atg’s:
-
Autophagy-related genes
- Aβ:
-
Amyloid-β
- BAG5:
-
Bcl-2 associated athanogene domain chaperone
- CMA:
-
Chaperone-mediated autophagy
- COR:
-
C-terminal-of-ROC
- DA:
-
Dopamine
- EndoA:
-
EndophilinA
- ERK:
-
Extracellular Signal-regulated Kinase
- GAK:
-
Cyclin-G-associated kinase
- GAPs:
-
GTPase activating proteins
- GCase:
-
Glucocerebrosidase
- GEFs:
-
Guanine nucleotide exchange factors
- GFAP:
-
Glial fibrillary acidic protein
- GWAS:
-
Genome-wide association studies
- JNK:
-
c-Jun N-terminal kinase
- LBs:
-
Lewy bodies
- LRRK2:
-
Leucine-Rich Repeat Kinase 2
- MAPK:
-
Mitogen-activated protein kinase
- MAPKKKs:
-
MAPK kinase kinases
- MAPKKs:
-
MAPK kinases
- MKK-3/6:
-
Mitogen-activated protein kinase kinases 3/6
- MKK-4/7:
-
Mitogen-activated protein kinase kinases 4/7
- MKP-1:
-
Mitogen-activated protein kinase/dual-specificity phosphatase 1
- MPP+ :
-
1-methyl-4-phenylpyridinium
- NGF:
-
Nerve growth factor
- NSF:
-
N-ethylmaleimide-sensitive factor
- PD:
-
Parkinson’s disease
- PFFs:
-
Pre-formed fibrils
- Plk2:
-
Polo-like kinase 2
- Rab:
-
Ras analog in brain
- ROC:
-
Ras-of-complex
- SNARE:
-
Soluble NSF Attachment Protein Receptor
- SNc:
-
Substantia Nigra pars compacta
- ULK:
-
Unc-51 Like Kinase
- UPS:
-
Ubiquitine proteasome system
- WT:
-
Wild-type
References
Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson's disease. Physiol Rev. 2011;91:1161–218.
Lang AE, Lozano AM. Parkinson's disease. Second of two parts. N Engl J Med. 1998;339:1130–43.
Lang AE, Lozano AM. Parkinson’s disease. First of two parts. N Engl J Med. 1998;339:1044–53.
Jellinger K. Overview of morphological changes in Parkinson’s disease. Adv Neurol. 1987;45:1–18.
Birkmayer W, Hornykiewicz O: [The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia]. Wien Klin Wochenschr 1961, 73:787-788.
Ascherio A, Schwarzschild MA. The epidemiology of Parkinson's disease: risk factors and prevention. Lancet Neurol. 2016;15:1257–72.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–7.
Fleming SM. Mechanisms of Gene-Environment Interactions in Parkinson's Disease. Curr Environ Health Rep. 2017;4:192–9.
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat Genet. 2014;46:989–93.
Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. 2017;49:1511–6.
Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16:R183–94.
Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain. 2013;136:2419–31.
Obeso JA, Rodriguez-Oroz MC, Lanciego JL, Rodriguez Diaz M. How does Parkinson's disease begin? The role of compensatory mechanisms. Trends Neurosci. 2004;27:125–7. author reply 127-128
Volta M, Milnerwood AJ, Farrer MJ. Insights from late-onset familial parkinsonism on the pathogenesis of idiopathic Parkinson's disease. Lancet Neurol. 2015;14:1054–64.
Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. 2008;7:583–90.
Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet. 2009;373:2055–66.
Cavaliere F, Cerf L, Dehay B, Ramos-Gonzalez P, De Giorgi F, Bourdenx M, Bessede A, Obeso JA, Matute C, Ichas F, Bezard E. In vitro alpha-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol Dis. 2017;103:101–12.
Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–62.
Hetz C, Glimcher LH. Protein homeostasis networks in physiology and disease. Curr Opin Cell Biol. 2011;23:123–5.
Devine MJ, Gwinn K, Singleton A, Hardy J. Parkinson's disease and alpha-synuclein expression. Mov Disord. 2011;26:2160–8.
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to alpha-synuclein inclusions. Neurobiol Dis. 2009;35:385–98.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5.
Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32.
Maday S, Holzbaur EL. Compartment-Specific Regulation of Autophagy in Primary Neurons. J Neurosci. 2016;36:5933–45.
Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–305.
Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24:9–23.
Soukup SF, Verstreken P. EndoA/Endophilin-A creates docking stations for autophagic proteins at synapses. Autophagy. 2017;13:971–2.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9.
Piras A, Collin L, Gruninger F, Graff C, Ronnback A. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun. 2016;4:22.
Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31.
Sasaki S. Autophagy in Spinal Cord Motor Neurons in Sporadic Amyotrophic Lateral Sclerosis. J Neuropathol Exp Neurol. 2011;70:349–59.
Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–97.
Sidransky E, Samaddar T, Tayebi N. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology. 2009;73:1424–5. author reply 1425-1426
Kinghorn KJ, Asghari AM, Castillo-Quan JI. The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson's disease. Neural Regen Res. 2017;12:380–4.
Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci. 2010;30:12535–44.
Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, Yuan J. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:14164–9.
Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95.
Ryu HW, Oh WK, Jang IS, Park J. Amurensin G induces autophagy and attenuates cellular toxicities in a rotenone model of Parkinson's disease. Biochem Biophys Res Commun. 2013;433:121–6.
Jang W, Kim HJ, Li H, Jo KD, Lee MK, Song SH, Yang HO. 1,25-Dyhydroxyvitamin D(3) attenuates rotenone-induced neurotoxicity in SH-SY5Y cells through induction of autophagy. Biochem Biophys Res Commun. 2014;451:142–7.
Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem. 2013;288:15194–210.
Button RW, Roberts SL, Willis TL, Hanemann CO, Luo S. Accumulation of autophagosomes confers cytotoxicity. J Biol Chem. 2017;292:13599–614.
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435.
Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain. 2008;131:1969–78.
Garcia-Garcia A, Anandhan A, Burns M, Chen H, Zhou Y, Franco R. Impairment of Atg5-dependent autophagic flux promotes paraquat- and MPP(+)-induced apoptosis but not rotenone or 6-hydroxydopamine toxicity. Toxicol Sci. 2013;136:166–82.
Biondi RM, Nebreda AR. Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J. 2003;372:1–13.
Kuma Y, Sabio G, Bain J, Shpiro N, Marquez R, Cuenda A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J Biol Chem. 2005;280:19472–9.
Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69.
Marinissen MJ, Chiariello M, Gutkind JS. Regulation of gene expression by the small GTPase Rho through the ERK6 (p38 gamma) MAP kinase pathway. Genes Dev. 2001;15:535–53.
Sakabe K, Teramoto H, Zohar M, Behbahani B, Miyazaki H, Chikumi H, Gutkind JS. Potent transforming activity of the small GTP-binding protein Rit in NIH 3T3 cells: evidence for a role of a p38gamma-dependent signaling pathway. FEBS Lett. 2002;511:15–20.
Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, Bokoch GM. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem. 1995;270:23934–6.
Oh-hashi K, Maruyama W, Yi H, Takahashi T, Naoi M, Isobe K. Mitogen-activated protein kinase pathway mediates peroxynitrite-induced apoptosis in human dopaminergic neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun. 1999;263:504–9.
Bhat NR, Zhang P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. J Neurochem. 1999;72:112–9.
Karunakaran S, Saeed U, Mishra M, Valli RK, Datt Joshi S, Meka DP, Seth P, Ravindranath V. Selective Activation of p38 Mitogen-Activated Protein Kinase in Dopaminergic Neurons of Substantia Nigra Leads to Nuclear Translocation of p53 in <span class="sc">1</span>-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Treated Mice. J Neurosci. 2008;28:12500–9.
Li R, Johnson AB, Salomons G, Goldman JE, Naidu S, Quinlan R, Cree B, Ruyle SZ, Banwell B, D'Hooghe M, et al. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol. 2005;57:310–26.
Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–20.
Tang G, Yue Z, Talloczy Z, Hagemann T, Cho W, Messing A, Sulzer DL, Goldman JE. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17:1540–55.
He Y, She H, Zhang T, Xu H, Cheng L, Yepes M, Zhao Y, Mao Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J Cell Biol. 2018;217:315–28.
Prick T, Thumm M, Haussinger D, Vom Dahl S. Deletion of HOG1 leads to Osmosensitivity in starvation-induced, but not rapamycin-dependent Atg8 degradation and proteolysis: further evidence for different regulatory mechanisms in yeast autophagy. Autophagy. 2006;2:241–3.
Webber JL, Tooze SA. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 2010;29:27–40.
Keil E, Hocker R, Schuster M, Essmann F, Ueffing N, Hoffman B, Liebermann DA, Pfeffer K, Schulze-Osthoff K, Schmitz I. Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ. 2013;20:321–32.
Schnöder L, Hao W, Qin Y, Liu S, Tomic I, Liu X, Fassbender K, Liu Y. Deficiency of Neuronal p38α MAPK Attenuates Amyloid Pathology in Alzheimer Disease Mouse and Cell Models through Facilitating Lysosomal Degradation of BACE1. J Biol Chem. 2016;291:2067–79.
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy Induction and Autophagosome Clearance in Neurons: Relationship to Autophagic Pathology in Alzheimer's Disease. J Neurosci. 2008;28:6926–37.
Taylor DM, Moser R, Regulier E, Breuillaud L, Dixon M, Beesen AA, Elliston L, Silva Santos Mde F, Kim J, Jones L, et al. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington's disease via additive effects of JNK and p38 inhibition. J Neurosci. 2013;33:2313–25.
Ferrer I, Blanco R, Carmona M, Puig B, Barrachina M, Gomez C, Ambrosio S. Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinson's disease and Dementia with Lewy bodies. J Neural Transm (Vienna). 2001;108:1383–96.
Choi WS, Eom DS, Han BS, Kim WK, Han BH, Choi EJ, Oh TH, Markelonis GJ, Cho JW, Oh YJ. Phosphorylation of p38 MAPK induced by oxidative stress is linked to activation of both caspase-8- and -9-mediated apoptotic pathways in dopaminergic neurons. J Biol Chem. 2004;279:20451–60.
Gomez-Lazaro M, Galindo MF, Concannon CG, Segura MF, Fernandez-Gomez FJ, Llecha N, Comella JX, Prehn JH, Jordan J. 6-Hydroxydopamine activates the mitochondrial apoptosis pathway through p38 MAPK-mediated, p53-independent activation of Bax and PUMA. J Neurochem. 2008;104:1599–612.
Hu X, Weng Z, Chu CT, Zhang L, Cao G, Gao Y, Signore A, Zhu J, Hastings T, Greenamyre JT, Chen J. Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J Neurosci. 2011;31:247–61.
Izumi Y, Yamamoto N, Matsuo T, Wakita S, Takeuchi H, Kume T, Katsuki H, Sawada H, Akaike A. Vulnerability to glutamate toxicity of dopaminergic neurons is dependent on endogenous dopamine and MAPK activation. J Neurochem. 2009;110:745–55.
Jeohn GH, Cooper CL, Wilson B, Chang RC, Jang KJ, Kim HC, Liu B, Hong JS. p38 MAP kinase is involved in lipopolysaccharide-induced dopaminergic neuronal cell death in rat mesencephalic neuron-glia cultures. Ann N Y Acad Sci. 2002;962:332–46.
Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–8.
Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841.
Nishioka K, Ross OA, Ishii K, Kachergus JM, Ishiwata K, Kitagawa M, Kono S, Obi T, Mizoguchi K, Inoue Y, et al. Expanding the clinical phenotype of SNCA duplication carriers. Mov Disord. 2009;24:1811–9.
Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di Iorio G, Sanges G, Stenroos ES, Pho LT, Schaffer AA, et al. Mapping of a gene for Parkinson's disease to chromosome 4q21-q23. Science. 1996;274:1197–9.
Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schule B, Langston JW, Middleton FA, Ross OA, Hulihan M, et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68:916–22.
Myhre R, Toft M, Kachergus J, Hulihan MM, Aasly JO, Klungland H, Farrer MJ. Multiple alpha-synuclein gene polymorphisms are associated with Parkinson’s disease in a Norwegian population. Acta Neurol Scand. 2008;118:320–7.
Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32.
Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–7.
Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–52.
Lotharius J, Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–42.
Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci. 2011;31:14508–20.
Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O'Connor C, et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One. 2013;8:e62143.
Klucken J, Poehler AM, Ebrahimi-Fakhari D, Schneider J, Nuber S, Rockenstein E, Schlotzer-Schrehardt U, Hyman BT, McLean PJ, Masliah E, Winkler J. Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy. 2012;8:754–66.
Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17.
Tofaris GK. Lysosome-dependent pathways as a unifying theme in Parkinson's disease. Mov Disord. 2012;27:1364–9.
Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, Lee HJ, Kim S, Masliah E, Sardi SP, Lee SJ. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat Commun. 2014;5:4755.
Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–6.
Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42.
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–51.
Bellingham SA, Guo BB, Coleman BM, Hill AF. Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front Physiol. 2012;3:124.
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211.
Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53.
Olanow CW, Brundin P. Parkinson's disease and alpha synuclein: is Parkinson's disease a prion-like disorder? Mov Disord. 2013;28:31–40.
Hasegawa T, Matsuzaki M, Takeda A, Kikuchi A, Akita H, Perry G, Smith MA, Itoyama Y. Accelerated alpha-synuclein aggregation after differentiation of SH-SY5Y neuroblastoma cells. Brain Res. 2004;1013:51–9.
Iwata A, Maruyama M, Kanazawa I, Nukina N. alpha-Synuclein affects the MAPK pathway and accelerates cell death. J Biol Chem. 2001;276:45320–9.
Inglis KJ, Chereau D, Brigham EF, Chiou SS, Schobel S, Frigon NL, Yu M, Caccavello RJ, Nelson S, Motter R, et al. Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J Biol Chem. 2009;284:2598–602.
Wang S, Xu B, Liou L-C, Ren Q, Huang S, Luo Y, Zhang Z, Witt SN. α-Synuclein disrupts stress signaling by inhibiting polo-like kinase Cdc5/Plk2. Proc Natl Acad Sci. 2012;109:16119–24.
Filloux F, Townsend JJ. Pre- and postsynaptic neurotoxic effects of dopamine demonstrated by intrastriatal injection. Exp Neurol. 1993;119:79–88.
Ben-Shachar D, Zuk R, Glinka Y. Dopamine neurotoxicity: inhibition of mitochondrial respiration. J Neurochem. 1995;64:718–23.
Tolo J, Taschenberger G, Leite K, Stahlberg MA, Spehlbrink G, Kues J, Munari F, Capaldi S, Becker S, Zweckstetter M, et al. Pathophysiological Consequences of Neuronal alpha-Synuclein Overexpression: Impacts on Ion Homeostasis, Stress Signaling, Mitochondrial Integrity, and Electrical Activity. Front Mol Neurosci. 2018;11:49.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34.
Bussi C, Peralta Ramos JM, Arroyo DS, Gaviglio EA, Gallea JI, Wang JM, Celej MS, Iribarren P. Autophagy down regulates pro-inflammatory mediators in BV2 microglial cells and rescues both LPS and alpha-synuclein induced neuronal cell death. Sci Rep. 2017;7:43153.
Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res. 2003;73:341–50.
Paisan-Ruiz C, Lewis PA, Singleton AB. LRRK2: cause, risk, and mechanism. J Parkinsons Dis. 2013;3:85–103.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7.
West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–7.
Tsika E, Moore DJ. Contribution of GTPase activity to LRRK2-associated Parkinson disease. Small GTPases. 2013;4:164–70.
Liao J, Wu CX, Burlak C, Zhang S, Sahm H, Wang M, Zhang ZY, Vogel KW, Federici M, Riddle SM, et al. Parkinson disease-associated mutation R1441H in LRRK2 prolongs the "active state" of its GTPase domain. Proc Natl Acad Sci U S A. 2014;111:4055–60.
Islam MS, Moore DJ. Mechanisms of LRRK2-dependent neurodegeneration: role of enzymatic activity and protein aggregation. Biochem Soc Trans. 2017;45:163–72.
Lichtenberg M, Mansilla A, Zecchini VR, Fleming A, Rubinsztein DC. The Parkinson's disease protein LRRK2 impairs proteasome substrate clearance without affecting proteasome catalytic activity. Cell Death Dis. 2011;2:e196.
Manzoni C, Lewis PA. LRRK2 and Autophagy. Adv Neurobiol. 2017;14:89–105.
Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S, Lorentzen E, Duddy G, Wilson S, et al. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. eLife. 2016;5:e12813.
Lis P, Burel S, Steger M, Mann M, Brown F, Diez F, Tonelli F, Holton JL, Ho PW, Ho SL, et al. Development of phospho-specific Rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson's disease kinase. Biochem J. 2018;475:1–22.
Piccoli G, Condliffe SB, Bauer M, Giesert F, Boldt K, De Astis S, Meixner A, Sarioglu H, Vogt-Weisenhorn DM, Wurst W, et al. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci. 2011;31:2225–37.
Cookson MR. Cellular functions of LRRK2 implicate vesicular trafficking pathways in Parkinson's disease. Biochem Soc Trans. 2016;44:1603–10.
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16:394–406.
Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O, Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet. 2009;18:4022–34.
Manzoni C, Mamais A, Dihanich S, Abeti R, Soutar MPM, Plun-Favreau H, Giunti P, Tooze SA, Bandopadhyay R, Lewis PA. Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim Biophys Acta. 1833;2013:2900–10.
Henry AG, Aghamohammadzadeh S, Samaroo H, Chen Y, Mou K, Needle E, Hirst WD. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet. 2015;24:6013–28.
MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–93.
MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77:425–39.
Hockey LN, Kilpatrick BS, Eden ER, Lin-Moshier Y, Brailoiu GC, Brailoiu E, Futter CE, Schapira AH, Marchant JS, Patel S. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J Cell Sci. 2015;128:232–8.
Schapansky J, Khasnavis S, DeAndrade MP, Nardozzi JD, Falkson SR, Boyd JD, Sanderson JB, Bartels T, Melrose HL, LaVoie MJ. Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble alpha-synuclein in neurons. Neurobiol Dis. 2018;111:26–35.
Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A. 2014;111:2626–31.
Chan D, Citro A, Cordy JM, Shen GC, Wolozin B. Rac1 protein rescues neurite retraction caused by G2019S leucine-rich repeat kinase 2 (LRRK2). J Biol Chem. 2011;286:16140–9.
Gloeckner CJ, Schumacher A, Boldt K, Ueffing M. The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J Neurochem. 2009;109:959–68.
Hsu CH, Chan D, Greggio E, Saha S, Guillily MD, Ferree A, Raghavan K, Shen GC, Segal L, Ryu H, et al. MKK6 binds and regulates expression of Parkinson's disease-related protein LRRK2. J Neurochem. 2010;112:1593–604.
Hsu CH, Chan D, Wolozin B. LRRK2 and the stress response: interaction with MKKs and JNK-interacting proteins. Neurodegener Dis. 2010;7:68–75.
Carballo-Carbajal I, Weber-Endress S, Rovelli G, Chan D, Wolozin B, Klein CL, Patenge N, Gasser T, Kahle PJ. Leucine-rich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cell sign. 2010;22:821–7.
Plowey ED, Cherra SJ 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–56.
Yuan Y, Cao P, Smith MA, Kramp K, Huang Y, Hisamoto N, Matsumoto K, Hatzoglou M, Jin H, Feng Z. Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. elegans. PLoS One. 2011;6:e22354.
Appelqvist H, Waster P, Kagedal K, Ollinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol. 2013;5:214–26.
Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson's disease. EMBO Mol Med. 2012;4:380–95.
Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109:9611–6.
Daher JP, Volpicelli-Daley LA, Blackburn JP, Moehle MS, West AB. Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc Natl Acad Sci U S A. 2014;111:9289–94.
Daher JP, Abdelmotilib HA, Hu X, Volpicelli-Daley LA, Moehle MS, Fraser KB, Needle E, Chen Y, Steyn SJ, Galatsis P, et al. Leucine-rich Repeat Kinase 2 (LRRK2) Pharmacological Inhibition Abates alpha-Synuclein Gene-induced Neurodegeneration. J Biol Chem. 2015;290:19433–44.
Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JP, Milnerwood AJ, Unni VK, Hirst WD, Yue Z, Zhao HT, et al. G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. J Neurosci. 2016;36:7415–27.
Zhao HT, John N, Delic V, Ikeda-Lee K, Kim A, Weihofen A, Swayze EE, Kordasiewicz HB, West AB, Volpicelli-Daley LA. LRRK2 Antisense Oligonucleotides Ameliorate alpha-Synuclein Inclusion Formation in a Parkinson's Disease Mouse Model. Mol Ther Nucleic Acids. 2017;8:508–19.
Hoenen C, Gustin A, Birck C, Kirchmeyer M, Beaume N, Felten P, Grandbarbe L, Heuschling P, Heurtaux T. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS One. 2016;11:e0162717.
Lee H, James WS, Cowley SA. LRRK2 in peripheral and central nervous system innate immunity: its link to Parkinson's disease. Biochem Soc Trans. 2017;45:131–9.
Manzoni C, Mamais A, Dihanich S, Soutar MPM, Plun-Favreau H, Bandopadhyay R, Abeti R, Giunti P, Hardy J, Cookson MR, et al. mTOR independent alteration in ULK1 Ser758 phosphorylation following chronic LRRK2 kinase inhibition. Biosci Rep. 2018;38(2).
Hong L, Sklar LA. Targeting GTPases in Parkinson's disease: comparison to the historic path of kinase drug discovery and perspectives. Front Mol Neurosci. 2014;7:52.
Abeliovich A, Gitler AD. Defects in trafficking bridge Parkinson's disease pathology and genetics. Nature. 2016;539:207–16.
Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–8.
Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC, McCaffery JM, et al. The Parkinson's disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A. 2008;105:145–50.
Kim H, Calatayud C, Guha S, et al. Mol Neurobiol. 2018. https://doi.org/10.1007/s12035-018-0881-7.
Pankratz N, Beecham GW, DeStefano AL, Dawson TM, Doheny KF, Factor SA, Hamza TH, Hung AY, Hyman BT, Ivinson AJ, et al. Meta-analysis of Parkinson's disease: identification of a novel locus, RIT2. Ann Neurol. 2012;71:370–84.
Shi GX, Cai W, Andres DA. Rit subfamily small GTPases: regulators in neuronal differentiation and survival. Cell Signal. 2013;25:2060–8.
Shi G-X, Andres DA. Rit Contributes to Nerve Growth Factor-Induced Neuronal Differentiation via Activation of B-Raf-Extracellular Signal-Regulated Kinase and p38 Mitogen-Activated Protein Kinase Cascades. Mol Cell Biol. 2005;25:830–46.
Zhou Q, Li J, Wang H, Yin Y, Zhou J. Identification of nigral dopaminergic neuron-enriched genes in adult rats. Neurobiol Aging. 2011;32:313–26.
Bossers K, Meerhoff G, Balesar R, van Dongen JW, Kruse CG, Swaab DF, Verhaagen J. Analysis of gene expression in Parkinson's disease: possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain pathol. 2009;19:91–107.
Cai W, Rudolph JL, Harrison SM, Jin L, Frantz AL, Harrison DA, Andres DA. An evolutionarily conserved Rit GTPase-p38 MAPK signaling pathway mediates oxidative stress resistance. Mol Biol Cell. 2011;22:3231–41.
Kitatani K, Wada M, Perry D, Usui T, Sun Y, Obeid LM, Yaegashi N, Grabowski GA, Hannun YA. Activation of p38 Mitogen-Activated Protein Kinase in Gaucher's Disease. PLoS One. 2015;10:e0136633.
Sreetama SC, Takano T, Nedergaard M, Simon SM, Jaiswal JK. Injured astrocytes are repaired by Synaptotagmin XI-regulated lysosome exocytosis. Cell Death Differ. 2016;23:596–607.
Mastrototaro L, Tietjen U, Sponder G, Vormann J, Aschenbach JR, Kolisek M. Insulin Modulates the Na+/Mg2+ Exchanger SLC41A1 and Influences Mg2+ Efflux from Intracellular Stores in Transgenic HEK293 Cells. J Nutr. 2015;145:2440–7.
Li Z, Zhu W, Xiong L, Yu X, Chen X, Lin Q. Role of high expression levels of STK39 in the growth, migration and invasion of non-small cell type lung cancer cells. Oncotarget. 2016;7:61366–77.
Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H, Hollinger HC, Hartley JA, Brockington A, Burness CE, et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One. 2010;5:e9872.
Krasniak CS, Ahmad ST. The role of CHMP2B(Intron5) in autophagy and frontotemporal dementia. Brain Res. 1649;2016:151–7.
Feng X, Zhao Z, Li Q, Tan Z. Lysosomal Potassium Channels: Potential Roles in Lysosomal Function and Neurodegenerative Diseases. CNS Neurol Disord Drug Targets. 2018;17(4):261–6.
Jinn S, Drolet RE, Cramer PE, Wong AH, Toolan DM, Gretzula CA, Voleti B, Vassileva G, Disa J, Tadin-Strapps M, Stone DJ. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc Natl Acad Sci U S A. 2017;114:2389–94.
Takahashi N, Nagamine M, Tanno S, Motomura W, Kohgo Y, Okumura T. A diacylglycerol kinase inhibitor, R59022, stimulates glucose transport through a MKK3/6-p38 signaling pathway in skeletal muscle cells. Biochem Biophys Res Commun. 2007;360:244–50.
Baldanzi G, Alchera E, Imarisio C, Gaggianesi M, Dal Ponte C, Nitti M, Domenicotti C, van Blitterswijk WJ, Albano E, Graziani A, Carini R. Negative regulation of diacylglycerol kinase theta mediates adenosine-dependent hepatocyte preconditioning. Cell Death Differ. 2010;17:1059–68.
Wang G, Pan J, Chen SD. Kinases and kinase signaling pathways: potential therapeutic targets in Parkinson's disease. Prog Neurobiol. 2012;98:207–21.
Kametaka S, Moriyama K, Burgos PV, Eisenberg E, Greene LE, Mattera R, Bonifacino JS. Canonical interaction of cyclin G associated kinase with adaptor protein 1 regulates lysosomal enzyme sorting. Mol Biol Cell. 2007;18:2991–3001.
Crawford TQ, Ndhlovu LC, Tan A, Carvidi A, Hecht FM, Sinclair E, Barbour JD. HIV-1 infection abrogates CD8+ T cell mitogen-activated protein kinase signaling responses. J Virol. 2011;85:12343–50.
Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, Wong JC, Satkunarajah M, Schweneker M, Chapman JM, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205:2763–79.
Zhang Y, Xu M, Xia M, Li X, Boini KM, Wang M, Gulbins E, Ratz PH, Li PL. Defective autophagosome trafficking contributes to impaired autophagic flux in coronary arterial myocytes lacking CD38 gene. Cardiovasc Res. 2014;102:68–78.
Xiong J, Xia M, Xu M, Zhang Y, Abais JM, Li G, Riebling CR, Ritter JK, Boini KM, Li PL. Autophagy maturation associated with CD38-mediated regulation of lysosome function in mouse glomerular podocytes. J Cell Mol Med. 2013;17:1598–607.
Inoue M, Yamada J, Aomatsu-Kikuchi E, Satoh K, Kondo H, Ishisaki A, Chosa N. SCRG1 suppresses LPS-induced CCL22 production through ERK1/2 activation in mouse macrophage Raw264.7 cells. Mol Med Rep. 2017;15:4069–76.
Hong C, Shen C, Ding H, Huang S, Mu Y, Su H, Wei W, Ma J, Zheng F. An involvement of SR-B1 mediated p38 MAPK signaling pathway in serum amyloid A-induced angiogenesis in rheumatoid arthritis. Mol Immunol. 2015;66:340–5.
Guo H, Zhang J, Zhang X, Wang Y, Yu H, Yin X, Li J, Du P, Plumas J, Chaperot L, et al. SCARB2/LIMP-2 Regulates IFN Production of Plasmacytoid Dendritic Cells by Mediating Endosomal Translocation of TLR9 and Nuclear Translocation of IRF7. J Immunol. 2015;194:4737–49.
Kigoshi Y, Tsuruta F, Chiba T. Ubiquitin ligase activity of Cul3-KLHL7 protein is attenuated by autosomal dominant retinitis pigmentosa causative mutation. J Biol Chem. 2011;286:33613–21.
Sasaki F, Kumagai K, Uto H, Takami Y, Kure T, Tabu K, Nasu Y, Hashimoto S, Kanmura S, Numata M, et al. Expression of glycoprotein nonmetastatic melanoma protein B in macrophages infiltrating injured mucosa is associated with the severity of experimental colitis in mice. Mol Med Rep. 2015;12:7503–11.
Tajima JY, Futamura M, Gaowa S, Mori R, Tanahashi T, Tanaka Y, Matsuhashi N, Takahashi T, Yamaguchi K, Miyazaki T, Yoshida K. Clinical Significance of Glycoprotein Non-metastatic B and Its Association with EGFR/HER2 in Gastrointestinal Cancer. J Cancer. 2018;9:358–66.
Bhattacharyya S, Feferman L, Tobacman JK. Inhibition of Phosphatase Activity Follows Decline in Sulfatase Activity and Leads to Transcriptional Effects through Sustained Phosphorylation of Transcription Factor MITF. PLoS One. 2016;11:e0153463.
Li B, Castano AP, Hudson TE, Nowlin BT, Lin SL, Bonventre JV, Swanson KD, Duffield JS. The melanoma-associated transmembrane glycoprotein Gpnmb controls trafficking of cellular debris for degradation and is essential for tissue repair. FASEB J. 2010;24:4767–81.
Itoh N, Ohta H. Roles of FGF20 in dopaminergic neurons and Parkinson's disease. Front Mol Neurosci. 2013;6:15.
Ohmachi S, Mikami T, Konishi M, Miyake A, Itoh N. Preferential neurotrophic activity of fibroblast growth factor-20 for dopaminergic neurons through fibroblast growth factor receptor-1c. J Neurosci Res. 2003;72:436–43.
Zhong W, Zhu H, Sheng F, Tian Y, Zhou J, Chen Y, Li S, Lin J. Activation of the MAPK11/12/13/14 (p38 MAPK) pathway regulates the transcription of autophagy genes in response to oxidative stress induced by a novel copper complex in HeLa cells. Autophagy. 2014;10:1285–300.
Rosati A, Basile A, D'Auria R, d'Avenia M, De Marco M, Falco A, Festa M, Guerriero L, Iorio V, Parente R, et al. BAG3 promotes pancreatic ductal adenocarcinoma growth by activating stromal macrophages. Nat Commun. 2015;6:8695.
Carra S, Seguin SJ, Landry J. HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy. 2008;4:237–9.
Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, Behl C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011;12:149–56.
Behl C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol Sci. 2016;37:672–88.
Guo ML, Xue B, Jin DZ, Mao LM, Wang JQ. Interactions and phosphorylation of postsynaptic density 93 (PSD-93) by extracellular signal-regulated kinase (ERK). Brain Res. 2012;1465:18–25.
Mao LM, Wang JQ. Synaptically Localized Mitogen-Activated Protein Kinases: Local Substrates and Regulation. Mol Neurobiol. 2016;53:6309–15.
Liu Y, Baumgardt SL, Fang J, Shi Y, Qiao S, Bosnjak ZJ, Vasquez-Vivar J, Xia Z, Warltier DC, Kersten JR, Ge ZD. Transgenic overexpression of GTP cyclohydrolase 1 in cardiomyocytes ameliorates post-infarction cardiac remodeling. Sci Rep. 2017;7:3093.
Chiarini A, Armato U, Pacchiana R, Dal Pra I. Proteomic analysis of GTP cyclohydrolase 1 multiprotein complexes in cultured normal adult human astrocytes under both basal and cytokine-activated conditions. Proteomics. 2009;9:1850–60.
Fullgrabe J, Heldring N, Hermanson O, Joseph B. Cracking the survival code: autophagy-related histone modifications. Autophagy. 2014;10:556–61.
Fullgrabe J, Klionsky DJ, Joseph B. Histone post-translational modifications regulate autophagy flux and outcome. Autophagy. 2013;9:1621–3.
Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, Joseph B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500:468–71.
Xue P, Zeng F, Duan Q, Xiao J, Liu L, Yuan P, Fan L, Sun H, Malyarenko OS, Lu H, et al. BCKDK of BCAA Catabolism Cross-talking With the MAPK Pathway Promotes Tumorigenesis of Colorectal Cancer. EBioMedicine. 2017;20:50–60.
Refojo D, Holsboer F. CRH signaling. Molecular specificity for drug targeting in the CNS. Ann N Y Acad Sci. 2009;1179:106–19.
Wang W, Ji P, Dow KE. Corticotropin-releasing hormone induces proliferation and TNF-alpha release in cultured rat microglia via MAP kinase signalling pathways. J Neurochem. 2003;84:189–95.
Sun Y, Xiao Q, Luo C, Zhao Y, Pu D, Zhao K, Chen J, Wang M, Liao Z. High-glucose induces tau hyperphosphorylation through activation of TLR9-P38MAPK pathway. Exp Cell Res. 2017;359:312–8.
Maphis N, Jiang S, Xu G, Kokiko-Cochran ON, Roy SM, Van Eldik LJ, Watterson DM, Lamb BT, Bhaskar K. Selective suppression of the alpha isoform of p38 MAPK rescues late-stage tau pathology. Alzheimers Res Ther. 2016;8:54.
Lauretti E, Pratico D. Glucose deprivation increases tau phosphorylation via P38 mitogen-activated protein kinase. Aging Cell. 2015;14:1067–74.
Ittner A, Chua SW, Bertz J, Volkerling A, van der Hoven J, Gladbach A, Przybyla M, Bi M, van Hummel A, Stevens CH, et al. Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer's mice. Science. 2016;354:904–8.
Inoue K, Rispoli J, Kaphzan H, Klann E, Chen EI, Kim J, Komatsu M, Abeliovich A. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener. 2012;7:48.
Pacheco CD, Elrick MJ, Lieberman AP. Tau deletion exacerbates the phenotype of Niemann-Pick type C mice and implicates autophagy in pathogenesis. Hum Mol Genet. 2009;18:956–65.
Cai W, Rudolph JL, Sengoku T, Andres DA. Rit GTPase regulates a p38 MAPK-dependent neuronal survival pathway. Neurosci Lett. 2012;531:125–30.
Shi GX, Han J, Andres DA. Rin GTPase couples nerve growth factor signaling to p38 and b-Raf/ERK pathways to promote neuronal differentiation. J Biol Chem. 2005;280:37599–609.
Zhang M, Zhu X, Zhang Y, Cai Y, Chen J, Sivaprakasam S, Gurav A, Pi W, Makala L, Wu J, et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 2015;22:1922–34.
Jimenez-Garcia L, Traves PG, Lopez-Fontal R, Herranz S, Higueras MA, de Las HB, Hortelano S, Luque A. 8,9-Dehydrohispanolone-15,16-lactol diterpene prevents LPS-triggered inflammatory responses by inhibiting endothelial activation. Biochem J. 2016;473:2061–71.
Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol. 2008;64:644–53.
Zang Y, Xin WJ, Pang RP, Li YY, Liu XG. Upregulation of Nav1.3 Channel Induced by rrTNF in Cultured Adult Rat DRG Neurons via p38 MAPK and JNK Pathways. Chin J Physiol. 2011;54:241–6.
Kuo TC, Chao CC. Hepatitis B virus X protein prevents apoptosis of hepatocellular carcinoma cells by upregulating SATB1 and HURP expression. Biochem Pharmacol. 2010;80:1093–102.
Lim CS, Kim SH, Jung JG, Kim JK, Song WK. Regulation of SPIN90 phosphorylation and interaction with Nck by ERK and cell adhesion. J Biol Chem. 2003;278:52116–23.
Liu H, Wang Z, Li Y, Yu G, Fu X, Wang C, Liu W, Yu Y, Bao F, Irwanto A, et al. Genome-Wide Analysis of Protein-Coding Variants in Leprosy. J Invest Dermatol. 2017;137:2544–51.
Lim EJ, Lee SH, Lee JG, Kim JR, Yun SS, Baek SH, Lee C. Toll-like receptor 9 dependent activation of MAPK and NF-kB is required for the CpG ODN-induced matrix metalloproteinase-9 expression. Exp Mol Med. 2007;39:239–45.
Xie L, Liu S, Cheng J, Wang L, Liu J, Gong J. Exogenous administration of mitochondrial DNA promotes ischemia reperfusion injury via TLR9-p38 MAPK pathway. Regul Toxicol Pharmacol. 2017;89:148–54.
Hayashi K, Taura M, Iwasaki A. The interaction between IKKalpha and LC3 promotes type I interferon production through the TLR9-containing LAPosome. Sci Signal. 2018;11:eaan4144.
Lim JS, Kim HS, Nguyen KC, Cho KA. The role of TLR9 in stress-dependent autophagy formation. Biochem Biophys Res Commun. 2016;481:219–26.
Franco LH, Fleuri AKA, Pellison NC, Quirino GFS, Horta CV, de Carvalho RVH, Oliveira SC, Zamboni DS. Autophagy downstream of endosomal Toll-like receptor signaling in macrophages is a key mechanism for resistance to Leishmania major infection. J Biol Chem. 2017;292:13087–96.
Aceros H, Farah G, Noiseux N, Mukaddam-Daher S. Moxonidine modulates cytokine signalling and effects on cardiac cell viability. Eur J Pharmacol. 2014;740:168–82.
Gstrein T, Edwards A, Pristoupilova A, Leca I, Breuss M, Pilat-Carotta S, Hansen AH, Tripathy R, Traunbauer AK, Hochstoeger T, et al. Mutations in Vps15 perturb neuronal migration in mice and are associated with neurodevelopmental disease in humans. Nat Neurosci. 2018;21:207–17.
Kzhyshkowska J, Gratchev A, Martens JH, Pervushina O, Mamidi S, Johansson S, Schledzewski K, Hansen B, He X, Tang J, et al. Stabilin-1 localizes to endosomes and the trans-Golgi network in human macrophages and interacts with GGA adaptors. J Leukoc Biol. 2004;76:1151–61.
Chen Y, Lohr M, Jesnowski R. Inhibition of ankyrin-B expression reduces growth and invasion of human pancreatic ductal adenocarcinoma. Pancreatology. 2010;10:586–96.
Lorenzo DN, Badea A, Davis J, Hostettler J, He J, Zhong G, Zhuang X, Bennett V. A PIK3C3-ankyrin-B-dynactin pathway promotes axonal growth and multiorganelle transport. J Cell Biol. 2014;207:735–52.
Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A, Marshall B, Schoenlein PV, Lebedyeva IO, Liu K. Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid-derived suppressor cells. Oncotarget. 2016;7:83907–25.
Han H, Li J, Feng X, Zhou H, Guo S, Zhou W. Autophagy-related genes are induced by histone deacetylase inhibitor suberoylanilide hydroxamic acid via the activation of cathepsin B in human breast cancer cells. Oncotarget. 2017;8:53352–65.
Nagakannan P, Eftekharpour E. Differential redox sensitivity of cathepsin B and L holds the key to autophagy-apoptosis interplay after Thioredoxin reductase inhibition in nutritionally stressed SH-SY5Y cells. Free Radic Biol Med. 2017;108:819–31.
Sun Y, Grabowski GA. Altered autophagy in the mice with a deficiency of saposin A and saposin B. Autophagy. 2013;9:1115–6.
Ribbens JJ, Moser AB, Hubbard WC, Bongarzone ER, Maegawa GH. Characterization and application of a disease-cell model for a neurodegenerative lysosomal disease. Mol Genet Metab. 2014;111:172–83.
Del Grosso A, Antonini S, Angella L, Tonazzini I, Signore G, Cecchini M. Lithium improves cell viability in psychosine-treated MO3.13 human oligodendrocyte cell line via autophagy activation. J Neurosci Res. 2016;94:1246–60.
Yan Q, Han C, Wang G, Waddington JL, Zheng L, Zhen X. Activation of AMPK/mTORC1-Mediated Autophagy by Metformin Reverses Clk1 Deficiency-Sensitized Dopaminergic Neuronal Death. Mol Pharmacol. 2017;92:640–52.
Valapala M, Wilson C, Hose S, Bhutto IA, Grebe R, Dong A, Greenbaum S, Gu L, Sengupta S, Cano M, et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/betaA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy. 2014;10:480–96.
Hsin IL, Sheu GT, Jan MS, Sun HL, Wu TC, Chiu LY, Lue KH, Ko JL. Inhibition of lysosome degradation on autophagosome formation and responses to GMI, an immunomodulatory protein from Ganoderma microsporum. Br J Pharmacol. 2012;167:1287–300.
Hinton A, Sennoune SR, Bond S, Fang M, Reuveni M, Sahagian GG, Jay D, Martinez-Zaguilan R, Forgac M. Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J Biol Chem. 2009;284:16400–8.
Morimura T, Fujita K, Akita M, Nagashima M, Satomi A. The proton pump inhibitor inhibits cell growth and induces apoptosis in human hepatoblastoma. Pediatr Surg Int. 2008;24:1087–94.
Acknowledgements
We are thankful to Christa Überbacher and Dr. Corrado Corti for helpful discussion and critical reading of the manuscript. The authors thank the Department of Educational Assistance, University and Research of the Autonomous Province of Bozen/Bolzano for covering the Open Access publication costs.
Funding
This work was supported by the Department of Educational Assistance, University and Research of the Autonomous Province of Bozen/Bolzano and partially by the DFG (FOR2488 Z2/Z1 to AAH and PPP). The funders had no role in the design, discussion and writing of the manuscript.
Author information
Authors and Affiliations
Contributions
JO, FG: equally contributed to literature search, writing and editing of the manuscript; PP, AH: read and commented the final version of the manuscript; MV: supervised literature search, discussion and writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Obergasteiger, J., Frapporti, G., Pramstaller, P.P. et al. A new hypothesis for Parkinson’s disease pathogenesis: GTPase-p38 MAPK signaling and autophagy as convergence points of etiology and genomics. Mol Neurodegeneration 13, 40 (2018). https://doi.org/10.1186/s13024-018-0273-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-018-0273-5